Back to Journals » International Journal of Nanomedicine » Volume 16
Cancer Cell Membrane Camouflaged Mesoporous Silica Nanoparticles Combined with Immune Checkpoint Blockade for Regulating Tumor Microenvironment and Enhancing Antitumor Therapy
Authors Zhao P ![]() , Qiu L, Zhou S, Li L, Qian Z, Zhang H
, Qiu L, Zhou S, Li L, Qian Z, Zhang H
Received 4 December 2020
Accepted for publication 27 February 2021
Published 12 March 2021 Volume 2021:16 Pages 2107—2121
DOI https://doi.org/10.2147/IJN.S295565
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Linlin Sun
Peiqi Zhao, Lihua Qiu, Shiyong Zhou, Lanfang Li, Zhengzi Qian, Huilai Zhang
Department of Lymphoma, Tianjin’s Clinical Research Center for Cancer, Key Laboratory of Cancer Prevention and Therapy, National Clinical Research Center for Cancer, Tianjin Medical University Cancer Institute and Hospital, Tianjin Medical University, Tianjin, 300060, People’s Republic of China
Correspondence: Huilai Zhang
Department of Lymphoma, Tianjin Medical University Cancer Institute and Hospital, 24 Huanhu West Road, Hexi District, Tianjin, 300060, People’s Republic of China
Tel +86-22-23340123
Email [email protected]
Purpose: Although anti-programmed cell death protein 1 antibody (aPD1) immunotherapy and chemotherapy has made much progress in the treatment of melanoma, the efficacy still needs to be further improved.
Methods: Cancer treatment has been greatly enhanced by the use of nanotechnology. Cancer cell membrane (CCM)-camouflaged nanoparticles have shown promising potential in tumor therapy due to their excellent homologous-targeting ability, long blood circulation and immune escape. This work presents a biocompatible and tumor acidic environmental responsive CCM-camouflaged mesoporous silica nanoparticle (CMSN) that is loaded with dacarbazine (DTIC) and combined with aPD1 to achieve better antitumor efficacy.
Results: In vitro cell experiments demonstrated that DTIC@CMSN exhibits a better anti-tumor killing efficiency and a stronger ability to promote the apoptosis of tumor cells than free DTIC. In vivo antitumor results demonstrated that combination therapy of DTIC@CMSN chemotherapy and aPD1 immunotherapy remarkably suppress the melanoma growth and prolong survival time due to highly selective tumor killing, activation of tumor-specific T cells, and regulation of the immunosuppressive tumor microenvironment. In addition, safety evaluation studies of DTIC@CMSN also demonstrate their increased tumor accumulation and decreased systemic toxicity.
Conclusion: This study provides a promising nano-platform for the combination of chemotherapy with immunotherapy, which is potentially useful for the treatment of melanoma.
Keywords: anti-PD-1, cancer cell membrane, cancer immunotherapy, melanoma, mesoporous silica nanoparticle
Introduction
Melanoma is the fifth most common type of cancer among men and sixth among women in the United States, with an estimated 100,350 new cases and 12,830 deaths in 2020.1 More than half of the patients were diagnosed with stage I, and these patients had a five-year survival rate of approximately 100%.2 By contrast, a small percentage of patients with advanced melanoma exhibited a 5-year survival rate of only 19%.2
Many treatments have been developed for the management of metastatic melanoma,3–8 among which Dacarbazine (DTIC) is the only chemotherapeutic drug approved by the US Food and Drug Administration (FDA); it is used as a first-line chemotherapy drug for melanoma.9 DTIC is a precursor of purine biosynthesis, that not only interferes with purine biosynthesis, but also inhibits the synthesis of RNA, protein and DNA. After entering the body, dacarbazine is demethylated by liver microsomes to form monomethyl compounds, which have direct cytotoxic effects.10 However, the following disadvantages of DTIC in the treatment of melanoma limit its further application.10–13 First, the efficacy of dacarbazine in the treatment of melanoma is generally poor, presenting an overall response rate of 5–20%, a rare complete response rate. It has been reported to present a median progression-free survival of 1.5–1.6 months.11–13 Second, DTIC is extremely unstable under light and heat. It is also unstable in water, and should be injected immediately after formulation.10 Third, DTIC requires intravenous injection and has a short half-life. Lastly, DTIC does not target melanoma cells and it does produce noticeable damage to liver and kidney function.
Cancer treatment has been greatly enhanced by the use of nanotechnology.14–16 Nanoparticles can overcome biological barriers and achieve enhanced uptake in cancer cells, thus improving the efficacy of chemotherapy drugs, reducing adverse reactions, and even reversing multidrug resistance in tumors.14,17–19 Therefore, we hypothesized that nanoparticle-based approaches might improve the efficacy of DTIC.
However, because drug-loaded nanoparticles passively target tumors via the enhanced permeability and retention (EPR) effect of leaky blood vessels in tumors, less than 1% of anticancer drugs reach the tumor site.20–22 In addition, unintended drug release of drug-loaded nanoparticles before they reach the tumor site causes damage to normal tissues and cells.23 Various targeting components, such as peptides, aptamers and antibodies, have been used to further improve the tumor-targeting efficiency of the nanocarriers.24–27 However, the integration of the targeting component requires multiple chemical reactions and formulation processes, that inevitably lead to problems of low stability, poor reproducibility and complex evaluation of the nanocarriers.28,29 Nevertheless, most of the nanocarriers remain excluded by the reticuloendothelial system during blood circulation, thus resulting in low targeting efficiency.30
Recently, a biomimetic technology has emerged in which nanoparticles are coated by natural cell membranes31–33 and demonstrates improved tumor targeting efficiency. In addition, the cancer cell membrane (CCM) coating on the nanoparticles results in homogenous (self-identifying) binding to primitive cancer cells.34,35 This homogenous affinity lowers systemic clearance, thereby prolonging blood circulation and effectively improving tumor targeting.35
The study protocol, as illustrated in Scheme 1, designed, synthesized and utilized mesoporous silica nanoparticles (MSN) that encapsulated DTIC in their cavities. The extrusion method was then used to coat CCM fragments on their surfaces, thereby generating the DTIC@CMSN that provided additional resistance to DTIC leakage. When injected intravenously into tumor-bearing mice with human melanoma cells (B16F10), nanoparticles can accumulate at tumor sites through the EPR effects and homologous tumor targeting in vivo.36 We hypothesized that when DTIC@CMSN arrives at the tumor site, it releases the chemotherapy drug DTIC, which directly kills tumor cells. Furthermore, we speculate that this action exposes tumor antigens and further triggers an anti-tumor immune response. The subsequent application of anti-programmed cell death protein 1 antibody (aPD1) resists the negative feedback pathway of the immune response during chemotherapy by blocking the PD-1/PD-L1 axis, thereby allowing activation of additional immune pathways. Thus, we hypothesized that the strategy of using DTIC@CMSN combined with aPD1 will display significant improvement in both primary tumor inhibition and reduction of systemic side effects.
 |
Scheme 1 (A) Schematic illustrations of the process for synthesizing the dacarbazine (DTIC) loaded cancer cell membrane camouflaged mesoporous silica nanoparticle (DTIC@CMSN). (B) Schematic diagram of antitumor immune response induced by DTIC@CMSN combined with anti-programmed cell death protein 1 antibody (aPD1). |
Materials and Methods
Materials
Cetyltrimethylammonium bromide (CTAB), dichloromethane (DCM), dimethyl sulfoxide (DMSO), tetraethylorthosilicate (TEOS), phosphate-buffered saline (PBS, pH 7.4; pH 6.5; pH 5.0), hematoxylin and eosin (H&E), methylthiazoletetrazolium (MTT), DTIC, fluorescein isothiocyanate (FITC) and Cy5.5 were purchased from Sigma-Aldrich. Annexin V-FITC/PI apoptosis assay kit, Membrane Protein Extraction Kit, the monoclonal antibody against CD47, E-cadherin and EpCAM were supplied by Thermo Fisher Scientific (USA). Melanoma cell line B16F10 cells were provided from American Type Culture Collection (ATCC, Manassas, VA, USA).
C57BL/6 mice (5–6 weeks old) were obtained by the Vital Laboratory Animal Center (Beijing, China). Mice were fed in our specific-pathogen-free facility at a temperature of 24°C. All animal experiments were approved by the Animal Ethical and Welfare Committee of Tianjin Medical University Cancer Institute and Hospital, and all animal studies were performed in compliance with the guidelines of the committee.
Preparation of CCM
To prepare CCM, we cultured B16-F10 cells in a 10 cm diameter culture dish at 37°C with 5% CO2, and then isolated the cells with a cell scraper. Next, the cells were isolated by centrifugation at 1000 g for 4 min. The collected cells were re-suspended in a precooled PBS buffer (pH = 7.4) and centrifuged again at 1000 g for 4 min. The collected cell pellets were re-suspended in a hypotonic lysing buffer containing the membrane protein extraction reagent. The above samples were then incubated in an ice bath for 10 min. Next, the cells in the solution were fractured by freeze-thaw and centrifuged at 1000 g for 7 min at 4°C. To remove excess cancer cell vesicles and collect cell membrane fragments, the supernatant was further centrifuged at 14,000 g for 30 min. Subsequently, we used an Avanti mini extruder to extrude 11 times. Finally, we centrifuge to remove the redundant cancer cell vesicles.
Preparation and Characterization of CMSN
MSN was synthesized according to a reported procedure.19 To implement the membrane coating, MSN was mixed with cancer cell membranes at a nanoparticle-to-membrane weight ratio of 2:1, and prepared cancer cell vesicles were mixed and resuspended in 1 mL of PBS. We then used an Avanti mini-extruder to extrude 11 times and centrifuged to remove the excess cancer cell vesicles. Finally, this preparation of CMSN was placed at 4 °C in 1× PBS overnight for further use.
The particle size, surface potential and polydispersity index of MSN and CMSN were determined by using the dynamic light scattering (DLS) method. The distribution of CCM at the surface of the MSN was examined under a Leica TCS SP5 CLSM (Heidelberg, Germany).
In brief, CCM was resuspended in sterile PBS. Then, FITC NHS ester at final concentration of 3 μM were added to the CCM and incubated gently on a shaker in the dark at room temperature for 1 h. The FITC-labeled CCM were then washed with PBS and collected by certification for further characterization. And then the CMSN were prepared using the method described above. A small drop of FITC labeled CMSN in water was deposited on microscope slides and visualized through a FITC (488 nm) channel filter. Representative images were taken and selected from a slice through the midsection of the CMSN.
Synthesis of DTIC@CMSN
One hundred-milligram CMSN and 30 mg DTIC were dispersed in 10 mL DMF by ultrasonic treatment, and the mixture was stirred at room temperature for 24 h. The obtained crude product was then centrifuged (8000 r/min, 10 min). The encapsulation efficiency of DTIC in CMSN was determined by reverse phase high-performance liquid chromatography (RP-HPLC, Agilent Technologies Inc., 1100S, USA) with a UV-vis detector at wavelength of 319 nm. Chromatographic separations were performed on a reversed phase-C18 column (Hypersil ODS2 C18 5 µm, 250 × 4.6 mm). Methanol/water (30/70, v/v) containing 0.1% trifluoroacetic acid was used as the mobile phase at a flow rate of 1 mL/min.
To determine in vivo targeting of CMSN, the fluorescent dye Cy5.5 was incorporated as the tracer fluorescent probe. To prepare Cy5.5-labeled CMSN, 50 μL of the Cy5.5 dye was dissolved in ethanol (1 mg/mL) and incorporated in place of drugs. Cy5.5 labeled CMSN was prepared in the same manner.
CCM Protein Characterization
First, membrane proteins of melanoma cell lysates, melanoma cell membrane fragments, and CMSN were extracted, and the total protein was measured using the BCA assay kit (Beyotime, China). Proteins were separated by SDS-polyacrylamide gel electrophoresis (10%) and then transferred to polyvinylidene fluoride (PVDF) membranes. Western blot experiments were performed to verify the presence of cell membrane proteins on the surface of CMSN. Specific proteins were detected using an enhanced chemiluminescence detection assay (Santa Cruz Biotechnology, Inc.) according to manufacturer’s instructions. The sample imprinting was analyzed using the Bio-RAD imaging system (Bio-RAD, Hercules, CA, USA).
In vitro Release of DTIC from CMSN and MSN
In vitro drug release of DTIC from DTIC@CMSN and DTIC@MSN was measured after time-controlled equilibration and centrifugation in PBS at pH 7.4, 6.5 and 5.0. Briefly, an amount of 15 mg DTIC@CMSN and DTIC@MSN were dispersed in a 10 mL release medium and placed in a rocking bath at 37°C at a speed of 100 rpm. At predetermined time intervals, the released samples were centrifuged, and the supernatant was collected for subsequent analysis. The same volume of fresh medium was immediately added to the test tube to enable continuous release with no change in dilution volume. The supernatant was then extracted and analyzed to detect DTIC by RP-HPLC.
Biocompatibility Experiment
To test the cytotoxicity of CMSN in vitro, B16F10 cells were first inoculated in 96-well plates at a density of 5×103 cells per well and cultured for 24 h. CMSN suspensions at different concentrations (1, 2, 5, 10, 20, 50, 100 and 200 μg/mL) were then added to each well and incubated with the cells for 24 and 48 h. Next 20 μL 3-(4,5-dimethyl-2-thiazolyl)-2,5-diphenyl-2-H-tetrazolium bromide (MTT) solution (5 mg/mL) was injected into each well and cultured for 4 h. The supernatant was then discarded and the crystals at the bottom were dissolved with 150 μL dimethyl sulfoxide (DMSO). Finally, the OD value at 490 nm was measured by a microplate reader to calculate the cell viability.
Cellular Uptake of CMSN
The intracellular distribution of CMSN was examined by CLSM. In brief, B16F10 cells (2×105 cells/well) were seeded on 22 mm round glass coverslips, placed in 6-well plates, and cultured overnight in DMEM. The nanoparticles were added to B16F10 cells and cultured at 37°C for 4 h. We used FITC to label CCM. Cells were washed 3 times with PBS and fixed with paraformaldehyde (PFA) for 30 min. Finally, we stained the nuclei using DAPI and then imaged them using CLSM. FITC and DAPI channels presented green and blue fluorescent images, respectively.
In vitro Anti-Tumor Activity of DTIC@CMSN
To gauge the effectiveness of DTIC@CMSN in vitro, B16F10 cells were transferred to 96-well plates (Corning Costar Corp, New York, USA) at a density of 1×104 cells per well and cultured for 24 h. Cells were then exposed to serial concentrations of DTIC, DTIC@MSN and DTIC@CMSN for 24 h and 48 h. To measure cell viability after these exposures, 20 μL of 5 mg/mL MTT solution was added to each well. The plates were incubated in darkness at 37°C for an additional 4 h. The rest of the experimental protocol was detailed in Section 2.8 describing the biocompatibility experiment.
Measurement of Cell Apoptosis
Apoptosis of B16F10 cells was detected using the Annexin V-FITC/PI apoptosis detection kit. Cells were first exposed to continuous concentrations of DTIC, DTIC@MSN and DTIC@CMSN for 24 h. After incubation, the cells were washed in PBS and then re-suspended in the binding buffer (1×106 cells per mL). The resulting cell suspensions (500 µL per sample) were incubated with 5 µL Annexin V-FITC and 10 µL PI at room temperature in the dark for 10 min. Flow cytometry was used to analyze apoptosis.
In vivo Antitumor Experiments
The C57BL/6 male mice model with subcutaneous B16F10 melanoma was established by implanting approximately 1×106 B16F10 cells into the right flank of the mice. After 10 days, mice were randomly divided into six groups (n = 8) and were given intravenous injections of the following agents: PBS, DTIC, aPD1, DTIC@MSN, DTIC@CMSN and DTIC@CMSN + aPD1 at a dose of 40 µg aPD1 and/or 10 mg/kg DTIC per mouse. Tumor volume was measured every two days and calculated based on the following formula: V= (long diameter × short diameter2)/2. The survival time of model mice was recorded from the day of tumor cell implantation. The Kaplan-Meier survival curve was plotted.
In vivo Biodistribution of CMSN
To validate the in vivo targeting of CMSN, we labeled CMSN with the fluorescent dye Cy5.5. Our test system comprised a mouse melanoma-bearing model according to the methods described above. The tumor-bearing mice were then injected with Cy5.5-labeled CMSN through the tail vein. Four hours after administration, the mice were killed by cervical dislocation. Tumors and major organs (heart, kidney, spleen, lung, and liver) were removed and then imaged using the IVIS in vivo imaging system.
Evaluation of Systematic Toxicity
Toxicity was evaluated in tumor-bearing mice after three does administration. Serum chemistry levels from collected blood in every group were measured according to manufacturer’s instructions. Organs (heart, liver, spleen, lung and kidney) and tumors were fixed in paraformaldehyde fixative solution for H&E staining.
Flow Cytometric Analysis of Immune Responses
In order to study the immune response in the tumor microenvironment, tumors were excised on the day following the administration of the last dose to collect infiltrating immune cells. The tumor was weighed and digested with collagenase A and DNase I. The cell suspension was then divided into a series of antibody staining solutions to label CD4 + T cells (CD3 + CD4 +), CD8 + T cells (CD3 + CD8 +) and regulatory T cells (CD4 + Foxp3 +).
Cytokine Analysis Assay
Equal weights of tumors were excised in every group to detect secretion of cytokines (TNF-α, TGF-β, IL-10 and IFN-γ) in the tumor microenvironment. The supernatant was subjected to high-speed homogenization and centrifugation and then tested by ELISA.
Statistical Analysis
All experiments must be repeated at least three times unless otherwise specified. All experimental data are expressed as mean ± standard deviation. The statistical differences between groups were determined by Tukey’s test. *P < 0.05 was considered as statistically different.
Results and Discussion
Design of DTIC@CMSN
The nanocomplexes were designed to achieve effective DTIC encapsulation and cell membrane cloaking. Our intent is to demonstrate effective and efficient melanoma cell killing. As shown in Scheme 1, the synthesis of the nanocomposite consists of three steps: i) MSN synthesis and loading with DTIC (DTIC@MSN), ii) cell membrane isolation from B16F10 cells, and iii) construction of DTIC@CMSN by self-assembly of CCM on the surface of DTIC@MSN by repeated extrusion cycles. Studies have shown that MSN is an ideal carrier for carrying and delivering chemotherapy drugs.33,37,38 The self-assembly of the cancer cell membrane on the MSN surface is mainly driven by thermodynamic considerations, and assisted by interactions between the amino groups on the MSN surface and membrane phospholipids and proteins.39,40 The targeted delivery of DTIC@CMSN is achieved through the EPR and homologous targeting.36 After uptake by melanoma cells, DTIC@CMSN is activated to decompose in the lysosomal acid environment. DTIC is consequently released into the cytoplasm, eventually leading to the DTIC binding to nuclear DNA and initiation of the apoptosis cascade.
Preparation and Characterization of the CMSN
CCM coatings confer excellent targeting capabilities by avoiding immune elimination and homologous adherence to nanoparticles. In our earlier work, MSN was synthesized according to a modified method previously reported.19,41 TEM images showed them to be uniformly dispersed and spherical with an average particle size of about 140 nm. We prepared natural cancer cell vesicles from B16-F10 cells and co-extruded the CCM and MSN, thereby generating CCM coated MSN. The hydrodynamic diameter of MSN measured by DLS increased by about 9 nm after encapsulation of cancer cell vesicles, which is consistent with other reports.33,42 The zeta potential measurements also suggest a successful coating, as the surface charges of the MSN cores increased to approximately that of the membrane vesicles after being coated (Table 1). In addition, MSN and CMSN samples showed high monodispersity in PBS (Table 1). In order to further verify that CCM is wrapped on MSN surface, we tagged the CCM with FITC dye. Figure 1A shows that a generally uniform green shell is observed in the CMSN, indicating that the MSN was successfully covered by CCM.
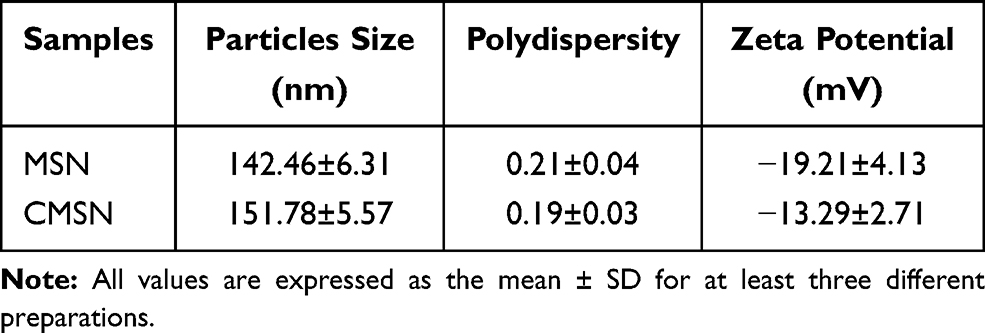 |
Table 1 Average Size, Polydispersity and Zeta Potential of MSN and CMSN |
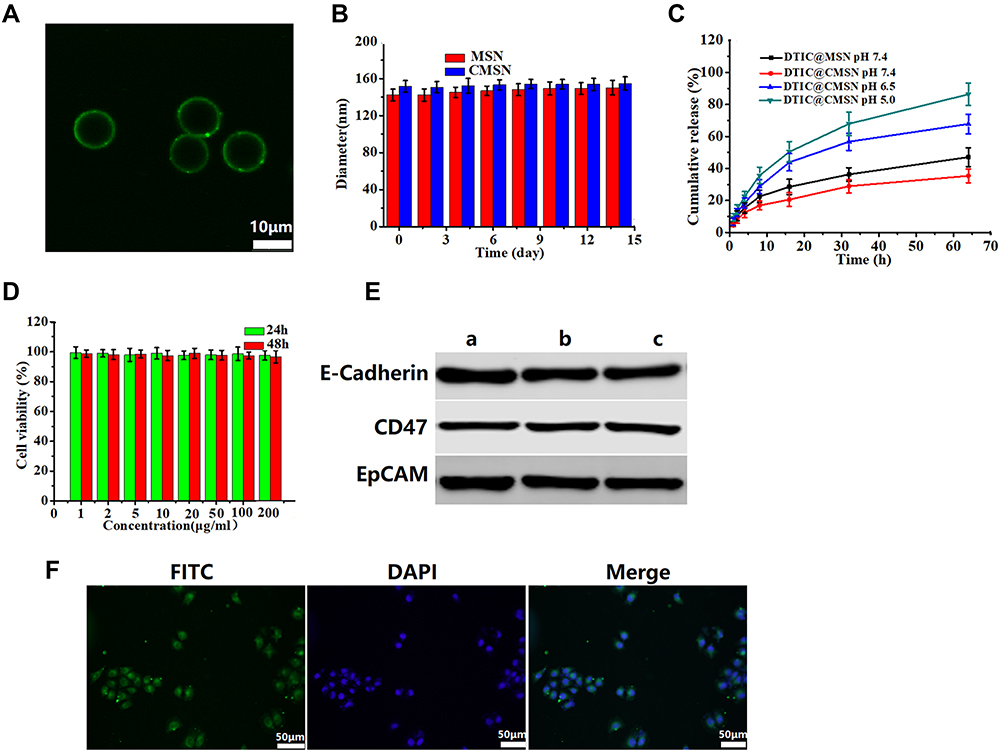 |
Figure 1 (A) Evidence for the coverage of cancer cell membranes coated on the surface of mesoporous silica nanoparticle by confocal microscopy. Scale bar represents 10 μm. (B) Stability of MSN and CMSN in PBS over time. (C) In vitro release profiles of DTIC from DTIC@MSN (pH 7.4) and DTIC@CMSN in PBS (at pH 7.4, pH 6.5 and pH 5.0). (D) Cell viability of B16F10 cells incubated with CMSN without drug at various concentrations measured at 24 h and 48 h. (E) The analysis of E-Cadherin, CD47 and EpCAM by Western blot. a: cancer cell lysate; b: cancer cell membranes; c: CMSN. (F) Confocal laser scanning microscopy images of B16F10 cell by cultured with FITC (green) labeled CMSN and DAPI (blue) used for staining cell nuclei. Scale bar is 50 μm. |
The stability of CMSN in simulated physiological media was also assessed by measuring particle size changes. Figure 1B shows that the particle size change of CMSN in PBS during 14 days is negligible, indicating that it has excellent stability in PBS.
To simulate the drug release behavior of DTIC@CMSN under physiological conditions, in tumor and lysosome microenvironments, we evaluated and mapped the drug release curve of DTIC@CMSN in vitro at pH 7.4, 6.5 and 5.0 (Figure 1C). At pH 7.4, only a small amount of DTIC was released from DTIC@CMSN. After incubation for 16 h, 20% of the original payload of DTIC was released from DTIC@CMSN compared to 30% DTIC released from DTIC@MSN. This suggest that CCM effectively prevents the premature release of the drug under physiological conditions. At pH 6.5, after incubation for only 8 hours, the amount of DTIC released by DTIC@CMSN increased sharply to about 30%. Notably, the greatest rate of drug release was observed at pH 5.0, with nearly 90% of the encapsulated drug released after 64 h. Drug release at the lowest pH from DTIC@CMSN was significantly greater than the release yield at pH 7.4 and 6.5. The pH sensitivity of the carrier is attributed to the membrane structure, surrounding the complex. Drug release has been shown to involve three steps: 1) acid environment enhances the release of DTIC absorbed on the surface of the membrane; 2) CCM is disengaged from MSN through acid-induced aqueous solution dissolution;43 and 3) excessive H + in the acidic solution weakens the interaction between DTIC and MSN through competitive adsorption.44
Biocompatibility is one of the most important indicators for evaluating the performance of nanoparticles.45 We first measured the cytotoxicity of CMSN based on MTT tests. As shown in Figure 1D, all cells displayed high viability after incubation with CMSN for 24 h and 48 h. Even at the maximum concentration of 200 μg/mL, the cell metabolic activity remained above 95%, indicating that the nanoparticles had good biocompatibility. Therefore, the low cytotoxicity of nanoparticles potentially qualifies them as a safe drug delivery system.
Considering that proteins on the cell membrane surface play crucial roles in the homologous adhesion characteristics of tumor cells,46 three major membrane proteins associated with cellular adhesion (CD47, E-cadherin and EpCAM) were investigated using Western blotting.47–49 The results showed that CMSN carrier had protein profiles similar to those of CCM and cancer cell lysates, indicating that their membrane proteins were not destroyed during cell membrane extraction and coating by extrusion methods on the nanoparticle surfaces (Figure 1E).
Next, we tested the endocytic function of B16F10 cancer cells with respect to their ability to internalize CMSN. We labeled the CCM with FITC. After incubation with CLSM, the cancer cells displayed FITC (green) signals surrounding the nuclei, indicating that CMSN has the ability to target B16F10 cells (Figure 1F). Figure 1F also shows that the core-shell structure of CMSN has been disrupted and FITC marker is distributed throughout the cell.
Cytotoxicity and Anti-Proliferative Effects of DTIC@CMSN in vitro
Next, the in vitro cytotoxicity of DTIC@CMSN to B16F10 cells was assessed. The cells treated with free DTIC (5 μg/mL DTIC) for 48 h resulted in about 20% of cell death, while the activity of the cells treated with DTIC@MSN (5 μg/mL DTIC) decreased to 75% (Figure 2), and DTIC@CMSN (5 μg/mL DTIC) induced up to 40% of cell death. These data also indicate that the anti-tumor efficacy is further enhanced by increasing the intracellular DTIC concentration due to the homologous cell targeting by DTIC@CMSN.
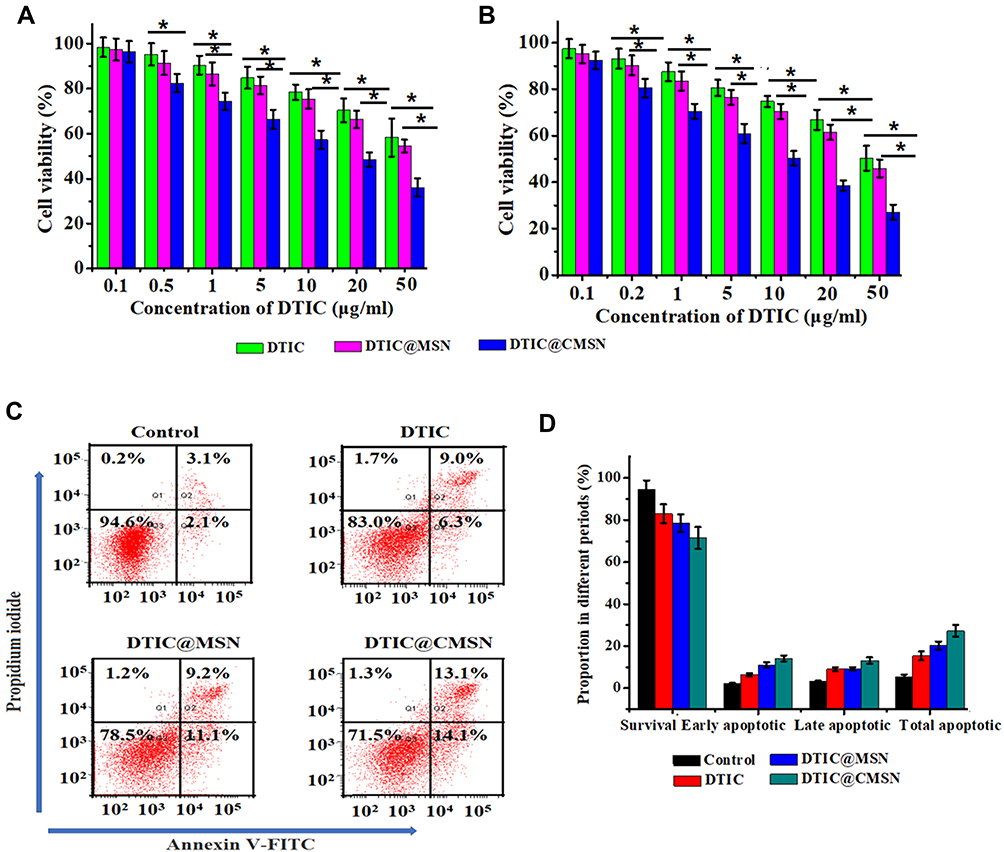 |
Figure 2 Cell viability of B16F10 cells treated with different concentrations of DTIC, DTIC@MSN and DTIC@CMSN for 24 h (A) or 48 h (B). (C) Apoptosis by flow cytometry assay in B16F10 cells induced by medium alone as negative control, DTIC, DTIC@MSN and DTIC@CMSN for 24 h. (D) Corresponding percentages of living, early apoptotic, late apoptotic, and necrotic tumor cells under different treatments. Error bars were calculated based on six samples. *P < 0.05. |
Apoptosis of B16F10 cells induced by DTIC, DTIC@MSN and DTIC@CMSN was studied using an Annexin V-PI staining assay (Figure 2C and D). After 24 h of exposure, B16F10 cells treated with DTIC@CMSN showed the highest apoptosis rate, about 27.2%, while the apoptosis rates of DTIC and DTIC@MSN on B16F10 cells were lower (15.3% and 20.3%, respectively). Annexin V-PI staining further demonstrated the excellent antitumor efficacy of DTIC@CMSN.
In vivo Anti-Tumor Activity
We evaluated the synergistic effect of DTIC@CMSN combined with systemic administration of aPD1 on melanoma in a mouse tumor-bearing model. Figure 3A shows the details of the treatment schedule. Melanoma mice were intravenously injected with PBS, aPD1, DTIC, DTIC @ MSN, DTIC @ CMSN and DTIC @ CMSN+aPD1 at a dose of 40 µg aPD1 and/or 10 mg/kg DTIC per mouse. After 20 days of treatment, we evaluated the therapeutic effect. As shown in Figure 3B, tumor volume in the control group increased rapidly and reached 3600 mm3 by the end of treatment. The tumor volume of mice in the DTIC and aPD1 groups increased to 2600 mm3 and 2400 mm3 respectively, indicating that the tumor inhibition ability by these agents was relatively weak. However, tumor volume was significantly reduced in the DTIC@MSN and DTIC@CMSN groups. After twenty days of treatment, tumor volumes in these groups were found to average 1376 mm3 and 798 mm3 respectively. The DTIC@CMSN combined with aPD1 group showed the best antitumor activity, with the inhibition rate of tumors in mice reaching 91.7%, while the inhibition rates in the DTIC, aPD1, DTIC@MSN and DTIC@CMSN groups were 27.8%, 33.3%, 61.8% and 77.8%, respectively (Figure 3C). The images of tumors in the six treatment groups shown in Figure 3D further indicate that the antitumor activity of the DTIC@CMSN combined with aPD1 group was greater than that for other groups. In addition, we assessed the overall survival rate of tumor-bearing mice in each group (Figure 3E). Only one animal in the group of DTIC@CMSN combined with aPD1 co-therapy died at day 55. The odds of survival were 50% in the DTIC@CMSN group. The PBS, DTIC and aPD1 groups had the worst survival, with nearly all the mice dying before day 40. As shown in Figure 3E, the combination of DTIC@CMSN with α-PD1 improved the survival rate of the tumor-bearing mice, further suggesting that the strategy of blocking PD-1 significantly improved the anti-tumor efficiency of DTIC@CMSN. During this experiment, we also monitored the weight of mice, and no significant weight loss was observed in any group. These data suggest that our carrier system, CMSN, has appropriate biocompatibility (Figure 3F).
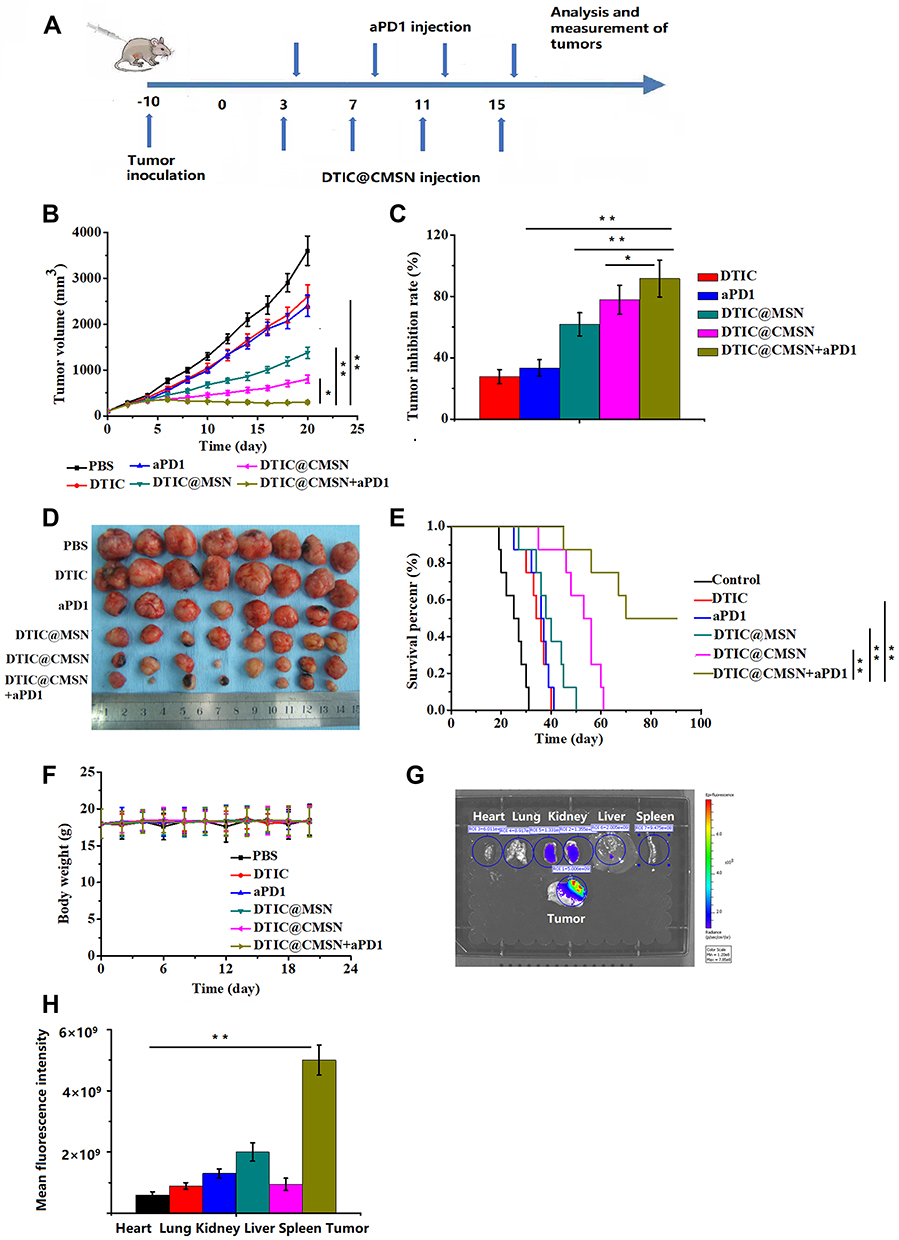 |
Figure 3 (A) Scheme of time axis showing the design of animal study. (B) The tumor growth curves under various treatments. (C) Tumor growth inhibition ratios of different groups at the 20th day of treatment. (D) Photographs of excised tumors from the tested groups after various treatments. (E) Survival curves of tumor-bearing mice after various treatments. (F) Body weight profiles of tumor-bearing mice at the end of the treatment period (day 20). (G) Images of major organs and tumors after tail vein injection of Cy5.5-labeled CMSN. (H) Quantification of the fluorescence intensity in the tumor, heart, liver, spleen, lung, and kidney at 4 h post injection. *P < 0.05, **P < 0.01. (mean ± SD, n =8). |
The targeting capability by CMSN was evaluated by in vivo imaging. First, a mouse tumor model bearing melanoma B16F10 was established for 10 days, and subsequently Cy5.5-labeled CMSN was injected intravenously. Mice were sacrificed at 4 h after injection and dissected for in vivo imaging. The results showed that Cy5.5 labeled CMSN showed brighter signals in mice tumors than in other tissues and organs, indicating that CMSN has good tumor targeting efficiency (Figure 3G and H). Fluorescence intensity of liver and kidney treated with Cy5.5 labeled CMSN was stronger than heart, lung and spleen, mainly due to the metabolism of CMSN in kidney and liver. These results show the degree to which the membrane coated nanoparticles are able to perform homologous targeting.
Evaluation of in vivo Systematic Toxicity
H&E staining further examined six treatment groups’ damage to five major organs (liver, spleen, lung, kidney, and heart) and tumors (Figure 4A). Staining showed that the liver of mice in the DTIC group was slightly damaged, which may occur because the liver is the major organ for metabolic breakdown of DTIC. The other treatment groups, including DTIC@MSN, DTIC@CMSN and DTIC@CMSN combined with aPD1 showed no significant damage to liver or other organs, indicating that the actual dosages provided good biocompatibility. DTIC@CMSN combined with aPD1 treatment showed the most severe tumor cell apoptosis in H&E stained tumor slices. Homologous targeting appears to be attributable to CCM layering upon MSN drug carriers. In addition, the synergistic anti-tumor action caused by accelerated DTIC release from targeted lysosomes, in combination with aPD1 administration over the course of treatment, appear to reduce the non-specific cytotoxic effects of chemotherapy drugs.
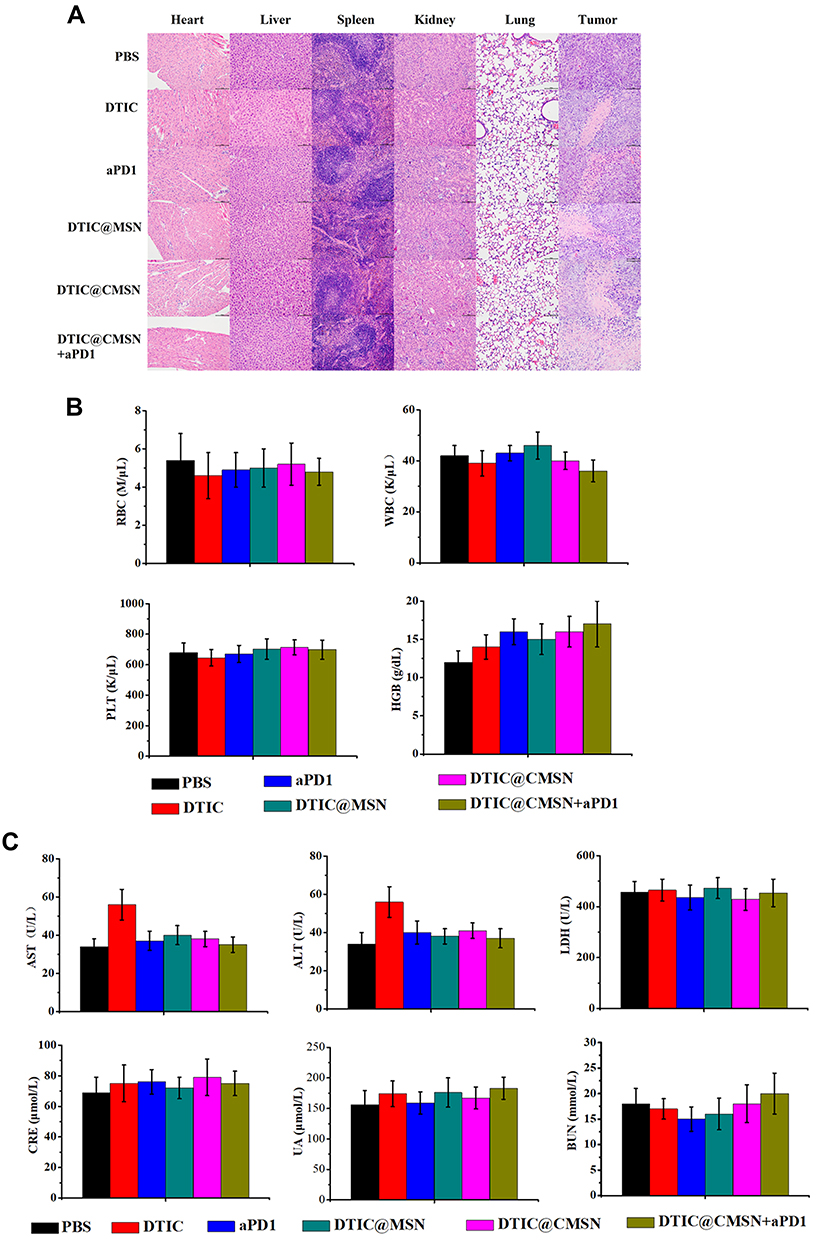 |
Figure 4 (A) Hematoxylin & eosin analysis of major organs of mice (heart, liver, spleen, lung, and kidney) and tumors under different treatments (PBS, DTIC, aPD1, DTIC@MSN, DTIC@CMSN, DTIC@CMSN+aPD1). Magnification 200×. (B) Serum levels of blood count (red blood cells (RBC), white blood cells (WBC), platelets (PLT) and hemoglobin (HGB)). (C) Serum levels of liver function biomarkers. Abbreviations: ALT, alanine transaminase; LDH, lactate dehydrogenase; AST, aspartate aminotransferase; CRE, renal function creatinine; BUN, blood urea nitrogen; UA, urea. |
To assess systemic toxicity, whole blood was collected 24 h post-injection. Hematological parameters largely showed no difference among the PBS, DTIC, aPD1, DTIC@MSN, DTIC@CMSN and DTIC@CMSN+aPD1 treatments (Figure 4B).
Hepatotoxicity was further investigated since DTIC is mainly metabolized by the liver. As shown in Figure 4C, serum levels of alanine aminotransferase (ALT) and aspartate aminotransferase (AST) significantly increased in DTIC treated mice, while ALT and AST levels were significantly lower in mice treated with aPD1, DTIC@MSN, DTIC@CMSN and DTIC@CMSN+aPD1. These serological indicators were consistent with histological assessments. Collectively, these results consistently demonstrated the good biocompatibility of DTIC@CMSN+aPD1. We speculate that encapsulation of DTIC in the nanoparticles may alleviate the hepatotoxicity of DTIC to some extent.
Mechanistic Analysis of the Immune Response
We analyzed the tumor-infiltrating lymphocytes in each treatment group using flow cytometry to better understand the mechanism of DTIC@CMSN combined with PD-1 immunotherapy. T cells were first collected from tumor tissues and co-stained with CD3, CD4, CD8 and Foxp3 for flow cytometry analysis. CD8 +T cells are cytotoxic T cells, and CD4 +T cells are helper T cells, both of which are functional for cell-mediated adaptive immunity. Studies have shown that the increase in the number of CD8 + and CD4 +T cells promotes immunotherapy against cancer.40 Our results show that the magnitude of infiltrates of CD8 +T cells in the DTIC@CMSN+ aPD1 treatment group is greater than those in other groups (Figure 5A). In contrast, regulatory T cells (Tregs) (CD4 + Foxp3 +) inhibit the antitumor immune response. As shown in Figure 5A, the proportion of Tregs (CD4 + Foxp3 +) in the DTIC@CMSN plus aPD1 group is significantly lower than in other groups.
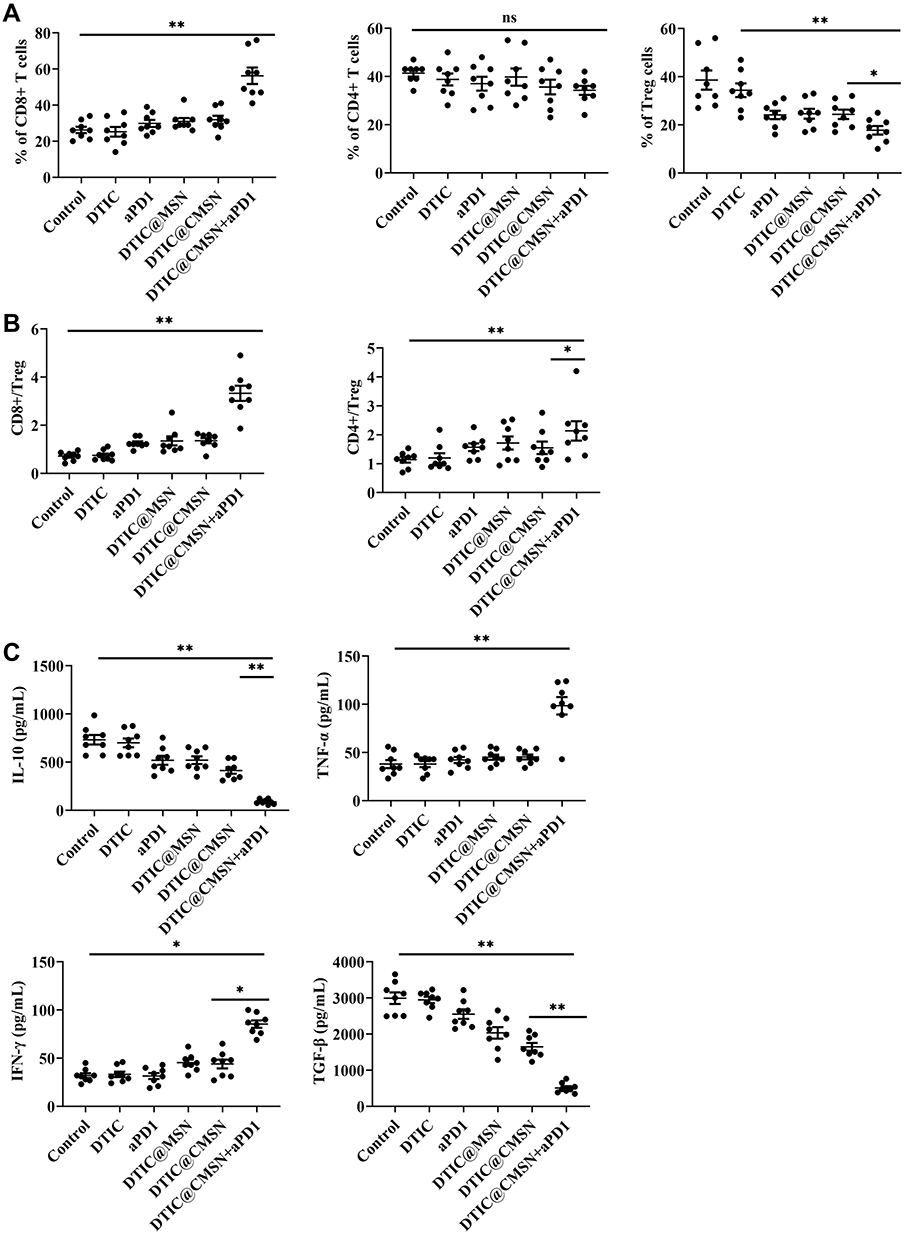 |
Figure 5 (A) Fractions of tumor-infiltrating CD4+, CD8+ and CD4+ FoxP3+ regulatory T cells (Treg cells) in the tumors with different treatments by flow cytometry. (B) CD8+ CTL-to-Treg ratios and CD4+ effecter T cells-to-Treg ratios in the tumors with different treatments. (C) Enzyme-linked immunosorbent assay (ELISA) analysis of IL-10, TNF-α, TGF-β and IFN-γ after the mice were injected with PBS, DTIC, aPD1, DTIC@MSN, DTIC@CMSN and DTIC@CMSN+aPD1. *P < 0.05, **P < 0.01. |
The group representing the DTIC@CMSN plus aPD1 treatment modality exhibited significantly higher CD8+ Teff-to-Treg and CD4+ Teff to-Treg ratios (Figure 5B). In addition, by comparing the aPD1 and DTIC@CMSN plus aPD1 treatments (Figure 5), we found that the combination therapy increases the effective T cell ratio (CD8+ T cells, CD8+ Teff-to-Treg ratio, and CD4+ Teff-to-Tregs ratio) and lowers the Tregs rate. This may be because these coated nanoparticles experienced immune evasion while they are capable of targeting cancer cells.
In recognition of the important role that cytokine secretion plays in the immune response, we collected serum from mice 24 h after different treatments to analyze the changes of IL-10, TNF-α, TGF-β and IFN-γ (Figure 5C). The release of these cytokines indicates acute inflammation, which is an important mechanism for inducing anti-tumor immunity.50 The secretion of TNF-α, and IFN-γ was significantly greater when induced by DTIC@CMSN+aPD1 than by the DTIC, aPD1, DTIC@MSN and DTIC@CMSN groups. The highest expression of both TNF-α and IFN-γ in the group of DTIC@CMSN+aPD1 in the tumor microenvironment also confirmed the enhanced immune response after aPD1 blockade (Figure 5C), due to the neutralization of the negative immune feedback pathway during chemotherapy. TNF-α, and IFN-γ are also key mediators of cellular immunity. Continuous elevation of these serum cytokines indicates that DTIC@CMSN + aPD1 activates cellular immunity. As the anti-tumor effect of aPD1 is mediated by cellular immunity, its continuous activation will be beneficial to further improve the efficacy of aPD1.51,52 The anti-tumor immune response of DTIC@CMSN combined with aPD1 was clarified by measuring levels of transforming growth factor-beta (TGF-β) and IL-10. The results in Figure 5C show that these immunosuppressive factors are significantly lower in the DTIC@CMSN combined with aPD1 group than in the other groups of the treatment series. These results confirm the existence of a significant shift from immunosuppression to immune activation.
Conclusion
In this study, we have successfully constructed a cancer cell membrane coated mesoporous silica nanoparticles that combine dacarbazine (DTIC@CMSN). CMSN exhibits high stability for drug retention, tumor acidic environmental responsiveness and good biocompatibility. For in vitro and in vivo validation studies, it demonstrated excellent homologous-targeting ability toward melanoma cells. When tested under the joint administration of DTIC@CMSN and aPD1, this carrier also showed excellent anti-tumor effects in a melanoma tumor model. Such effects were evidenced by a significant delay in tumor growth, thus prolonging the survival time, as well as displaying no obvious systemic toxicity. Effective killing is ascribed to three synergistic effects: the induction of tumor apoptosis by the chemotherapeutic drug DTIC, the regulation of the immunosuppressive tumor microenvironment and the enhancement of antitumor immunity. Our results show that the combined treatment strategy of “DTIC@CMSN and aPD1” has significant potential to provide superior chemoimmunotherapy in patients with melanoma.
Acknowledgments
The authors gratefully acknowledge National Natural Science Foundation of China (Grant 81402575). We thank LetPub for its linguistic assistance and scientific consultation during the preparation of this manuscript.
Disclosure
The authors declare no competing financial interest.
References
1. Siegel RL, Miller KD, Jemal A. Cancer statistics, 2020. CA Cancer J Clin. 2020;70(1):7–30. doi:10.3322/caac.21590
2. Miller KD, Nogueira L, Mariotto AB, et al. Cancer treatment and survivorship statistics, 2019. CA Cancer J Clin. 2019;69(5):363–385. doi:10.3322/caac.21565
3. Rosenberg SA, Yang JC, Topalian SL, et al. Treatment of 283 consecutive patients with metastatic melanoma or renal cell cancer using high-dose bolus interleukin 2. JAMA. 1994;271(12):907–913. doi:10.1001/jama.1994.03510360033032
4. Serrone L, Zeuli M, Sega FM, et al. Dacarbazine-based chemotherapy for metastatic melanoma: thirty-year experience overview. J Exp Clin Cancer Res. 2000;19(1):21–34.
5. Hodi FS, O’Day SJ, McDermott DF, et al. Improved survival with ipilimumab in patients with metastatic melanoma. N Engl J Med. 2010;363(8):711–723. doi:10.1056/NEJMoa1003466
6. Robert C, Schachter J, Long GV, et al. Pembrolizumab versus ipilimumab in advanced melanoma. N Engl J Med. 2015;372(26):2521–2532. doi:10.1056/NEJMoa1503093
7. Long GV, Hauschild A, Santinami M, et al. Adjuvant dabrafenib plus trametinib in stage III BRAF-mutated melanoma. N Engl J Med. 2017;377(19):1813–1823. doi:10.1056/NEJMoa1708539
8. Weber J, Mandala M, Del Vecchio M, et al. Adjuvant nivolumab versus ipilimumab in resected stage III or IV melanoma. N Engl J Med. 2017;377(19):1824–1835. doi:10.1056/NEJMoa1709030
9. Tarhini AA, Agarwala SS. Cutaneous melanoma: available therapy for metastatic disease. Dermatol Ther. 2006;19(1):19–25. doi:10.1111/j.1529-8019.2005.00052.x
10. Tagne JB, Kakumanu S, Nicolosi RJ. Nanoemulsion preparations of the anticancer drug dacarbazine significantly increase its efficacy in a xenograft mouse melanoma model. Mol Pharm. 2008;5(6):1055–1063. doi:10.1021/mp8000556
11. Bajetta E, Del Vecchio M, Bernard-Marty C, et al. Metastatic melanoma: chemotherapy. Semin Oncol. 2002;29(5):427–445. doi:10.1053/sonc.2002.35238
12. Avril MF, Aamdal S, Grob JJ, et al. Fotemustine compared with dacarbazine in patients with disseminated malignant melanoma: a Phase III study. J Clin Oncol. 2004;22(6):1118–1125. doi:10.1200/JCO.2004.04.165
13. Eggermont AM, Kirkwood JM. Re-evaluating the role of dacarbazine in metastatic melanoma: what have we learned in 30 years? Eur J Cancer. 2004;40(12):1825–1836. doi:10.1016/j.ejca.2004.04.030
14. Brown SD, Nativo P, Smith JA, et al. Gold nanoparticles for the improved anticancer drug delivery of the active component of oxaliplatin. J Am Chem Soc. 2010;132(13):4678–4684. doi:10.1021/ja908117a
15. Shi J, Kantoff PW, Wooster R, et al. Cancer nanomedicine: progress, challenges and opportunities. Nat Rev Cancer. 2017;17(1):20–37. doi:10.1038/nrc.2016.108
16. Song W, Anselmo AC, Huang L. Nanotechnology intervention of the microbiome for cancer therapy. Nat Nanotechnol. 2019;14(12):1093–1103. doi:10.1038/s41565-019-0589-5
17. Bertrand N, Wu J, Xu X, et al. Cancer nanotechnology: the impact of passive and active targeting in the era of modern cancer biology. Adv Drug Deliv Rev. 2014;66:2–25. doi:10.1016/j.addr.2013.11.009
18. Xue J, Zhao Z, Zhang L, et al. Neutrophil-mediated anticancer drug delivery for suppression of postoperative malignant glioma recurrence. Nat Nanotechnol. 2017;12(7):692–700. doi:10.1038/nnano.2017.54
19. Zhao P, Li L, Zhou S, et al. TPGS functionalized mesoporous silica nanoparticles for anticancer drug delivery to overcome multidrug resistance. Mater Sci Eng C Mater Biol Appl. 2018;84:108–117. doi:10.1016/j.msec.2017.11.040
20. Arnold RD, Mager DE, Slack JE, et al. Effect of repetitive administration of doxorubicin-containing liposomes on plasma pharmacokinetics and drug biodistribution in a rat brain tumor model. Clin Cancer Res. 2005;11(24):8856–8865. doi:10.1158/1078-0432.CCR-05-1365
21. Lingappa M, Song H, Thompson S, et al. Immunoliposomal delivery of 213Bi for α-emitter targeting of metastatic breast cancer. Cancer Res. 2010;70(17):6815–6823. doi:10.1158/0008-5472.CAN-09-4548
22. Wong AD, Ye M, Ulmschneider MB, et al. Quantitative analysis of the enhanced permeation and retention (EPR) effect. PLoS One. 2015;10(5):e0123461. doi:10.1371/journal.pone.0123461
23. Molinaro R, Corbo C, Martinez JO, et al. Biomimetic proteolipid vesicles for targeting inflamed tissues. Nat Mater. 2016;15(9):1037–1046. doi:10.1038/nmat4644
24. Zhao P, Wang H, Yu M, et al. Paclitaxel-loaded, folic-acid-targeted and TAT-peptide-conjugated polymeric liposomes: in vitro and in vivo evaluation. Pharm Res. 2010;27(9):1914–1926. doi:10.1007/s11095-010-0196-5
25. Zhao P, Wang H, Yu M, et al. Paclitaxel loaded folic acid targeted nanoparticles of mixed lipid-shell and polymer-core: in vitro and in vivo evaluation. Eur J Pharm Biopharm. 2012;81(2):248–256. doi:10.1016/j.ejpb.2012.03.004
26. Lao YH, Phua KK, Leong KW. Aptamer nanomedicine for cancer therapeutics: barriers and potential for translation. ACS Nano. 2015;9(3):2235–2254. doi:10.1021/nn507494p
27. Srinivasarao M, Galliford CV, Low PS. Principles in the design of ligand-targeted cancer therapeutics and imaging agents. Nat Rev Drug Discov. 2015;14(3):203–219. doi:10.1038/nrd4519
28. Teli MK, Mutalik S, Rajanikant GK. Nanotechnology and nanomedicine: going small means aiming big. Curr Pharm Des. 2010;16(16):1882–1892. doi:10.2174/138161210791208992
29. Tinkle S, McNeil SE, Muhlebach S, et al. Nanomedicines: addressing the scientific and regulatory gap. Ann N Y Acad Sci. 2014;1313:35–56. doi:10.1111/nyas.12403
30. Peer D, Karp JM, Hong S, et al. Nanocarriers as an emerging platform for cancer therapy. Nat Nanotechnol. 2007;2(12):751–760. doi:10.1038/nnano.2007.387
31. Rao L, Bu LL, Cai B, et al. Cancer cell membrane-coated upconversion nanoprobes for highly specific tumor imaging. Adv Mater. 2016;28(18):3460–3466. doi:10.1002/adma.201506086
32. Rao L, Cai B, Bu LL, et al. Microfluidic electroporation-facilitated synthesis of erythrocyte membrane-coated magnetic nanoparticles for enhanced imaging-guided cancer therapy. ACS Nano. 2017;11(4):3496–3505. doi:10.1021/acsnano.7b00133
33. Xie W, Deng WW, Zan M, et al. Cancer cell membrane camouflaged nanoparticles to realize starvation therapy together with checkpoint blockades for enhancing cancer therapy. ACS Nano. 2019;13(3):2849–2857. doi:10.1021/acsnano.8b03788
34. Chen Z, Zhao P, Luo Z, et al. Cancer cell membrane-biomimetic nanoparticles for homologous-targeting dual-modal imaging and photothermal therapy. ACS Nano. 2016;10(11):10049–10057. doi:10.1021/acsnano.6b04695
35. Wang D, Dong H, Li M, et al. Erythrocyte-cancer hybrid membrane camouflaged hollow copper sulfide nanoparticles for prolonged circulation life and homotypic-targeting photothermal/chemotherapy of melanoma. ACS Nano. 2018;12(6):5241–5252. doi:10.1021/acsnano.7b08355
36. Cabral H, Matsumoto Y, Mizuno K, et al. Accumulation of sub-100 nm polymeric micelles in poorly permeable tumours depends on size. Nat Nanotechnol. 2011;6(12):815–823. doi:10.1038/nnano.2011.166
37. Kong M, Tang J, Qiao Q, et al. Biodegradable hollow mesoporous silica nanoparticles for regulating tumor microenvironment and enhancing antitumor efficiency. Theranostics. 2017;7(13):3276–3292. doi:10.7150/thno.19987
38. Liu CM, Chen GB, Chen HH, et al. Cancer cell membrane-cloaked mesoporous silica nanoparticles with a pH-sensitive gatekeeper for cancer treatment. Colloids Surf B Biointerfaces. 2019;175:477–486. doi:10.1016/j.colsurfb.2018.12.038
39. Nel AE, Madler L, Velegol D, et al. Understanding biophysicochemical interactions at the nano-bio interface. Nat Mater. 2009;8(7):543–557. doi:10.1038/nmat2442
40. Parodi A, Quattrocchi N, van de Ven AL, et al. Synthetic nanoparticles functionalized with biomimetic leukocyte membranes possess cell-like functions. Nat Nanotechnol. 2013;8(1):61–68. doi:10.1038/nnano.2012.212
41. Radu DR, Lai C-Y, Wiench JW, et al. Gatekeeping layer effect: a poly(lactic acid)-coated mesoporous silica nanosphere-based fluorescence probe for detection of amino-containing neurotransmitters. J Am Chem Soc. 2004;126(6):1640–1641. doi:10.1021/ja038222v
42. Li J, Zhen X, Lyu Y, et al. Cell membrane coated semiconducting polymer nanoparticles for enhanced multimodal cancer phototheranostics. ACS Nano. 2018;12(8):8520–8530. doi:10.1021/acsnano.8b04066
43. Zhao Y, Luo Z, Li M, et al. A preloaded amorphous calcium carbonate/doxorubicin@silica nanoreactor for pH-responsive delivery of an anticancer drug. Angew Chem Int Ed Engl. 2015;54(3):919–922. doi:10.1002/anie.201408510
44. Ganta S, Devalapally H, Shahiwala A, et al. A review of stimuli-responsive nanocarriers for drug and gene delivery. J Control Release. 2008;126(3):187–204. doi:10.1016/j.jconrel.2007.12.017
45. Luo D, Cui S, Liu Y, et al. Biocompatibility of magnetic resonance imaging nanoprobes improved by transformable gadolinium oxide nanocoils. J Am Chem Soc. 2018;140(43):14211–14226. doi:10.1021/jacs.8b08118
46. Xia J, Cheng Y, Zhang H, et al. The role of adhesions between homologous cancer cells in tumor progression and targeted therapy. Expert Rev Anticancer Ther. 2017;17(6):517–526. doi:10.1080/14737140.2017.1322511
47. Naor D, Sionov RV, Ish-Shalom D. CD44: structure, function, and association with the malignant process. Adv Cancer Res. 1997;71:241–319. doi:10.1016/s0065-230x(08)60101-3
48. Majeti R, Chao MP, Alizadeh AA, et al. CD47 is an adverse prognostic factor and therapeutic antibody target on human acute myeloid leukemia stem cells. Cell. 2009;138(2):286–299. doi:10.1016/j.cell.2009.05.045
49. Yu Z, Zhou P, Pan W, et al. A biomimetic nanoreactor for synergistic chemiexcited photodynamic therapy and starvation therapy against tumor metastasis. Nat Commun. 2018;9(1):5044. doi:10.1038/s41467-018-07197-8
50. Castano AP, Mroz P, Wu MX, et al. Photodynamic therapy plus low-dose cyclophosphamide generates antitumor immunity in a mouse model. Proc Natl Acad Sci U S A. 2008;105(14):5495–5500. doi:10.1073/pnas.0709256105
51. Pardoll DM. The blockade of immune checkpoints in cancer immunotherapy. Nat Rev Cancer. 2012;12(4):252–264. doi:10.1038/nrc3239
52. Ribas A, Dummer R, Puzanov I, et al. Oncolytic virotherapy promotes intratumoral T cell infiltration and improves Anti-PD-1 immunotherapy. Cell. 2017;170(6):1109–1119e10. doi:10.1016/j.cell.2017.08.027
 © 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.