")
Back to Journals » Drug Design, Development and Therapy » Volume 16
Canagliflozin Attenuates Hepatic Steatosis and Atherosclerosis Progression in Western Diet-Fed ApoE-Knockout Mice
Authors Zuo Q , Zhang G, He L, Ma S , Ma H, Zhai J, Wang Z, Zhang T, Wang Y, Guo Y
Received 5 September 2022
Accepted for publication 21 November 2022
Published 6 December 2022 Volume 2022:16 Pages 4161—4177
DOI https://doi.org/10.2147/DDDT.S388823
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Manfred Ogris
Qingjuan Zuo,1,2 Guorui Zhang,1,3 Lili He,2 Sai Ma,4 Huijuan Ma,5 Jianlong Zhai,6 Zhongli Wang,7 Tingting Zhang,2 Yan Wang,2 Yifang Guo1,2
1Department of Internal Medicine, Hebei Medical University, Shijiazhuang, People’s Republic of China; 2Department of Geriatric Cardiology, Hebei General Hospital, Shijiazhuang, People’s Republic of China; 3Department of Cardiology, the Third Hospital of Shijiazhuang City Affiliated to Hebei Medical University, Shijiazhuang, People’s Republic of China; 4Department of Internal Medicine, Hebei General Hospital, Shijiazhuang, People’s Republic of China; 5Department of Endocrinology, Hebei General Hospital, Shijiazhuang, People’s Republic of China; 6Department of Cardiology, Hebei General Hospital, Shijiazhuang, People’s Republic of China; 7Department of Physical Examination Center, Hebei General Hospital, Shijiazhuang, People’s Republic of China
Correspondence: Yifang Guo, Department of Geriatric Cardiology, Hebei General Hospital, No. 348, Heping West Road, Xinhua District, Shijiazhuang, Hebei, 050051, People’s Republic of China, Tel +86-15100189182, Email [email protected]
Purpose: To investigate the effect of canagliflozin (20 mg/kg) on hepatic steatosis and atherosclerosis, and further to explore its possible mechanism.
Methods: Blood glucose, blood lipid, oxidative stress response and inflammatory cytokines were examined by intraperitoneal glucose tolerance test and ELISA assay. HE and Oil Red O staining were used to estimate the extent of hepatic steatosis and atherosclerosis. RNA-seq and qRT-PCR were used to further investigate the potential mechanism. The effects of canagliflozin on autophagy were detected using transmission electron microscopy and Western blotting. The endothelial function-related markers were determined by qRT-PCR.
Results: Canagliflozin notably alleviated the elevation in blood glucose and insulin resistance in western diet-fed ApoE-/- mice. In ApoE-/-+Cana group, ApoE-/- mice had lower levels of TG, TC, LDL-C, TNF-α, IL-6, IL-1β, and MCP-1. HE and Oil Red O staining presented that canagliflozin restrained the atherosclerotic plaque development and lipid accumulation. RNA-seq showed that 87 DEGs were relevant to improvement of hepatic steatosis and atherosclerosis by canagliflozin. Among them, CPS1, ASS1, ASL, ARG1, MATLA, GLS2, GOT1, SREBP1, Plin5, Retreg1, and C/EBPβ were verified. KEGG enrichment analysis indicated that DEGs were mainly involved in amino acid metabolism. Besides, we observed that canagliflozin reduced the contents of aspartic acid and citrulline in liver. Western blotting showed that ASS1 and p-AMPK/AMPK was remarkably elevated after administration of canagliflozin. Correspondingly, canagliflozin down-regulated SREBP1, FAS, ACC1, HMGCR, p-mTOR/m-TOR, p-ULK1/ULK1 and p62, but up-regulated CPT1, Beclin 1 and LC3 II/LC3I. TEM showed that canagliflozin reduced the number of lipid droplets and increased the autophagosomes. Moreover, we found that canagliflozin elevated the aortic endothelial function-associated markers including ASS1, ASL and eNOS.
Conclusion: Canagliflozin may attenuate hepatic steatosis by improving lipid metabolism, enhancing autophagy, and reducing inflammatory response through ASS1/AMPK pathway. Besides, canagliflozin further effectively improves the aortic endothelial function, thereby suppressing atherosclerosis development.
Keywords: canagliflozin, hepatic steatosis, atherosclerosis, RNA-seq, dyslipidemia, inflammatory response, autophagy
Introduction
Atherosclerosis is a chronic inflammatory disease of arteries driven by dyslipidemia, characterized by endothelial dysfunction, inflammatory infiltration in the vascular wall, lipid accumulation and plaque formation.1 In general, atherosclerosis may lead to many serious cardiovascular and cerebrovascular diseases, such as myocardial infarction, coronary heart disease, cerebral infarction and cerebral hemorrhage.2 To date, atherosclerotic cardiovascular disease (ASCVD) is still the main cause of morbidity and mortality worldwide, imposing a significant economic burden on individuals and families.3 However, preventative means to lower the risk of the disease remain elusive.
It has been recognized that nonalcoholic fatty liver disease (NAFLD) is an early risk factor for the development of atherosclerosis.4 NAFLD is a clinical syndrome characterized by hepatic cell steatosis and fat storage, ranging from simple steatosis to nonalcoholic steatohepatitis (NASH), cirrhosis, and hepatocellular carcinoma (HCC).5 In cases of obesity, NAFLD often co-occurs with insulin resistance, metabolic syndrome, and atherosclerosis.6 Moreover, a previous study attempting to link atherosclerosis to NAFLD suggests that atherosclerotic lesions are broadly impacted by NAFLD-induced dyslipidemia, oxidative stress, inflammatory response and secretion of hepatic factors.7 Those findings suggest that prevention and treatment of NAFLD may slow progression of atherosclerosis.
Sodium glucose co-transporter-2 (SGLT-2) inhibitors are a new type of anti-hyperglycemic drugs that reduce glucose reabsorption in renal tubules and increase urinary glucose excretion by selectively inhibiting SGLT-2.8 In addition to hypoglycemic effects, accumulating studies have confirmed that SGLT-2 inhibitors exert protective effects on cardiovascular and kidney.9,10 Canagliflozin, as a member of SGLT-2 inhibitors, is believed to have a certain preventive and therapeutic effect on atherosclerosis.11,12 Another study has demonstrated that canagliflozin can regulate cholesterol metabolism and improve blood lipids.13 Moreover, Shib et al14 have demonstrated that canagliflozin relieves the symptoms of or even delays the onsets of NASH and HCC. However, how canagliflozin alleviates the developments of hepatic steatosis and atherosclerosis has not yet been fully understood.
In the current study, we firstly disclosed the potential mechanism of canagliflozin on improving hepatic steatosis as well as atherosclerosis in western diet-fed ApoE-/- mice based on RNA-Seq analysis. We discovered that canagliflozin may increase ASS1 activity, enhance ornithine cycle, and promote phosphorylation of AMPK, thereby improving hepatic steatosis, as well as preventing the development of atherosclerotic lesions.
Materials and Methods
Animals and Treatment
Male ApoE-/- mice (6–8 weeks old) and wild-type C57BL/6J mice were purchased from Beijing Charles River Laboratory Animal Technology Co., Ltd (Beijing, China). All mice were acclimated in a specific-pathogen free-grade room for 1-week prior to the start of the experiments. At the end of the week, ApoE-/- mice were randomly divided into western diet group (ApoE-/- group, n=10) and canagliflozin intervention group (ApoE-/-+Cana group, n=10). Specifically, canagliflozin (Janssen-Cilag International NV, Xi’an, China) was dissolved in 0.5% hydroxypropyl methylcellulose (0.5% HPMC, Coolaber, Beijing, China). Mice in the ApoE-/-+Cana group were fed with western diet (D12079B, 41% fat, 1.5‰ cholesterol) and given intragastric administration of canagliflozin (20 mg/kg) per day. Mice in the ApoE-/- were also fed with western diet (D12079B, 41% fat, 1.5‰ cholesterol) and received the same dose of 0.5% HPMC as did those in the ApoE-/-+Cana group. C57BL/6J mice (Control group, n=10) continued to eat normal diet and were given equal volume of 0.5% HPMC once daily. At the beginning of the 14th week, all mice were anesthetized with an intraperitoneal injection of pentobarbital sodium (40 mg/kg) and slaughtered. Blood samples and liver, heart, and aorta tissues were collected. The liver index was calculated by the formula as follows: wet weight of liver (g)/body weight (g)×100%. The present study was approved by the ethics committee of Hebei General Hospital (NO.2022056). All procedures were in strict accordance with the recommendations in the Guide for the Care and Use of Laboratory Animals by the National Institutes of Health.
Measurement of Blood Glucose and Insulin Resistance
After 13 weeks of treatment, all mice were fasted for 8 hours and then intraperitoneal glucose tolerance test (IPGTT) was conducted. In brief, blood glucose at 0, 15, 30, 60, and 120 min was measured by glucometer after intraperitoneal injections of glucose (2 g/kg).
Insulin levels were detected by mouse insulin ELISA kit (Taicang, Shanghai, China). Homeostasis model assessment of insulin resistance (HOMA-IR) was defined by following formula: (HOMA-IR = Glucose (mmol/L) × Insulin (mIU/L)/22.5).
Measurement of Blood Lipid, Oxidative Stress Response and Inflammatory Cytokines
Blood collected from the orbital vascular plexus was centrifuged at 3000×g for 8 min to obtain the serum. Serum levels of TG, TC, HDL, LDL, ALT, and AST were measured using colorimetric kits (Nanjing Jiancheng, Nanjing, China). The concentrations of inflammatory cytokines in serum including MCP-1, IL-6, IL-1β and TNF-α were detected by ELISA kits (Lianke, Hangzhou, China). The supernatant of liver tissue was also prepared, and the levels of TC, TG, FFA and MDA were measured by colorimetric kits (Nanjing Jiancheng, Nanjing, China).
Oil Red O Staining
Aorta and liver were carefully collected and fixed with 4% paraformaldehyde. For atherosclerosis evaluation, 10 μm thick sections of whole aorta and frozen aortic root were made and stained with Oil Red O. For lipid deposition evaluation, similar sections were prepared for frozen liver tissues. Finally, the percentage of plaque area and lipid deposition area was quantified with Image-Pro Plus software (Version 6.0, Media Cybernetics, Rockville, MD, USA).
HE Staining
Liver and aortic root tissues were fixed with 4% paraformaldehyde, embedded in paraffin, and sliced into 4 µm sections. Subsequently, the slices were dewaxed, rehydrated, and stained with hematoxylin–eosin (HE). Images were captured by microscope (ECLIPSE Ci-L, Nikon, Tokyo, Japan) and visualized by Image-Pro Plus software (Version 6.0, Media Cybernetics, Rockville, MD, USA).
Immunohistochemistry Assay
Briefly, paraffin-embedded liver slides (4 µm) were treated with 3% hydrogen peroxide and incubated in blocking solution (10% goat serum). After washing, the slides were incubated with anti-F4/80 antibody (GB113373, 1:800, Affinity, Wuhan, China) overnight, and then with anti-rabbit IgG-HRP (1:200, GB23303, Servicebio, Wuhan, China). Next, the sections were stained with 3,3’-diaminobenzidine (DAB) kit (ZLI-9017, Zhongshan, China). Images were captured by microscopy (ECLIPSE Ci-L, Nikon, Tokyo, Japan), and the relative F4/80 level was quantified by Image-Pro Plus software (Version 6.0, Media Cybernetics, Rockville, MD, USA).
Hepatic Transcriptome Analysis
Liver tissue samples were sequenced on the machine to create an image file, which was transformed by the software of the sequencing platform to generate the raw data of FASTQ, that is, the off-line data. The raw data were filtered to remove some low-quality reads with connectors, so as to avoid great interference caused by subsequent information analysis. The filtered reads were compared with reference genome to obtain gene information using HISAT2 software. RSeQC software was utilized to evaluate the quality control of RNA-seq experiments. The gene expression was normalized as fragments per kilobase per million (FPKM) fragments using the HTseq package. Differential expression analysis was done using DESeq software package. Genes with fold change greater than 1.5 and p-value less than 0.05 (deemed as differentially expressed genes (DEG)) were selected for further KEGG enrichment analysis.
Analysis of Amino Acid Metabolism
The contents of hepatic amino acid were detected by liquid chromatography-mass spectrometry. Samples of liver tissue (100 mg) homogenized with 500 µL of water and 500 µL of methanol underwent centrifugation at 12,000 × g for 5 min at 4°C. Twenty microliters of the supernatant was mixed with 5 µL of internal standard and 40 µL of isopropanol with 1% formic acid in a vortex mixer for 60 min. Then, 10 µL of the suspension, to which 70 µL of buffer salt solution and 20 µL of derivatization reagent were added, was kept at 50°C for 10 min to undergo derivation reactions. Diluted 5 times, the samples were fed into the LC-MS equipment to determine the concentration of amino acids including aspartic acid (Asp) and citrulline (Cit).
Quantitative Real-Time PCR Assay
Total RNA was extracted from the aorta and liver samples with TRIzol Universal Reagent (Tiangen, Beijing, China), and RNase-free DNase I (Tiangen, Beijing, China) was utilized to remove DNA. After assessing the quality and quantity, cDNA was synthesized with ReverTra Ace reverse transcriptase (Tiangen, Beijing, China), and real-time PCR was conducted by SuperReal PreMix Plus (Tiangen, Beijing, China). β-Actin was termed as internal control for quantification of mRNA levels. Primer sequences for aorta and liver tissues are listed in Table 1. The 2−ΔΔCt method was applied to calculate relative mRNA expression levels.
 |
Table 1 Primer Sequences |
Western Blotting Assay
Liver tissues were lysed with lysate containing protease inhibitor and phosphatase inhibitor, and the concentration of total protein was measured with BCA kit (Solarbio, Beijing, China). Fifty micrograms of the tissue was mixed with SDS and separated using SDS-PAGE. Separated proteins were electrophoretically transferred into a PVDF membrane, which was then blocked with 5% skimmed milk at room temperature for 3 h. The blotted membranes were then incubated overnight at 4°C with the following primary antibodies successively: anti-ASS1 (ab170952-40, 1:20,000), anti-SREBP1 (ab28481, 1:3000), and anti-p62 (ab109012, 1:1000) (Abcam, Cambridge, UK); anti-Plin5 (26,951-1-AP, 1:1000), anti-ACC1 (21,923-1-AP-50, 1:2000), CPT1 (15,184-1-AP-50, 1:2000) and anti-β-actin (1:5000) (Proteintech, Wuhan, China); anti-Retreg1 (125,727–50, 1:1000), anti-HMGCR (384,588–20, 1:1000), anti-AMPK Antibody (383,314.5, 1:1000), anti-p-AMPK (Thr172) (381,164–5, 1:1000), anti-C/EBPβ (380,893–50, 1:1000), and anti-FAS (200,194–25, 1:1000) (Zhengneng, Chengdu, China); anti-mTOR (2983S, 1:1000), anti-p-mTOR (Ser2448) (5536S, 1:1000), anti-ULK1 (8054S, 1:1000), anti-p-ULK1 (Ser757) (14202T, 1:1000), anti-Beclin1 (3495S, 1:1000), anti-LC3 I/II (4108S, 1:1000) (Cell Signaling Technology, Beverly, MA, USA). After washing, the membranes were incubated with either anti-rabbit IgG-HRP (MF094-01, 1:8000, Mei5 Biotechnology, Beijing, China) or anti-Mouse IgG-HRP (MF093-01, 1:5000, Mei5 Biotechnology, Beijing, China) at room temperature for 2 h in a decolorizing shaking table. Immunoblots were visualized using the enhanced chemiluminescence (ECL) kit and imaged using ImageQuant LAS500 (GE, Chicago, IL, USA). Band gray values were read in ImageJ (NIH) to determine the relative protein levels.
Transmission Electron Microscopy
Liver tissues were collected from mice and immediately fixed in 2.5% glutaraldehyde for 48 h at 4°C. After washing, the tissues were postfixed in 2% osmium tetroxide, dehydrated in ethanol/propylene oxide and infiltrated in EMbed 812. Then, liver tissues were cut at 70nm thickness using ultramicrotome (ULTURECUT R, Leica, Germany), and next stained with uranyl acetate and lead citrate solution. After the slices dried, images were captured by transmission electron microscope (H-7650, Hitachi, Tokyo, Japan).
Statistical Analysis
IBM SPSS 23.0 (IBM Corp., Armonk, USA) was used for the statistical analysis. One-way analysis of variance (ANOVA) followed by least significant difference (LSD) test or Tamhane’s T2 test was conducted to assess the differences between the groups. Differences with P-value less than 0.05 are interpreted as statistically significant.
Results
Canagliflozin Reduced Glucose and Insulin Resistance in Western Diet-Fed ApoE-/- Mice
To examine whether canagliflozin can ameliorate western diet-associated metabolic disorders of ApoE-/- mice, we conducted IPGTT on mice in all three groups. IPGTT results indicated that blood glucose of the Control group reached the peak value at 15 min, while the peak values in the ApoE-/- group and ApoE-/-+Cana group were delayed to about 30 min (Figure 1A). Consistent with this observation, the glucose area under the curve (AUC) was increased in the western diet groups compared to the Control group, but in the ApoE-/-+Cana group the increase was attenuated by canagliflozin (P<0.05) (Figure 1B). We also found that canagliflozin reduced the fasting insulin levels (FINS) (P<0.05) (Figure 1C). Besides, HOMA-IR results were largely consistent with IPGTT results (Figure 1D), suggesting that canagliflozin can reduce glucose and insulin resistance in western diet-fed ApoE-/- mice.
 |
Figure 1 Canagliflozin reduced glucose tolerance and insulin resistance in western diet-fed ApoE-/- mice. (A) IPGTT test among the three groups, n=5; (B) Area under the curve of IPGTT test, n=5; (C) The fasting insulin level, n=5; (D) The HOMA-IR level, n=5; (B-D, ANOVA, LSD test); *, **P<0.05, P<0.01 versus Control group; #, ##P<0.05, P<0.01 versus ApoE-/- group. Abbreviations: IPGTT, intraperitoneal glucose tolerance test, HOMA-IR, homeostasis model assessment of insulin resistance. |
Canagliflozin Ameliorated the Blood Lipid Profile and Reduced Inflammation in Western Diet-Fed ApoE-/- Mice
As shown in Figure 2A-D, the levels of TC, TG, and LDL-C in ApoE-/- group were notably increased compared to those in the Control group, while the HDL level decreased (P<0.01). After administration of canagliflozin, the contents of TC, TG, and LDL-C in the ApoE-/-+Cana group were reduced compared to those in the Model group. However, there was no significant difference in HDL between the ApoE-/- group and the ApoE-/-+Cana groups (P>0.05). ELISA results revealed that western diet-fed ApoE-/- mice developed severe inflammatory responses, as demonstrated by serum contents of TNF-α, IL-6, IL-1β and MCP-1. However, the inflammatory response was effectively relieved following intervention with canagliflozin (Figure 2D-H).
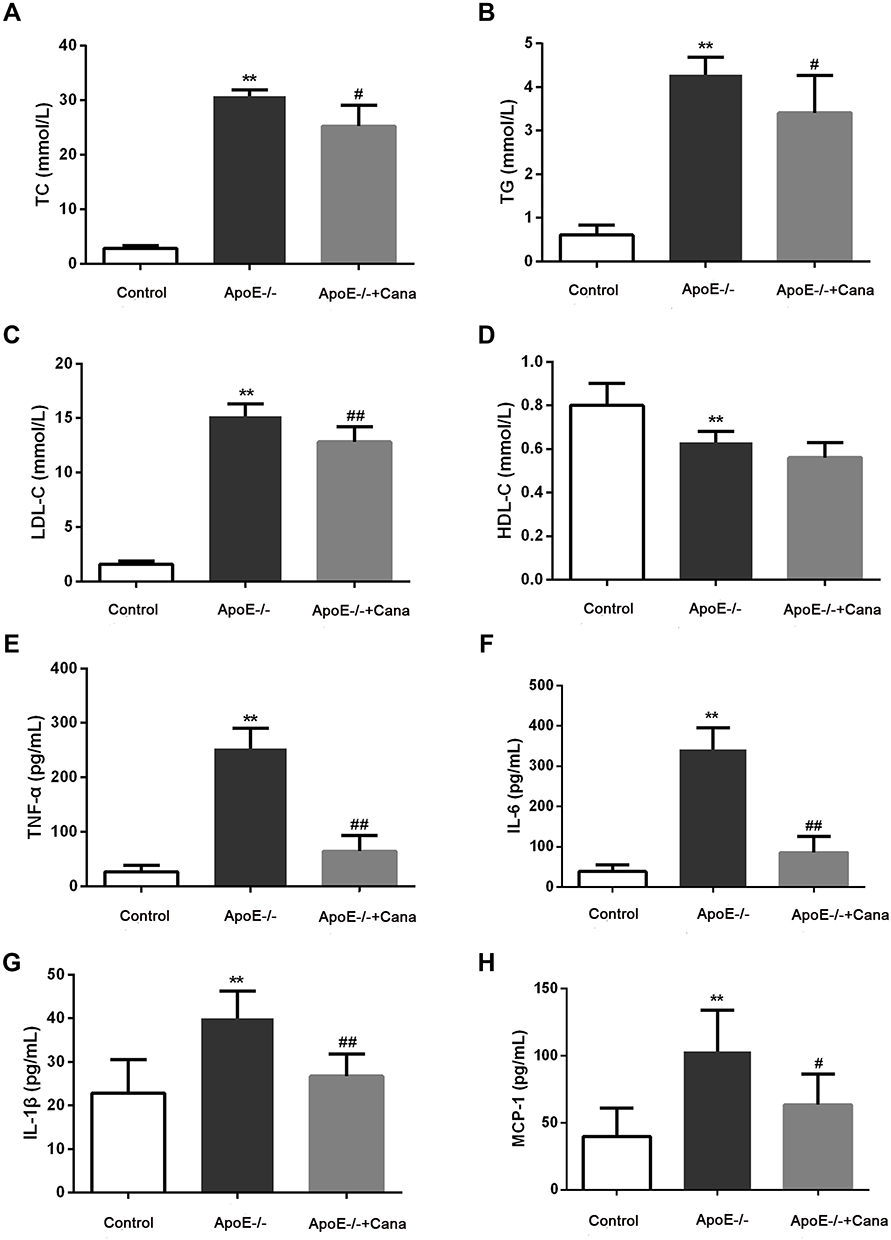 |
Figure 2 Canagliflozin ameliorated the blood lipid profile and reduced inflammation in western diet-fed ApoE-/- mice. The levels of TC (A), TG (B), LDL-C (C) and HDL (D) level, n=10; The serum contents of (E) TNF-α (n=8), (F) IL-6 (n=8), (G) IL-1β (n=8), and (H) MCP-1 (n=6) were tested by ELISA assay; (A-C, E and F), ANOVA, Tamhane’s T2 test; (D and G-H), ANOVA, LSD test); **P<0.01 versus Control group; #, ##P<0.05, P<0.01 versus ApoE-/- group. |
Canagliflozin Alleviated Atherosclerosis in Western Diet-Fed ApoE-/- Mice
As presented in Figure 3A-D, ApoE-/- mice of ApoE-/- group exhibited atherosclerotic plaque development, with lipid deposits scattered throughout the aorta and the aortic roots. However, no obvious atherosclerotic plaque in the Control group was detected. Compared with the ApoE-/- group, the percentage of plaque area in ApoE-/-+Cana group was decreased by almost 50%, which was statistically significant (P<0.01). Consistently, HE staining showed that the degree of atherosclerotic lesions in ApoE-/- mice was obviously alleviated following intervention with canagliflozin (Figure 3E). Immunohistochemistry staining showed that the percentage of F4/80 positive cells was considerably elevated in the ApoE-/- group compared with the Control group (P<0.01), though the elevation was not as significant in the ApoE-/-+Cana group (P<0.05) (Figure 3F and G). qRT-PCR results indicated higher levels of MCP-1 and MMP-9 expression in the ApoE-/- group than in the Control group (P<0.01), whereas these elevated levels of expression were attenuated in mice in the ApoE-/-+Cana group (P<0.01), as shown in Figure 3H and I.
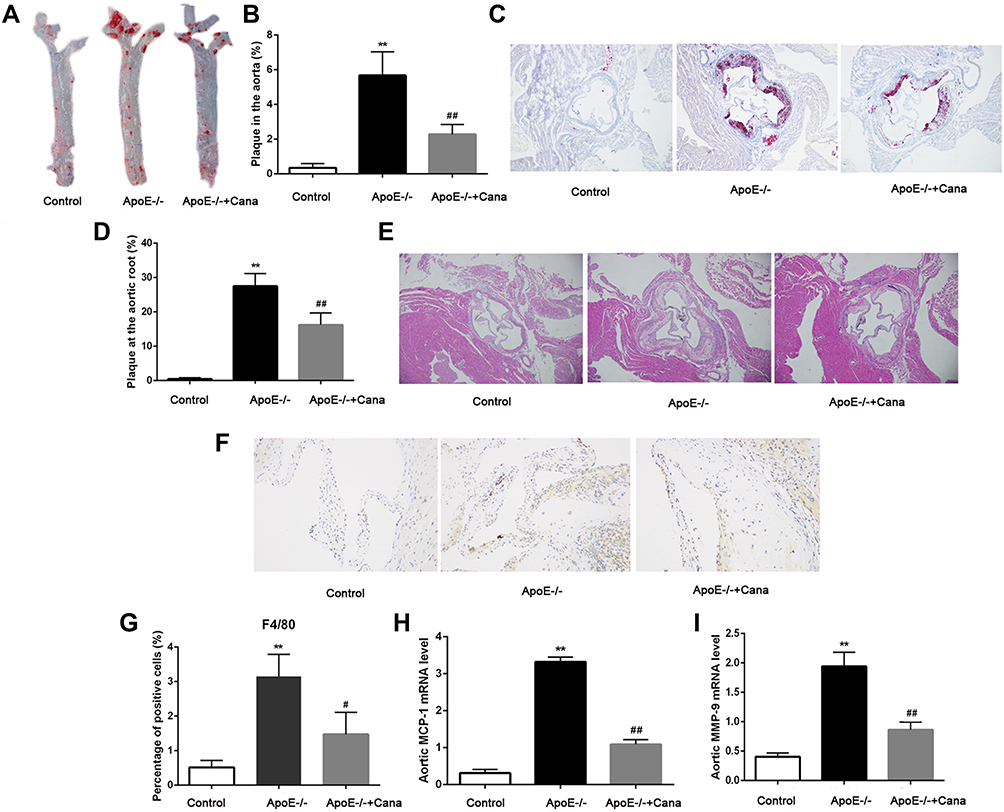 |
Figure 3 Canagliflozin alleviated atherosclerosis in western diet-fed ApoE-/- mice. (A and B) Representative images of aorta and ratio of plaque area stained with Oil Red O (×40 magnification, n=5); (C and D) Representative images of aortic roots and ratio of plaque area stained with Oil Red O (×40 magnification, n=5); (E) Representative images of aortic roots stained with HE (×40 magnification); (F and G) The percentage of F4/80 positive cells in aortic root plaque area was determined by immunohistochemistry assay (×200 magnification, n=4); The relative mRNA levels of (H) MCP-1 (n=4) and (I) MMP-9 (n=4) in aorta was tested by qRT-PCR assay; (A-D and F-G), ANOVA, Tamhane’s T2 test; (H-I), ANOVA, LSD test); **P<0.01 versus Control group; #, ##P<0.05, P<0.01 versus ApoE-/- group. |
Canagliflozin Alleviated Hepatic Steatosis in Western Diet-Fed ApoE-/- Mice
As shown in Figure 4A, mice in the Control group had a regular hepatic structure and no steatosis. By comparison, hepatic structure in the ApoE-/- group was disorganized and more hepatocytes appeared balloon like. In the ApoE-/-+Cana group, we observed that liver structure tended to be normal and the number of hepatocytes with balloon like was decreased. Oil O red staining of liver tissue indicated that the lipid deposition in the ApoE-/- group was aggravated, whereas canagliflozin treatment remarkably reduced the lipid accumulation (Figure 4B and C). Similarly, canagliflozin was responsible for the difference in liver index between the two western diet groups (P<0.05) (Figure 4D). The same pattern was also seen in lipid components in liver tissues (TG, TC, and FFA) as measured by ELISA, that is, canagliflozin attenuated all three hepatic lipid contents (P<0.01) (Figure 4E and F). In terms of oxidative stress, we found that the content of MDA in the liver tissue was notably lower following intervention with canagliflozin (P<0.01) (Figure 4G). Moreover, canagliflozin relieved liver injury, as evidenced by the reduction of ALT and AST level in serum (P<0.05) (Figure 4H).
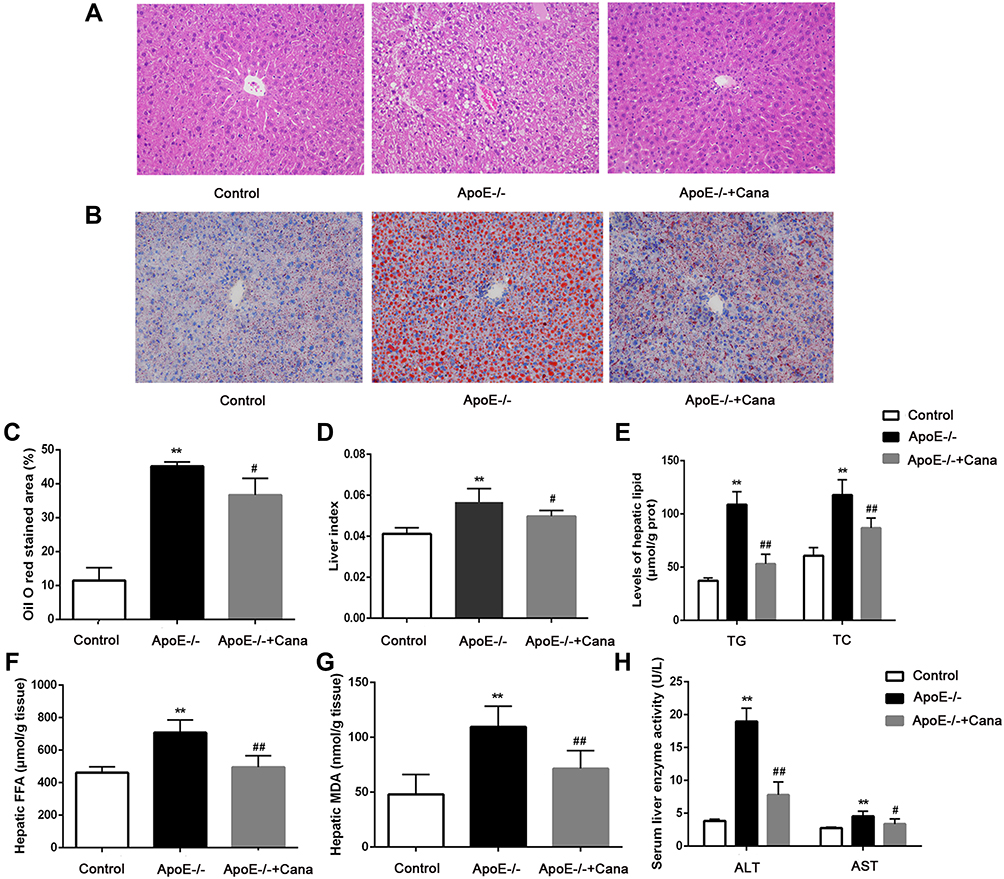 |
Figure 4 Canagliflozin alleviated hepatic steatosis in western diet-fed ApoE-/- mice. (A) Representative image of the liver section stained with HE (×200 magnification); (B) Representative image of the liver section stained with Oil Red O (×200 magnification); (C) Percentage of positive area of liver stained with Oil Red O, n=4; (D) The liver index, n=10; (E) The content of hepatic TG and TC, n=6; (F) The content of hepatic FFA, n=6; (G) The content of hepatic MDA, n=6; (H) ALT and AST level in serum, n=10; ((C, D, E) (TG), and H (ALT), ANOVA, Tamhane’s T2 test; € (TC), (F, G and H) (AST), ANOVA, LSD test)**P<0.01 versus Control group; #, ##P<0.05, P<0.01 versus ApoE-/- group. |
Effects of Canagliflozin on Liver Transcriptomics in Western Diet-Fed ApoE-/- Mice
To further identify the mechanism of action of canagliflozin on hepatic steatosis and injury, we conducted an RNA-Seq analysis of liver tissues from wild-type C57BL/6J mice and from western diet-fed ApoE-/- mice with or without canagliflozin intervention. Results revealed that compared to the Control group, the ApoE-/- group had 1303 up-regulated genes and 569 down-regulated genes, and that relative to the ApoE-/- group, the ApoE-/-+Cana group saw 154 genes up-regulated and 159 gene down-regulated. (Figure 5A). Further analysis showed that 44 of the up-regulated genes in the ApoE-/- group were down-regulated in the ApoE-/-+Cana group, and 43 of the down-regulated genes in ApoE-/- group were also restored by canagliflozin (Figure 5B). Aforementioned results indicated that these 87 DEGs may be related to improvement of hepatic steatosis and atherosclerosis by canagliflozin. KEGG enrichment analysis stated that those DEGs were mainly involved in amino acid metabolism (Figure 5C). To verify the involvement of these DEGs in metabolism, inflammation, and oxidative stress, we performed qRT-PCR and found 11 genes strongly regulated by canagliflozin, consistent with our RNA-Seq results (Figure 5D). Briefly, CPS1, ASS1, ASL, ARG1, MATLA, GLS2, and GOT1 genes are known to involved in amino acid metabolism (of which CPS1, ASS1, ASL and ARG1 are ornithine cycling-related enzymes); SREBP1 is related to lipid metabolism and insulin resistance; and Plin5, Retreg1 and C/EBPβ have effects on inflammation, autophagy and endoplasmic reticulum stress.
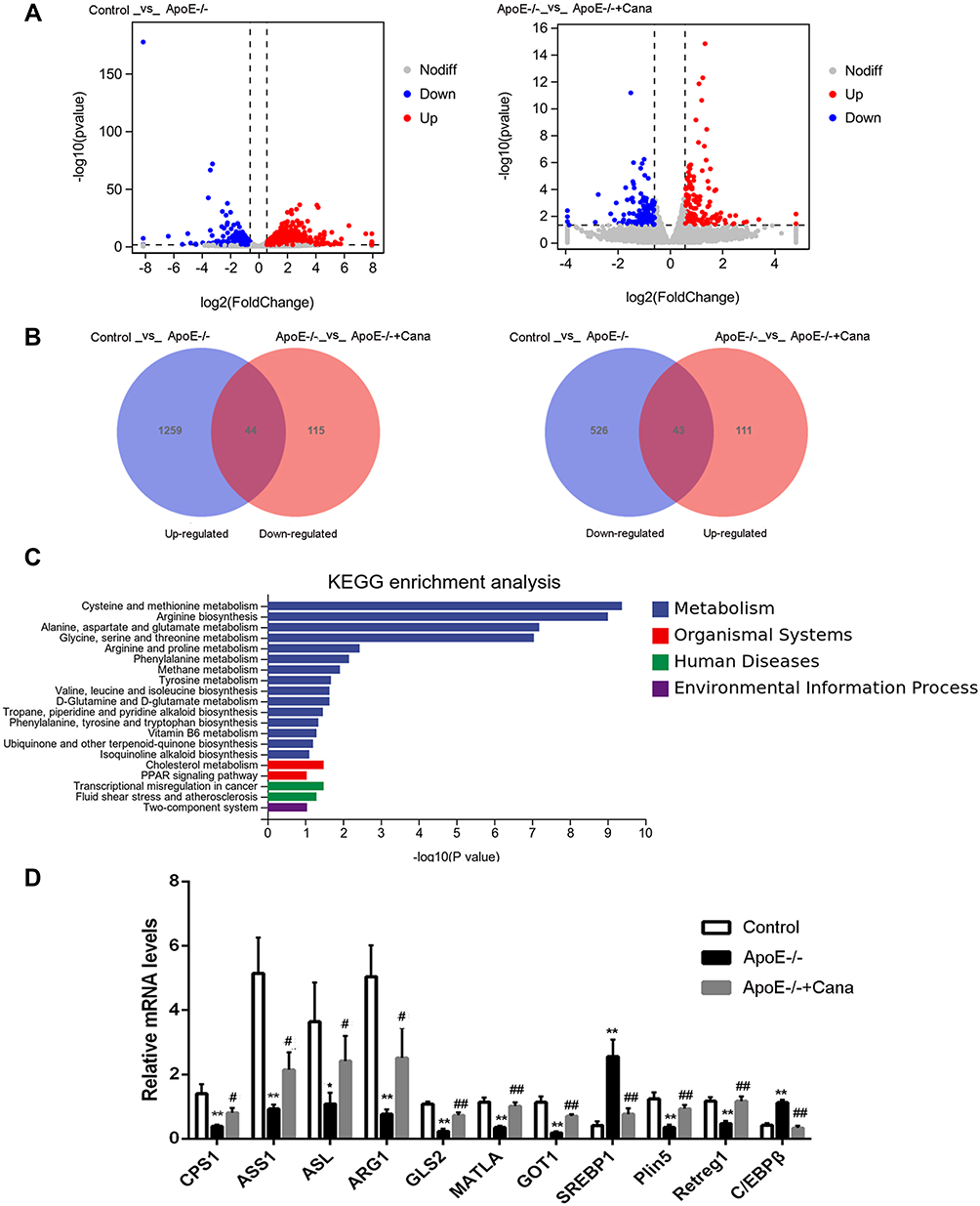 |
Figure 5 Effects of canagliflozin on liver transcriptomics in western diet-fed ApoE-/- mice. (A) The volcano plots of DEGs with fold change > 1.5 and p value < 0.05 in the mice of Control vs ApoE-/- and ApoE-/- vs ApoE-/-+Cana groups; (B) Venn diagram of the DEGs in the mice of Control vs ApoE-/- and ApoE-/- vs ApoE-/-+Cana groups; (C) 87 DEGs was enriched into top 20 pathways in KEGG enrichment analysis; (D) 11 DEGs in RNA-Seq were validated by qRT-PCR, n=5; ((D) (CPS1, ASS1, ASL, ARG1, GOT1 and SREBP1), ANOVA, Tamhane’s T2 test; (D) (CPS1, ASS1, ASL, ARG1, GOT1 and SREBP1), ANOVA, LSD test); *,**P<0.05, P<0.01 versus Control group; #, ##P<0.05, P<0.01 versus ApoE-/- group; DEGs, differentially expressed genes. |
Canagliflozin Increased AMPK Activity via Modulation of ASS1 in Western Diet-Fed ApoE-/- Mice
In terms of amino acid metabolism, we determined that the levels of Asp and Cit in liver of ApoE-/-+Cana group were down-regulated by comparison with those of the ApoE-/- group (P<0.05) (Figure 6A and B). It is well known that Asp and Cit are considered as the substrate of ASS1.15 Western blotting indicated that the expression of ASS1 protein in the liver of ApoE-/- group was also outstandingly down-regulated, while canagliflozin treatment caused the increases of ASS1 level (P<0.01) (Figure 6C and D). Correspondingly, p-AMPK/AMPK protein levels in the ApoE-/- group were lower than that in the Control group (P<0.05). In comparison with that of the ApoE-/- group, the p-AMPK/AMPK level in ApoE-/-+Cana group was markedly increased, indicating that canagliflozin could activate AMPK (P<0.01) (Figure 6C and E). Based on those data and RNA-seq results, we speculated that canagliflozin may increase AMPK activity via modulating ASS1, thereby affecting hepatic lipid metabolism.
 |
Figure 6 Canagliflozin increased AMPK activity via modulating ASS1 in western diet-fed ApoE-/- mice. (A and B) Effects of canagliflozin on the concentration of Asp and Cit in liver tissues; (C-E) Western blotting was utilized to test the levels of ASS1, AMPK and p-AMPK in liver tissues, n=6; (A, C-E), ANOVA, LSD test; (B), ANOVA, Tamhane’s T2 test); *, **P<0.05, P<0.01 versus Control group; #, ##P<0.05, P<0.01 versus ApoE-/- group. |
Canagliflozin Inhibited de Novo Lipogenesis and Cholesterol Synthesis, and Increased Fatty Acid Oxidation in Liver of Western Diet-Fed ApoE-/- Mice via AMPK
AMPK, a mediator for lipid metabolism, is known to phosphorylate and inactivate serial enzymes including SREBP-1 and HMGCR.16 Here, Western blotting showed that SREBP1 and its downstream proteins including FAS and ACC1 in the liver of ApoE-/- group was highly expressed, whereas these levels were notably decreased after treatment with canagliflozin (P<0.01) (Figure 7A-D). The opposite pattern, however, was observed for CPT-1 expression (Figure 7A and E). Besides, administration of canagliflozin was found to inhibit cholesterol synthesis, as evidenced by a reduction of HMGCR level in the ApoE-/-+Cana group as compared to the ApoE-/- group (P<0.01) (Figure 7A and F). Taken together, our findings that canagliflozin inhibited de novo lipogenesis, fatty acid oxidation, and cholesterol synthesis in the liver of the ApoE-/-+Cana group mice suggested that canagliflozin can regulate hepatic lipid metabolism via the activation of AMPK.
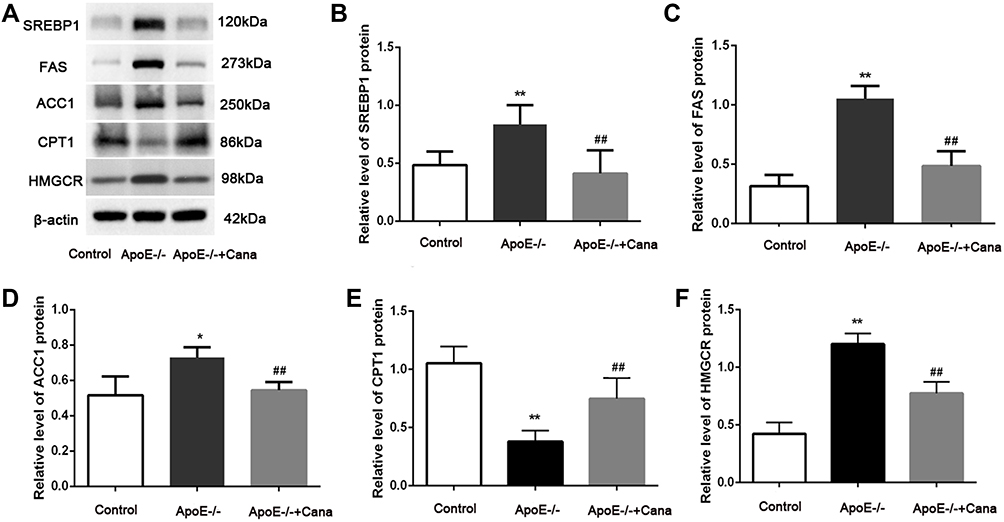 |
Figure 7 Canagliflozin restrained de novo lipogenesis and cholesterol synthesis, and increased fatty acid oxidation in liver of western diet-fed ApoE-/- mice via AMPK. (A-F) Western blotting assay displayed that the SREBP1, FAS, ACC1, CPT1 and HMGCR in liver tissues, n=5; (ANOVA, LSD test); *,** P<0.05, P<0.01 versus Control group; ##P<0.01 versus ApoE-/- group. |
Canagliflozin Induced Autophagy in Liver of Western Diet-Fed ApoE-/- Mice via AMPK
To our knowledge, AMPK activity plays a crucial role in the modulation of autophagy through interactions with ULK1 and mTOR.17 Accordingly, we investigated the autophagic alterations in the western diet-fed ApoE-/- mice with or without canagliflozin treatment. TEM images of hepatocytes showed that canagliflozin intervention attenuated the increase in the number of lipid droplets, as observed in the ApoE-/- group, but increased the count of autophagosomes (Figure 8A). Additionally, canagliflozin apparently down-regulated the levels of p-mTOR/mTOR, p-ULK1/ULK1 and P62, but up-regulated the levels of Beclin 1 and LC3 II/LC3I in the liver of ApoE-/- mice fed with western diet (P<0.01) (Figure 8B and C). Consistent with the results of RNA-seq and mRNA analyses, the expression of autophagy-related proteins including Plin5 and Retreg1 in the ApoE-/- group was decreased, whereas that of C/EBPβ was increased. After administration of canagliflozin, the levels of Plin5, Retreg1 and C/EBPβ were restored in the ApoE-/- group (Figure 8D and E). These results suggest that canagliflozin might promote hepatic autophagy via activating AMPK.
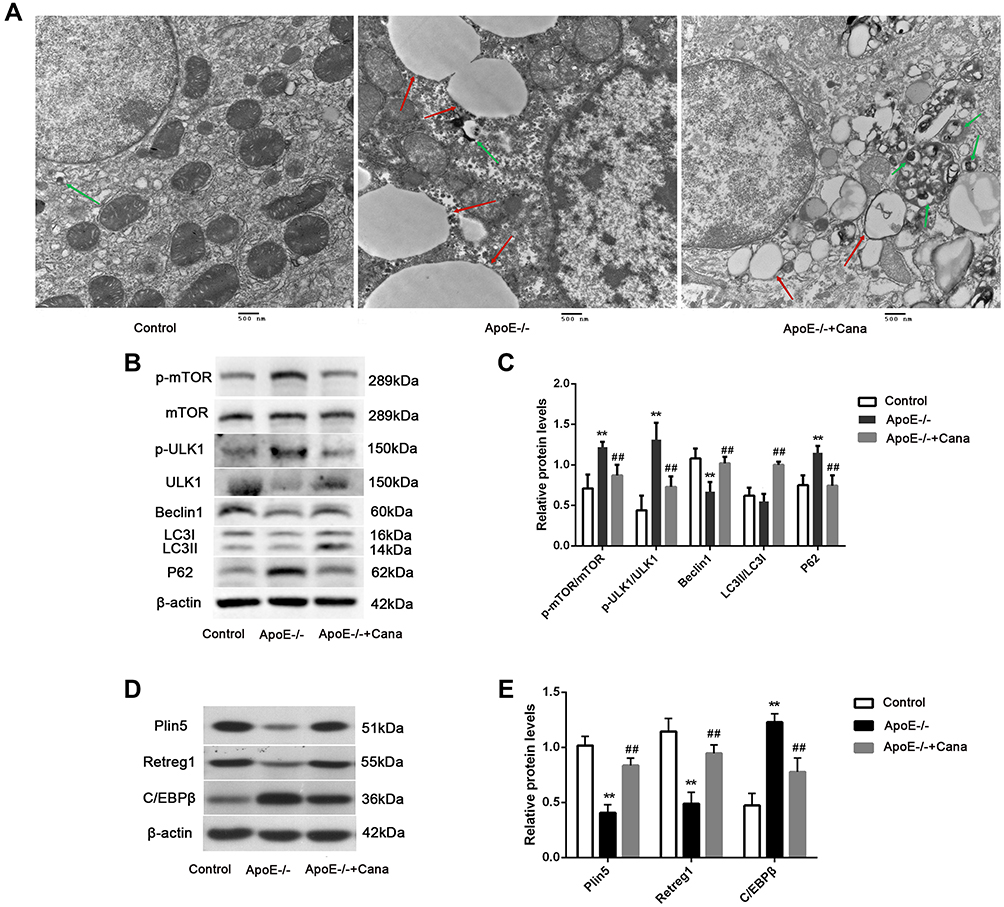 |
Figure 8 Canagliflozin induced autophagy in liver of western diet-fed ApoE-/- mice via AMPK. (A) TEM images showed the lipid droplets (red arrows) and autophagosome (green arrows) (Scar bar=500 nm); (B and C) Western blotting displayed that the levels of p-mTOR/mTOR, p-ULK1/ULK1, Beclin1, LC3II/LC3I and P62 in liver tissues; (D and E) Quantitative analysis of Plin5, Retreg1 and C/EBPβ protein level were examined by Western blotting, n=5; (ANOVA, LSD test); **P<0.01 versus Control group; ##P<0.01 versus ApoE-/- group. |
Canagliflozin Improved the Aortic Endothelial Function in Western Diet-Fed ApoE-/- Mice
NO, an endothelium-derived relaxing factor, serves vital functions in atherosclerosis development.18 In the current study, we found that several key components of NO production including ASS1, ASL and eNOS were lowly expressed in the ApoE-/- group compared with the Control group (P<0.01). However, the mRNA expressions of ASS1, ASL and eNOS were significantly increased following treatment with canagliflozin (P<0.05) (Figure 9). Taken together, the evidence suggests that, canagliflozin can effectively improve the aortic endothelial function, thereby suppressing atherosclerosis development.
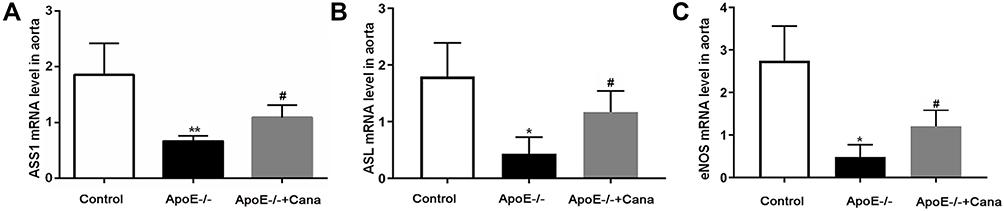 |
Figure 9 Canagliflozin improved the aortic endothelial function in western diet-fed ApoE-/- mice. The mRNA levels of ASS1 (A), ASL (B) and eNOS (C) in aorta tissues were determined by qRT-PCR, n=5; ((A and B), ANOVA, Tamhane’s T2 test; (B), ANOVA, LSD test). *,** P<0.05, P<0.01 versus Control group; #P<0.05 versus ApoE-/- group. |
Discussion
Atherosclerosis and NAFLD are common diseases worldwide and are frequent causes of mortality in developed countries.19 Canagliflozin, approved by FDA in 2013, is used to improve glycemic control in patients with type 2 diabetes.20,21 In two preclinical studies, the beneficial effects of canagliflozin on lipid metabolism and atherosclerosis have also been reported.11,22 Inspired by these developments, we undertook this study to understand physiological and cellular mechanisms underlying these effects. Here, we proposed that canagliflozin could elevate ASS1 activity, enhance ornithine circulation, and promote AMPK phosphorylation, thereby improving lipid metabolism, enhancing autophagy, and reducing inflammation. Importantly, we suggested that the prevention of atherosclerotic lesions by canagliflozin was possibly attributed to improvement in endothelial function, as evidenced by increased levels of ASS1, ASL, and eNOS.
Western diet-induced atherosclerosis, hyperlipidemia, and NAFLD in ApoE-/- mice are widely utilized and well-recognized models.23 To our knowledge, impaired lipid clearance due to abnormal lipid metabolism pathways is possibly implicated in the development of hepatic steatosis and atherosclerosis.24 As presented in this study, the levels of TC, TG and LDL-C in the ApoE-/- mice fed with western diet were obviously increased, whereas HDL-C decreased. However, canagliflozin treatment reversed the TC, TG and LDL-C levels. This observation was in line with changes in lipid profiles characteristic of anti-atherosclerotic effects.25 On the other hand, previous studies have shown that insulin resistance and inflammation are two common risk factors for atherosclerosis and NAFLD.4,26 Fortunately, we also observed that canagliflozin alleviated the insulin resistance and reduced the expression of TNF-α, IL-6, IL-1β and MCP-1, which was consistent with the effects of SGILT-2 inhibitors.27,28 Collectively, these findings indicated that canagliflozin may alleviate the progression of hepatic steatosis and atherosclerosis by modulating lipid metabolism and inflammatory response.
In order to further explore the mechanism of canagliflozin in the prevention and treatment of atherosclerosis and NAFLD, we conducted a liver transcriptome analysis. In our study, canagliflozin was shown to prevent NAFLD in the ApoE-/-+Cana group by restoring the expression of metabolism-related genes including CPS1, ASS1, ASL, ARG1, MATLA, GOT1, GLS2, SREBP1, Plin5, Retreg1 and C/EBPβ, many of which are involved in the ornithine cycle and have been shown to be inhibited in mice on a high-fat diet.29 This has led us to speculate that the protective effect of canagliflozin is mediated partly through the ornithine cycle. Besides, we also observed that the levels of Asp and Cit in the liver of the ApoE-/- group were up-regulated by comparison with the Control group. These two amino acids, Asp and Cit, as the substrate of ASS1, are involved in the synthesis of Argininosuccinate. On the relationship between ASS1 and type I citrullinemia, previous studies have established that ASS1-deficiency leads to high levels of Cit in clinical settings.30 It has been found that ASS1, a major site of AMP generation in the liver, increases AMP/ATP ratio and activates AMPK by phosphorylating threonine 172 on its subunit.31 Tellingly, in our study, both upregulation of ASS1 expression and acceleration of the phosphorylation of AMPK were observed in the ApoE-/-+Cana group. Furthermore, AMPK can trigger metabolic switches that reduce the activity of anabolic pathways and enhance catabolic processes.32 Based on these findings, we believe that canagliflozin may increase AMPK activity via modulation of ASS1, thereby affecting hepatic lipid metabolism and atherosclerosis.
SREBP is a family of transcriptional activators required for lipid homeostasis, which is divided into two subtypes of SREBP1 and SREBP2.33 The molecule highlighted by our results is SREBP1, a type of transcriptional activators primarily involved in the synthesis of fatty acid and TG lipogenic organs, essential for lipid homeostasis.34 We found that levels of SREPB1 expression were elevated in the ApoE-/- group but reduced in the ApoE-/-+Cana group, compared to those in the Control group. Similar differences in expression level were also observed in fatty acid synthesis-related proteins including ACC1 and FAS as well as in TG level. Incidentally, ACC1 inactivation in the ApoE-/-+Cana group could probably explain the observed increases in their CPT1 level, for CPT1, a fatty acid β oxidation rate-limiting enzyme, is allosterically regulated by malonyl-CoA synthesized by ACC1.35 Importantly, Li et al36 have found that AMPK suppresses SREBP1 activity, thereby reducing lipogenesis and lipid accumulation. Another study has also determined that canagliflozin protects against the lipid synthesis and modulates SRERP1 expression.22 Thus, our findings suggested that canagliflozin alleviated hepatic steatosis in our western diet-fed ApoE-/- mice by restraining de novo lipogenesis and increasing fatty acid oxidation through an ASS1-AMPK-SREBP1 pathway.
AMPK has been shown to influence heavily the regulation of autophagy and cholesterol metabolism.37 In the current study, we found that HMGCR expression was increased following intervention with canagliflozin. Tang et al38 have concluded that the regulation of HMGCR via AMPK is key to cholesterol synthesis, and in the present study we found that HMGCR expression increased following intervention with canagliflozin. This result supports our hypothesis that canagliflozin regulates cholesterol metabolism via activation of AMPK. Impaired hepatic autophagy flux is known to be closely linked to the development of hepatic steatosis.39 Previous evidence has supported the involvement of AMPK in autophagy through interactions with ULK1 and mTOR.17 Our results showed that the levels of p-mTOR/mTOR and p-ULK1/ULK1 in the liver of ApoE-/- mice decreased after the treatment with canagliflozin. Incidentally, the induction of autophagy in the ApoE-/-Cana group was confirmed by increases in Beclin 1 level and decreases in p62 level. Furthermore, the differential genes identified by our RNA-seq results (namely Plin5 and Retreg1, and C/EBPβ) have been reported to regulate autophagy.40,41 Similarly, previous studies have pointed out that SGLT2 inhibitors can enhance hepatic autophagy via AMPK/mTOR pathway.42,43 Taken together, our evidence suggested that canagliflozin may alleviate the hepatic steatosis by restoring cholesterol metabolism and autophagy via ASS1/AMPK pathway.
It is worth noting that the influence of NAFLD on risks of atherosclerosis has been confirmed, and the association between the two has been established.44 On the one hand, increased lipolysis, VLDL secretion, small dense LDL fractions, and reduction in HDL fractions are all known to substantially contribute to the atherogenic risk of NAFLD.45 On the other hand, the secretion of inflammatory markers (such as IL-6, TNF-α, Fetuin-A, C reactive protein, and fibrinogen) induced by oxidative stress and fat accumulated in the liver. More specifically, Fetuin-A is a protein secreted by the liver through which lipids induce insulin resistance.46 Increased C reactive protein promotes inflammation and accelerates atherosclerosis by increasing the expression of plasminogen activator inhibitor-1 and adhesion molecules in endothelial cells, inhibiting nitric oxide formation, and increasing LDL uptake into macrophages.47 At the onset and in the development of atherosclerosis, endothelial dysfunction is an early event, mainly characterized as a decrease of NO, an endodermal relaxation factor.18 Here, we found that core components for NO production including ASS1, ASL and eNOS were lowly expressed in the ApoE-/- group compared with the Control group. Following treatment with canagliflozin, the mRNA expressions of ASS1, ASL and eNOS were significantly increased. Additionally, our results showed that canagliflozin reduced the aortic proatherogenic plaque stability-associated markers, including F4/80, MCP-1, and MMP-9, which was consistent with previous studies.11,48 These results suggested that canagliflozin can effectively improve the aortic endothelial function, and thereby suppress atherosclerosis development. Based on existing data, we have demonstrated that canagliflozin may reduce hepatic steatosis by modulating the ASS1/AMPK pathway. Given the potential link between NAFLD and ASS1, and the effect of ASS1 on eNOS, we speculated that canagliflozin may also inhibit atherosclerosis through the ASS1/AMPK pathway. Of course, more precise conclusions need to be confirmed by AMPK expression in aorta and sequencing data.
Conclusion
Our results suggested that canagliflozin may attenuate hepatic steatosis by improving lipid metabolism, enhancing autophagy, and reducing inflammatory response through ASS1/AMPK pathway. Besides, we have found that canagliflozin further effectively improves the aortic endothelial function, thereby suppressing atherosclerosis development, and we have inferred that the protective effect was likely mediated by the ASS1/AMPK pathway. This study has some limitations. First, this study was that it was designed using ApoE-/- mouse in vivo model. Further investigations are needed to investigate the changes of the ASS1/AMPK pathway in vitro studies. Secondly, this study lacked a group of C57 mice on a Western diet. Thirdly, there were partial defects in the aorta tissue samples.
Abbreviations
TG, triglyceride; TC, total cholesterol; LDL-C, low-density lipoprotein cholesterin; FFA, free fatty acid; MDA, malondialdehyde; TNF-α, tumor necrosis factor-α; IL-6, interleukin-6; IL-1β, interleukin-1 beta; MCP-1, monocyte chemotactic protein 1; CPS1, carbamoyl-phosphate synthase 1; ASS1, argininosuccinate synthase 1; ASL, argininosuccinate lyaseL; ARG1, arginase 1; GLS2, glutaminase 2, GOT1, glutamic oxaloacetic transaminase 1; SREBP1, sterol-regulatory element-binding protein 1; Plin5, perilipin 5; Retreg1, reticulophagy regulator 1; AMPK, adenosine 5’-monophosphate-activated protein kinase; FAS, fatty acid synthase; ACC1, acetyl-CoA carboxylase 1; HMGCR, hydroxy-3-methylglutaryl-CoA reductase; mTOR, mammalian target of rapamycin; ULK1, unc-51 like autophagy activating kinase 1; CPT1, carnitine palmitoyltransferase 1; eNOS, endothelial nitric oxide synthase.
Data Sharing Statement
The data used to support the findings of this study are available from the corresponding author upon request.
Funding
The study was supported by The 2019 Hebei Science and Technology Project (No. 19277787D), The 2019 Hebei Innovation Capability Promotion Project (No. 199776249D), the 2020 medical science research project of Hebei Provincial Health Commission (No. 20200734) and the 2022 government funded the provincial medical Talents Project.
Disclosure
All authors declare that they have no conflict of interest.
References
1. Marchio P, Guerra-Ojeda S, Vila JM, Aldasoro M, Victor VM, Mauricio MD. targeting early atherosclerosis: a focus on oxidative stress and inflammation. Oxidative Med Cell Longevity. 2019;2019:8563845. doi:10.1155/2019/8563845
2. Barquera S, Pedroza-Tobías A, Medina C, et al. Global overview of the epidemiology of atherosclerotic cardiovascular disease. Arch Med Res. 2015;46(5):328–338. doi:10.1016/j.arcmed.2015.06.006
3. Herrington W, Lacey B, Sherliker P, Armitage J, Lewington S. Epidemiology of atherosclerosis and the potential to reduce the global burden of atherothrombotic disease. Circ Res. 2016;118(4):535–546. doi:10.1161/CIRCRESAHA.115.307611
4. Tarantino G, Caputi A. JNKs, insulin resistance and inflammation: a possible link between NAFLD and coronary artery disease. World j Gastroenterol. 2011;17(33):3785–3794. doi:10.3748/wjg.v17.i33.3785
5. Cohen JC, Horton JD, Hobbs HH. Human fatty liver disease: old questions and new insights. Science. 2011;332(6037):1519–1523. doi:10.1126/science.1204265
6. Stols-Gonçalves D, Hovingh GK, Nieuwdorp M, Holleboom AG. NAFLD and atherosclerosis: two sides of the same dysmetabolic coin? Trends Endocrinol Metab. 2019;30(12):891–902. doi:10.1016/j.tem.2019.08.008
7. Zhang L, She ZG, Li H, Zhang XJ. Non-alcoholic fatty liver disease: a metabolic burden promoting atherosclerosis. Clin Sci. 2020;134(13):1775–1799. doi:10.1042/CS20200446
8. Kalra S. Sodium glucose co-transporter-2 (SGLT2) inhibitors: a review of their basic and clinical pharmacology. Diabetes Therapy. 2014;5(2):355–366. doi:10.1007/s13300-014-0089-4
9. Zhang N, Feng B, Ma X, Sun K, Xu G, Zhou Y. Dapagliflozin improves left ventricular remodeling and aorta sympathetic tone in a pig model of heart failure with preserved ejection fraction. Cardiovascular Diabetology. 2019;18(1):107. doi:10.1186/s12933-019-0914-1
10. Vallon V, Verma S. Effects of SGLT2 inhibitors on kidney and cardiovascular function. Annu Rev Physiol. 2021;83(1):503–528. doi:10.1146/annurev-physiol-031620-095920
11. Nasiriansari Ν, Dimitriadis GK, Agrogiannis G, et al. Canagliflozin attenuates the progression of atherosclerosis and inflammation process in APOE knockout mice. Cardiovasc Diabetol. 2018;17(1):106. doi:10.1186/s12933-018-0749-1
12. Rahadian A, Fukuda D, Salim HM, et al. Canagliflozin prevents diabetes-induced vascular dysfunction in ApoE-deficient mice. J Atheroscler Thromb. 2020;27(11):1141–1151. doi:10.5551/jat.52100
13. Zhao Y, Li Y, Liu Q, et al. Canagliflozin facilitates reverse cholesterol transport through activation of AMPK/ABC transporter pathway. Drug Des Devel Ther. 2021;15:2117–2128. doi:10.2147/DDDT.S306367
14. Shiba K, Tsuchiya K, Komiya C, et al. Canagliflozin, an SGLT2 inhibitor, attenuates the development of hepatocellular carcinoma in a mouse model of human NASH. Scientific Reports. 2018;8(1):2362. doi:10.1038/s41598-018-19658-7
15. Ratner S. Enzymes of arginine and urea synthesis. Adv Enzymol Relat Areas Mol Biol. 1973;39:1–90. doi:10.1002/9780470122846.ch1
16. Garcia D, Mihaylova MM, Shaw RJ. AMPK: central Regulator of glucose and lipid metabolism and target of type 2 diabetes therapeutics. Biol Pathobiol. 2020;1:472–484.
17. Kim J, Kundu M, Viollet B, Guan KL. AMPK and mTOR regulate autophagy through direct phosphorylation of Ulk1. Nat Cell Biol. 2011;13(2):132–141. doi:10.1038/ncb2152
18. Bauer V, Sotníková R. Nitric oxide--The endothelium-derived relaxing factor and its role in endothelial functions. Gen Physiol Biophys. 2010;29(4):319–340. doi:10.4149/gpb_2010_04_319
19. Lloyd-Jones DM. Cardiovascular risk prediction: basic concepts, current status, and future directions. Circulation. 2010;121(15):1768–1777. doi:10.1161/CIRCULATIONAHA.109.849166
20. Lamos EM, Younk LM, Davis SN. Canagliflozin, an inhibitor of sodium–glucose cotransporter 2, for the treatment of type 2 diabetes mellitus. Expert Opin Drug Metab Toxicol. 2013;9(6):763–775. doi:10.1517/17425255.2013.791282
21. Stenlöf K, Cefalu WT, Kim KA, et al. Efficacy and safety of canagliflozin monotherapy in subjects with type 2 diabetes mellitus inadequately controlled with diet and exercise. Diabetes Obes Metab. 2013;15(4):372–382. doi:10.1111/dom.12054
22. Day EA, Ford RJ, Lu JH, et al. The SGLT2 inhibitor canagliflozin suppresses lipid synthesis and interleukin-1 beta in ApoE deficient mice. Biochem J. 2020;477(12):2347–2361. doi:10.1042/BCJ20200278
23. Kolovou G, Anagnostopoulou K, Mikhailidis DP, Cokkinos DV. Apolipoprotein E knockout models. Curr Pharm Des. 2008;14(4):338–351. doi:10.2174/138161208783497769
24. Liu K, Czaja MJ. Regulation of lipid stores and metabolism by lipophagy. Cell Death Differ. 2013;20(1):3–11. doi:10.1038/cdd.2012.63
25. Jia Q, Cao H, Shen D, et al. Quercetin protects against atherosclerosis by regulating the expression of PCSK9, CD36, PPARγ, LXRα and ABCA1. Int J Mol Med. 2019;44(3):893–902. doi:10.3892/ijmm.2019.4263
26. Khan R, Spagnoli V, Tardif JC, L’Allier PL. Novel anti-inflammatory therapies for the treatment of atherosclerosis. Atherosclerosis. 2015;240(2):497–509. doi:10.1016/j.atherosclerosis.2015.04.783
27. Raj H, Durgia H, Palui R, et al. SGLT-2 inhibitors in non-alcoholic fatty liver disease patients with type 2 diabetes mellitus: a systematic review. World J Diabetes. 2019;10(2):114–132. doi:10.4239/wjd.v10.i2.114
28. Liu Y, Wu M, Xu B, Kang L. Empagliflozin alleviates atherosclerosis progression by inhibiting inflammation and sympathetic activity in a normoglycemic mouse model. J Inflamm Res. 2021;14:2277–2287. doi:10.2147/JIR.S309427
29. Liao CC, Lin YL, Kuo CF. Effect of high-fat diet on hepatic proteomics of hamsters. J Agric Food Chem. 2015;63(6):1869–1881. doi:10.1021/jf506118j
30. Häberle J, Pauli S, Schmidt E, Schulze-Eilfing B, Berning C, Koch HG. Mild citrullinemia in Caucasians is an allelic variant of argininosuccinate synthetase deficiency (citrullinemia type 1). Mol Genet Metab. 2003;80(3):302–306. doi:10.1016/j.ymgme.2003.08.002
31. Madiraju AK, Alves T, Zhao X, et al. Argininosuccinate synthetase regulates hepatic AMPK linking protein catabolism and ureagenesis to hepatic lipid metabolism. Proc Natl Acad Sci U S A. 2016;113(24):E3423–3430. doi:10.1073/pnas.1606022113
32. Ruderman NB, Carling D, Prentki M, Cacicedo JM. AMPK, insulin resistance, and the metabolic syndrome. J Clin Invest. 2013;123(7):2764–2772. doi:10.1172/JCI67227
33. Eberlé D, Hegarty B, Bossard P, Ferré P, Foufelle F. SREBP transcription factors: master regulators of lipid homeostasis. Biochimie. 2004;86(11):839–848. doi:10.1016/j.biochi.2004.09.018
34. Brown MS, Goldstein JL. The SREBP pathway: regulation of cholesterol metabolism by proteolysis of a membrane-bound transcription factor. Cell. 1997;89(3):331–340. doi:10.1016/S0092-8674(00)80213-5
35. Savage DB, Choi CS, Samuel VT, et al. Reversal of diet-induced hepatic steatosis and hepatic insulin resistance by antisense oligonucleotide inhibitors of acetyl-CoA carboxylases 1 and 2. J Clin Invest. 2006;116(3):817–824. doi:10.1172/JCI27300
36. Li Y, Xu S, Mihaylova MM, et al. AMPK phosphorylates and inhibits SREBP activity to attenuate hepatic steatosis and atherosclerosis in diet-induced insulin-resistant mice. Cell Metab. 2011;13(4):376–388. doi:10.1016/j.cmet.2011.03.009
37. Li Y, Chen Y. AMPK and Autophagy. Adv Exp Med Biol. 2019;1206:85–108.
38. Tang H, Yu R, Liu S, Huwatibieke B, Li Z, Zhang W. Irisin inhibits hepatic cholesterol synthesis via AMPK-SREBP2 signaling. EBioMedicine. 2016;6:139–148. doi:10.1016/j.ebiom.2016.02.041
39. Mao Y, Yu F, Wang J, Guo C, Fan X. Autophagy: a new target for nonalcoholic fatty liver disease therapy. Hepat Med. 2016;8:27–37. doi:10.2147/HMER.S98120
40. Zhang E, Cui W, Lopresti M, et al. Hepatic PLIN5 signals via SIRT1 to promote autophagy and prevent inflammation during fasting. J Lipid Res. 2020;61(3):338–350. doi:10.1194/jlr.RA119000336
41. Mookherjee D, Das S, Mukherjee R, Bera M. RETREG1/FAM134B mediated autophagosomal degradation of AMFR/GP78 and OPA1 -a dual organellar turnover mechanism. Autophagy. 2021;17(7):1729–1752. doi:10.1080/15548627.2020.1783118
42. Meng Z, Liu X, Li T, et al. The SGLT2 inhibitor empagliflozin negatively regulates IL-17/IL-23 axis-mediated inflammatory responses in T2DM with NAFLD via the AMPK/mTOR/autophagy pathway. Int Immunopharmacol. 2021;94:107492. doi:10.1016/j.intimp.2021.107492
43. Yu T, Li G, Wang C, et al. MIR210HG regulates glycolysis, cell proliferation, and metastasis of pancreatic cancer cells through miR-125b-5p/HK2/PKM2 axis. RNA Biol. 2021;18(12):2513–2530. doi:10.1080/15476286.2021.1930755
44. Wójcik-Cichy K, Koślińska-Berkan E, Piekarska A. The influence of NAFLD on the risk of atherosclerosis and cardiovascular diseases. Clin exp hepatol. 2018;4(1):1–6. doi:10.5114/ceh.2018.73155
45. Gaggini M, Morelli M, Buzzigoli E, DeFronzo RA, Bugianesi E, Gastaldelli A. Non-alcoholic fatty liver disease (NAFLD) and its connection with insulin resistance, dyslipidemia, atherosclerosis and coronary heart disease. Nutrients. 2013;5(5):1544–1560. doi:10.3390/nu5051544
46. Pal D, Dasgupta S, Kundu R, et al. Fetuin-A acts as an endogenous ligand of TLR4 to promote lipid-induced insulin resistance. Nat Med. 2012;18(8):1279–1285. doi:10.1038/nm.2851
47. Chapman MJ, Ginsberg HN, Amarenco P, et al. Triglyceride-rich lipoproteins and high-density lipoprotein cholesterol in patients at high risk of cardiovascular disease: evidence and guidance for management. Eur Heart J. 2011;32(11):1345–1361. doi:10.1093/eurheartj/ehr112
48. Nakatsu Y, Kokubo H, Bumdelger B, et al. The SGLT2 inhibitor luseogliflozin rapidly normalizes aortic mRNA levels of inflammation-related but not lipid-metabolism-related genes and suppresses atherosclerosis in diabetic ApoE KO mice. Int J Mol Sci. 2017;18(8):1704. doi:10.3390/ijms18081704
 © 2022 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2022 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.