")
Back to Journals » Clinical Epidemiology » Volume 16
Can Lipid-Lowering Drugs Reduce the Risk of Cholelithiasis? A Mendelian Randomization Study
Authors Dong H, Chen R, Xu F, Cheng F
Received 10 September 2023
Accepted for publication 9 February 2024
Published 22 February 2024 Volume 2024:16 Pages 131—141
DOI https://doi.org/10.2147/CLEP.S439642
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 6
Editor who approved publication: Professor Henrik Sørensen
Hao Dong,1,* Rong Chen,2,* Fang Xu,3,* Fang Cheng4
1Department of Gastroenterology and Hepatology, The First Medical Center of Chinese PLA General Hospital, Beijing, 100853, People’s Republic of China; 2Department of Rehabilitation Medicine, the First Affiliated Hospital of Sun Yat-sen University, Guangzhou, 510080, People’s Republic of China; 3Clinical Medical Laboratory Center, Taizhou People’s Hospital, Taizhou, Jiangsu, 225300, People’s Republic of China; 4Department of Gastroenterology, Wuhan Jinyintan Hospital, Tongji Medical College of Huazhong University of Science and Technology, Wuhan, 430023, People’s Republic of China
*These authors contributed equally to this work
Correspondence: Fang Cheng, Department of Gastroenterology, Wuhan Jinyintan Hospital, Tongji Medical College of Huazhong University of Science and Technology, No. 1 Yintan Road, Dongxihu District, Wuhan, 430023, Hubei Province, People’s Republic of China, Email [email protected]
Background and Aims: Cholelithiasis etiology intricately involves lipid metabolism. We sought to investigate the plausible causal link between genetically proxied lipid-lowering medications—specifically HMGCR inhibitors, PCSK9 inhibitors, and NPC1L1 inhibitors—and cholelithiasis risk.
Methods: Our study utilized two genetic instruments for exposure to lipid-lowering drugs. These instruments encompassed genetic variants linked to low-density lipoprotein (LDL) cholesterol within or in proximity to drug target genes, along with loci governing gene expression traits of these targets. Effect estimates were derived through Inverse-variance-weighted MR (IVW-MR) and summary-data-based MR (SMR) methods.
Results: Higher HMGCR-mediated LDL cholesterol levels (IVW-MR, OR = 2.15, 95% CI = 1.58– 2.94; P = 0.000) and increased HMGCR expression (SMR, OR = 1.19, 95% CI = 1.04– 1.37; P = 0.014) are linked to elevated cholelithiasis risk, suggesting potential benefits of HMGCR inhibition. In contrast, higher PCSK9-mediated LDL cholesterol levels (IVW-MR, OR = 0.72, 95% CI = 0.56– 0.94; P = 0.015) and increased PCSK9 expression (SMR, OR = 0.90, 95% CI = 0.82– 0.99; P = 0.035) both correlate with lower cholelithiasis risk, indicating that PCSK9 inhibition may elevate this risk. Nevertheless, no substantial link emerged between NPC1L1-mediated LDL cholesterol or NPC1L1 expression and cholelithiasis in both IVW-MR and SMR analyses.
Conclusion: This MR investigation affirms the causal link between the utilization of HMGCR inhibitors and a diminished risk of cholelithiasis. Additionally, it indicates a causal link between PCSK9 inhibitors use and increased cholelithiasis risk. However, no significant correlation was found between NPC1L1 inhibitors use and cholelithiasis risk.
Keywords: cholelithiasis, lipid-lowering drugs, Mendelian randomization analysis, HMGCR inhibitors, PCSK9 inhibitors, NPC1L1 inhibitors
Introduction
Cholelithiasis represents a significant global public health concern, affecting an estimated 10–20% of adults worldwide.1,2 Over 20% of individuals with cholelithiasis encounter symptoms or complications, including potential life-threatening conditions like gallstone-related pancreatitis, acute cholecystitis, and acute cholangitis.1 These stones can be categorized into two main types based on composition: pigment stones and cholesterol stones.3 In Western nations, approximately 80% to 90% of gallstones extracted through surgery consist of cholesterol stones.4 The formation of cholesterol stones is often linked to cholesterol supersaturation in the bile.4 Previous research has explored the relationship between plasma cholesterol levels and gallstones, yielding varying results. Some studies suggest a positive correlation,5–7 while others propose a negative correlation,8,9 and there are studies indicating no significant correlation.10
Lipid-lowering medications, including 3-hydroxy-3-methylglutaryl–CoA reductase (HMGCR) inhibitors commonly referred to as statins, Niemann-PickC1–like1 (NPC1L1) inhibitors such as ezetimibe, and proprotein convertase subtilisin/kexin type 9 (PCSK9) inhibitors like alirocumab, are crucial for primary and secondary prevention of cardiovascular diseases and are widely used worldwide.11–13 These medications not only lower serum low-density lipoprotein cholesterol concentrations but also affect cholesterol metabolism in the liver and/or its secretion into the bile.11–14
In a recent Mendelian randomization (MR) study, a link emerged between reduced serum total cholesterol levels and heightened cholelithiasis risk.8 This finding has raised concerns regarding the potential risk of cholelithiasis among individuals taking lipid-lowering drugs, considering the large population using these medications. While numerous prior observational studies have indicated a potential connection between the utilization of statins and a lowered cholelithiasis risk, these studies are susceptible to residual confounding and the possibility of reverse causation.15–17 For example, individuals taking statins often exhibit various characteristics associated with cardiovascular disease.17,18 Additionally, traditional observational studies investigating the impact of statins on cholelithiasis can be influenced by factors such as dosage and duration of medication use.16 Limited research exists on the effects of NPC1L1 inhibitors on gallstones. Animal experiments suggest that ezetimibe may prevent cholesterol gallstone formation by inhibiting intestinal cholesterol absorption and promoting gallstone dissolution.19 However, a single-sample MR study simulating ezetimibe effects through genetic variations in NPC1L1 indicates an elevated likelihood of symptomatic gallstones.20 Currently, the correlation between PCSK9 inhibitors and cholelithiasis risk lacks research evidence.
MR studies employ single-nucleotide polymorphisms (SNPs) as instruments to investigate causal effects between exposures and outcomes.21 By mimicking random assignment in controlled trials, MR studies minimize common confounding biases observed in observational studies, given that genetic variants are arbitrarily assigned during conception. Furthermore, MR studies can overcome issues of reverse causality since the genetic variants are determined before disease onset.22 By analyzing the natural variations in the gene responsible for encoding the protein drug target, we can gain valuable insights into the potential clinical effects associated with these drugs.23 The primary aim of this research is to employ a two-sample MR strategy to explore the possible causal link between cholelithiasis risk and three genetically proxied lipid-lowering medications: HMGCR inhibitors, NPC1L1 inhibitors, and PCSK9 inhibitors.
Methods
Study Design
This two-sample MR investigation relies on openly accessible summary-level data sourced from genome-wide association studies (GWASs) and studies involving expression quantitative trait loci (eQTLs) (Supplementary Table S1). Ethical approval has been granted by the relevant institutional review boards for all of these studies, with participants giving informed consent. In our MR study, we utilized two different sources of genetic instruments as proxies for lipid-lowering drugs (Figure 1).
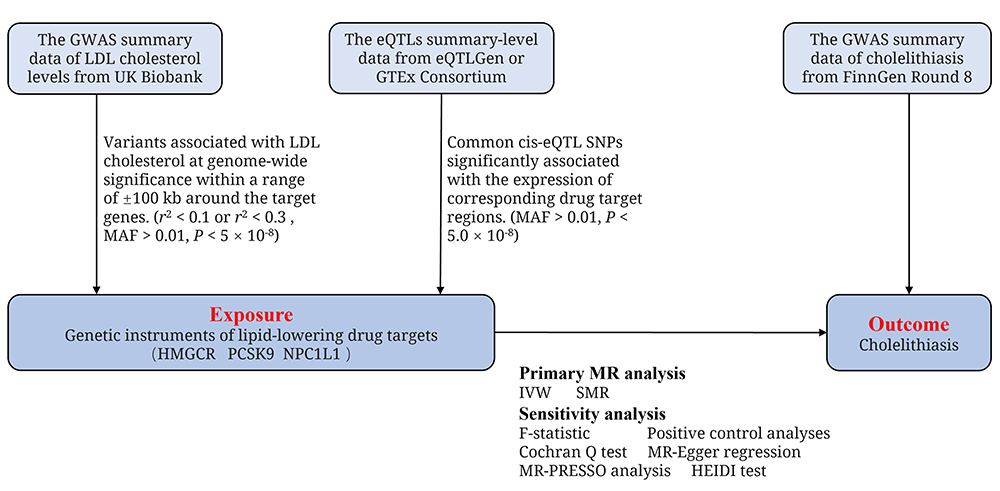 |
Figure 1 Two-Sample MR study design for the association between lipid-lowering drugs and cholelithiasis. Abbreviations: GWAS, genome-wide association study; eQTLs, expression quantitative trait loci; LDL, low-density lipoprotein; HMGCR, 3-hydroxy-3-methylglutaryl CoA reductase; PCSK9, proprotein convertase subtilisin/kexin type 9; NPC1L1, Niemann-Pick C1–like 1; IVW, multiplicative random effects inverse variance weighted method; SMR, summary-data-based mendelian randomization; HEIDI, heterogeneity in dependent instruments; MR-PRESSO, Mendelian Randomization Pleiotropy RESidual Sum and Outlier; MAF, minor allele frequency. |
Genetic GWAS Data of Cholelithiasis
The GWAS summary statistics employed in the MR analysis for cholelithiasis were acquired from FinnGen Round 8, accessible at https://r8.finngen.fi/. The cholelithiasis GWAS dataset comprised 32,894 cases and 301,383 controls. Cholelithiasis in FinnGen R8 was defined as “The presence of calculi in the gallbladder” and corresponded to ICD codes: ICD-10: K80, ICD-9: 574, ICD-8: 574. Additional details can be found at https://risteys.finregistry.fi/endpoints/K11_CHOLELITH. A total of 20,175,454 variants were analyzed in this GWAS dataset. The association analysis utilized the mixed-model logistic regression method SAIGE (v.0.35.8.8), and the analysis accounted for covariates including sex, age, genotyping batch, and principal components (PCs).24
Genetic Proxies for Lipid-Lowering Drugs
MR analysis relies on three key assumptions for valid instrumental variables in ensuring causal inference.22 Firstly, selected variants should strongly associate with the studied exposures. We assessed instrument strength using F statistics calculated as the ratio of the squared β coefficient to the squared standard error,25,26 with an F value greater than 10 indicating sufficient strength.27 The second assumption necessitates that the chosen variants remain independent of any potential confounding factors. To address this, we utilized a curated genotype-phenotype database, namely PhenoScanner, to screen for variants associated with confounding factors. In the present study, factors previously recognized as risk factors for cholelithiasis, such as obesity-related indicators, diabetes, metabolic syndrome, and parenteral nutrition, are considered potential confounding variables.1–3 Lastly, selected variants should uniquely influence the outcome solely via their influence on the exposure. We performed sensitivity analyses to confirm this condition.
Low-Density Lipoprotein (LDL) Cholesterol-Based Genetic Instruments
For the first set of genetic instruments employed to represent the three lipid-lowering medications, we selected LDL cholesterol as the biomarker, as previous research has demonstrated that each of the three lipid-lowering drugs effectively reduces LDL cholesterol levels.11–13 To identify relevant SNPs, we utilized GWAS summary data of LDL cholesterol levels from a UK Biobank study, which consisted of 440,546 individuals of European ancestry.28 Only common SNPs possessing a minor allele frequency (MAF) surpassing 1% were considered for inclusion in the analysis. The GWAS dataset is available at https://gwas.mrcieu.ac.uk/datasets/ieu-b-110/.28
For instrumental variable selection, we focused on variants linked to LDL cholesterol at genome-wide significance (r2 < 0.1; P < 5 × 10−8) within a range of ±100 kb around the target genes. Specifically, the HMGCR gene (build GRCh37/hg19: chromosome 5: 74632993-74657941) was used as an instrument for statins, the NPC1L1 gene (chromosome 7: 44552134-44580929) for ezetimibe, and the PCSK9 gene (chromosome 1: 55505221-55530525) for PCSK9 inhibitors like alirocumab or evolocumab.29 Limited SNP availability (r2 < 0.1) impeded sensitivity analysis in the MR study for NPC1L1 inhibitors. To ensure an adequate number of SNPs, instrumental SNPs for NPC1L1 inhibitors were permitted to exhibit limited linkage disequilibrium (r2 < 0.3) with one another.29
What’s more, we utilized PhenoScanner (http://www.phenoscanner.medschl.cam.ac.uk/),30 a curated genotype-phenotype resource, to investigate associations between variants instrumental for specific drug targets and additional traits that might indicate pleiotropic pathways. We identified and excluded four SNPs from the HMGCR gene that were associated with body fat (P < 1 × 10−5), considering body fat as a known risk factor for cholelithiasis.8 Additionally, two SNPs from the PCSK9 gene were excluded as they were associated with the use of statin drugs. No SNPs from the NPC1L1 gene used as proxies for NPC1L1 inhibitors were excluded. (Supplementary Table S2).
eQTL-Based Genetic Instruments
To validate the causal associations derived from LDL cholesterol-based genetic instruments, we employed the available eQTLs associated with the target genes of each drug as proxies for the drug exposure.
We utilized eQTLs summary-level data obtained from two sources. The eQTLs summary-level data for the HMGCR gene were acquired from the eQTLGen Consortium (https://www.eqtlgen.org/). Additionally, the eQTLs summary-level data for the PCSK9 and NPC1L1 genes were obtained from the GTEx Consortium V8 (https://gtexportal.org/).
In our analysis, we identified common SNPs of eQTLs with an MAF greater than 1%. These SNPs were found to be significantly (P < 5.0×10−8) associated with the expression levels of three genes: HMGCR and PCSK9 in blood, and NPC1L1 in adipose subcutaneous tissue. Notably, no eQTLs for NPC1L1 were available in blood or other tissues at a significant level.29
We focused solely on cis-eQTLs, which are eQTLs located within a 1 Mb region on either side of the encoded gene. We identified a total of 921 common cis-eQTLs (MAF > 1%) in blood for the HMGCR gene, with the top SNP being rs6453133. Furthermore, we identified 24 common cis-eQTLs in blood for the PCSK9 gene, with the top SNP being rs472495. Additionally, we found 11 common cis-eQTLs in adipose subcutaneous tissue for the NPC1L1 gene, with the top SNP being rs41279633 (Supplementary Table S3).
Statistical Analyses
Primary MR Analysis
We employed the Inverse-variance-weighted MR (IVW-MR) approach to amalgamate effect estimations when utilizing genetic variants linked to LDL cholesterol levels as instruments. To assess the robustness of the results against horizontal pleiotropy, we also implemented MR Egger, weighted median, and weighted mode methods. These analyses were conducted using the TwoSampleMR package in R software, version 4.3.0.
Additionally, we employed the Summary-data-based MR (SMR) method with eQTLs as instruments. This approach enabled the exploration of the relationship between gene expression levels and the outcome of interest through the utilization of summary-level data from GWAS and eQTL studies.31 We performed the analysis using the SMR software, version 1.3.1 (https://cnsgenomics.com/software/smr/#Overview).
Sensitivity Analysis
Validation of the genetic instruments employed in the study involved performing positive control analyses. To validate the instrument derived from LDL cholesterol GWAS, we investigated the connection between the studied exposures and coronary heart disease (CHD), given that CHD is the primary indication for lipid-lowering medications. The CHD GWAS datasets were obtained from https://gwas.mrcieu.ac.uk/datasets/finn-b-I9_CHD/. The association between exposures of interest and LDL cholesterol levels was investigated for the instrument derived from eQTLs, given the established role of lipid-lowering drugs in reducing LDL cholesterol. The LDL cholesterol GWAS datasets were sourced from https://gwas.mrcieu.ac.uk/datasets/ieu-b-110/.
In the IVW-MR approach, we assessed heterogeneity through the Cochran Q test, where a P value below 0.05 indicated the presence of heterogeneity.32 To gauge directional horizontal pleiotropy, MR Egger regression was applied, focusing on the intercept term with P < 0.05 indicating its presence.33 Furthermore, we conducted Mendelian Randomization Pleiotropy RESidual Sum and Outlier (MR-PRESSO) analysis to identify and adjust for horizontal pleiotropic outliers, with a P < 0.05 for the Global test indicating their presence.34 In the SMR approach, we utilized the heterogeneity in dependent instruments (HEIDI) test via the SMR software,31 with a significant threshold of P < 0.01, indicating a likely association due to linkage.35
To address multiple testing, we adjusted significance thresholds using Bonferroni correction. We consideredP < 0.017 as strong evidence (three exposures and one outcome) and 0.017 ≤ P < 0.05 as suggestive evidence.
Results
In the IVW-MR analysis, we selected a total of seven, 14, and six SNPs near the HMGCR, PCSK9, and NPC1L1 genes, respectively, from the UK Biobank GWAS summary data on LDL cholesterol levels. The F-Statistics of all instrument variants exceeded 10, indicating minimal weak instrument bias in our study (Supplementary Table S2). The IVW-MR analysis provided robust evidence linking HMGCR-mediated LDL cholesterol (equivalent to a 1 mmol/l increase) with cholelithiasis risk (OR = 2.15, 95% CI = 1.58–2.94; P = 0.000), implying a potential protective role of HMGCR inhibitors against cholelithiasis. Furthermore, compelling evidence emerged for a connection between PCSK9-mediated LDL cholesterol and cholelithiasis risk (OR = 0.72, 95% CI = 0.56–0.94; P = 0.015), hinting at a potential unfavorable impact of PCSK9 inhibitors on cholelithiasis development. Nonetheless, the IVW-MR analysis yielded no indications of a link between NPC1L1-mediated LDL cholesterol and cholelithiasis. Heterogeneity was evaluated through the Cochran Q test, and none of the reported outcomes displayed evidence of heterogeneity (all P > 0.05). Furthermore, neither the intercept term in MR-Egger regression nor the MR-PRESSO analysis demonstrated significant evidence of overall horizontal pleiotropy (all P > 0.05) (Figure 2; Table 1).
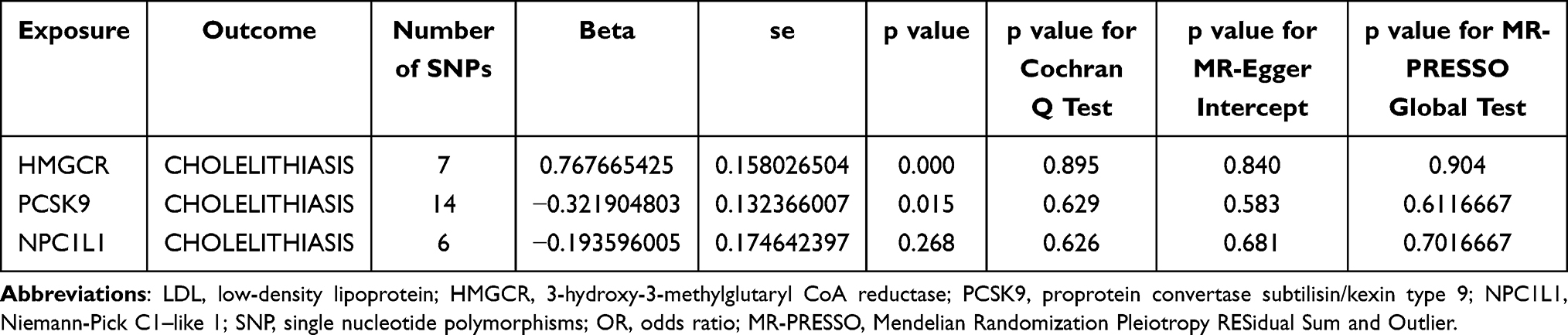 |
Table 1 Associations Between LDL Cholesterol Mediated by Gene HMGCR, PCSK9, or NPC1L1 and Cholelithiasis Risk |
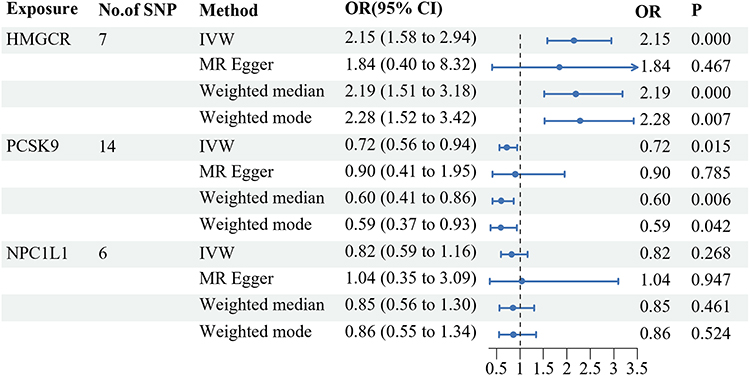 |
Figure 2 Associations between low-density lipoprotein (LDL) cholesterol mediated by gene HMGCR, PCSK9, or NPC1L1 and cholelithiasis risk. Abbreviations: HMGCR, 3-hydroxy-3-methylglutaryl CoA reductase; PCSK9, proprotein convertase subtilisin/kexin type 9; NPC1L1, Niemann-Pick C1–like 1; SNP, single nucleotide polymorphisms; OR, odds ratio; IVW, inverse variance weighted. |
The SMR analysis revealed 921, 24, and 11 cis-eQTLs from eQTLGen or GTEx Consortium related to the respective target genes HMGCR, PCSK9, and NPC1L1. The utmost significant cis-eQTL SNP for each drug’s target gene was designated as the genetic instrument, all exhibiting F-Statistics exceeding 10 (Supplementary Table S3). Our results from the SMR analysis provided strong evidence that higher blood expression levels of the HMGCR gene (equivalent to a one standard deviation increase) are strongly linked to an increased cholelithiasis risk (OR = 1.19, 95% CI = 1.04–1.37; P = 0.014), further suggesting that HMGCR inhibitors may reduce the risk of cholelithiasis. Furthermore, we uncovered suggestive evidence suggesting a negative link between PCSK9 expression and cholelithiasis risk (OR = 0.90, 95% CI = 0.82–0.99; P = 0.035), further supporting the possibility of adverse effects of PCSK9 inhibitors against cholelithiasis. However, there was no notable association detected between NPC1L1 expression and cholelithiasis, aligning with the outcomes of the IVW-MR analysis. Moreover, the HEIDI test within the SMR analysis suggested that all identified associations, except for the link between HMGCR expression and cholelithiasis (P = 0.009), were not influenced by linkage (P > 0.01) (Figure 3; Table 2).
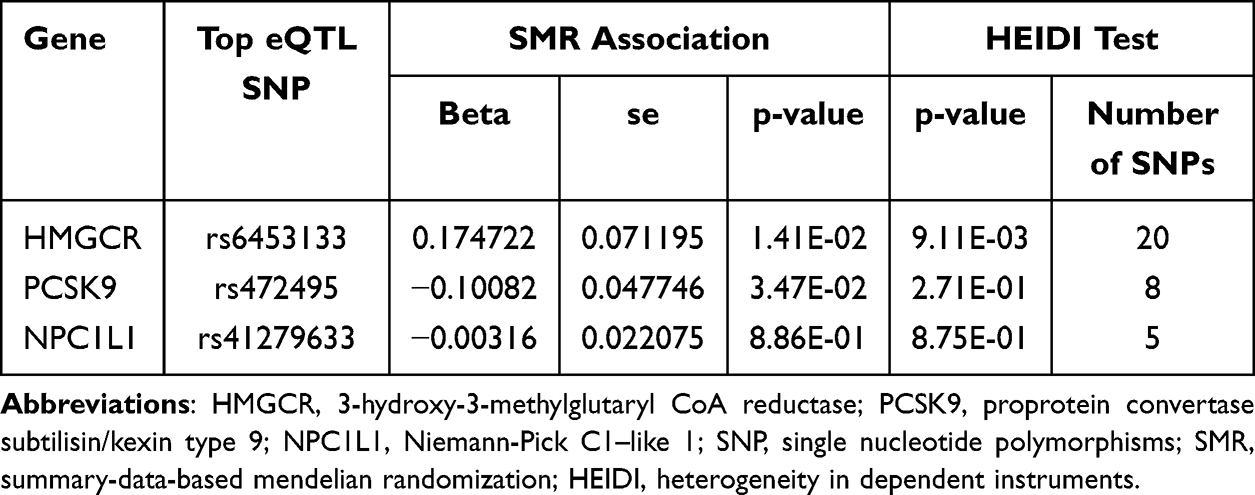 |
Table 2 SMR Association Between Expression of Gene HMGCR, PCSK9, or NPC1L1 and Cholelithiasis Risk |
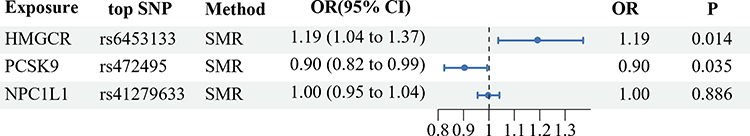 |
Figure 3 Summary-data-based Mendelian randomization (SMR) association between expression of gene HMGCR, PCSK9, or NPC1L1 and cholelithiasis risk. Abbreviations: HMGCR, 3-hydroxy-3-methylglutaryl CoA reductase; PCSK9, proprotein convertase subtilisin/kexin type 9; NPC1L1, Niemann-Pick C1–like 1; OR, odds ratio; SMR, summary-data-based mendelian randomization. |
In the positive control investigation, substantial links were identified between each drug exposure and coronary heart disease when utilizing genetic instruments derived from LDL cholesterol GWAS (Table 3). Similarly, we identified significant links between drug exposure and LDL cholesterol levels by employing genetic instruments based on eQTL (Table 4). These results provide further validation and assurance regarding the efficacy of the selected genetic instruments in our MR study.
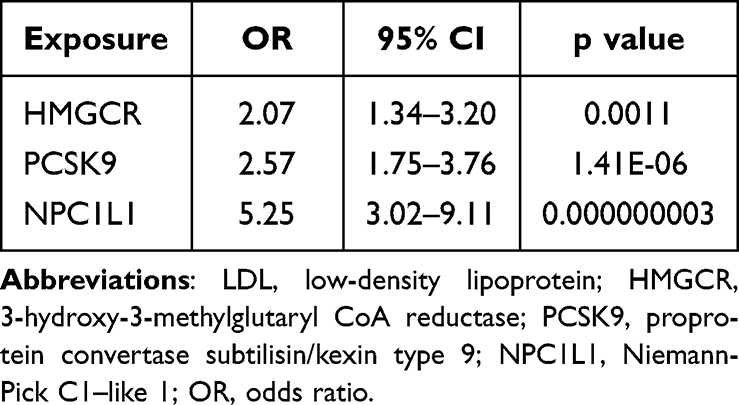 |
Table 3 Associations Between LDL Cholesterol Mediated by Gene HMGCR, PCSK9, or NPC1L1 and Coronary Heart Disease |
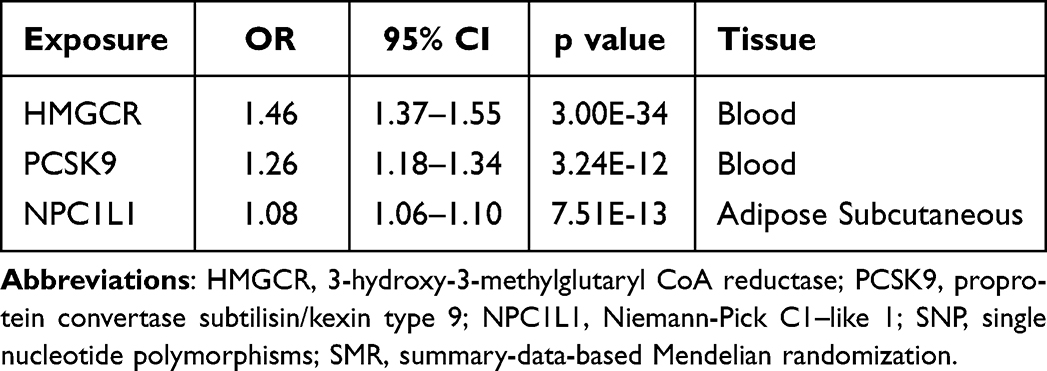 |
Table 4 SMR Association Between Expression of Gene HMGCR, PCSK9, or NPC1L1 and LDL Cholesterol Level |
Discussion
The current MR study provides strong evidence supporting a positive association between HMGCR-mediated LDL cholesterol levels and HMGCR expression with the risk of cholelithiasis. This implies that HMGCR inhibitors may potentially offer a protective effect against cholelithiasis. Conversely, our observations provide strong evidence suggesting a negative correlation between PCSK9-mediated LDL cholesterol levels and the risk of cholelithiasis. Moreover, there is suggestive evidence pointing to a negative association between PCSK9 expression and cholelithiasis risk. In summary, these findings imply that the use of PCSK9 inhibitors may elevate the risk of cholelithiasis. No evidence supported an association between NPC1L1-mediated LDL cholesterol levels or NPC1L1 expression with cholelithiasis risk. To the best of our knowledge, this study represents the first MR investigation of lipid-lowering drugs in relation to cholelithiasis. Furthermore, it is the first study to explore the potential impact of PCSK9 inhibitors on the risk of cholelithiasis. Our research results may offer insights into the influence of cholesterol-lowering drugs on the formation of cholelithiasis.
Previous observational studies have suggested a potential decrease in the risk of cholelithiasis with the use of statin drugs.16,36 However, observational studies are susceptible to lingering confounding variables and the possibility of reverse causation. Our current study utilized MR to establish a causal link between statin use and cholelithiasis risk.
A recent MR study has proposed that reduced total cholesterol levels could represent an autonomous risk factor for cholelithiasis.8 This raises concerns that the use of lipid-lowering drugs to decrease cholesterol levels could potentially increase the risk of cholelithiasis. The authors of that MR study speculate that one of the mechanisms behind this association may involve the liver’s compensatory synthesis of cholesterol and an enhanced cholesterol secretion into bile, both of which could increase the risk of cholelithiasis.37 Interestingly, HMGCR is an essential enzyme in cholesterol synthesis,14 and statin use not only lowers serum total cholesterol but also inhibit cholesterol synthesis by suppressing the activity of HMGCR, thus interrupting the aforementioned mechanism. The protective effect of statin drugs on the risk of cholelithiasis does not seem to be achieved by reducing serum cholesterol levels but may be related to a decrease in the secretion of cholesterol from the liver into bile.38,39 These two processes occur simultaneously after statin-mediated HMGCR inhibition. Therefore, the previous observational studies and the findings of our MR study regarding the decreased risk of cholelithiasis with statin use are not contradictory to the conclusions of that MR study.8
This study is the first to propose a potential increase in the risk of cholelithiasis associated with the use of PCSK9 inhibitors. Interestingly, this finding contradicts the findings of this study regarding the reduced risk of cholelithiasis with HMGCR inhibitors. This discrepancy may be perplexing, but we speculate that the reason for the effect of lipid-lowering drugs on the risk of cholelithiasis may not be due to their ability to lower serum cholesterol and then affect the saturation of cholesterol in bile.38,39 Instead, it may be due to different mechanisms by which different lipid-lowering drugs lower serum cholesterol.40,41
In the case of PCSK9 inhibitors, their mechanism of action involves inhibiting PCSK9 activity, thereby enhancing the expression and function of LDL receptors (LDLR).42 This, in turn, promotes increased metabolism of LDL cholesterol in the liver. Previous studies have suggested that hepatic fat accumulation may increase the secretion of PCSK9 by the liver,43,44 resulting in reduced hepatic uptake of LDL cholesterol. However, the use of PCSK9 inhibitors allows the liver to continue taking up LDL cholesterol, even if it is already accumulated in the hepatic cholesterol pool. Our hypothesis is that this augmented hepatic metabolism leads to an accumulation of cholesterol in the intrahepatic cholesterol pool, resulting in elevated cholesterol secretion into the bile and subsequently increasing the risk of cholelithiasis.14
Another possible reason is that the accumulation of cholesterol in the liver may inhibit the activity of Cholesterol 7α-hydroxylase (CYP7A1),45 which is the rate-limiting step in the synthesis of bile acids in the liver. This reduction in bile acid synthesis leads to a decrease in bile acid levels in the bile, resulting in cholesterol supersaturation in the bile and an increased risk of gallstone formation.46 Currently, it remains uncertain whether PCSK9 inhibition directly affects the cholesterol saturation index in bile. Thus, further research is required to validate it.
The association between PCSK9 inhibition and an increased risk of gallstones can also be observed in patients with hyperthyroidism. Some studies have found that serum PCSK9 levels decrease in patients with hyperthyroidism,47,48 while other studies have found an increased risk of gallstones in patients or mice with hyperthyroidism.49,50 Our study findings may provide insights into the underlying mechanisms that contribute to the increased risk of gallstones in patients with hyperthyroidism.
While our study suggests an increased risk of cholelithiasis associated with PCSK9 inhibitors, the specific dosage and duration of PCSK9 inhibitor use that leads to this risk are still unknown. Nevertheless, considering that lipid-lowering therapy for primary or secondary prevention of cardiovascular diseases is often lifelong, it is essential not to overlook this potential risk when clinically administering PCSK9 inhibitors.
In our study, we did not observe any correlation between NPC1L1 inhibitors and the risk of cholelithiasis, despite previous contradictory findings in NPC1L1 inhibitors research.19,20 An animal experiment suggested that ezetimibe effectively reduces intestinal cholesterol absorption, thereby preventing gallstones and reducing bile saturation in mice, and even promoting gallstone dissolution.19 However, a different study utilizing single-sample MR suggested that genetic variations in NPC1L1, resembling the effects of ezetimibe, are linked to an elevated risk of symptomatic cholelithiasis.20
The conclusions of both studies may be challenged. The former study’s conclusion is based on experimental mice receiving a high-cholesterol diet, which is not representative of the common diet in human populations. In addition, while NPC1L1 is predominantly expressed in the small intestine of rodents,51 humans exhibit significant NPC1L1 expression in the liver as well.52 The latter study’s conclusion may also be questioned because single-sample MR studies are prone to the “winner’s curse” effect, where the effect estimates of ezetimibe on gallstone risk may be exaggerated and higher than the actual situation.53 Additionally, the latter study did not undertake sensitivity analyses to test for potential bias due to pleiotropy.54
Based on our observations, we speculate that the lack of causal relationship between NPC1L1 inhibitors and cholelithiasis risk may be due to their dual effects.2 NPC1L1 inhibitors reduce intestinal cholesterol absorption by inhibiting NPC1L1 in the intestines, leading to decreased liver cholesterol secretion, which may lower the risk of cholelithiasis.19 However, NPC1L1 inhibitors also inhibit NPC1L1 in the liver, reducing the uptake of biliary cholesterol by the liver and potentially increasing cholesterol content in bile, which may increase the risk of cholelithiasis.20 Considering the combined effects of these two mechanisms, NPC1L1 inhibitors may neither increase nor decrease the risk of cholelithiasis.
This study has several limitations. Firstly, although MR and randomized controlled trials share the common goal of providing reliable evidence for causal relationships, it is not appropriate to directly compare their estimated treatment effects. MR estimates capture the enduring consequences of modulating drug targets, which may differ from interventions given at specific timeframes and their impacts over shorter durations. However, MR estimates still serve a valuable purpose in indicating the presence and direction of causal effects.23,55 Secondly, when verifying the link between HMGCR-mediated LDL cholesterol and the risk of cholelithiasis using the eQTL of target genes of HMGCR inhibitors as a proxy for HMGCR inhibition exposure, the HEIDI test suggested that the detected correlation between HMGCR gene expression and the outcome could potentially result from a linkage scenario. Nevertheless, in our main MR analysis of HMGCR-mediated LDL cholesterol levels, we found no evidence of pleiotropy. Thirdly, despite conducting multiple sensitivity analyses to scrutinize the MR study assumptions and employing PhenoScanner to identify proxy SNPs associated with confounding factors, we were still unable to completely eliminate the potential for confounding bias and/or horizontal pleiotropy. Fourthly, it is important to note that the eQTL and GWAS data used in this study primarily originated from European populations, where cholelithiasis is predominantly cholesterol-based. Hence, it’s important to approach the extrapolation of these findings to different populations with caution. Fifthly, the HMGCR inhibitor and PCSK9 inhibitor primarily target the liver, whereas the NPC1L1 inhibitor affects both the liver and the intestines. Regrettably, there are presently no eQTLs accessible specifically for these target genes in the liver. Using eQTLs obtained from blood or adipose subcutaneous tissue as substitutes for liver eQTLs could potentially compromise the accuracy of the genetic instruments in reflecting the mechanisms of action of these drugs in the liver. To address this concern, positive control analyses were performed to authenticate the genetic instruments employed in this study.
Conclusions
In summary, this MR study supports a causal association between HMGCR inhibitors use and reduced cholelithiasis risk. Additionally, it indicates a causal link between PCSK9 inhibitors use and increased cholelithiasis risk. However, no significant correlation was found between NPC1L1 inhibitors use and cholelithiasis risk.
Abbreviations
MR, Mendelian randomization; GWAS, genome-wide association study; eQTLs, expression quantitative trait loci; LDL, low-density lipoprotein; HMGCR, 3-hydroxy-3-methylglutaryl CoA reductase; PCSK9, proprotein convertase subtilisin/kexin type 9; NPC1L1, Niemann-Pick C1–like 1; IVW, multiplicative random effects inverse variance weighted method; SNP, single nucleotide polymorphisms; OR, odds ratio; SMR, summary-data-based mendelian randomization; HEIDI, heterogeneity in dependent instruments; MR-PRESSO, Mendelian Randomization Pleiotropy RESidual Sum and Outlier; MAF, minor allele frequency.
Ethical Approval and Informed Consent
This two-sample Mendelian randomization (MR) study relies on publicly available summary-level data from GWASs (Supplementary Table S1). These studies have received ethical clearance from their respective institutional review boards, and participants have provided informed consent for their involvement. Given that our research exclusively utilized publicly accessible datasets to conduct MR, consultation with the Medical Ethics Committee of Wuhan Jinyintan Hospital confirmed that no further ethics approval was necessary.
Acknowledgments
The authors extend their sincere gratitude to the UKB and FinnGen consortia for granting access to the GWAS summary data utilized in this study. Additionally, we would like to convey our heartfelt appreciation to the patients and investigators whose contributions were instrumental in the eQTLGen Consortium and GTEx Consortium.
Disclosure
The authors declare no conflicts of interest in this work.
References
1. Lammert F, Gurusamy K, Ko CW, et al. Gallstones. Nat Rev Dis Primers. 2016;2(1):16024. doi:10.1038/nrdp.2016.24
2. European Association for the Study of the Liver (EASL). Electronic address: [email protected]. EASL Clinical Practice Guidelines on the prevention, diagnosis and treatment of gallstones. J Hepatol. 2016;65(1):146–181. doi:10.1016/j.jhep.2016.03.005
3. Sanders G, Kingsnorth AN. Gallstones. BMJ. 2007;335(7614):295–299. doi:10.1136/bmj.39267.452257.AD
4. Portincasa P, Moschetta A, Palasciano G. Cholesterol gallstone disease. Lancet. 2006;368(9531):230–239. doi:10.1016/S0140-6736(06)69044-2
5. Atamanalp SS, Keles MS, Atamanalp RS, Acemoglu H, Laloglu E. The effects of serum cholesterol, LDL, and HDL levels on gallstone cholesterol concentration. Pak J Med Sci. 2013;29(1):187–190. doi:10.12669/pjms.291.2798
6. Halldestam I, Kullman E, Borch K. Incidence of and potential risk factors for gallstone disease in a general population sample. Br J Surg. 2009;96(11):1315–1322. doi:10.1002/bjs.6687
7. Bertomeu A, Ros E, Zambón D, et al. Apolipoprotein E polymorphism and gallstones. Gastroenterology. 1996;111(6):1603–1610. doi:10.1016/S0016-5085(96)70023-9
8. Chen L, Yang H, Li H, He C, Yang L, Lv G. Insights into modifiable risk factors of cholelithiasis: a Mendelian randomization study. Hepatology. 2022;75(4):785. doi:10.1002/hep.32183
9. Attili AF, Capocaccia R, Carulli N, et al. Factors associated with gallstone disease in the MICOL experience. Multicenter Italian Study on Epidemiology of Cholelithiasis. Hepatology. 1997;26(4):809–818. doi:10.1002/hep.510260401
10. Stender S, Frikke-Schmidt R, Benn M, Nordestgaard BG, Tybjærg-Hansen A. Low-density lipoprotein cholesterol and risk of gallstone disease: a Mendelian randomization study and meta-analyses. J Hepatol. 2013;58(1):126–133. doi:10.1016/j.jhep.2012.08.013
11. Stein EA, Raal FJ. Lipid-lowering drug therapy for CVD prevention: looking into the future. Curr Cardiol Rep. 2015;17(11):104. doi:10.1007/s11886-015-0659-8
12. Pirillo A, Catapano L, A D, Norata G. Niemann-Pick C1-Like 1 (NPC1L1) inhibition and cardiovascular diseases. Curr Med Chem. 2016;23(10):983–999. doi:10.2174/0929867323666160229114111
13. Dadu RT, Ballantyne CM. Lipid lowering with PCSK9 inhibitors. Nat Rev Cardiol. 2014;11(10):563–575. doi:10.1038/nrcardio.2014.84
14. Zanlungo S, Rigotti A. Determinants of transhepatic cholesterol flux and their relevance for gallstone formation. Liver Int. 2009;29(3):323–330. doi:10.1111/j.1478-3231.2009.01972.x
15. Bodmer M, Brauchli YB, Krähenbühl S, Jick SS, Meier CR. Statin use and risk of gallstone disease followed by cholecystectomy. JAMA. 2009;302(18):2001–2007. doi:10.1001/jama.2009.1601
16. Erichsen R, Frøslev T, Lash TL, Pedersen L, Sørensen HT. Long-term statin use and the risk of gallstone disease: a population-based case-control study. Am J Epidemiol. 2011;173(2):162–170. doi:10.1093/aje/kwq361
17. Kan H-P, Guo W-B, Tan Y-F, Zhou J, Liu C-D, Huang Y-Q. Statin use and risk of gallstone disease: a meta-analysis. Hepatol Res. 2015;45(9):942–948. doi:10.1111/hepr.12433
18. McEvoy JW. Lifetime risks of cardiovascular disease. N Engl J Med. 2012;366:1642; author reply 1642–1643.
19. Wang HH, Portincasa P, Mendez-Sanchez N, Uribe M, Wang DQ-H. Effect of ezetimibe on the prevention and dissolution of cholesterol gallstones. Gastroenterology. 2008;134(7):2101–2110. doi:10.1053/j.gastro.2008.03.011
20. Lauridsen BK, Stender S, Frikke-Schmidt R, Nordestgaard BG, Tybjærg-Hansen A. Genetic variation in the cholesterol transporter NPC1L1, ischaemic vascular disease, and gallstone disease. Eur Heart J. 2015;36(25):1601–1608. doi:10.1093/eurheartj/ehv108
21. Davey Smith G, Hemani G. Mendelian randomization: genetic anchors for causal inference in epidemiological studies. Hum Mol Genet. 2014;23(R1):R89–R98. doi:10.1093/hmg/ddu328
22. Davies NM, Holmes MV, Davey Smith G. Reading Mendelian randomisation studies: a guide, glossary, and checklist for clinicians. BMJ. 2018;362:k601. doi:10.1136/bmj.k601
23. Gill D, Georgakis MK, Walker VM, et al. Mendelian randomization for studying the effects of perturbing drug targets. Wellcome Open Res. 2021;6:16. doi:10.12688/wellcomeopenres.16544.1
24. Kurki MI, Karjalainen J, Palta P, et al. FinnGen provides genetic insights from a well-phenotyped isolated population. Nature. 2023;613(7944):508–518. doi:10.1038/s41586-022-05473-8
25. Li B, Martin EB. An approximation to the F distribution using the chi-square distribution. Comput Stat Data Anal. 2002;40(1):21–26. doi:10.1016/S0167-9473(01)00097-4
26. Zhao SS, Yiu ZZN, Barton A, Bowes J. Association of lipid-lowering drugs with risk of psoriasis: a mendelian randomization study. JAMA Dermatol. 2023;159(3):275–280. doi:10.1001/jamadermatol.2022.6051
27. Burgess S, Thompson SG; CRP CHD Genetics Collaboration. Avoiding bias from weak instruments in Mendelian randomization studies. Int J Epidemiol. 2011;40(3):755–764. doi:10.1093/ije/dyr036
28. Richardson TG, Sanderson E, Palmer TM, et al. Evaluating the relationship between circulating lipoprotein lipids and apolipoproteins with risk of coronary heart disease: a multivariable Mendelian randomisation analysis. PLoS Med. 2020;17(3):e1003062. doi:10.1371/journal.pmed.1003062
29. Huang W, Xiao J, Ji J, Chen L. Association of lipid-lowering drugs with COVID-19 outcomes from a Mendelian randomization study. Elife. 2021;10:e73873. doi:10.7554/eLife.73873
30. Kamat MA, Blackshaw JA, Young R, et al. PhenoScanner V2: an expanded tool for searching human genotype-phenotype associations. Bioinformatics. 2019;35(22):4851–4853. doi:10.1093/bioinformatics/btz469
31. Zhu Z, Zhang F, Hu H, et al. Integration of summary data from GWAS and eQTL studies predicts complex trait gene targets. Nat Genet. 2016;48(5):481–487. doi:10.1038/ng.3538
32. Higgins JPT, Thompson SG, Deeks JJ, Altman DG. Measuring inconsistency in meta-analyses. BMJ. 2003;327(7414):557–560. doi:10.1136/bmj.327.7414.557
33. Burgess S, Thompson SG. Interpreting findings from Mendelian randomization using the MR-Egger method. Eur J Epidemiol. 2017;32(5):377–389. doi:10.1007/s10654-017-0255-x
34. Verbanck M, Chen C-Y, Neale B, Do R. Detection of widespread horizontal pleiotropy in causal relationships inferred from Mendelian randomization between complex traits and diseases. Nat Genet. 2018;50(5):693–698. doi:10.1038/s41588-018-0099-7
35. Chauquet S, Zhu Z, O’Donovan MC, Walters JTR, Wray NR, Shah S. Association of antihypertensive drug target genes with psychiatric disorders: a mendelian randomization study. JAMA Psychiatry. 2021;78(6):623–631. doi:10.1001/jamapsychiatry.2021.0005
36. Tsai C-J, Leitzmann MF, Willett WC, Giovannucci EL. Statin use and the risk of cholecystectomy in women. Gastroenterology. 2009;136(5):1593–1600. doi:10.1053/j.gastro.2009.01.042
37. Everhart JE. Contributions of obesity and weight loss to gallstone disease. Ann Intern Med. 1993;119(10):1029–1035. doi:10.7326/0003-4819-119-10-199311150-00010
38. Kallien G, Lange K, Stange EF, Scheibner J. The pravastatin-induced decrease of biliary cholesterol secretion is not directly related to an inhibition of cholesterol synthesis in humans. Hepatology. 1999;30(1):14–20. doi:10.1002/hep.510300119
39. Hoogerbrugge-vd Linden N, de Rooy FW, Jansen H, van Blankenstein M. Effect of pravastatin on biliary lipid composition and bile acid synthesis in familial hypercholesterolaemia. Gut. 1990;31(3):348–350. doi:10.1136/gut.31.3.348
40. Brookes ZLS, McGown CC, Reilly CS. Statins for all: the new premed? Br J Anaesth. 2009;103(1):99–107. doi:10.1093/bja/aep149
41. Hess CN, Low Wang CC, Hiatt WR. PCSK9 inhibitors: mechanisms of action, metabolic effects, and clinical outcomes. Ann Rev Med. 2018;69(1):133–145. doi:10.1146/annurev-med-042716-091351
42. Seidah NG, Prat A. The multifaceted biology of PCSK9. Endocr Rev. 2022;43(3):558–582. doi:10.1210/endrev/bnab035
43. Emma MR, Giannitrapani L, Cabibi D, et al. Hepatic and circulating levels of PCSK9 in morbidly obese patients: relation with severity of liver steatosis. Biochim Biophys Acta Mol Cell Biol Lipids. 2020;1865(12):158792. doi:10.1016/j.bbalip.2020.158792
44. Ruscica M, Ferri N, Macchi C, et al. Liver fat accumulation is associated with circulating PCSK9. Ann Med. 2016;48(5):384–391. doi:10.1080/07853890.2016.1188328
45. Henkel AS, Anderson KA, Dewey AM, Kavesh MH, Green RM. A chronic high-cholesterol diet paradoxically suppresses hepatic CYP7A1 expression in FVB/NJ mice. J Lipid Res. 2011;52(2):289–298. doi:10.1194/jlr.M012781
46. Sedaghat A, Grundy SM. Cholesterol crystals and the formation of cholesterol gallstones. N Engl J Med. 1980;302(23):1274–1277. doi:10.1056/NEJM198006053022302
47. Yildirim AM, Koca AO, Beyan E, et al. Association of serum proprotein convertase Subtilisin/Kexin Type 9 (PCSK9) level with thyroid function disorders. Eur Rev Med Pharmacol Sci. 2021;25(17):5511–5517. doi:10.26355/eurrev_202109_26662
48. Bonde Y, Breuer O, Lütjohann D, Sjöberg S, Angelin B, Rudling M. Thyroid hormone reduces PCSK9 and stimulates bile acid synthesis in humans. J Lipid Res. 2014;55(11):2408–2415. doi:10.1194/jlr.M051664
49. Song ST, Shi J, Wang XH, et al. Prevalence and risk factors for gallstone disease: a population-based cross-sectional study. J Digest Dis. 2020;21(4):237–245. doi:10.1111/1751-2980.12857
50. Wang Y, Yu X, Zhao Q, et al. Thyroid dysfunction, either hyper or hypothyroidism, promotes gallstone formation by different mechanisms. J Zhejiang Univ Sci B. 2016;17(7):515–525. doi:10.1631/jzus.B1500210
51. Altmann SW, Davis HR, Zhu L-J, et al. Niemann-Pick C1 Like 1 protein is critical for intestinal cholesterol absorption. Science. 2004;303(5661):1201–1204. doi:10.1126/science.1093131
52. Yu L, Bharadwaj S, Brown JM, et al. Cholesterol-regulated translocation of NPC1L1 to the cell surface facilitates free cholesterol uptake. J Biol Chem. 2006;281(10):6616–6624. doi:10.1074/jbc.M511123200
53. Lawlor DA. Commentary: two-sample Mendelian randomization: opportunities and challenges. Int J Epidemiol. 2016;45(3):908–915. doi:10.1093/ije/dyw127
54. Bowden J, Davey Smith G, Burgess S. Mendelian randomization with invalid instruments: effect estimation and bias detection through Egger regression. Int J Epidemiol. 2015;44(2):512–525. doi:10.1093/ije/dyv080
55. Haycock PC, Burgess S, Wade KH, Bowden J, Relton C, Davey Smith G. Best (but oft-forgotten) practices: the design, analysis, and interpretation of Mendelian randomization studies. Am J Clin Nutr. 2016;103(4):965–978. doi:10.3945/ajcn.115.118216
 © 2024 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms.php
and incorporate the Creative Commons Attribution
- Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2024 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms.php
and incorporate the Creative Commons Attribution
- Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.