Back to Journals » Cancer Management and Research » Volume 11
Cadherin-11 is inactivated due to promoter methylation and functions in colorectal cancer as a tumour suppressor
Authors Yuan S, Li L, Xiang S, Jia H, Luo T
Received 12 November 2018
Accepted for publication 28 February 2019
Published 28 March 2019 Volume 2019:11 Pages 2517—2529
DOI https://doi.org/10.2147/CMAR.S193921
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Beicheng Sun
Shiyun Yuan,1 Lin Li,1 Shili Xiang,2 Hexun Jia,3 Tao Luo1
1Department of Geriatrics, The First Affiliated Hospital of Chongqing Medical University, Chongqing 400016, People’s Republic of China; 2Department of Critical Care Medicine, The First Affiliated Hospital of Chongqing Medical University, Chongqing 400016, People’s Republic of China; 3Office of academic, The First Affiliated Hospital of Chongqing Medical University, Chongqing 400016, People’s Republic of China
Background: The cadherin-11 (CDH11, OB-cadherin) gene is a member of the cadherin family and is located on chromosome 16q22.1. Previous studies have revealed that cadherins play significant roles in the development of many human malignancies. Increasing evidence has identified CDH11 as a functional tumour suppressor, which is commonly silenced by promoter methylation, but the functions of this gene in colorectal cancer (CRC) have been unclear.
Methods: The CDH11 expression in primary CRC tissues and cell lines was investigated by qRT-PCR, RT-PCR and immunohistochemistry. The promoter methylation status of CDH11 was measured by methylation-specific PCR (MSP). Cell proliferation assay, colony formation assay, flow cytometry analysis, wound-healing assay, transwell assay and in vivo experiments were used to investigate the function of CDH11 in CRC. The mechanisms of CDH11 also were explored by western blots.
Results: Our study suggests that CDH11 downregulation in CRC due to its promoter methylation and induced cell cycle arrest in G0/G1 phase and apoptosis, suppressing tumor cell proliferation, colony formation, migration and invasion by affecting the NF-kB signaling pathway.
Conclusion: Overall, CDH11 may be considered as a functional tumour suppressor gene (TSG) in CRC, CDH11 has the potential to serve as a valuable prognostic marker for colorectal cancer.
Keywords: cadherin 11, colorectal cancer, methylation, NF-kB, tumour suppressor
Introduction
Colon and rectum cancer (colorectal cancer, CRC) is the third most-common cancer, and CRC accounting for 6.1% (1,800,977) of all cancer cases and 9.2% (881,000) of cancer deaths in 2018, according to the latest global cancer statistics from the International Agency for Research on Cancer.1 Mounting more evidence has suggested that screening and early diagnosis is associated with a reduction in CRC. However, the relevant methodologies are still limited, underlining the importance of finding a reliable biomarker for an early detection of this disease.2 Six classical cadherin family genes (six cadherin clusters) are located on 16q22.1-16q24.3 and comprise of six cell-cell adhesion molecules mediating intercellular adhesion by Ca2+-dependent homophilic interactions. The family members include CDH1(E-cadherin), CDH3(P-cadherin), CDH5(VE-cadherin), CDH8, CDH11(OB-cadherin) and CDH13(H-cadherin).3 CDH1 and CDH13 have been identified as functional tumour suppressors, which inhibit cell proliferation and invasiveness in most cancer cell lines and reducing tumour growth in in vivo models.4,5
The specific mechanism of CRC tumourigensis involves the activation of an oncogene and the inactivation of a tumour suppressor gene. The tumour suppressor gene inactivation is closely related to epigenetic changes.6 A previous study has identified DNA copy number aberrations and an ~1 Mb hemizygous deletion at 16q21-22.1 in tumour cell lines by performing 1-Mb array comparative genomic hybridization (aCGH), indicating that CDH11, the only known gene located at this deletion, could be 16q21-22.1 deletion-related candidate TSG.3 Researchers have found that CDH11 is downregulated and frequently methylated in multiple types of tumours.7–10 However the roles of CDH11 in CRC have not been elucidated. Therefore, in this study, we investigated the CDH11 expression, promoter methylation status, biological functions and related molecular mechanisms in CRC.
Material and methods
Cells lines and tumour samples
Six human colorectal cancer cell lines (ie, LoVo, HCT116, SW480, HT-29, CaCo-2, RKO) were used. The CRC cell lines HT-29 and HCT116 were provided by Professor Q.Tao at the Chinese University of Hong Kong, and the SW480, LoVo, CaCo-2, RKO cell lines were purchased from the Chinese Academy of Sciences. And the use of the gifted cell lines was approved by the ethics committee of the First Affiliated Hospital of Chongqing Medical University. The cell lines were cultured in RPMI-1640 medium (Gibco-BRL, Karlsruhe, Germany) with 10% fetal bovine serum (FBS) (ExCell Bio, Shanghai, China), and maintained at a humidified atmosphere of 5% CO2 at 37 °C.11 The CRC tissues and their corresponding adjacent tissues were collected from surgical patients at the First Affiliated Hospital of Chongqing Medical University (Chongqing,China), and all tissues were diagnosed and verified at the Pathology Department, Chongqing Medical University (Chongqing,China). The samples were immediately snap-frozen in liquid nitrogen and then stored at −80 °C until analysis. The study protocol was authorized under the guidelines of the ethics committee of the First Affiliated Hospital of Chongqing Medical University. This study was conducted in accordance with the Declaration of Helsinki and all patients signed an informed consent form.
Nucleic acid and protein extraction
Total RNA was extracted from tissues and cell lines using TRIzol® reagent (LifeTechnologies, Carlsbad, CA, USA). Genomic DNA was isolated from tissues and cell lines using the QIAamp DNAMini kit (Qiagen, Hilden, Germany), according to the manufacturer’s protocol. The DNA and RNA were stored at −80 °C after measuring their concentrations of them using a NanoDrop 2000 spectrophotometer (Thermo Scientific, Rockford, IL, USA).The experimental and control groups of the HCT116 and LoVo cells were lysed using a protein extraction reagent (Thermo Scientific) that contained the protease inhibitors, phenylmethane sulfonyl fluoride, and a phosphatase inhibitor cocktail (Sigma-Aldrich, St. Louis, MO, USA), and the lysate was then homogenized using an Ultrasonic Cell Grinder (Scientz, Ningbo). The supernatant was collected after centrifugation, and protein concentration of protein in the supernatant was determined using the BCA protein kit (Thermo Scientific).12
Semiquantitative polymerase chain reaction (PCR) and quantitative RT-PCR (qRT-PCR)
Semiquantitative PCR and quantitative PCR were used to determine expression of CDH11 in CRC cells and tissues. The RNA (1 μg) was reverse transcribed to 20 μg of cDNA using the Reverse Transcription system (Promega, Madison, WI, USA). For semiquantitative PCR, the CDH11 gene was amplified using GoTaq DNA polymerase (Promega). The protocol included an initial denaturation at 95 °C for 2 min, followed by 34 amplification reaction cycles (95 °C for 30 s, 55 °C for 30 s and 72 °C for 30 s), with a final extension at 72 °C for 3 min, and used glyceraldehyde-3-phosphate dehydrogenase (GAPDH) as an internal control. The primer sequences are listed in Table 1. Quantitative PCR was performed with an ABI 7500 Real-Time PCR system (Applied Biosystems, Foster City, CA, USA), using SYBR® Green PCR Master Mix (Thermo Fisher Scientific, Hong Kong, China) with GAPDH as a control.11 Each sample was tested in triplicate.
 | Table 1 The primers used in this study |
Bisulfite treatment and methylation-specific PCR (MSP) analyses of CDH11
Bisulfite modification of DNA and methylation-specific PCR (MSP) were carried out in accordance with previously described methods.13 The primers used for MSP are listed in Table 1. MSP for the CDH11 promoter was performed using AmpliTaq®-Gold DNA Polymerase (Applied Biosystems) mixed with bisulfite-treated DNA, MgCl2 and deoxynucleotide triphosphates for the MSP amplification reaction. The reaction conditions were as follows: initial denaturation for 10 min at 95 °C, then 40 cycles consisting of 95 °C for 30 s, 60 °C (methylated reactions) or 58 °C (unmethylated reactions) for 30 s, 72 °C for 30 s, and a final extension for 5 min at 72 °C. The final products were identified on a 2% agarose gel containing 100 bp DNA markers (MBI Fermentas Vilnius, Lithuania), and then recorded using a Molecular Imager (Bio-Rad, Hercules, CA, USA).11,12
5-Aza-2ʹ-deoxycytidine (Aza) and trichostatin a (TSA) treatment
As previously reported,12,14 the cells were treated with a DNA methyltransferase (DNMT) inhibitor,5-Aza-2ʹ-deoxycytidine (Aza) (Sigma-Aldrich, Steinheim, Germany), in the dark at a final concentration of 10 mmol/l for 3 days, and further treated with 100 nmol/l trichostatin A(TSA) (Cayman Chemical, Ann Arbor, MI, USA), a histone deacetylase inhibitor, for another 1 day. Other groups had cells treated only with Aza (Sigma-Aldrich) in the dark at a final concentration of 10 mmol/l for 3 days or TSA at a final concentration of 100 nmol/l for 1 day. Cells were then collected for RNA and DNA extraction.
Construction of CDH11-overexpressing LoVo and HCT116 cell lines
CRC cell lines were transfected with pcDNA3.1-CDH11or pcDNA3.1 (+) vectors using Lipofectamine® 2000 (Invitrogen; Thermo Fisher Scientific, Inc.) in serum-free RPMI-1640 medium; 4–6 h later, the medium was replaced with fresh growth medium containing 10% FBS for 48 h. Then, CRC LoVo and HCT116 cells were selected in the medium containing 10% FBS with G418 (Invitrogen/Gibco) at a final concentration of 5 μl/ml or 3 μl/ml approximately 20 days later. RT-PCR and western blot analyses were confirmed stable overexpression of CDH11. CDH11-expressingplasmid was provided by Professor Qian Tao at the Chinese University of Hong Kong.
Cell proliferation assay
Cell proliferation was assessed by CCK-8 assay. LoVo and HCT116 cells stably expressing CDH11 or pcDNA 3.1 were seeded in 96-well plates (2x103 cells/well) with 100 μl/ml of complete culture medium. Then, the medium in each well was replaced with 100 μl RPMI-1640 (10% FBS) containing 10 μl CCK-8 solution and incubated at 37 °C for 2 h in the dark. The absorbance was measured at 450 nm using a microplate reader (Multiskan MK3; ThermoFisher Scientific, former Fermentas, Schwerte, Germany) at 24, 48 and 72 h. All experiments were repeated three times.
Colony formation assay
Stably expressing cells were planted in 6-well plates at 1×103 cells/well and cultured for 14 days, then fixed in 4% paraformaldehyde for 30 min, and stained with Gentian Violet (ICM Pharma, Singapore, Singapore) for 30 min. Finally, the cells were scanned using a CanoScan8800F (Canon, Tokyo, Japan). Surviving colonies (>50 cells/colony) were counted using the ImageJ (V.1.8.0) software (National Institutes of Health, Bethesda, MD, USA) for the analysis. All experiments were assessed in triplicate.
Flow cytometry analysis of cell cycle and apoptosis
Stable HCT116 and LoVo cells were collected, digested, rinsed with phosphate-buffered saline (PBS) and centrifuged at 800 rpm for 5 min, then fixed with ice-cold 70% ethanol at 4 °C overnight. Then the cells were stained with 50 mg/l propidium iodide (PI) (Beyotime) for 30 min at 4 °C in the dark. For the cell apoptosis measurements, cells were washed, collected, resuspended in PBS, stained with Annexin V-FITC (BD Pharmingen, San, Jose, CA, USA) and PI in the dark for 15 min, and analysed using a flow cytometer (BD Biosciences, Franklin Lakes, NJ, USA). Data were analysed using the Cell Quest software (BD Biosciences, San Jose, CA, USA).
Wound-healing assays
Stably expressing cells were planted in six-well plates until 95% confluent. Asterile P-20 pipette tip was used to scratch wounds across each well. Cells were then washed three times with 1 ml/well PBS to carefully remove cell debris and cultured in serum-free media. The cell-free wound area was photographed at 0, 24, 48 and 72 in LoVo culture, and the images of the wound closure areas of HCT116 cells were observed at 0, 24, 48 and 60 h using a light microscope (CTR4000; Leica, Germany) at 100× magnification.
Transwell assays
Transwell chambers (8 μm pore size; Corning, New York, NY, USA) with or without a Matrigel (BD Biosciences) barrier added to the top chamber were used to detect the migratory and invasive abilities of CRC cells.15 HCT116 and LoVo cells stably expressing CDH11 or pcDNA 3.1 were collected, washed twice in serum-free medium, then2×104 cells suspended in 100 μl were added into the upper transwell chamber, and 800 μl of medium with 10% FBS was added to the lower chamber. After incubation at 37 °C and 5% CO2 for 48 h, cells at the lower side of the inserts were fixed in 4% paraformaldehyde for 30 min and stained with crystal violet (DC079; Genview, Beijing, China) for 20 min at room temperature. Non-migratory cells on the upper side of the chamber were wiped off with a cotton bud. Cells from 5 random fields were counted under a microscope at 400× magnification (CTR4000; Leica, Wetzlar, Germany).
Western blot analysis
A total of 40 mg of protein lysate was subjected to western blotting as previously described.16 The primary antibodies used were as follows: CDH11 (1:1,000, ab151302; Abcam, Cambridge, UK), cleaved-caspase 3 (1:500, WL03089; Wanleibio, Shenyang, China), cleaved-caspase 9 (1:500, WL01838; Wanleibio, Shenyang, China), cleaved-PARP (1:1,000, 7851; Cell Signaling Technology), BAX (5023; Cell Signaling Technology), NF-κB (ab16502; Abcam, Cambridge, UK), phospho-NF-κB (3031s; Cell Signaling Technology), Bcl-2 (2872T; Cell Signaling Technology), Bcl-XL (2762S; Cell Signaling Technology), and β-actin (LK-ab008-100; Liankebio, China) was used as control. Anti-mouse IgG (7076, 1:3,000 Cell Signaling Technology) and anti-rabbit IgG (7074, 1:2,000 Cell Signaling Technology) horseradish peroxidase conjugate secondary antibodies were used. Protein blots were analysed using an enhanced chemiluminescence detection kit (ECL; Amersham Pharmacia Biotech, Piscataway, NJ, USA), and the membranes were visualized with a Las-4,000 Imaging System (Medical Systems, Fujifilm Global, Tokyo, Japan).
In vivo tumourigenicity
HCT116 cells stably expressing CDH11 or pcDNA 3.1 was (5×106 cells in 0.15 ml serum-free medium) were injected subcutaneously into six4-week-old nude mice (purchased from the Experimental Animal Center of Chongqing Medical University, China). The length and width were measured every 3 days using a microcaliper, and the tumour volume (mm3) was calculated using the following equation: volume =0.5× length × width2 When the tumour length reached 1.5 cm, the tumours were removed, and mice were euthanatized. All removed tumor were weighed immediately before fixing in 4% paraformaldehyde, dehydrated and embedded in paraffin for hematoxylin/eosin (HE) staining and immunohistochemistry. And Instructive notions with respect to caring for laboratory animals (which is released by the Ministry of Science and Technology of the People’s Republic of China in September 30th, 2006.) were followed for the welfare of the animals. The animal experiments were authorized by the Animal Ethics Committee of the Experimental Animal Center of the Chongqing Medical University, Chongqing, China.
Immunohistochemical staining
The tumour tissues from nude mice, CRC and the corresponding adjacent human tissues were fixed in 4% paraformaldehyde, dehydrated and embedded in paraffin. The tissue samples were sectioned into 4 μm slices, and dewaxed in a 60 °Cincubatorfor 2 h. Then, the sections were rinsedin xylene I (10 min), xylene II (10 min), xylene III (10 min), xylene IV (10 min) in sequence, and rehydrated through graded alcohol in the following order: absolute ethyl alcohol (7 min), 95%, 80%, and 70% (ethyl alcohol (5 min each)). After that, slides were boiled in citrate buffer solution for 20–25 min for antigen retrieval followed by cooling at room temperature (approximately 2–3 h), then washed three times with PBS (5 min each). The slides were then incubated in 3% hydrogen peroxide for 10 min to neutralize endogenous peroxidase activity, and washed three times with PBS, and then blocked with 5% FBS–PBS solution at room temperature for 15 min without washing. Then, the slides were incubated with the primary antibody at 4 °C overnight. The next day, the slides were warmed to room temperature for 1 h, then washed three times with PBS (5 min each) and incubated with a secondary antibody for 30 min at 37 °C. Then the slices were washed three times with PBS (5 min each) and incubated with horseradish peroxidase-labeled streptomycin anti-biotin antibody for 30 min at 37 °C.The slides were washed three times, followed by colour development with DAB (30 s).The slides were washed with water, and the cell nuclei were dyed using hematoxylin (5 s). The slides were washed with water (15–30 min), followed by observation using light microscopy. Images were observed under a microscope at 400× light magnification.12
Statistical analysis
SPSS 19.0 statistical software (SPSS Inc. Chicago, IL, USA) was performed for statistical analyses. Data were analyzed with a Student’s t-test (independent-samples t-test) and the chi-square (also termed ×2) test as appropriate. Differences were considered statistically significant with a value of p<0.05.
Results
CDH11 expression is downregulated in CRC cell lines and tissues
The expression levels of CDH11 in HCT116, HT-29, LoVo, SW480, CaCo-2 and RKO colorectal tumour cell lines and normal colorectal tissues were determined using RT-PCR. CDH11 expression was distinctly suppressed in 5 of the 6 CRC cell lines, and CDH11 was significantly expressed in the CaCo-2 cell line (Figure 1A). The results also showed strongly expression of CDH11 in the 4 normal colorectal tissues (Figure 1A). In addition, the messenger RNA (mRNA) expression of CDH11 in 16 pairs of CRC and para-carcinoma tissues was measured by quantitative RT-PCR (qRT-PCR). Obviously, the expression of CDH11 was downregulated in tumour tissues compared with the corresponding adjacent tissue (p<0.05) (Figure 1D). CDH11 was located predominantly in the cell membrane. Furthermore, protein expression of CDH11 was examined in 21 paired CRC tissues and appropriate surgical-margin tissues (adjacent tissues) by immunohistochemistry (IHC). We observed that the CDH11 expression level was higher in appropriate surgical-margin tissues than in carcinoma tissues in 15 of the 21 paired samples (Figure 1F). The CDH11 expression levels and the clinicopathological characteristics of CRC patients are shown in Table 2.
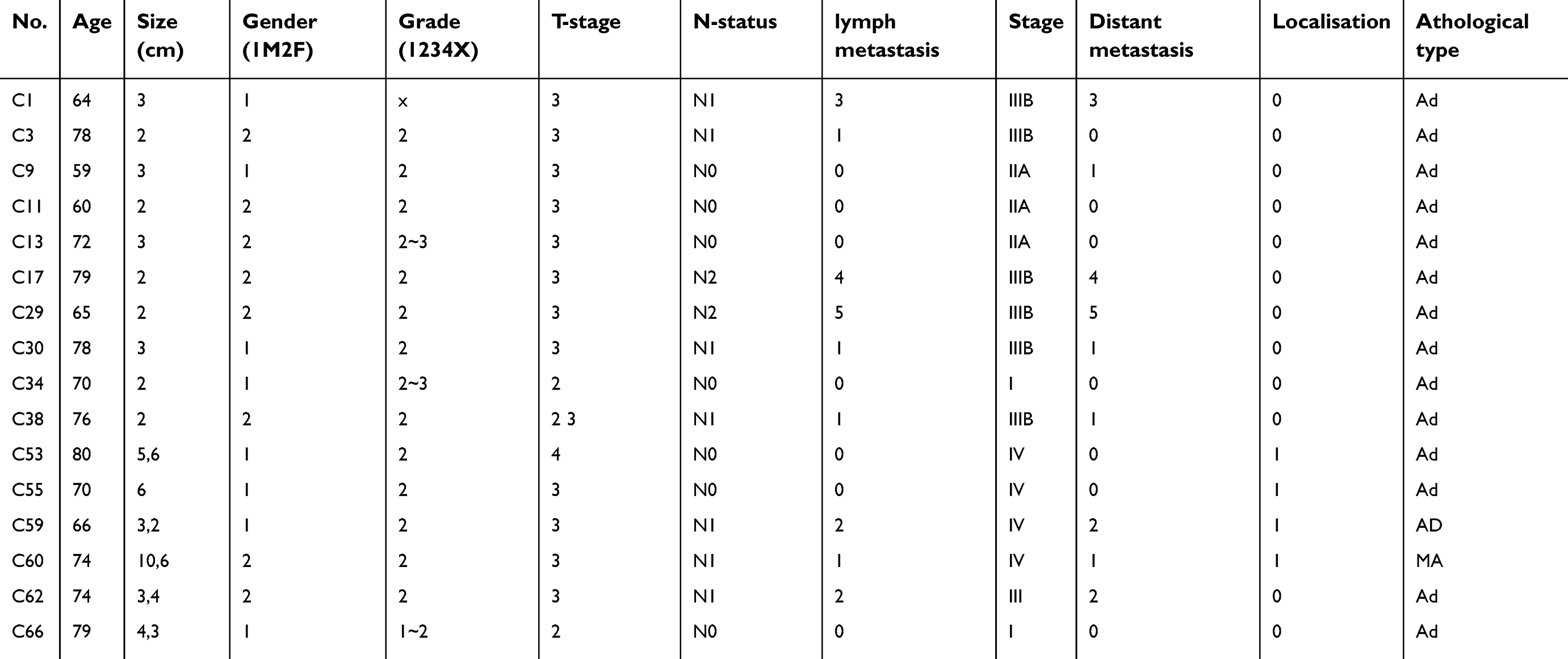 | Table 2 The clinical information of colorectal carcinoma tissues |
 | Figure 1 The expression and the methylation status of CDH11 in CRC cell lines, primary colorectal and normal colorectal tissues. (A) Expression levels of CDH11 mRNA in CRC cell lines and normal colorectal tissues and the methylation status of CDH11 in CRC cell lines. (B) Restoration of CDH11 expression by TSA, Aza and the combination CRC cell line LoVo and HCT116, and performed by RT-PCR. “M” indicates methylated CDH11; “U” indicates unmethylated CDH11. (C) Representative methylation of CDH11 in primary colorectal tumour. (D) CDH11 expression levels in LoVo and HCT116 cells stably expressing CDH11 or pcDNA 3.1 and 16 pairs of CRC and adjacent tissues were measured by quantitative RT-PCR (qRT-PCR). (E) Representative images of CDH11 IHC in paried CRC tissues and adjacent tissues (magnification ×200 and ×400).Abbreviations: CA, colorectal tumor adjacent tissues; CRC, colorectal cancer tissues. |
The CDH11 expression in CRC was downregulated due to promoter methylation and restored by its demethylation
Previous analysis showed that CDH11contains a typical CpG island, and suggested that CDH11 was a likely subject of methylation-mediated silencing.3 To identify whether CDH11 silencing or downreglation in CRC was due to promoter methylation, CRC cell lines LoVo and HCT-116 were treated with TSA, Aza and their combination, and then RT-PCR and MSP were performed. The results showed that its expression was restored together with increasing unmethylated alleles of the CDH11 promoter (Figure 1B). Furthermore, we analysed the methylation statues of CDH11 by MSP in 64CRC tissues. The results of MSP showed that CDH11 was methylated in 75.00% (48/64) of CRC tissues (Figure 1C), and revealed that the CDH11 methylation level was specifically high in the bulk of the CRC tissues.
Overexpression of CDH11 suppresses CRC cell proliferation and colony formation
The CCK-8 and the colony formation assays were used to determine the viability-inhibiting effect of stably expressing CDH11 in HCT116 and LoVo CRC cell lines. Overexpression of CDH11 in HCT116 and LoVo cell lines was ascertained by RT-PCR and western blotting (Figure 2A and B).CCK-8 assay showed that overexpression of CDH11 in HCT116 and LoVo cell lines resulted in a significant decrease in cell proliferation at 24, 48 and 72 h compared with the controls (p<0.05) (Figure 2C). Furthermore, the colony formation assay showed that cell transfection with CDH11 resulted in markedly fewer and smaller colonies, and the colony numbers were reduced by 50–55% compared with transfection with the empty vector in HCT116 and LoVo cells (p<0.001) (Figure 2D and F). These results indicated that CDH11 inhibited growth and viability in colorectal cancer.
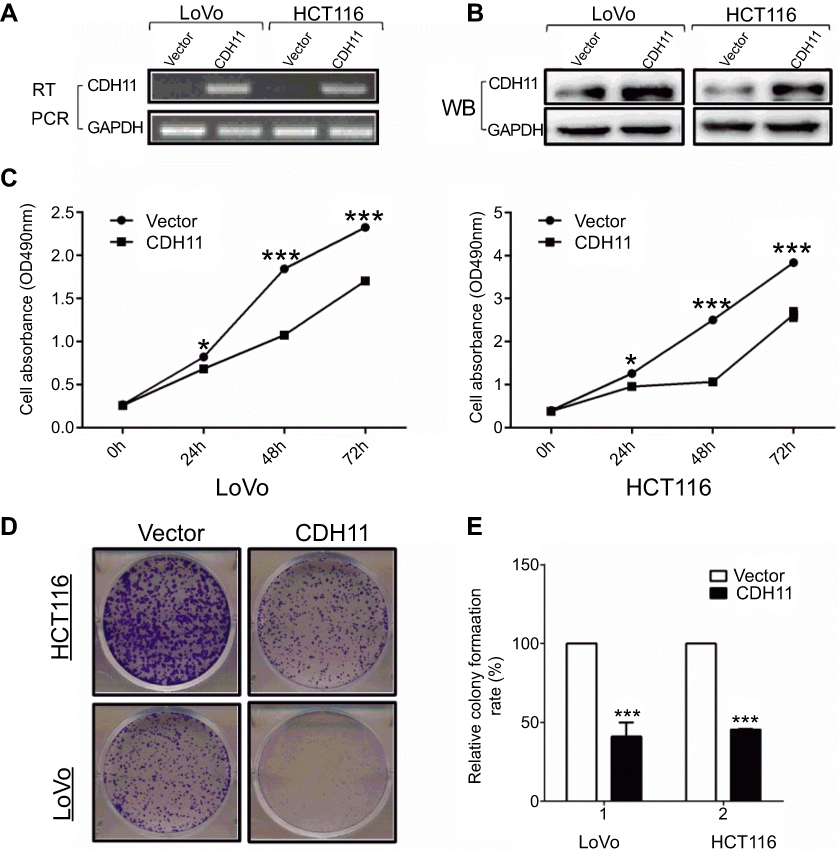 | Figure 2 Overexpression of CDH11 inhibits the cell proliferation and colony formation in LoVo andHCT116 cell lines. (A) Expression of CDH11 in vector- and CDH11-transfected cells by RT-PCR. (B) Expression of CDH11by western blot analysis in vector- and CDH11-transfected in CRC cells. (C) CCK-8 assay for cell proliferation of vector-and CDH11-infected CRC cell lines. ***p<0.001, *p<0.05. (D) Representative colony formation assay in LoVo and HCT116 cells infected with the control vector and CDH11-expressing vector. (E) Quantitative analysis of colony formation (***p<0.001). |
CDH11 induces cell cycle arrest in the G0/G1 phase and apoptosis in CRC cell lines
To determine how CDH11 affects cell proliferation in CRC, the cell cycle and apoptosis analysis were performed by flow cytometry. The result shows a significantly increased number of CDH11-expressing cells accumulating in the G0/G1 phase of the cell cycle compared with the control cells (p<0.01, p<0.001) (Figure 3A), indicating that the inhibition of cell proliferation by CDH11 was likely mediated by a cell cycle delay at theG0/G1 phase. Subsequently, the rate of apoptosis was detected using Annexin V-FITC/PI staining assay. The flow cytometry results confirmed that the number of Annexin V-PI-positive cells markedly increased in CDH11-transfected cells compared with controls (p<0.001 and p<0.05) (Figure 3B). Overall, these results suggested that the inhibition of cell proliferation by CDH11 is due to its mediation of the cell cycle arrest at the G0/G1 phase and induction of apoptosis.
 | Figure 3 Flow cytometric analysis. (A) Overexpression of CDH11induced LoVo and HCT116 cell cycle arrest at G0-G1. Left: Representative distribution of cell cycles. Right: statistical summary of flow cytometry data. Values were assessed by Student’s t-test. The graph displays the mean ± SD of three independent experiments (**p<0.01); (***p<0.001). (B) The apoptosis rate of control vector andectopic expression of CDH11 in LoVo and HCT116 cell was detected by flow cytometric analysis with Annexin V-FITC and PI-staining (***p<0.001). (D) Western blot analysis of four classic markers of apoptosis, cleaved-PARP, Bax, cleaved-caspase 3, cleaved-caspase 9 in vector- and CDH11-transfectedLoVo and HCT116 cells, β-actin as a control. |
CDH11 inhibits cell migration and invasion in colorectal tumour cells
Wound-healing and transwell assays were performed to further investigate the effects of CDH11 on cell migration and invasion in CRC. The results of wound-healing assay showed that, compared with the vector-transfected cells, CDH11 stablye-expressing cells migrated along the wound borderline significantly slower, as observed for cells at LoVo 0 h, 24 h, 48 h and 72 h (p<0.001) and HCT116 cells at 0 h, 24 h,48 h and 60 h (p<0.05) (Figure 4A and B). The transwell assay both with and without a Matrigel barrier further illustrated that the numbers of migratedor invaded CDH11-overexpressing in LoVo and HCT116 cells were significantly reduced compared with control groups(p<0.001) (Figure 4C and D). These results determined that CDH11 inhibits cell migration and invasion in colorectal tumour cells.
 | Figure 4 Ectopic expression of CDH11 inhibited the cell motility in LoVo and HCT116 cells. (A and B) Representative images of wound healing assay were captured at 0, 48 or 72 h, and quantitative analysis of migration length of vector- and CDH11-transfered LoVo and HCT116 cells, shown as means ± SD. ***p<0.001, **p<0.01, *p<0.05. And the suppressive effects of CDH11 on cell migration and invasion in CRC cells were investigated by transwell assays. Representative images of transwell (C) invation and (D) migration assays (magnification ×400) and quantitative analysis of the number of migrating and invasion cells (***p<0.001). |
CDH11 suppresses the growth of tumuor xenografts in nude mice
To further identify if CDH11 inhibits the CRC growth, in vivo tumourigenicity test were carried out. Stably-transfected and vector-transfected HCT116 were injected to form tumours in nude mice (Figure 5A). Tumours were removed from nude mice 19 days after injection (Figure 5B), the measurement results showed that the mean sizes and weights of tumours formed CDH11 stable cells were markedly lower compared with the control groups (p<0.001, p<0.05) (Figure 5C and D). Hematosylin & eosin (H&E) staining and immunohistochemistry (IHC) were performed to analyse the tumour features and expression of CDH11 protein and proliferation-associated protein Ki-67 (Figure 5E). Ki-67 expression was significantly downregulatied in tumours overexpressing CDH11compared with control. These data confirmed that CDH11acts as a TSG in CRC tumourigenesis in vivo.
 | Figure 5 CDH11 suppresses the growth of tumor xenografts in nude mice. (A and B) Images of tumor xenografts in nude mice. (C) Tumor growth curve for vector-and CDH11-infected tumors in nude mice (*p<0.05). (D) Tumor weight of CDH11-infected cells in nude mice compared with control tumors (*p<0.05). (E) Representative images of IHC analysis of the expression of Ki67and HE staining in tumours from nude mice (magnification ×400). |
CDH11 antagonized the NF-kB signaling pathways
A previous study has shown that CDH11 functions as a pro-apoptotic tumour suppressor by antagonizing the Wnt/β-catenin and AKT/Rho A signaling pathway.3 Several reports have suggested that NF-kB signaling is frequently affected in CRC.17–20 And Bcl-2 and Bcl-XL are downstream target genes of the NF-kB pathway. Here, we investigated whether CDH11 functions as a tumour suppressor through the NF-kB signaling pathway. Our results showed that phospho-NF-kB were markedly downregulated by overexpressing CDH11. Furthermore, overexpression CDH11 resulted in an obvious reduction of Bcl-2 and Bcl-XL. All of those indicated that CDH11 suppressed the CRC carcinogenic process by inactiving NF-kB signaling pathway (Figure 6).
 | Figure 6 Overexpression of CDH11 antagonized NF-kB signaling pathway. Western blot was performed using antibodies against NF-kB, phospho-NF-kB and its downstream targets Bcl-2 and Bcl-XL; β-actin was used as a control. |
Discussion
Cadherin-11 expression has been studied in many solid tumors. But CDH11 in CRC has not been still well elucidated. Increasing evidence has revealed that CDH11 plays a momentous role in the development of a broad series of human malignancies, and identified CDH11 as a functional tumour suppressor which is commonly silenced by promoter methylation.3 Previous studies have shown that CDH11 inhibits invasion and proliferation in head and neck cancer,21 osteosarcoma,8,22 glioma,23 melanoma24 and bladder cancer.25 The correlation between the function of CDH11 and related mechanisms in colorectal cancer remained unclear. In this study, we discovered that CDH11 was expressed in normal colon tissues, but frequently silenced or downregulated in CRC cell lines. These above results suggested that the expression of CDH11 is downregulated in CRC.
It is known that promoter methylation is a primary mechanism involved in gene changes such as gene absent or gene downregulation, which has been found in multiple human tumours. The results of this study suggested that the expression of CDH11 in CRC is epigenetically inactivated by promoter-specific methylation in CRC. The results of MSP revealed that the CDH11 methylation level was specifically high in the bulk of the CRC tissues. And the demethylation treatment A + T assay showed that its expression was restored together with increasing unmethylated alleles of the CDH11 promoter.
Previous studies reported that CDH11 inhibited tumour cell clonogenicity and induced apoptosis.3 We investigated the biological functions of CDH11 by both in vivo and in vitro assays. The results of the present study ascertain that overexpression of CDH11 induces cell cycle arrest in the G0/G1 phase and apoptosis and suppresses CRC cell proliferation and colony formation in both HCT116 and LoVo CRC cell lines. In addition, it has been reported that ectopic-expression of CDH11 inhibits invasion in head and neck cancer.21 Consistent with these previous reports, we found that ectopic-expression of CDH11 inhibited migration and invasion in colorectal tumour cells. Subsequently, an in vivo tumourigenicity test in nude mice was carried out to confirm the inhibitory effect of CDH11 on CRC growth. In short, all these results together strongly suggest that CDH11 functions as a tumour suppressor in CRC cells.
Previous study found that CDH11 functions as a TSG through antagonizing Wnt/β-catenin and AKT/Rho A signaling.3 And some early studies showed that the re-expression of CDH1, one member of cadherin superfamily, may depend on NF-kB activation, and NF-kB promotes migration and invasion by re-expression of CDH1 in cholangiocarcinoma cells.26 Whether CDH11, the member of cadherin superfamily, performs by a similar mechanism that acts on the NF-kB pathway in CRC remains unclear. In this study, we investigated whether CDH11 functions as a tumor suppressor in CRC through the NF-kB signaling pathway.
Nuclear factor kappa B (NF-kB), as a pleotropic transcription factor, regulates expression of a number of genes that promote multiple cancer cells growth, survival and neoplastic transformation.27–30 In this signaling pathway, transcriptionally competent NF-kB is a heterodimer composed of p65 (NF-kB) and p50 subunits,31,32 and this heterodimer is insulated in the cytoplasm by p65(NF-kB) bound IkBα27,33 in unstimulated cells. Phospho-NF-kB is a key component in the NF-kB pathway, where it suppresses cell survival and growth, and Bcl-2 and Bcl-XL are downstream target genes of the NF-kB pathway.34,35 Our results showed that when compared to the empty vector group, the expression levels of phospho-NF-kB were markedly downregulated in the CDH11 group, and CDH11 resulted in an obvious reduction of Bcl-2 and Bcl-XL, indicating that CDH11 may affect the CRC carcinogenic process by inactiving NF-kB signaling pathway. Further rescue t trials are needed to identify the role of NF-kB signaling pathway in the CDH11 acting mechanism.
However, the target genes of CDH11 were not identified in this study, which may therefore assist in identifying its role in the progression of CRC. And the number of clinical samples of CRC used in this study was insufficient, so we could not determine the possible relationship between CDH11 methylation and the pathological features and survival prognoses of CRC. Further studies of more clinical samples are warranted to explore the mechanism of CDH11 and to confirm that CDH11 is a prognostic marker for CRC.
Conclusion
In summary, our study suggests that CDH11 downregulation in CRC results from promoter methylation. And CDH11 induces cell cycle arrest in the G0/G1 phase and apoptosis to suppressing CRC cell proliferation and colony formation, thus suppressing tumour cell proliferation, migration and invasion, probably by affecting the NF-kB signaling pathway. Overall, CDH11 may be considered as a functional TSG in CRC, and has the potential to serve as a valuable prognostic marker for colorectal cancer.
Abbreviation list
CDH11, cadherin-11; CRC, colorectal cancer; qRT-PCR, quantitative RT-PCR; MSP, methylation-specific PCR; TSG, tumor suppressor gene; aCGH, array comparative genomic hybridization; mRNA, messenger RNA; IHC, Immunohistochemistry; H&E, Hematosylin & eosin; NFkB, Nuclear factor kappa B; GAPDH, glyceraldehyde-3-phosphate dehydrogenase; DNMT, DNA methyltransferase; Aza, 5-Aza-2ʹ-deoxycytidine; TSA, trichostatin A; PBS, phosphate-buffered saline; PI, propidium iodide; WB, Western blot.
Acknowledgments
The authors thank Professor Qian Tao (the Chinese University of Hong Kong, Hong Kong, China) for generously providing CRC cell lines, plasmid and primers, and thank Professor Tingxiu Xiang technical assistance, and help with experimental design. This research was funded by National Natural Science Foundation of China, grant number 3142010391.
Disclosure
The authors report no conflicts of interest in this work.
References
1. Bray F, Ferlay J, Soerjomataram I, Siegel RL, Torre LA, Jemal A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin. 2018;394–424.
2. Rabeneck L, Horton S, Zauber AG, Earle C. Colorectal cancer. In: Cancer. Disease Control Priorities.
3. Li L, Ying J, Li H, et al. The human cadherin 11 is a pro-apoptotic tumor suppressor modulating cell stemness through Wnt/beta-catenin signaling and silenced in common carcinomas. J Cell Sci. 2012;31(34):3901–3912.
4. Margulis A, Zhang W, Alt-Holland A, Crawford HC, Fusenig NE, Garlick JA. E-cadherin suppression accelerates squamous cell carcinoma progression in three-dimensional, human tissue constructs. Cancer Res. 2005;65(5):1783–1791. doi:10.1158/0008-5472.CAN-04-3399
5. Andreeva AV, Kutuzov MA. Cadherin 13 in cancer. Genes Chromosomes Cancer. 2010;49(9):775–790.
6. Grady WM, Carethers JM. Genomic and epigenetic instability in colorectal cancer pathogenesis. Gastroenterology. 2008;135(4):1079–1099. doi:10.1053/j.gastro.2008.07.076
7. Assefnia S, Dakshanamurthy S, Guidry Auvil JM, et al. Cadherin-11 in poor prognosis malignancies and rheumatoid arthritis: common target, common therapies. Oncotarget. 2014;5(6):1458–1474. doi:10.18632/oncotarget.1538
8. Deng Z, Niu G, Cai L, Wei R, Zhao X. The prognostic significance of CD44V6, CDH11, and beta-catenin expression in patients with osteosarcoma. Biomed Res Int. 2013;2013:496193. doi:10.1155/2013/496193
9. Yao J, Deng B, Zheng L, Dou L, Guo Y, Guo K. miR-27b is upregulated in cervical carcinogenesis and promotes cell growth and invasion by regulating CDH11 and epithelial-mesenchymal transition. Oncol Rep. 2016;35(3):1645–1651. doi:10.3892/or.2015.4500
10. Carmona FJ, Villanueva A, Vidal A, et al. Epigenetic disruption of cadherin-11 in human cancer metastasis. J Pathol. 2012;228(2):230–240. doi:10.1002/path.4011
11. Wang C, Yue Y, Shao B, et al. Dickkopf-related protein 2 is epigenetically inactivated and suppresses colorectal cancer growth and tumor metastasis by antagonizing Wnt/β-catenin signaling. Cell Physiol Biochem. 2017;41(5):1709–1724. doi:10.1159/000471861
12. Xiang S, Xiang T, Xiao Q, Li Y, Shao B, Luo T. Zinc-finger protein 545 is inactivated due to promoter methylation and functions as a tumor suppressor through the Wnt/beta-catenin, PI3K/AKT and MAPK/ERK signaling pathways in colorectal cancer. Int J Oncol. 2017;51(3):801–811. doi:10.3892/ijo.2017.4064
13. Tao Q, Huang H, Geiman TM, et al. Defective de novo methylation of viral and cellular DNA sequences in ICF syndrome cells. Hum Mol Genet. 2002;11(18):2091–2102.
14. Pei L, He X, Li S, et al. KRAB zinc-finger protein 382 regulates epithelial-mesenchymal transition and functions as a tumor suppressor, but is silenced by CpG methylation in gastric cancer. Int J Oncol. 2018. doi:10.3892/ijo.2018.4446
15. Zhao L, Li S, Gan L, et al. Paired box 5 is a frequently methylated lung cancer tumour suppressor gene interfering beta-catenin signalling and GADD45G expression. J Cell Mol Med. 2016;20(5):842–854. doi:10.1111/jcmm.12768
16. Li C, Tang L, Zhao L, et al. OPCML is frequently methylated in human colorectal cancer and its restored expression reverses EMT via downregulation of smad signaling. Am J Cancer Res. 2015;5(5):1635–1648.
17. Elshaer M, Chen Y, Wang XJ, Tang X. Resveratrol: an overview of its anti-cancer mechanisms. Life Sci. 2018;207:340–349. doi:10.1016/j.lfs.2018.06.028
18. Hai Ping P, Feng BT, Li L, Nan Hui Y, Hong Z. IL-1beta/NF-kb signaling promotes colorectal cancer cell growth through miR-181a/PTEN axis. Arch Biochem Biophys. 2016;604:20–26. doi:10.1016/j.abb.2016.06.001
19. Puvvada SD, Funkhouser WK, Greene K, et al. NF-kB and Bcl-3 activation are prognostic in metastatic colorectal cancer. Oncology. 2010;78(3–4):181–188. doi:10.1159/000313697
20. De Simone V, Franze E, Ronchetti G, et al. Th17-type cytokines, IL-6 and TNF-alpha synergistically activate STAT3 and NF-kB to promote colorectal cancer cell growth. Oncogene. 2015;34(27):3493–3503. doi:10.1038/onc.2014.286
21. Piao S, Inglehart RC, Scanlon CS, Russo N, Banerjee R, D’Silva NJ. CDH11 inhibits proliferation and invasion in head and neck cancer. J Oral Pathol Med. 2017;46(2):89–97. doi:10.1111/jop.12471
22. Azarsina S, Otoukesh B, Taheriazam A, et al. Diagnostic investigations of PLA2G16 and CDH11 expression levels as independent prognostic markers of human osteosarcoma. Arch Med Sci. 2017;13(6):1347–1351. doi:10.5114/aoms.2016.59710
23. Delic S, Lottmann N, Jetschke K, Reifenberger G, Riemenschneider MJ. Identification and functional validation of CDH11, PCSK6 and SH3GL3 as novel glioma invasion-associated candidate genes. Neuropathol Appl Neurobiol. 2012;38(2):201–212. doi:10.1111/j.1365-2990.2011.01207.x
24. Bosserhoff AK, Ellmann L, Quast AS, Eberle J, Boyle GM, Kuphal S. Loss of T-cadherin (CDH-13) regulates AKT signaling and desensitizes cells to apoptosis in melanoma. Mol Carcinog. 2014;53(8):635–647. doi:10.1002/mc.22018
25. Lin YL, Gui SL, Ma JG. Aberrant methylation of CDH11 predicts a poor outcome for patients with bladder cancer. Oncol Lett. 2015;10(2):647–652. doi:10.3892/ol.2015.3337
26. Zhang K, Zhaos J, Liu X, et al. Activation of NF-B upregulates Snail and consequent repression of E-cadherin in cholangiocarcinoma cell invasion. Hepato-gastroenterology. 2011;58(105):1–7.
27. Jana A, Krett NL, Guzman G, et al. NFkB is essential for activin-induced colorectal cancer migration via upregulation of PI3K-MDM2 pathway. Oncotarget. 2017;8(23):37377–37393. doi:10.18632/oncotarget.16343
28. Wang S, Liu Z, Wang L, Zhang X. NF-kappaB signaling pathway, inflammation and colorectal cancer. Cell Mol Immunol. 2009;6(5):327–334. doi:10.1038/cmi.2009.43
29. Voboril R, Weberova-Voborilova J. Constitutive NF-kappaB activity in colorectal cancer cells: impact on radiation-induced NF-kappaB activity, radiosensitivity, and apoptosis. Neoplasma. 2006;53(6):518–523.
30. Baldwin AS
31. Ghosh S, Karin M. Missing pieces in the NF-kappaB puzzle. Cell. 2002;109(Suppl):S81–96.
32. Hassanzadeh P. Colorectal cancer and NF-kappaB signaling pathway. Gastroenterol Hepatol Bed Bench. 2011;4(3):127–132.
33. Hoffmann A, Baltimore D. Circuitry of nuclear factor kappaB signaling. Immunol Rev. 2006;210:171–186. doi:10.1111/j.0105-2896.2006.00375.x
34. Deeb D, Gao X, Liu YB, Pindolia K, Gautam SC. Pristimerin, a quinonemethide triterpenoid, induces apoptosis in pancreatic cancer cells through the inhibition of pro-survival Akt/NF-κB/mTOR signaling proteins and anti-apoptotic Bcl-2. Int J Oncol. 2014;44(5):1707–1715. doi:10.3892/ijo.2014.2325
35. Ghaderian SB, Hayati F, Shayanpour S, Beladi Mousavi SS. Diabetes and end-stage renal disease; a review article on new concepts. J Renal Inj Prev. 2015;4(2):28–33. doi:10.12861/jrip.2015.07
 © 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.