")
Back to Journals » The Application of Clinical Genetics » Volume 12
c.259A>C in the fibrinogen gene of alpha chain (FGA) is a fibrinogen with thrombotic phenotype
Authors Salomon O, Barel O, Eyal E, Shnerb Ganor R , Kleinbaum Y, Shohat M
Received 12 October 2018
Accepted for publication 7 January 2019
Published 28 February 2019 Volume 2019:12 Pages 27—33
DOI https://doi.org/10.2147/TACG.S190599
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Prof. Dr. Martin Maurer
Ophira Salomon,1 Ortal Barel,2 Eran Eyal,2 Reut Shnerb Ganor,3 Yeroham Kleinbaum,4 Mordechai Shohat2
1Institute of Thrombosis and Hemostasis, Sheba Medical Center, Sackler Faculty of Medicine, Tel Aviv University, Tel Aviv, Israel; 2Cancer Research Center, Wohl Institute of Translational Medicine, Sheba Medical Center, Sackler Faculty of Medicine, Tel Aviv University, Tel Aviv, Israel; 3The Bert W. Strassburger Lipid Center, Sheba Medical Center, Sackler Faculty of Medicine, Tel Aviv University, Tel Aviv, Israel; 4Diagnostic Imaging, Department of Radiology, Sheba Medical Center, Sackler Faculty of Medicine, Tel Aviv University, Tel Aviv, Israel
Introduction: Dysfibrinogenemia is a rare inherited disease that results from mutation in one of the three fibrinogen genes. Diagnosis can be misleading since it may present as a bleeding tendency or thrombosis and a specific coagulation test for diagnosis is not routinely available
Aim: To search for a new candidate gene of thrombophilia in a family with three generations of arterial and venous thrombosis.
Methods: Whole exome sequencing followed by Sanger validation and segregation analysis was carried out. In addition, structural modeling was performed. Screening for thrombophilia along with blood counts, prothrombin time, activated partial thromboplastin, thrombin, reptilase time, and fibrinogen was done in each patient.
Results and discussion: A missense c.259A>C, p.K87Q (g.chr4: 155510050A-C) (rs764281241) in FGA gene was found in all three siblings without any other known thrombophilia marker to explain thrombosis in all three siblings. It is expected to be damaging by six out of seven prediction programs and is very rare in the entire population with Exac=0.000008.
Conclusion: The occurrence of the c.259A>C mutation in FGA may well explain the thrombosis phenotype of the affected family and is suggested as a new marker for thrombophilia phenotype.
Keywords: dysfibrinogenemia, thrombophilia, thrombosis, exome sequencing, structural modeling
Introduction
Fibrinogen is a soluble plasma glycoprotein that circulates as a dimer and plays a central role in coagulation.1 It converts into fibrin by thrombin upon activation of coagulation pathway through tissue factor pathway or contact activation and is polymerized to form the main backbone of the clot. The fibrinogen is secreted by the hepatocytes as a hexamer Aα2Bβ2ɤ2 encoded by three different fibrinogen genes (FG) FGA, FGB, and FGG, respectively.2
Dysfibrinogenemia is a rare qualitative disorder, inherited most often as an autosomal dominant disease and caused by mutations in one of the three FG.3 Diagnosis of dysfibrinogenemia can be misleading since there is no specific coagulation test available for diagnosis up to now. The usual tests such as prothrombin time, partial thromboplastin time, thrombin time, and reptilase time can be intact in patients with dysfibrinogenemia. The thromboelastometry and rotational thromboelastometry, which are part of the global coagulation assays evaluating the kinetics of clot formation, maximum clot firmness, and rate of fibrinolysis, are also not specific tests even though they may point to the presence of hypofibrinogenemia. In addition, the turbidometric assay test, which assesses the kinetics of fibrin polymerization and clot lysis, is not a useful tool for diagnosis of dysfibrinogenemia.2 Besides, difficulties in the diagnosis of dysfibrinogenemia are due to the fact that patients can be asymptomatic and present with bleeding or thrombosis. Indeed, the variable clinical phenotype could be explained by the various mutations resulting in changes in the structure of fibrinogen, clot strength, stability, and/or tendency to fibrinolysis.4
Dysfibrinogenemia as a risk factor for thrombosis is quite rare and estimated to account for <1% of inherited thrombophilia.5 One of the mechanisms contributing to thrombosis is defective binding of thrombin to altered fibrin molecule resulting in an excess of free thrombin in the circulation.6 Another assumption is decreased exposure to tissue plasminogen activator binding site attributed to conformational change in dysfibrinogenemia resulting in reduced fibrinolysis.7
We herein report a family with a history of venous and arterial thrombosis associated with heterozygous missense mutation in α chain of FG. Modeling analysis and homologous sequence alignment predict a change in the function of fibrinogen. The site of the mutation may well explain the impact of the specific mutation on thrombosis phenotype.
Materials and methods
Family pedigree
The family pedigree is presented in Figure 1. In this family, three siblings (IV-3, IV-4, IV-5) (“probands”) had a history of thrombosis.
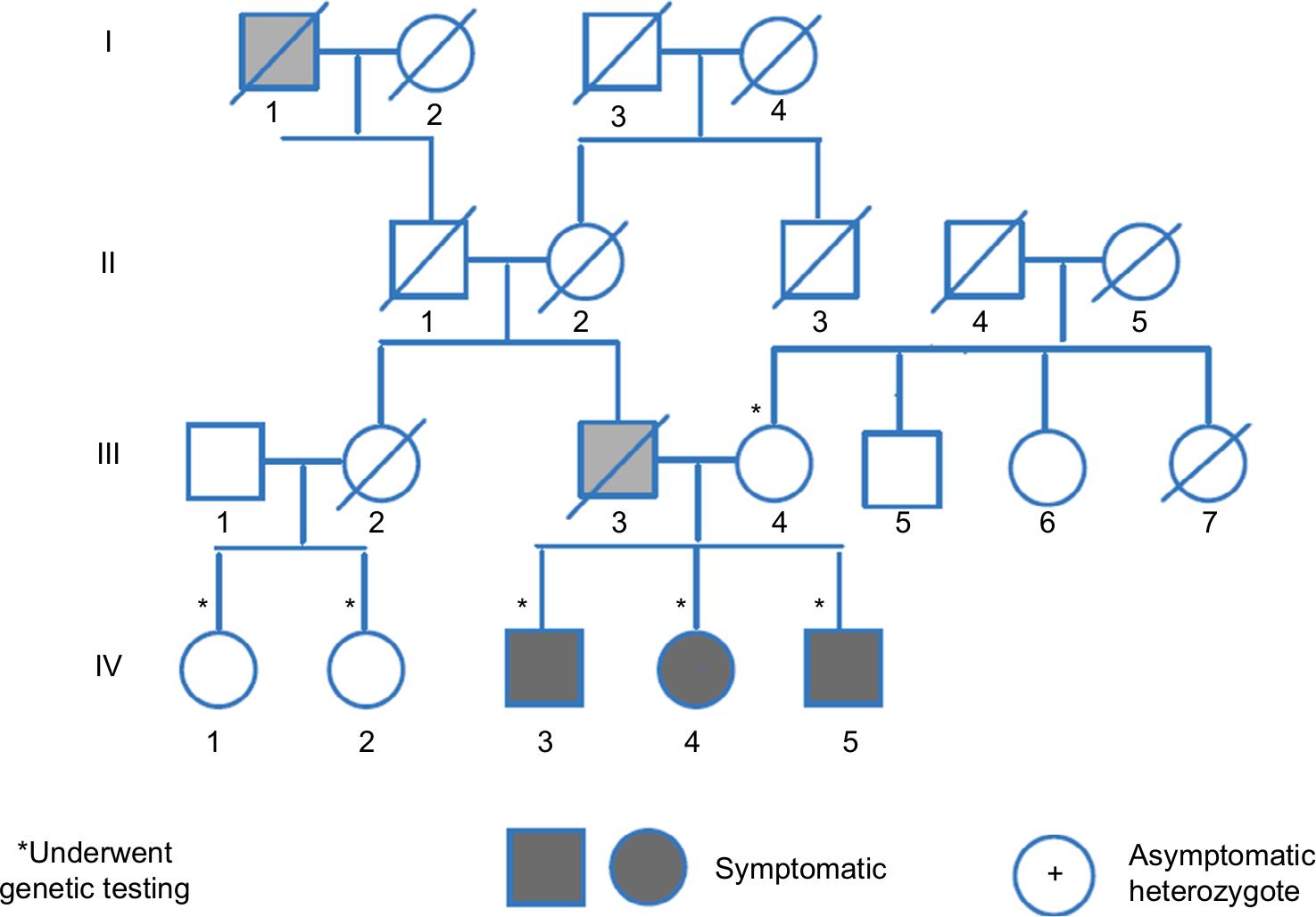 | Figure 1 Family pedigree. Notes: Roman numbers to the left of the figure indicate generations. The black symbol is for family members who experienced thrombosis. The asterisk next to the Arabic numbers indicates the family member tested for fibrinogen variant. Deceased family member is indicated by a slash thought the symbol. |
The probands’ father (III-3) had several myocardial infarctions beginning at the age of 39 as well as a severe peripheral vascular disease. He was a heavy smoker since the age 14, but did not have any additional risk factors for atherosclerotic vascular disease. The mother is healthy.
The probands’ paternal great-grandfather (I-1) had amputation of his leg at young age. Individual IV-5 had his first deep vein thrombosis (DVT) aged 30 years following a transatlantic flight. About 6 years later, he had recurrent DVT, and ultrasound Doppler study at that time revealed a superficial femoral, popliteal, and tibial vein thrombosis of left leg. He was then treated with coumadin for 6 months once ultrasound Doppler study turned normal. Two years later, he developed superficial vein thrombosis at the arm at the site where a venflon was put on. He was treated with enoxaparin for 4 weeks, and 2 months later, he had pain in his left leg along with elevated D-dimer. Repeat ultrasound Doppler revealed recurrence of DVT. Since then he is being treated with coumadin.
His brother (individual IV-3) at the age of 39 experienced the first DVT in his right leg involving similarly the superficial femoral, popliteal, and tibial veins. He was treated for 6 months with coumadin and rigorously kept prothrombin time-international normalized ratio at therapeutic levels. Three months later, he had a recurrent DVT, and coumadin was resumed.
Their sister (individual IV-4) had one incident of left popliteal and tibial vein thrombosis at age 30 while she was on contraceptives.
Thrombophilia screening in all three siblings was negative, and blood count was normal.
The only living adult paternal relatives are two first cousins (IV-1, IV-2) who have been asymptomatic for clotting or bleeding events.
The whole exome sequencing was approved by the ethics committee at the Sheba Medical Center and written informed consent was obtained according to the Declaration of Helsinki. Sheba Medical Center has provided waiver of approval for publication of one case study. The family has provided written informed consent for the publication of the family case details.
Whole exome sequencing
Whole exome sequencing was carried out on the three affected siblings (IV-3, IV-4, IV-5). Genomic DNA extraction, exome enrichment, and sequencing were completed at BGI Corporation. Exome enrichment was achieved by the Agilent Exome Kit following manufacturer’s guidelines. Sequencing was performed using the Illumina HiSeq technology to generate paired-end reads with average coverage of 120×. The data obtained by whole exome sequencing were analyzed using our own software and using additional online databases such as dbSNP 132.
All significant changes present in all three sibs were further investigated.
Validation and segregation analyses
For validation and segregation analyses, PCR primers were designed to amplify the regions flanking the variant in FGA gene. PCR products were sequenced using forward and backward internal primers to determine the noted regions. Segregation analysis of the mutation was performed on all members of the extended kindred by cycle sequencing (Thermo Fisher Scientific, Waltham, MA, USA). PCR verification was performed on the mother (III-4) and two first cousins (IV-1, IV-2).
Structural modeling
Structural modeling was done using the I-TASSER server8 by fold recognition, with PDB 3zlc9 found to be the best template.
Secondary structure prediction was done using PROFsec method by the PredictProtein server.10 Disorder prediction was conducted by the DISprot program11 and supported by B-factor flexibility predictions PRFbval.12 Functional predictions data of Polyphen2,13 Sift,14 Mutation Assessor,15 Mutation Taster,16 PROVEAN,17 and LRT were collected from the dbNFSP database.18 Jmol19 was used for molecular graphics and creation of 3D images. Consurf20 was used to obtain conservation scores.
Blood tests
Blood samples were evaluated for blood counts, prothrombin time, activated partial thromboplastin, thrombin and reptilase times, fibrinogen antigen (latex immunoassay), and fibrinogen activity (Claus method).
Thrombophilia screening was performed and search for antithrombin, protein C, protein S, factor V Leiden, prothrombin G20210A, and antiphospholipid antibodies (lupus anticoagulant, anticardiolipin antibody, and anti β2 glycoprotein 1) as described previously.21
Results
We have identified a missense c.259A>C, p.K87Q (g.chr4:155510050A-C) (rs 764281241) in the FGA gene. This variant was found in all three siblings with normal antigenic and activity of fibrinogen. The mutation is predicted to be damaging by six out of seven programs and is very rare in the general population with Exac=0.000008. There were no other variants that may explain the thrombotic phenotype and common to all three sibs. In addition, we analyzed the data for each affected sib separately. Based on this information, we classified the variant as a pathogenic or likely pathogenic.
The effect of the mutation on the protein is not entirely clear by the current knowledge. The mutation is located in the N-terminal region well within the elongated coiled-coil region. Following signal peptide cleavage, position 87 becomes position AαLys68 in the mature protein. According to the 3PGP crystal structure, the wild-type lysine side chain is facing outside and does not participate in significant intraprotein interactions. This is also reflecting in the results of stability changes prediction, which suggests only marginal changes in thermostability. The local sequence NRINKLKNSL does not match the classical heptad repeat of the coiled-coil proteins (hxxhcxc where c is charged residue and h hydrophobic), as two aspargines in positions –1 and –4 relative to the mutated lysine take the place of hydrophobic residues. Most function prediction tools, however, suggest that the mutation is damaging (Table 1). Most likely, the mutation in this elongated exposed region affects interactions with other factors. Several pathogenic mutations were already reported in the immediate vicinity of the current variant (Figure 2), including mutation in the adjacent AαLeu69. Such clustering of mutations in a nonglobular region may also suggest that this region participates in intermolecular interactions. The region of the mutation is relatively conserved (Figure 2), and position 87 (AαLys68 on the mature protein) gets score of 8/9 by Consurf.
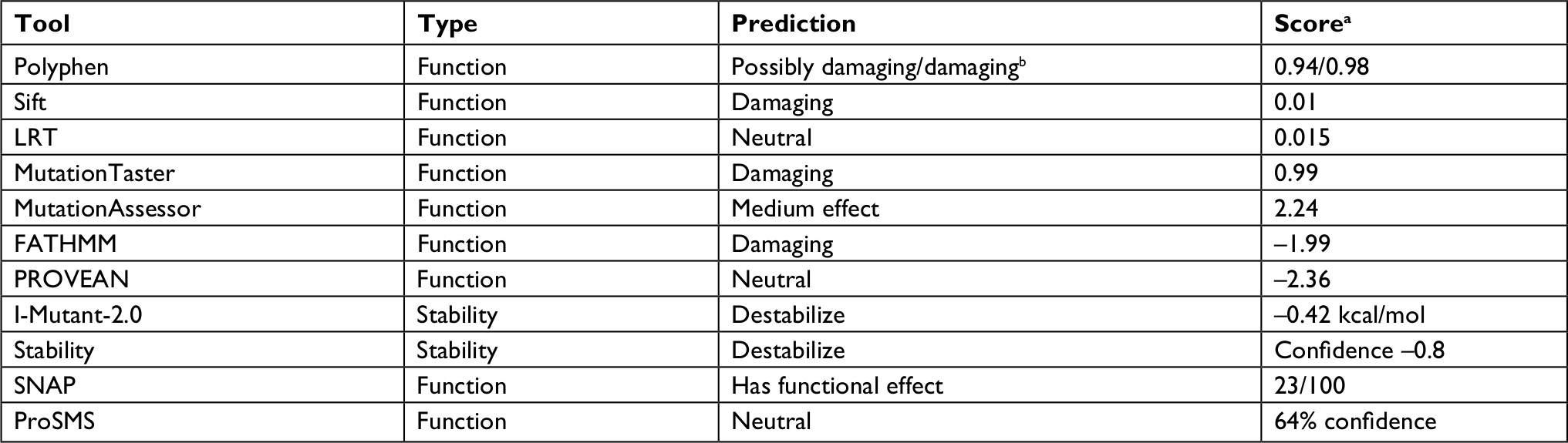 | Table 1 Prediction of functional consequence and stability change caused by the Lys87Gln mutant Notes: aThe score is specific to each program. bPolyphen provides different predictions based on the exact classifier and transcript used. |
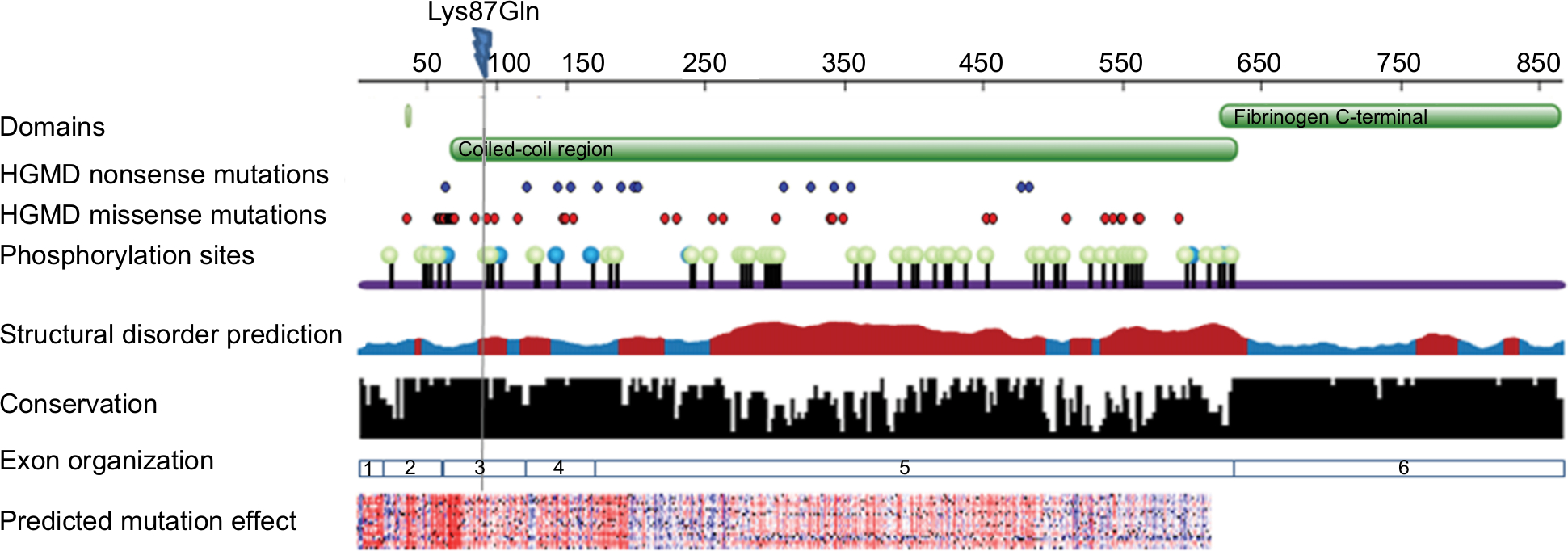 | Figure 2 FGA sequence and structural information. Notes: Sequence panel showing domain organization, known pathological mutations (from HGMD), phosphorylation sites (from phosphosite), disorder prediction (brown indicates disordered regions and blue ordered regions), conservation pattern (Consurf), exons organization and prediction of functional effect of all possible amino acid substitutions (SNAP, red indicates deleterious changes and blue benign changes). Lys87Gln (indicated on top) is located in a conserved region and is expected to have a functional effect. |
The mutation was not found in the mother of these three siblings, and we concluded that it was from the paternal side. Test done on the two first paternal cousins demonstrated the mutation in one of them. Both of them had Doppler ultrasound to the lower limbs to rule out asymptomatic DVT, and the results were normal.
The fibrinogen variants with mutations in the FGA gene and thrombotic phenotype that had been described are presented in Table 2. Notably, compound heterozygosity in the FGA gene or with FGB or FGC were excluded, likewise combined mutations with other thrombophilic markers.
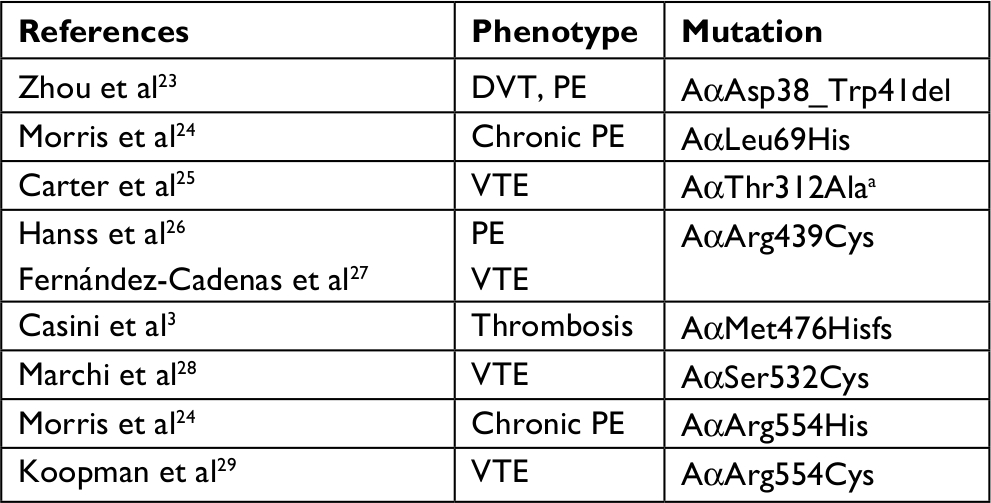 | Table 2 Variant of fibrinogens with mutations in p.FGA and thrombotic phenotype Note: aCommon variant. Abbreviations: DVT, deep vein thrombosis; PE, pulmonary embolism; VTE, venous thromboembolism. |
We then sought to analyze whether the mutated proteins share structural characteristics that could theoretically explain structural dysfunction. Unfortunately, we could only study the mutated Leu 69 since the PDB file 3ghg contains only 200 amino acids. AαLeu69 and AαLys68 are adjacent, indicating the importance of this local region on the coiled-coil (Figure 3). Their function might be related to stability of the coil (likely the role of Leu69) or interactions with transfactors (as might be the role of AαLys68 whose side chain is facing outside).
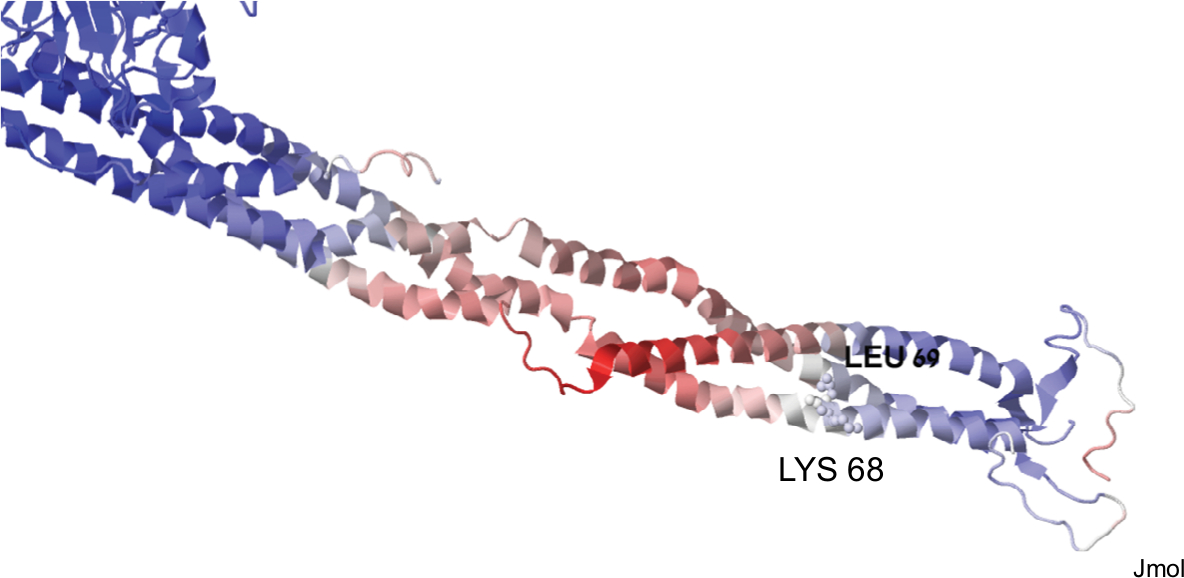 | Figure 3 A graphic representation of structural information on the two mutations of p. FGA with thrombotic phenotype based on PDB file 3ghg. |
Discussion
Detecting dysfibrinogenemia is sometimes difficult since it cannot be identified by standard coagulation assays and family members that are carrying the same genotype do not express the same phenotype.
The family described carry the mutation c.259A>C in the fibrinogen gene with normal antigenic and activity of fibrinogen and without any other thrombophilia parameters to explain thrombosis in all three siblings (IV-3, IV-4, IV-5). The first thrombotic events occurred in the young, and two of the three siblings are on permanent anticoagulants since an attempt to stop anticoagulants resulted in recurrence of vein thrombosis.
The variant c.259A>C found in the three affected siblings and their affected father seems to be damaging based on several popular prediction programs such as Polyphen, Sift, and MutationTaster (Table 1). In addition, we did not find any variant with any significant pathogenicity that may be associated with clotting/bleeding tendency in all three siblings even when the analysis was done in each one of them separately. Furthermore, this variant is distributed in this family according to the phenotype distribution, except a yet unaffected first cousin. This unaffected cousin with this mutation can be explained by the known wide variability in expression and penetrance as well as in the age of onset that is so typical to mutations in almost all autosomal dominantly expressed genes causing predisposition of common complications/diseases in adults. Besides, clinical phenotype is frequently modified by coincident risk factors for bleeding or thrombosis causing variable penetrance and expressivity.22
The pathogenicity in this case is also supported by the rarity of this variant. The molecular mechanism, which causes the pathogenicity, is not entirely clear, but we suggest that the variant affects intermolecular interactions with other factors, which bind to this coil-coiled region. Based on all these characteristics, we think this variant is “pathogenic”, resulting in a clotting tendency causing mutation in this family. Since the antigenic and activity of fibrinogen were in accordance, the mutation c.259A>C in the fibrinogen gene does not result in dysfibrinogenemia but with thrombotic phenotype.
Up to now, in the majority of the mutations reported in the FGA gene with thrombotic phenotype, there is replacement of cysteine and histidine but not in the mutation we describe. Looking for the structural context of these positions, we could only study AαLeu69, an immediate neighbor of AαLys68. This local region is apparently nontolerant to variations, which severely affect fibrinogen function. It is still left to explore the exact mechanism, either by disruption of transinteractions or by affecting the coiled-coil stability.
Conclusion
Whole exome sequencing along with validation and segregation and structural modeling greatly expands the opportunities for revealing new candidates of thrombophilia in private families.
Keypoints
- Whole exome sequencing with segregation analysis and structural modeling is an important toolbox for searching for new thrombophilic candidate genes.
- In families with severe thrombotic events and negative thrombophilic screening, such an approach should be applied.
- Structural modeling could help to differentiate between bleeding and thrombophilia in genes with biphenotype presentations.
Acknowledgments
This study was supported by the Adler Chair for Pediatric Cardiology, Sackler Faculty of Medicine, Tel Aviv University. The authors thank Professor Gideon Rechavi for review and critical and constructive comments on the manuscript. We would like to thank the family for their contribution to medical knowledge and allowing publication of the family case details.
Disclosure
The authors report no conflicts of interest in this work.
References
Ariëns RA. Fibrin(ogen) and thrombotic disease. J Thromb Haemost. 2013;11(Suppl):294–305. | ||
Neerman-Arbez M, de Moerloose P, Casini A. Laboratory and genetic investigation of mutations accounting for congenital fibrinogen disorders. Semin Thromb Hemost. 2016;42(4):356–365. | ||
Casini A, Blondon M, Lebreton A, et al. Natural history of patients with congenital dysfibrinogenemia. Blood. 2015;125(3):553–561. | ||
Casini A, de Moerloose P. Can the phenotype of inherited fibrinogen disorders be predicted? Haemophilia. 2016;22(5):667–675. | ||
Tarumi T, Martincic D, Thomas A, et al. Familial thrombophilia associated with fibrinogen Paris V: Dusart syndrome. Blood. 2000;96(3):1191–1193. | ||
Liu CY, Nossel HL, Kaplan KL. The binding of thrombin by fibrin. J Biol Chem. 1979;254(20):10421–10425. | ||
Yonekawa O, Voskuilen M, Nieuwenhuizen W. Localization in the fibrinogen gamma-chain of a new site that is involved in the acceleration of the tissue-type plasminogen activator-catalysed activation of plasminogen. Biochem J. 1992;283(1):187–191. | ||
Yang J, Zhang Y. I-TASSER server: new development for protein structure and function predictions. Nucleic Acids Res. 2015;43(W1):W174–W181. | ||
Biterova EI, Svärd M, Possner DD, Guy JE. The crystal structure of the lumenal domain of Erv41p, a protein involved in transport between the endoplasmic reticulum and Golgi apparatus. J Mol Biol. 2013;425(12):2208–2218. | ||
Yachdav G, Kloppmann E, Kajan L, et al. PredictProtein – an open resource for online prediction of protein structural and functional features. Nucleic Acids Res. 2014;42(W1):W337–W343. | ||
Sickmeier M, Hamilton JA, Legall T, et al. DisProt: the database of disordered proteins. Nucleic Acids Res. 2007;35:D786–D793. | ||
Schlessinger A, Yachdav G, Rost B. PROFbval: predict flexible and rigid residues in proteins. Bioinformatics. 2006;22(7):891–893. | ||
Adzhubei I, Jordan DM, Sunyaev SR. Predicting functional effect of human missense mutations using PolyPhen-2. Curr Protoc Hum Genet. 2013;Chapter 7:Unit7.20. | ||
Ng PC, Henikoff S. Predicting deleterious amino acid substitutions. Genome Res. 2001;11(5):863–874. | ||
Reva B, Antipin Y, Sander C. Predicting the functional impact of protein mutations: application to cancer genomics. Nucleic Acids Res. 2011;39(17):e118. | ||
Schwarz JM, Cooper DN, Schuelke M, Seelow D. MutationTaster2: mutation prediction for the deep-sequencing age. Nat Methods. 2014;11(4):361–362. | ||
Choi Y, Sims GE, Murphy S, Miller JR, Chan AP. Predicting the functional effect of amino acid substitutions and indels. PLoS One. 2012;7(10):e46688. | ||
Liu X, Wu C, Li C, Boerwinkle E. dbNSFP v3.0: a one-stop database of functional predictions and annotations for human nonsynonymous and splice-site SNVs. Hum Mutat. 2016;37(3):235–241. | ||
Jmol: an open-source HTML5 viewer for chemical structures in 3D. Available from: http://Jmol.sourceforge.net. Accessed February 12, 2019. | ||
Ashkenazy H, Abadi S, Martz E, et al. ConSurf 2016: an improved methodology to estimate and visualize evolutionary conservation in macromolecules. Nucleic Acids Res. 2016;44(W1):W344–W350. | ||
Salomon O, Steinberg DM, Zivelin A, et al. Single and combined prothrombotic factors in patients with idiopathic venous thromboembolism: prevalence and risk assessment. Arterioscler Thromb Vasc Biol. 1999;19(3):511–518. | ||
Miesbach W, Scharrer I, Henschen A, Neerman-Arbez M, Spitzer S, Galanakis D. Inherited dysfibrinogenemia: clinical phenotypes associated with five different fibrinogen structure defects. Blood Coagul Fibrinolysis. 2010;21(1):35–40. | ||
Zhou J, Ding Q, Chen Y, et al. Clinical features and molecular basis of 102 Chinese patients with congenital dysfibrinogenemia. Blood Cell Mol Dis. 2015;55(4):308–315. | ||
Morris TA, Marsh JJ, Chiles PG, et al. High prevalence of dysfibrinogenemia among patients with chronic thromboembolic pulmonary hypertension. Blood. 2009;114(9):1929–1936. | ||
Carter AM, Catto AJ, Kohler HP, Ariëns RA, Stickland MH, Grant PJ. Alpha-fibrinogen Thr312Ala polymorphism and venous thromboembolism. Blood. 2000;96(3):1177–1179. | ||
Hanss M, Vergnes C, Rugeri L, Ffrench P, de Mazancourt P. A new electrophoretic variant of fibrinogen associated with venous thromboembolism, fibrinogen Bordeaux a Arg439Cys. J Thromb Haemost. 2008;6(8):1422–1424. | ||
Fernández-Cadenas I, Penalba A, Boada C, et al. Exome sequencing and clot lysis experiments demonstrate the R458C mutation of the alpha chain of fibrinogen to be associated with impaired fibrinolysis in a family with thrombophilia. J Atheroscler Thromb. 2016;23(4):431–440. | ||
Marchi R, Lundberg U, Grimbergen J, et al. Fibrinogen Caracas V, an abnormal fibrinogen with an Aalpha 532 Ser-->Cys substitution associated with thrombosis. Thromb Haemost. 2000;84(2):263–270. | ||
Koopman J, Haverkate F, Grimbergen J, et al. Molecular basis for fibrinogen Dusart (A alpha 554 Arg-->Cys) and its association with abnormal fibrin polymerization and thrombophilia. J Clin Invest. 1993;91(4):1637–1643. |
 © 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.