Back to Journals » Journal of Inflammation Research » Volume 15
C-X-C-Chemokine-Receptor-Type-4 Inhibitor AMD3100 Attenuates Pulmonary Inflammation and Fibrosis in Silicotic Mice
Authors Sun Q, Tao X, Li B, Cao H, Chen H, Zou Y ![]() , Tao H, Mu M, Wang W, Xu K
, Tao H, Mu M, Wang W, Xu K
Received 11 May 2022
Accepted for publication 20 September 2022
Published 11 October 2022 Volume 2022:15 Pages 5827—5843
DOI https://doi.org/10.2147/JIR.S372751
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr Zili You
Qixian Sun,1 Xinrong Tao,1– 4 Bing Li,1 Hangbing Cao,1 Haoming Chen,1 Yuanjie Zou,1 Huihui Tao,1– 4 Min Mu,1– 4 Wenyang Wang,1 Keyi Xu1– 4
1Center for Medical Research, Medical School, Anhui University of Science and Technology, Huainan, People’s Republic of China; 2Key Laboratory of Industrial Dust Control and Occupational Health, Ministry of Education, Anhui University of Science and Technology, Huainan, People’s Republic of China; 3Key Laboratory of Industrial Dust Deep Reduction and Occupational Health and Safety, Anhui Higher Education Institutes, Anhui University of Science and Technology, Huainan, People’s Republic of China; 4Engineering Laboratory of Occupational Safety and Health, Anhui Province, Anhui University of Science and Technology, Huainan, People’s Republic of China
Correspondence: Xinrong Tao, Medical School, Anhui University of Science and Technology, Huainan, People’s Republic of China, Email [email protected]
Background: Silicosis is a severe pulmonary disease caused by inhaling dust containing crystalline silica. The progression of silicosis to pulmonary fibrosis is usually unavoidable. Recent studies have revealed positivity for the overexpression of C-X-C chemokine receptor type 4 (CXCR4) in pulmonary fibrosis and shown that the CXCR4 inhibitor AMD3100 attenuated pulmonary fibrosis after bleomycin challenge and paraquat exposure. However, it is unclear whether AMD3100 reduces crystalline silica-induced pulmonary fibrosis.
Methods: C57BL/6 male mice were instilled intranasally with a single dose of crystalline silica (12 mg/60 μL) to establish an acute silicosis mouse model. Twelve hours later, the mice were injected intraperitoneally with 5 mg/kg AMD3100 or control solution. Then, the mice were weighed daily and sacrificed on day 7, 14, or 28 to collect lung tissue and peripheral blood. Western blotting was also applied to determine the level of CXCR4, while different histological techniques were used to assess pulmonary inflammation and fibrosis. In addition, the level of B cells in peripheral blood was measured by flow cytometry.
Results: CXCR4 and its ligand CXCL12 were upregulated in the lung tissues of crystalline silica-exposed mice. Blocking CXCR4 with AMD3100 suppressed the upregulation of CXCR4/CXCL12, reduced the severity of lung injury, and prevented weight loss. It also inhibited neutrophil infiltration at inflammatory sites and neutrophil extracellular trap formation, as well as reduced B-lymphocyte aggregates in the lung. Additionally, it decreased the recruitment of circulating fibrocytes (CD45+collagen I+CXCR4+) to the lung and the deposition of collagen I and α-smooth muscle actin in lung tissue. AMD3100 also increased the level of B cells in peripheral blood, preventing circulating B cells from migrating to the injured lungs.
Conclusion: Blocking CXCR4 with AMD3100 delays pulmonary inflammation and fibrosis in a silicosis mouse model, suggesting the potential of AMD3100 as a drug for treating silicosis.
Keywords: CXCR4, silicosis, pulmonary fibrosis, inflammation, AMD3100, mice
Introduction
The inhalation of dust containing crystalline silica (CS) can cause incurable silicosis with irreversible pulmonary fibrosis, and thousands of people worldwide die from CS-induced silicosis every year.1,2 Unfortunately, fatal CS exposure in industrial workers is increasing,3 but treatments for silicosis are still limited because the pathogenesis is complex. Therefore, defining the molecular pathogenesis of silicosis would aid the development of efficient therapeutic approaches to prevent inflammation and fibrosis.
C-X-C chemokine receptor type 4 (CXCR4) is a seven-transmembrane G protein-coupled receptor that plays a crucial role in maintaining lung homeostatic. Overexpression of CXCR4 is associated with early mortality in patients with idiopathic pulmonary fibrosis (IPF).4 CXCR4 can be downregulated by small-molecule antagonists, including AMD3100, AMD070, and BL-8040.5,6 The FDA has approved AMD3100 for treating HIV-1 infection and mobilizing hematopoietic stem cells.7 In addition, AMD3100 has recently been shown to reduce pulmonary fibrosis after exposure to bleomycin (BLM) and paraquat.8,9 This background prompted an examination of the use of AMD3100 for treating silicosis.
CXCR4 and its ligand CXCL12 promote inflammation and fibrosis in the lung.10,11 They regulate the migration of bone marrow-derived cells to the lung in pulmonary diseases.12–14 They also recruit circulating fibrocytes (CFs) to the lung, which release extracellular matrix proteins, collagen I, and transforming growth factor β1 (TGF-β1) to promote the proliferation of fibroblasts and their differentiation into myofibroblasts, thereby promoting pulmonary fibrosis.15–17 In addition, CXCL12/CXCR4 regulates the immune response and attracts neutrophils, T cells, B lymphocytes, monocytes, and dendritic cells.18 Neutrophils produce enzymes that release damage mediators, such as reactive oxygen species and myeloperoxidase (MPO), and form neutrophil extracellular traps (NETs). NETs induce DNA injury, delay healing, promote scar formation, and promote the differentiation of fibroblasts into myofibroblasts during lung fibrosis.19,20 In addition, CXCR4 signaling affects the migration, retention, maturity, and survival of B cells.21,22 The expression of CXCR4 promotes B-cell survival under hypoxic conditions, and an inhibitor of hypoxia-inducible factor (HIF) downregulates CXCR4 and further decreases the number of B cells.21 Moreover, B cells secrete proinflammatory and profibrotic proteins in fibrotic disease, which promote fibroblast migration and activation.23,24 Besides, IL-10-producing regulatory B cells (B 10) were found to inhibit Th1 reaction and modulate Th balance, alleviating pulmonary inflammation and promoting fibrosis in silicosis mice.25 These findings suggest that CXCR4 promotes pulmonary fibrosis by regulating multiple cells and that it may represent a suitable target for treating silicosis with fibrosis.
However, to the best of our knowledge, no studies have investigated the role of CXCR4 in the pathology of silicosis and its suitability as a therapeutic target. Therefore, the present study was established to examine whether CXCR4 affects silicosis progression and whether the CXCR4 inhibitor AMD3100 alleviates pulmonary inflammation and fibrosis after CS exposure in a mouse model.
Materials and Methods
Animals and Treatments
Male C57BL/6 mice (10–12 weeks) were purchased from Changzhou Cavion Experimental Animal Co., Ltd. [license No. SCXY (Su) 20110003]. All mice were given food and water at a temperature of 23°C ± 1°C and humidity of 50%, under a 12-h light-dark cycle (lights on 8:00 a.m. until 8:00 p.m.). All procedures followed the Guide for the Care and Use of Laboratory Animals (NIH Publication No. 8023, revised 1978) and were approved by the Institutional Animal Care and Ethics Committee of Anhui University of Science and Technology.
After acclimation for 2 weeks, all mice were randomly separated into three groups: a control group (n = 21), a silica group (CS group) (n = 21), and a treatment group (AMD3100 group) (n = 21). At 8:00 p.m., the mice were intranasally instilled with CS (12 mg/60 μL) diluted in PBS (silica and treatment groups) or with PBS (control group). Twelve hours later (the next day at 8:00 a.m.), the mice in the treatment group were injected with AMD3100 (HY50912, MCE, 5 mg/kg, i.p.) dissolved in PBS daily for 1 week, while the control group and the silica group underwent daily injection of PBS for 1–7 days. The specific dose of AMD3100 was based on published papers and company recommendations.9,10 Since AMD3100 has been used in preclinical and clinical studies, we did not establish an AMD3100-only group.26 Seven mice were sacrificed for analysis on each of days 7, 14, and 28. The mice were weighed every day.
Antibodies
For immunohistochemical (IHC) staining, we used the following primary antibodies: collagen type I (14695-1-AP, Proteintech), CXCL12 (17402-1-AP, Proteintech), α-smooth muscle actin (α-SMA, ab124964, Abcam), MPO (ab9535, Abcam), CXCR4 (ab1670, Abcam), Ki67 (ab15580, Abcam), and CD45R (sc19597, Santa Cruz Biotechnology). The secondary antibodies included horseradish peroxidase (HRP)-conjugated donkey anti-goat IgG H&L (ab6885, Abcam), alkaline phosphatase (AP)-conjugated donkey anti-rabbit IgG H&L (ab6803, Abcam), HRP-conjugated goat anti-rabbit IgG H&L (ab6721, Abcam), and HRP-conjugated rabbit anti-rat IgG H&L (ab6734, Abcam).
CXCR4 (sc-53534, Santa Cruz Biotechnology) and GAPDH (10494-1-AP, Proteintech) were used for Western blotting. In addition, HRP-conjugated goat anti-mouse IgG (SA00001-1, Proteintech) and HRP-conjugated goat anti-rabbit IgG H&L (ab6721, Abcam) were used as the secondary antibodies.
For immunofluorescence (IF) staining, we used the following primary antibodies: CD45R (sc19597, Santa Cruz Biotechnology), myeloperoxidase (sc-390109, Santa Cruz Biotechnology), histone H3 (citrulline R2+R8+R17) (ab5103, Abcam), CXCR4 (ab1670, Abcam), Ki67 (ab15580, Abcam), collagen I (14695-1-AP, Proteintech), CD45 (60287-1-lg, Proteintech), pro-SPC (ab211326, Abcam), and CD3 (17617-1-AP, Proteintech). The secondary antibodies were: FITC-TSA (GB22401, Servicebio), CY3-TSA (GB21404, Servicebio), CY5-TSA (GB23303, Servicebio), Alexa Fluor 488-conjugated anti-rat lgG H&L (4416S, Cell Signaling), and Alexa Fluor 594-conjugated goat anti-rabbit (AS039, Abclonal).
For flow cytometry, we used FITC-conjugated anti-CD3 (100204, Biolegend) and PerCP-Cy5.5-conjugated CD19 (12478, BD Biosciences) antibodies.
Flow Cytometry
Peripheral blood was obtained from the orbital sinus of anesthetized mice. Mononuclear cells were isolated from peripheral blood using erythrocyte lysate. FITC-conjugated anti-CD3 (Biolegend) and PerCP-Cy5.5-conjugated CD19 (BD Biosciences) antibodies were used to label B cells. For labeling, 105 cells were incubated with antibodies at 4°C for 1 h, washed twice, and centrifuged (1000 rpm, 10 min, 4°C). Labeled cells were analyzed on a FACSCanto II flow cytometer using Cell QuestPro acquisition software. In addition, data were analyzed using FlowJo 7.6.1 software.
Histopathological Staining
The left lobes of the lungs were harvested, fixed in 4% paraformaldehyde, embedded in paraffin, and sectioned into 4-µm slices. The slices were heated at 60°C for 3 h, dewaxed with xylene, rehydrated through a graded alcohol series, and washed with ddH2O. Hematoxylin and eosin (HE) staining and Masson’s trichrome staining were used to determine the pathological score and assess fibrosis.
Immunohistochemical Staining
For IHC staining, sections were boiled in antigen retrieval buffer for 20 min and endogenous peroxidases were blocked in 3% H2O2 for 15 min. Tissue sections were then blocked in 5% BSA for 1 h at room temperature (RT) to prevent nonspecific binding, followed by incubation at 4°C overnight with primary antibodies. After rewarming for 30 min, the sections were washed with PBS and incubated with HRP-conjugated secondary antibodies for 1 h at RT. The sections were then stained with AP substrate (Vector Laboratories, SK-5105) or DAB (Vector Laboratories, SK-4103) and counterstained with HE. Isotype control was included as a negative control. Photos were acquired with a BX50 microscope (Olympus, Tokyo, Japan). IHC images were analyzed using Image-Pro Plus 6.0 software. The pulmonary fibrosis and pathology scores were calculated using the Ashcroft scale and Roderick J. criteria.27,28 Under blinded conditions, slides were analyzed and five fields at 400× magnification was randomly chosen to determine pathological scores.
Immunofluorescence Staining
For immunofluorescence staining, slides were heated and deparaffinized as described above. They were then permeabilized with 0.3% Triton X-100 in PBS (PBST) for 1 h at RT before incubation with primary antibodies overnight at 4°C in the dark. After three washes in PBST for 5 min each, sections were incubated with Alexa Fluor 488-conjugated anti-rat IgG (H+L) and Alexa Fluor 594-conjugated donkey anti-rabbit secondary antibodies for 1 h at RT. After three washes with PBST, the nuclei were stained with DAPI (1002, Beyotime Biotechnology) for 10 min. Tissue sections were imaged using a laser scanning confocal microscope (FV3000, Olympus). CFs were labeled by triple immunofluorescence using primary antibodies against CD45, collagen I, and CXCR4. After incubation with primary antibodies at 4°C overnight, the secondary antibody was added (FITC-TSA, CY3-TSA, or CY5-TSA). Immunofluorescence sections were visualized using MIDI:3Dhistech (Pannoramic).
Western Blotting
Total protein was isolated from lung tissue in RIPA buffer (P0013B; Beyotime Biotechnology). Protein concentration was determined using a bicinchoninic acid protein assay kit. Proteins were separated by SDS-PAGE and then transferred onto polyvinylidene fluoride membranes. Membranes were blocked with 5% non-fat milk and incubated with anti-CXCR4 antibody (overnight, 4°C). After washing in TBST, the membranes were incubated in secondary antibodies (1 h, RT). Protein bands were detected under an Amersham ImageQuant 800 and bands were analyzed using Image J. Experiments were repeated three times.
Statistical Analysis
Data were analyzed using IBM SPSS Statistics. Graphs were created using GraphPad Prism 5.0. Results are presented as mean ± standard error of the mean. The significance of differences between groups was tested using one-way analysis of variance (ANOVA), and least significant difference (LSD)was used to compare the differences between groups when data was homogeneous. The significance of differences between groups was also analyzed using unpaired t-tests. Weight loss was analyzed using two-way repeated measures ANOVA, followed by Bonferroni post hoc test. p < 0.05 was considered to indicate statistical significance.
Results
CXCL12/CXCR4 Expression is Upregulated in the Mouse Lung and Lung Architecture is Damaged After CS Exposure
Twenty-eight days after exposure to CS, the lung parenchyma was infiltrated by inflammatory cells such as neutrophils, macrophages, and lymphocytes. This infiltration damaged the alveolar architecture. Fibrous masses, lymphoid clusters, silicotic nodules, and thickening of the bronchiolar wall were also observed. No changes were observed in the lung tissue of control mice (Figure 1A). Concomitant with these changes, CXCL12 and CXCR4 expression increased in the lung after CS exposure (Figure 1B). The bar graph showed that the CXCL12/CXCR4 expression elevated significantly in the CS group (p < 0.001, Figure 1C and D). There were also more CXCR4+ cells in the lung tissue of CS mice, including MPO+ neutrophils, pro-SPC+ epithelial cells, and CD45R+ lymphocytes, as revealed by double-immunofluorescence staining (Supplemental Figure 1).
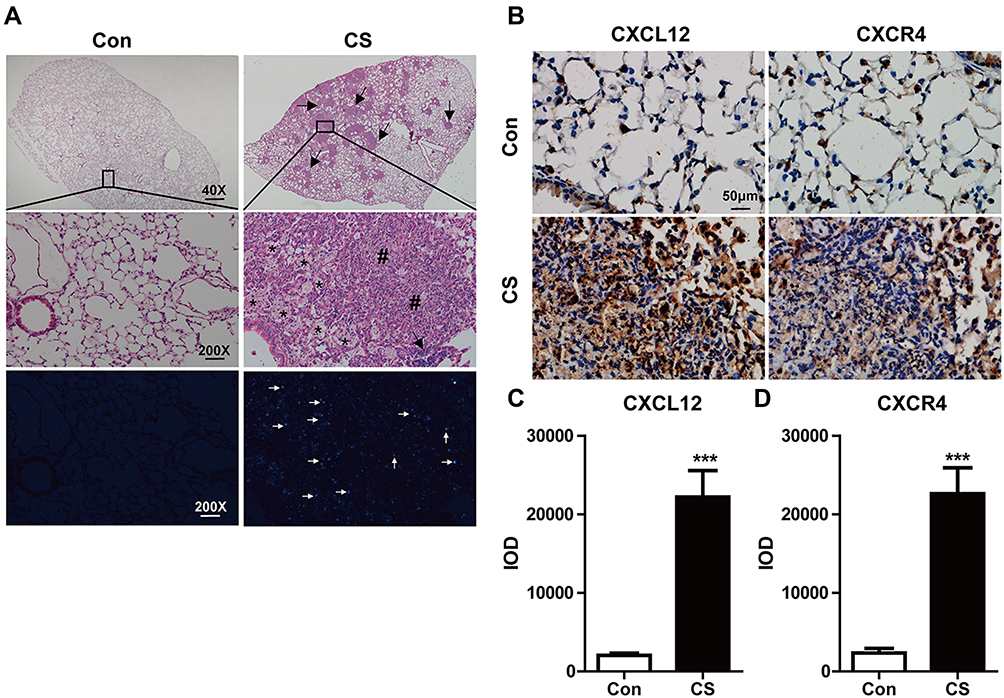 |
Figure 1 Silica particles induced pathological changes and increased CXCL12/CXCR4 expression in the mouse lung. (A) Represent histological sections of mouse lung on day 28 in CS-exposed group versa control one. Mice were administrated intranasally with 12 mg CS in 60 µL PBS and 60 µL PBS as the vehicle control. The low magnification (40×) of CS-exposed lung showed extensive silica nodule formation compared with the control in upper images. The large magnification (200×) showed lung architecture is normal in the control group (N=6), and the CS group (N=6) showed obvious trachea and alveolar wall thickening, inflammatory cells infiltration, and the formation of silicotic nodules in the middle images. Arrows indicate lymphoid clusters. Asterisk indicates reactive inflammatory cells. Pound signs indicate silicotic nodules. The bottom images showed crystalline silica under a polarizing light microscope. White arrows indicate CS. (B) The expressions of CXCL12/CXCR4 in the lung were assessed after CS exposure by immunohistochemistry staining. The quantitative analyses of CXCL12 (C) and CXCR4 (D) expression in the lung were shown. IOD: integrated optical density. Scale bar: 50 µm. ***P < 0.001 vs the control group. |
Blocking CXCR4 Reduces Lung Injury and Weight Loss After CS Exposure
CS exposure damaged the lung architecture and significantly reduced our mouse model’s body weight. To determine whether blocking CXCR4 can reduce CS-induced lung injury and weight loss, we treated mice with the CXCR4 inhibitor AMD3100. The experimental scheme was shown in Figure 2A. AMD3100 significantly reduced inflammatory cell infiltration and alveolar wall thickening at 7 and 14 days after CS exposure and delayed nodule formation (Figure 2B). These histological findings were confirmed by a lower pathology score in the AMD3100 group than in the CS group on days 7, 14, and 28 (p < 0.001, p < 0.001, p < 0.001, CS group vs control group; p < 0.01, p < 0.01, p < 0.01, CS group vs AMD3100 group, Figure 2C). Silicosis mice lost more than 20% of their body weight by day 4, and this weight loss remained until day 28 (p < 0.01), but the body weight of mice treated with AMD3100 gradually recovered and returned to its original level by day 28 (p < 0.01, Figure 2D).
 |
Figure 2 Blocking CXCR4 expression alleviated CS-induced lung injury and bodyweight loss. (A) 10–12 weeks C57BL/6 mice were randomly divided into three groups: the control group (60 μL PBS), the CS group (12 mg in 60 μL PBS), and the CS+AMD3100 group. Mice in the CS+AMD3100 group were treated with the following step. 12 mg CS in 60 μL PBS were instilled intranasally into the mouse 12 hours before AMD3100 treatment. Following CS exposure, AMD3100 (5 mg/kg) was injected intraperitoneally once a day for a week. Mice were sacrificed on days 7,14 and 28. Samples were collected and analyzed. (B) Representative presentation of HE staining of the whole lung tissue section on days 7, 14, and 28. (C) Quantitative analysis of pathological scores in the lung among three groups was presented. Scale bar: 500 μm. N=4 per group. (D) Bodyweight was measured daily for each group. Weight loss is significant in the CS group (red line) when compared with the control group (blue line), but CXCR4 antagonist AMD3100 attenuated weight loss in the CS+AMD3100 group (green line), shown in the graph. N=7 per group. *p < 0.05, **p < 0.01, ***P < 0.001 vs the control group. #P < 0.05, ##P < 0.01 vs the CS group. |
In addition, serial sections from the same lung tissue were stained for CXCR4 and CXCL12 separately. IHC staining showed that CXCR4+ and CXCL12+ cells were increased in number in the lung 28 days after CS exposure. Interestingly, CXCR4+ cells were located at the periphery of fibrotic regions, while CXCL12+ cells were found in their center (Figure 3A). Furthermore, the CXCR4+ cells included CD45R+ B lymphocytes, MPO+ neutrophils, and pro-SPC+ alveolar epithelial type II cells (Supplemental Figure 1). IHC staining also showed that AMD3100 significantly reduced the CXCR4 (p < 0.01) and CXCL12 (p < 0.05) expression in lung tissues (Figure 3B). Finally, Western blotting confirmed that AMD3100 dramatically reduced CXCR4 expression in the lungs on day 7 (p < 0.05) after CS exposure (Figure 3C). These results suggest that inhibiting CXCR4 with AMD3100 effectively reduced CXCR4/CXCL12 expression and prevented CS-induced changes in lung architecture and weight in mice.
 |
Figure 3 AMD3100 effectively reduced CXCR4 and CXCL12 expression in the silicotic lung. (A and B) Representative images show CXCR4 (upper) and CXCL12 (bottom) expression in lung sections determined by immunohistochemical staining analysis on day 28. Red arrows indicate CXCR4+ cells. Green arrows indicate CXCL12+ cells. DAB-labeled tissues were analyzed using Image-Pro Plus 6.0 software, as described in the material and methods. IOD: integrated optical density. Scale bar: 100 μm. (C) The expression of CXCR4 in the lungs was analyzed by Western blot on day 7. N=3 per group. **p < 0.01, ***p < 0.001 vs the control group. #P < 0.05, ##P < 0.01 vs the CS group. |
Blocking CXCR4 Reduces MPO Expression and NET Release in Lung Tissue After CS Exposure
Neutrophils secrete MPO, which can be used as a marker to determine the number of neutrophils in lung tissue.29 In this study, MPO staining revealed that the number of neutrophils increased on days 7, 14, and 28 in the CS group (p < 0.001, p < 0.01, p < 0.001, respectively) and that neutrophils were distributed throughout the lung tissue and that they accumulated around the trachea and in the center of silicotic nodules. IHC staining of MPO also showed that AMD3100 reduced neutrophil infiltration in lung tissue 7, 14, and 28 days after CS exposure (p < 0.01, p < 0.05, p < 0.01, respectively). Meanwhile, MPO expression was low in control lung tissue (Figure 4A and B).
 |
Figure 4 CXCR4 blocking alleviated MPO expression and NET formation in the lung of silicosis mice. (A and B) Immunohistochemical analysis of MPO staining was performed in the lung after CS exposure on days 7.14, and 28. Representative images and quantitative analysis of MPO were shown. (C and D) Representative double-staining immunofluorescence images showing NETs, defined as colocalized MPO (Red) and citH3 (Green) in the lung after CS-challenged at 28 days. Quantitative analysis of MPO+citH3+ cells/area (mm2) in the lung tissue was shown. White arrows indicate the NETs. N=4 per group. IOD: integrated optical density. Scale bar: 50 μm (A and C). **p < 0.01, ***p < 0.001 vs the control group. #P < 0.05, ##p < 0.01, ###p < 0.001 vs the CS group. |
We also performed citH3 IF to stain NETs and found that NETs formed in lung tissue after CS exposure, but that AMD3100 reduced the number of NETs 28 days after CS exposure (Figure 4C and D). These results suggest that AMD3100 reduces neutrophil infiltration and NET formation in lung tissue after CS exposure in mice, which may alleviate pulmonary inflammation and fibrosis.
Blocking CXCR4 Reduces B-Lymphoid Aggregate Formation in the Lung and Increases B Cells in Peripheral Blood After CS Exposure in Mice
After CS exposure, HE staining of lung tissue revealed lymphoid aggregates (LAs) in the peribronchial regions. AMD3100 treatment reduced the number and size of these LAs (Figure 5A). Next, we performed double staining of CD3-positive T cells and CD45R-positive B cells and showed that LAs contained both B cells and T cells, but primarily B cells, 28 days after CS exposure. The number of B cells was significantly lower in mice treated with AMD3100 (Figure 5B and C). We also showed that B cells produced collagen I in lung tissue after CS exposure, which was deposited around the LAs (Figure 5D). In addition, B-LAs were close to the fibroblastic foci after CS treatment. These findings show that inhibiting CXCR4 with AMD3100 reduces LA formation in lung tissue after CS exposure, which may alleviate pulmonary fibrosis.
 |
Figure 5 Blocking CXCR4 reduced B-lymphoid aggregate (LA) formation and collagen-I secretion by B-cells in the lung tissue after CS exposure. (A) HE staining showed LA formation 28 days after CS exposure; AMD3100 treatment reduced the number of LAs. Black arrows indicate LAs. (B) Double immunofluorescence analyses showed CD45R (B cells, green) and CD3 (T cells, red) staining in the lung after CS exposure on days 7, 14, and 28. White asterisks indicate B cells; white arrows point to T cells. (C) Quantitative analysis of CD45R+ cells and CD3+ cells/area (mm2) in the lung tissue was shown. (D) CD45R+ cells were stained with DAB (brown), and collagen-I+ cells were colored with AP (red). The brown arrowhead indicates CD45R positive collagen-I negative cells, the red arrowhead indicates CD45R negative collagen-I positive cells, and the yellow arrowhead indicates CD45R positive collagen-I positive cells. Scale bar: 100 μm (A), 50 μm (B and D). ***p < 0.001 vs control group. #P < 0.05 vs CS group. |
Flow cytometry was used to measure the level of CD19+ B cells in the blood. Figure 6A describes the gating strategy for CD19+ cells. The results showed that the proportion of peripheral B cells was significantly lower in the CS group than in the control group 7 and 14 days after CS exposure (p < 0.05, p < 0.01). The proportion of B cells in peripheral blood increased in the AMD3100 group 7 and 14 days after CS exposure (p < 0.05, p < 0.05, respectively). However, the proportion of peripheral B cells was significantly higher in the CS group than in the other groups 28 days after CS exposure. No significant changes were observed in the control group, and no differences were observed between the control group and the AMD3100 group (Figure 6B and C).
 |
Figure 6 AMD3100 up-regulated the level of CD19+ (B cells) in the peripheral blood after CS-challenged. (A) The gating strategy was shown to identify CD19+ (B cells) in the peripheral blood. A representative flow cytometry histogram of B cells (B) and bar graph of the level of B cells (C) in the peripheral blood on days 7.14, and 28 were presented. *p < 0.05, **p < 0.01 vs control group. #P < 0.05 vs CS group. |
We also quantified the number of proliferating B cells in the lungs using Ki67 and CD45R co-staining. Proliferating B cells were not detected in lung tissue 7, 14, and 28 days after CS exposure in both CS and AMD3100 groups. Notably, Ki67 staining was absent in the LAs, although some epithelial cells in the trachea and alveoli were positive for Ki67 (Supplemental Figure 2). These results show that AMD3100 increases the level of peripheral B cells and blocks the migration of circulating B cells to lung tissue after CS exposure, but that it does not activate B-cell proliferation.
Blocking CXCR4 Improves Pulmonary Fibrosis in Silicosis Mice
To determine whether AMD3100 can prevent lung fibrosis, we observed collagen deposition by Masson staining. As shown in Figure 7A, Masson staining revealed reduced collagen deposition in the lung of mice treated with AMD3100 compared with that in untreated mice. The Ashcroft score, which reflects the severity of fibrosis in lung tissue, was also significantly different among the groups, indicating more severe fibrosis in the CS group than in the control groups on days 7, 14, and 28 (p < 0.001, p < 0.001, p < 0.001, respectively, Figure 7B), but AMD3100 treatment reduced pulmonary fibrosis scores in silicotic mice on days 7, 14, and 28 (p < 0.05, p < 0.01, p < 0.05, respectively, Figure 7B).
 |
Figure 7 Blockade CXCR4 attenuated pulmonary fibrosis in silicosis mice. (A) Representative presentation of Masson’s trichrome staining of the whole lung tissue section on days 7, 14, and 28. (B) Quantitative analysis of Ashcroft score in the lung among three groups was presented. N=4 per group. (C) Immunohistochemical staining of collagen I and α-SMA were shown on days 14 and 28 after CS exposure. Quantitative analysis of collagen I and α-SMA immunohistochemical staining was shown in (D) after CS-challenged. IOD: integrated optical density. Scale bar: 50 µm. *P < 0.05, **P < 0.01, ***P < 0.001 vs the control group. #P < 0.05, ##P < 0.01 vs the CS group. |
To further study the effect of AMD3100 on CS-induced fibrogenesis, we performed IHC staining for collagen I and α-SMA in lung tissue 14 and 28 days after CS exposure (Figure 7C and D). We observed gradual increases in collagen I (p < 0.05, p < 0.001) and α-SMA (p < 0.001, p < 0.01) expression after CS exposure, while this expression was mainly detected in the fibrotic foci and silicotic nodules. In contrast, in control mice, collagen I and α-SMA expression was only observed around the trachea and blood vessels. AMD3100 treatment markedly reduced these fibrotic lesions along with collagen I and α-SMA expression. These results suggest that AMD3100 reduces the numbers of myofibroblasts (which produce α-SMA) and fibroblasts (which produce collagen I) in lung tissue after CS exposure, which may attenuate pulmonary fibrosis.
Inhibiting CXCR4 Prevents the Migration of CFs to the Lung After CS Exposure
After tissue injury, CFs are recruited to the site of damage through the CXCL12/CXCR4 axis, where they participate in tissue remodeling and promote fibrosis. We evaluated the recruitment of CFs to the lung after CS exposure by immunofluorescent staining of CD45, collagen I, and CXCR4. Immunofluorescent staining showed that CFs infiltrated the lung parenchyma after CS treatment and that AMD3100 treatment reduced this infiltration on days 7 and 28 (p < 0.001, p < 0.001, respectively, CS group vs AMD3100 group, Figure 8B). No CFs were found in the lung tissue of control mice (Figure 8A and B). These results show that AMD3100 blocked the migration of CD45+/collagen I+/CXCR4+ CFs to the lung after CS exposure.
 |
Figure 8 Blocking of CXCR4 reduced intrapulmonary recruitment of CFs to CS-injured sites. (A and B) Representative confocal images of CD45+/collagen-I+/CXCR4+ CFs assessed by immunofluorescent staining in the lung section were shown. Lung sections in control, CS, and CS+AMD3100 groups were stained with CD45-FITC(Green), CXCR4-Cy3(Red), and collagen I-Cy5(Purple). The cell nuclei were counterstained with DAPI(Blue). White arrows indicate CFs with triple colors of CD45+/collagen-I+/CXCR4+. Photos were taken under 3Dhistech (Pannoramic). Scale bar: 20 μm. *P < 0.05, **P < 0.01, ***P < 0.001 vs the control group. ###P < 0.001 vs the CS group. |
 |
Figure 9 CXCR4 blocking reduced pulmonary fibrosis in silicosis mice. CXCR4 and its ligand CXCL12 expression were up-regulated in silicosis mice, through which the CFs, neutrophils, and circulating B cells were recruited in the silicotic lung. After recruiting pro-inflammatory and pro-fibrotic cells traffic into the silicotic lung, CFs differentiated into fibroblasts, and further formed fibroblastic foci; B cells formed lymphoid aggregates and secreted collagen. In addition, neutrophils accumulated and released myeloperoxidase, forming neutrophil extracellular traps (NETs) in the inflammatory sites, and then NETs activated myofibroblasts to promote lung fibrosis. AMD3100 inhibited CXCR4 expression, which delayed the progression of pulmonary fibrosis by blocking the recruitment of multiple CXCR4 positive cells in silicosis mice. |
Discussion
In this study, blocking CXCR4 with AMD3100 reduced neutrophil retention, NET formation, and LA neogenesis in the silicotic lung after CS exposure. AMD3100 also inhibited CF migration to lung tissue and delayed pulmonary fibrosis in mice exposed to CS (Figure 9). Our findings suggest that CXCR4 may represent a molecular target for reducing silicosis progression.
AMD3100 is an FDA-approved CXCR4 inhibitor that mobilizes hematopoietic stem cells for bone marrow transplantation.30 In one recent study, it was speculated that AMD3100 might be suitable for treating silicosis,31 but its therapeutic effect remains unknown even at the whole-animal level. In this study, we used AMD3100 to treat silicosis mice and found that inhibiting CXCR4 ameliorated CS-induced weight loss and lung injury. We next noted that the CXCL12/CXCR4 axis is the critical signal promoting the formation of silicotic nodules. We also found that CXCR4- and CXCL12-positive cells are located at the periphery and center of silicotic nodules, respectively. A previous study confirmed that CXCR4-positive cells (fibroblasts, epithelial cells, etc.) migrated and were recruited along CXCL12 gradients.32 Therefore, the distribution of CXCR4/CXCL12-positive cells contributes to the formation of fibroblast foci and the exacerbation of pulmonary fibrosis in silicosis. Against this background, we next focused on the fate of CXCR4-positive immune cells (neutrophils, B cells, fibrocytes) after AMD3100 treatment in silicosis.
The accumulation of neutrophils in lung tissue is associated with CS-induced pulmonary inflammation. The CXCL12/CXCR4 axis is involved in the migration of neutrophils in acute lung injury.33 We showed that CS exposure increases neutrophil infiltration into the lung, but inhibiting CXCR4 with AMD3100 reduces this infiltration. Once CS is inhaled into lung tissue, neutrophils are quickly recruited into lung tissue and are further sustained by positive inflammatory feedback loops. Neutrophils eliminate invading molecules and repair damaged tissues,34 but an excess of neutrophils can induce pulmonary inflammation and fibrosis. Controlling neutrophil numbers via apoptosis can alleviate tissue injury.35 Neutrophils secrete MPO to delay their own apoptosis and promote their accumulation through the ERK and Akt pathways.36,37 We showed that MPO expression is upregulated in lung tissue after CS exposure and that this CS-induced upregulation is reversed by AMD3100 treatment. This suggests that blocking CXCR4 can reduce the accumulation of neutrophils in lung tissue by promoting neutrophil apoptosis. However, AMD3100 treatment did not stop neutrophil migration to inflammatory sites in an acute peritonitis model.38 This might be explained by differences in experimental procedures, drug administration, and sampling time points. Therefore, studying how AMD3100 affects neutrophil dynamics in CS-induced fibrosis would be worthwhile.
Neutrophils, as inflammatory cells, play a pro-fibrotic role in silicosis, as demonstrated by this study. We found that AMD3100 reduced NET formation in lung tissue after CS exposure. NETs are formed when activated neutrophils release DNA and antimicrobial proteins like neutrophil elastase into the extracellular environment to trap pathogens.39 In agreement with our finding that CS induces NET formation, NETs were found to be increased in lipopolysaccharide-injured mouse lungs. Other in vitro studies support our results by showing that NETs are formed in response to sterile inflammation. In addition, NETs were reported to induce epithelial and endothelial cell death and cause lung destruction,40 promoting the activation of fibroblasts and their differentiation into myofibroblasts, which can contribute to pulmonary fibrosis.20,41 Therefore, neutrophils play pro-inflammatory and pro-fibrotic roles in silicosis.
Although the role of B cells in silicosis is poorly understood, they have been implicated in several lung fibrosis diseases. For example, B-cell recruitment in lung tissue was shown to promote the progression of pulmonary fibrosis after BLM challenge.42 It was also shown that antifibrotics alleviated fibrosis by inhibiting B-cell activation and B-cell-mediated migration of pulmonary fibroblasts in patients with IPF.43 Moreover, regulatory B cells were shown to produce IL-10, promoting inflammation in vitro after CS challenge.44 We also showed that CXCL12/CXCR4-recruited B cells are involved in the pathogenesis of CS-induced pulmonary fibrosis. LAs have been reported in peribronchial regions in IPF patients and lupus-prone mice with acute silicosis.42,45 The number, size, and distribution sites of these LAs were found to be significantly related to the severity of pulmonary fibrosis.43,46 In agreement with this, we observed LAs near the bronchi and lymphatic vessels of mice exposed to CS and showed that these LAs were significantly reduced in number and size by AMD3100 treatment. These LAs were composed predominantly of B cells, which implicates B cells in the pathogenesis of silicosis and suggests that they may be a suitable target for effective fibrosis treatment. Notably, AMD3100 mobilized B cells from the bone marrow to the peripheral blood, increasing the number of circulating B cells in mice.47 We showed that more B cells migrated to the lung from the peripheral blood after CS exposure and that AMD3100 inhibited this. These findings suggest that AMD3100 inhibits the migration of circulating B cells to the lung via the CXCL12/CXCR4 axis.
We also observed that B cells accumulating in the lung after CS exposure did not express the proliferation marker Ki67. This is consistent with the findings of Cargnoni et al and Xue et al in BLM-injured and IPF lung tissues.42,48 These results indicate that inhibiting CXCR4 with AMD3100 promotes the migration of bone marrow-derived B cells into the peripheral blood and reduces B-cell infiltration into the lung, further reducing the number and size of LAs in the lung tissues after CS exposure. LAs have been shown to be associated with the severity of IPF. One study showed that B cells from patients with fibroinflammatory disease secreted profibrotic molecules and activated the production of collagen by fibroblasts in vitro.23 In contrast, B-cell deletion decreased the expression of TGF-β and IL-6, reduced collagen deposition, and suppressed fibroblast migration and activation, thereby alleviating fibrosis.43,49 We found that B cells produce collagen I after CS exposure, but that the level of collagen-I+ B cells decreased upon AMD3100 treatment. Further study is needed to determine the mechanism underlying this. Blocking CXCR4-mediated chemotaxis might be an effective strategy to ameliorate pulmonary fibrosis mediated by B cells in the lungs of individuals with silicosis.
Recently, increasing interest has focused on bone marrow-derived CFs in pulmonary fibrosis. We observed that the CXCL12/CXCR4 axis is involved in the migration of CD45+collagen-I+CXCR4+ CFs to silicosis-affected lungs. The infiltrating fibrocytes may promote fibrosis progression and predict a poor prognosis in individuals with acute lung injury, acute respiratory distress syndrome, and IPF.50,51 Li et al showed that fibrocytes co-expressing CD45 and collagen I were increased in the blood and lung of rats exposed to CS.52 Here, we further confirmed that CFs co-expressing CD45, collagen I, and CXCR4 were increased in the lungs of mice after CS exposure, but that AMD3100 reduced the recruitment of CFs to lung tissue in mice. Fibrocytes can differentiate into fibroblasts and myofibroblasts, obtaining the capacity to secrete collagen and other extracellular matrix proteins to promote wound healing and pathologic remodeling.53 We assumed that CXCR4 overexpression increased the numbers of fibroblasts (producing collagen I) and myofibroblasts (producing α-SMA) in CS-exposed lungs, which mediated fibrosis, but AMD3100 reduced these pro-fibrotic cells in the silicotic lung. In short, this study showed that CXCR4 is important for the migration and differentiation of CFs in the silicotic lung, and AMD3100 can slow the progression of pulmonary fibrosis in individuals with silicosis.
Conclusions
CXCR4+ cells (CFs, neutrophils, B lymphocytes) are involved in the progression of pulmonary inflammation and fibrosis in silicosis mice. Our data suggest that targeting CXCR4 may effectively prevent silicosis progression as it reduces inflammation and fibrosis. Therefore, AMD3100 may be a suitable candidate drug for patients with silicosis.
Data Sharing Statement
All data reported in this study are available from the corresponding authors upon reasonable.
Ethics Approval
The study protocol was approved by the Ethics Committee of Anhui University of Science and Technology (NO.2021018).
Author Contributions
All authors made a significant contribution to the work reported, whether that is in the conception, study design, execution, acquisition of data, analysis and interpretation, or in all these areas; they took part in drafting, revising, or critically reviewing the article; gave final approval of the version to be published; have agreed on the journal to which the article has been submitted; and agree to be accountable for all aspects of the work.
Funding
This work was supported by the University Synergy Innovation Program of Anhui Province (GXXT-2021-077).
Disclosure
The authors have declared that this work has no conflicts of interest.
References
1. Barnes H, Goh NSL, Leong TL., et al. Silica-associated lung disease: an old-world exposure in modern industries. Respirology. 2019;24(12):1165–1175. doi:10.1111/resp.13695
2. Feng F, Li N, Cheng P, et al. Tanshinone IIA attenuates silica-induced pulmonary fibrosis via inhibition of TGF-β1-Smad signaling pathway. Biomed Pharmacother. 2020;121:109586. doi:10.1016/j.biopha.2019.109586
3. Hoy RF, Chambers DC. Silica-related diseases in the modern world. Allergy. 2020;75(11):2805–2817. doi:10.1111/all.14202
4. Derlin T, Jaeger B, Jonigk D, et al. Clinical Molecular Imaging of Pulmonary CXCR4 Expression to Predict Outcome of Pirfenidone Treatment in Idiopathic Pulmonary Fibrosis. Chest. 2021;159(3):1094–1106. doi:10.1016/j.chest.2020.08.2043
5. Abraham M, Pereg Y, Bulvik B, et al. Single Dose of the CXCR4 Antagonist BL-8040 Induces Rapid Mobilization for the Collection of Human CD34+ Cells in Healthy Volunteers. Clin Cancer Res. 2017;23(22):6790–6801. doi:10.1158/1078-0432.CCR-16-2919
6. Harms M, Gilg A, Ständker L, et al. Microtiter plate-based antibody-competition assay to determine binding affinities and plasma/blood stability of CXCR4 ligands. Sci Rep. 2020;10(1):16036. doi:10.1038/s41598-020-73012-4
7. Wang J, Tannous BA, Poznansky MC, et al. CXCR4 antagonist AMD3100 (plerixafor): from an impurity to a therapeutic agent. Pharmacological Res. 2020;159:105010. doi:10.1016/j.phrs.2020.105010
8. Song JS, Kang CM, Kang HH, et al. Inhibitory effect of CXC chemokine receptor 4 antagonist AMD3100 on bleomycin induced murine pulmonary fibrosis. Exp Mol Med. 2010;42(6):465–472. doi:10.3858/emm.2010.42.6.048
9. Zhang H, Xiao B, Jiang L, et al. Inhibition of mesenchymal stromal cells’ chemotactic effect to ameliorate paraquat-induced pulmonary fibrosis. Toxicol Lett. 2019;307:1–10. doi:10.1016/j.toxlet.2019.01.005
10. Konrad FM, Meichssner N, Bury A, et al. Inhibition of SDF-1 receptors CXCR4 and CXCR7 attenuates acute pulmonary inflammation via the adenosine A2B-receptor on blood cells. Cell Death Dis. 2017;8(5):e2832. doi:10.1038/cddis.2016.482
11. Li F, Xu X, Geng J, et al. The autocrine CXCR4/CXCL12 axis contributes to lung fibrosis through modulation of lung fibroblast activity. Exp Ther Med. 2020;19(3):1844–1854. doi:10.3892/etm.2020.8433
12. Peng Y, Wu Q, Tang H, et al. NLRP3 Regulated CXCL12 Expression in Acute Neutrophilic Lung Injury. J Inflamm Res. 2020;13:377–386. doi:10.2147/JIR.S259633
13. Zhao FY, Cheng TY, Yang L, et al. G-CSF Inhibits Pulmonary Fibrosis by Promoting BMSC Homing to the Lungs via SDF-1/CXCR4 Chemotaxis. Sci Rep. 2020;10(1):10515. doi:10.1038/s41598-020-65580-2
14. Ghosh MC, Makena PS, Gorantla V, et al. CXCR4 regulates migration of lung alveolar epithelial cells through activation of Rac1 and matrix metalloproteinase-2. Am J Physiol Lung Cell Mol Physiol. 2012;302(9):L846–856. doi:10.1152/ajplung.00321.2011
15. Chiang HY, Chu PH, Lee TH. R1R2 peptide ameliorates pulmonary fibrosis in mice through fibrocyte migration and differentiation. PLoS One. 2017;12(10):e0185811. doi:10.1371/journal.pone.0185811
16. Griffiths K, Habiel DM, Jaffar J, et al. Anti-fibrotic Effects of CXCR4-Targeting i-body AD-114 in Preclinical Models of Pulmonary Fibrosis. Sci Rep. 2018;8(1):3212. doi:10.1038/s41598-018-20811-5
17. Rajasingh J, Li C, Li X, et al. Circulating Fibrocytes Are Increased in Neonates with Bronchopulmonary Dysplasia. PLoS One. 2016;11(6):1–12. doi:10.1371/journal.pone.0157181
18. Sun W, Shi H, Yuan Z, et al. Prognostic Value of Genes and Immune Infiltration in Prostate Tumor Microenvironment. Front Oncol. 2020;10:584055. doi:10.3389/fonc.2020.584055
19. Rodrigues DAS, Prestes EB, Gama AMS, et al. CXCR4 and MIF are required for neutrophil extracellular trap release triggered by Plasmodium-infected erythrocytes. PLoS Pathog. 2020;16(8):e1008230. doi:10.1371/journal.ppat.1008230
20. Chrysanthopoulou A, Mitroulis I, Apostolidou E, et al. Neutrophil extracellular traps promote differentiation and function of fibroblasts. J Pathol. 2014;233(3):294–307. doi:10.1002/path.4359
21. Jang JW, Thuy PX, Lee JW, et al. CXCR4 promotes B cell viability by the cooperation of nuclear factor (erythroid-derived 2)-like 2 and hypoxia-inducible factor-1alpha under hypoxic conditions. Cell Death Dis. 2021;12(4):330. doi:10.1038/s41419-021-03615-w
22. Becker M, Hobeika E, Jumaa H, et al. CXCR4 signaling and function require the expression of the IgD-class B-cell antigen receptor. Proc Natl Acad Sci U S A. 2017;114(20):5231–5236. doi:10.1073/pnas.1621512114
23. Della-Torre E, Rigamonti E, Perugino C, et al. B lymphocytes directly contribute to tissue fibrosis in patients with IgG4-related disease. J Allergy Clin Immunol. 2020;145(3):968–981.e914. doi:10.1016/j.jaci.2019.07.004
24. Eleftheriadis T, Pissas G, Zarogiannis S, et al. Crystalline silica activates the T-cell and the B-cell antigen receptor complexes and induces T-cell and B-cell proliferation. Autoimmunity. 2019;52(3):136–143. doi:10.1080/08916934.2019.1614171
25. Liu F, Dai W, Li C, et al. Role of IL-10-producing regulatory B cells in modulating T-helper cell immune responses during silica-induced lung inflammation and fibrosis. Sci Rep. 2016;6:28911. doi:10.1038/srep28911
26. Barwinska D, Oueini H, Poirier C, et al. AMD3100 ameliorates cigarette smoke-induced emphysema-like manifestations in mice. Am J Physiol Lung Cell Mol Physiol. 2018;315(3):L382–86. doi:10.1152/ajplung.00185.2018
27. Szarka RJ, Wang N, Gordon L, Nation PN, Smith RH. A murine model of pulmonary damage induced by lipopolysaccharide via intranasal instillation. J Immunol Methods. 1997;202:49–57. doi:10.1016/s0022-1759(96)00236-0.
28. Ashcroft T, Simpson JM, Timbrell V. Simple method of estimating severity of pulmonary fibrosis on a numerical scale. J Clin Pathol. 1988;41(4):467–470. doi:10.1136/jcp.41.4.467
29. Nie Y, Wang Z, Chai G, et al. Dehydrocostus Lactone Suppresses LPS-induced Acute Lung Injury and Macrophage Activation through NF-kappaB Signaling Pathway Mediated by p38 MAPK and Akt. Molecules. 2019;24(8):1510. doi:10.3390/molecules24081510
30. De Clercq E. Mozobil(R) (Plerixafor, AMD3100), 10 years after its approval by the US Food and Drug Administration. Antivir Chem Chemother. 2019;27:2040206619829382. doi:10.1177/2040206619829382
31. Pang J, Luo Y, Wei D, et al. Comparative Transcriptome Analyses Reveal a Transcriptional Landscape of Human Silicosis Lungs and Provide Potential Strategies for Silicosis Treatment. Front Genet. 2021;12:652901. doi:10.3389/fgene.2021.652901
32. Garg B, Giri B, Modi S, et al. NFkappaB in Pancreatic Stellate Cells Reduces Infiltration of Tumors by Cytotoxic T Cells and Killing of Cancer Cells, via Up-regulation of CXCL12. Gastroenterology. 2018;155(3):880–891 e888. doi:10.1053/j.gastro.2018.05.051
33. Sercundes MK, Ortolan LS, Debone D, et al. Targeting Neutrophils to Prevent Malaria-Associated Acute Lung Injury/Acute Respiratory Distress Syndrome in Mice. PLoS Pathog. 2016;12(12):e1006054. doi:10.1371/journal.ppat.1006054
34. Scalerandi MV, Peinetti N, Leimgruber C, et al. Inefficient N2-Like Neutrophils Are Promoted by Androgens During Infection. Front Immunol. 2018;9:1980. doi:10.3389/fimmu.2018.01980
35. Sugimoto MA. Anti-Inflammatory Potential of 1-Nitro-2-Phenylethylene. Molecules. 2017;22(11):1–12. doi:10.3390/molecules22111977
36. Perl M, Lomas-Neira J, Chung CS, et al. Epithelial cell apoptosis and neutrophil recruitment in acute lung injury-a unifying hypothesis? What we have learned from small interfering RNAs. Mol Med. 2008;14(7–8):465–475. doi:10.2119/2008-00011.Perl
37. Aratani Y. Myeloperoxidase: its role for host defense, inflammation, and neutrophil function. Arch Biochem Biophys. 2018;640:47–52. doi:10.1016/j.abb.2018.01.004
38. Liu Q, Li Z, Gao J-L, et al. CXCR4 antagonist AMD3100 redistributes leukocytes from primary immune organs to secondary immune organs, lung, and blood in mice. Eur J Immunol. 2015;45(6):1855–1867. doi:10.1002/eji.201445245
39. El-Kashef DH. Nicorandil ameliorates pulmonary inflammation and fibrosis in a rat model of silicosis. Int Immunopharmacol. 2018;64:289–297. doi:10.1016/j.intimp.2018.09.017
40. Alessandro M, Conticini E, Bergantini L, et al. Neutrophil Extracellular Traps in ANCA-Associated Vasculitis and Interstitial Lung Disease: a Scoping Review. Life. 2022;12(2):317. doi:10.3390/life12020317
41. Brinkmann V, Goosmann C, Kuhn LI, et al. Automatic quantification of in vitro NET formation. Front Immunol. 2012;3:413. doi:10.3389/fimmu.2012.00413
42. Cargnoni A, Romele P, Bonassi Signoroni P, et al. Amniotic MSCs reduce pulmonary fibrosis by hampering lung B‐cell recruitment, retention, and maturation. Stem Cells Transl Med. 2020;9(9):1023–1035. doi:10.1002/sctm.20-0068
43. Ali MF, Egan AM, Shaughnessy GF, et al. Antifibrotics Modify B-Cell–induced Fibroblast Migration and Activation in Patients with Idiopathic Pulmonary Fibrosis. Am J Respir Cell Mol Biol. 2021;64(6):722–733. doi:10.1165/rcmb.2020-0387OC
44. Lu Y, Liu F, Li C, et al. IL-10-Producing B Cells Suppress Effector T Cells Activation and Promote Regulatory T Cells in Crystalline Silica-Induced Inflammatory Response in vitro. Mediators Inflamm. 2017;2017:8415094. doi:10.1155/2017/8415094
45. Chauhan PS, Wagner JG, Benninghoff AD, et al. Rapid Induction of Pulmonary Inflammation, Autoimmune Gene Expression, and Ectopic Lymphoid Neogenesis Following Acute Silica Exposure in Lupus-Prone Mice. Front Immunol. 2021;12:635138. doi:10.3389/fimmu.2021.635138
46. Peng M, Wang W, Qin L, et al. Association between nonspecific interstitial pneumonia and presence of CD20+ B lymphocytes within pulmonary lymphoid follicles. Sci Rep. 2017;7(1):16912. doi:10.1038/s41598-017-17208-1
47. Winkler IG, Bendall LJ, Forristal CE, et al. B-lymphopoiesis is stopped by mobilizing doses of G-CSF and is rescued by overexpression of the anti-apoptotic protein Bcl2. Haematologica. 2013;98(3):325–333. doi:10.3324/haematol.2012.069260
48. Xue J, Kass DJ, Bon J, et al. Plasma B lymphocyte stimulator and B cell differentiation in idiopathic pulmonary fibrosis patients. J Immunol. 2013;191(5):2089–2095. doi:10.4049/jimmunol.1203476
49. Mo F, Luo Y, Yan Y, et al. Are activated B cells involved in the process of myocardial fibrosis after acute myocardial infarction? An in vivo experiment. BMC Cardiovasc Disord. 2021;21(1):5. doi:10.1186/s12872-020-01775-9
50. Tai W, Zhou Z, Zheng B, et al. Inhibitory effect of circulating fibrocytes on injury repair in acute lung injury/acute respiratory distress syndrome mice model. J Cell Biochem. 2018;119(10):7982–7990. doi:10.1002/jcb.26664
51. Heukels P, van Hulst JAC, van Nimwegen M, et al. Fibrocytes are increased in lung and peripheral blood of patients with idiopathic pulmonary fibrosis. Respir Res. 2018;19(1):90. doi:10.1186/s12931-018-0798-8
52. Li J, Yao W, Hou JY, et al. The Role of Fibrocyte in the Pathogenesis of Silicosis. Biomed Environ Sci. 2018;31(4):311–316. doi:10.3967/bes2018.040
53. Desai O, Winkler J, Minasyan M, et al. The Role of Immune and Inflammatory Cells in Idiopathic Pulmonary Fibrosis. Front Med. 2018;5:43. doi:10.3389/fmed.2018.00043
 © 2022 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2022 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.