Back to Journals » OncoTargets and Therapy » Volume 16
c-Kit Receptors as a Therapeutic Target in Cancer: Current Insights
Authors Abdellateif MS, Bayoumi AK, Mohammed MA
Received 18 June 2023
Accepted for publication 19 September 2023
Published 27 September 2023 Volume 2023:16 Pages 785—799
DOI https://doi.org/10.2147/OTT.S404648
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Arseniy Yuzhalin
Mona S Abdellateif,1 Ahmed K Bayoumi,2,3 Mohammed Aly Mohammed1
1Medical Biochemistry and Molecular Biology, Cancer Biology Department, National Cancer Institute, Cairo University, Cairo, 11796, Egypt; 2Paediatric Oncology Department, National Cancer Institute, Cairo University, Cairo, 11796, Egypt; 3Children’s Cancer Hospital 57357, Cairo, 11617, Egypt
Correspondence: Mona S Abdellateif, Medical Biochemistry and Molecular Biology, Cancer Biology Department, National Cancer Institute, Cairo University, Cairo, 11796, Egypt, Fax +20 2 23644720, Email [email protected]
Abstract: c-Kit is a type III receptor tyrosine kinase (RTK) that has an essential role in various biological functions including gametogenesis, melanogenesis, hematopoiesis, cell survival, and apoptosis. c-KIT aberrations, either overexpression or loss-of-function mutations, have been implicated in the pathogenesis and development of many cancers, including gastrointestinal stromal tumors, mastocytosis, acute myeloid leukemia, breast, thyroid, and colorectal cancer, making c-KIT an attractive molecular target for the treatment of cancers. Therefore, a lot of effort has been put into investigating the utility of tyrosine kinase inhibitors for the management of c-KIT mutated tumors. This review of the literature illustrates the role of c-KIT mutations in many cancers, aiming to provide insights into the role of TKIs as a therapeutic option for cancer patients with c-KIT aberrations. In conclusion, c-KIT is implicated in different types of cancer, and it could be a successful molecular target; however, proper detection of the underlying mutation type is required before starting the appropriate personalized therapy.
Keywords: c-KIT, SCF, cancer, RTKs, TKIs, therapy
Introduction
c-Kit is a classical proto-oncogene that encodes receptor tyrosine kinases (RTKs), which is expressed in nearly all tissues in the body.1 RTKs respond to stem cell factor (SCF), which is crucial for the self-renewal potency, differentiation, and maintenance of stem cells and many other progenitor cells.2 Hence, c-KIT has an essential role in many vital functions in the human body, including fertility, homeostasis, hematopoiesis, and melanogenesis.3 The RTK is formed of three distinct domains: an extracellular ligand-binding domain, a hydrophobic transmembrane domain, and the cytoplasmic or intracellular domain, with conserved tyrosine kinase activity,4 The intracellular domain consists of a juxtamembrane region, a tyrosine kinase (TK) domain, and a flexible carboxy-terminal (C-terminal) tail.1,4
Activation of the RTK occurs following binding to its receptor-specific ligands, which results in dimerization of the extracellular domain. This conformational change leads to trans-autophosphorylation of the tyrosine residue in the juxtamembrane domain, with the release of ADP, and subsequent autophosphorylation of the activation loop and the C-terminal tail.5–7 This results in the activation of downstream signaling pathways, including the phosphatidylinositol 3-kinase/protein kinase B (PI3K/AKT), mitogen-activated protein kinase/extracellular signal-regulated kinase (MAPK/ERK), Src kinase, and Janus kinase/signal transducer and activator of transcription (JAK/STAT) pathways.8 The PI3K/AKT pathway has an important role in maintaining cell survival, proliferation, regulation of the actin cytoskeleton, and avoidance of apoptosis,9 while the MAPK/ERK pathway is essential for the regulation of gene transcription and cell proliferation.10 Activation of the JAK/STAT pathway results in the stimulation of different gene promoters required for cell proliferation, differentiation, and apoptosis.11,12 The Src kinase pathway is essential for many cellular biological functions, such as cell survival, proliferation, motility, and angiogenesis.13 Therefore, dysregulation of c-Kit function, caused by either overexpression or mutations, leads to the development and progression of various human cancers, including gastrointestinal stromal tumors (GISTs), mastocytosis (MC), acute myeloid leukemia (AML), seminomas, and some types of melanoma.14–18 The constitutive activation of RTKs in human cancers occurs as a result of gain-of-functions mutations, genomic amplification, chromosomal rearrangement, and/or autocrine activation.19 On the other hand, c-Kit loss-of-function mutations have been observed in tumors such as melanoma, thyroid carcinoma (TC), and breast cancer (BC).20–22
The two main approaches that are commonly used for targeting and treating c-KIT mutated tumors are small molecule inhibitors and monoclonal antibodies (mAbs). Regarding small molecule inhibitors, imatinib mesylate was the first tyrosine kinase inhibitor (TKI) to be developed and approved for the treatment of hematological malignancies with aberrant TK activity.23 It was then approved as the first-line treatment for patients with advanced metastatic GIST.24 Subsequently, other TKIs were developed, including sunitinib, sorafenib, dasatinib, regorafenib, ripretinib, avapritinib, nilotinib, amuvatinib, and tivozanib, which were used to target c-KIT in various tumors in which c-KIT is the main driver oncogene.25
Regarding mAbs, they are used to target or inhibit dysregulated c-Kit to overcome the resistance developed in certain wild-type or mutant c-Kit-positive cancer treated with TKIs.26 Moreover, antibody–drug conjugates (ADCs) were designed by conjugating mAbs with different therapeutic agents such as chemotherapeutic drugs, TKIs, or immune checkpoint inhibitors (ICIs). These ADCs could be a successful modality that is able to induce a potent cytotoxic effect on cancer cells while reducing toxicity to normal tissues, and therefore, improving patient survival and outcomes.27
In the current review, we aim to illustrate the role of c-KIT mutations in the development and progression of cancer from a clinical point of view. We also evaluate the therapeutic efficacy of TKIs in different types of tumor, including GIST, AML, melanoma, BC, TC, renal cell carcinoma (RCC), colorectal cancer (CRC), seminoma, germ cell tumors (GCTs), and lung cancer (Table 1). This was performed by investigating the exact role of c-KIT aberrations in each type of cancer individually. Each type has its specific pathogenesis and therefore the role of c-kit varies according to the type of the tumor. In addition, this review discusses the therapeutic significance and limitations of each TKI recommended for the aforementioned cancer types. This will help to open up avenues for more research on c-KIT as a molecular target for different cancer types when used alone or in combination with other treatment strategies.
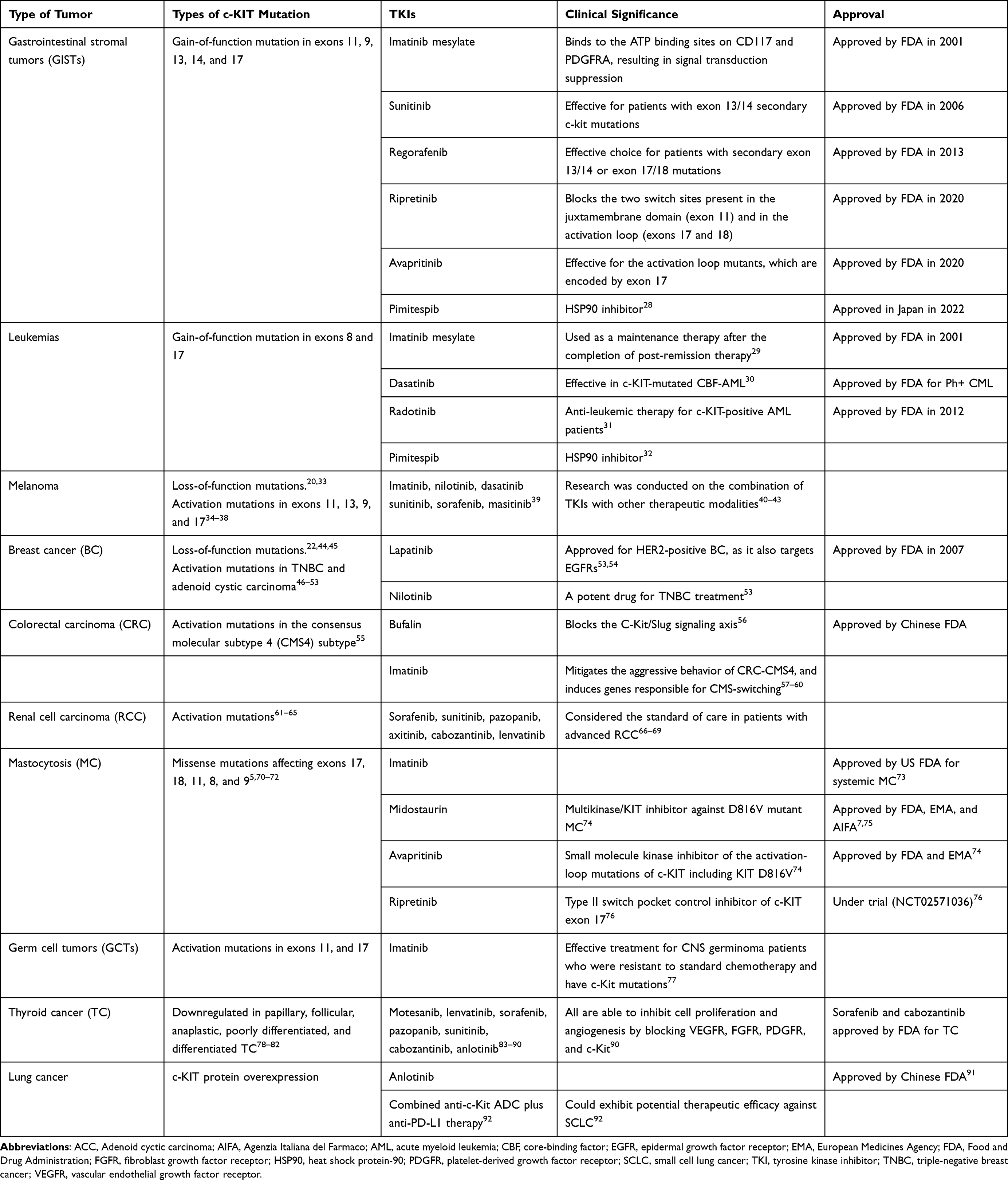 |
Table 1 c-KIT Mutations and the Clinical Significance of Some TKIs in Different Types of Cancer |
Gastrointestinal Stromal Tumors (GISTs)
Gastrointestinal stromal tumors (GISTs) are the main malignant mesenchymal neoplasm originating from the gastrointestinal tract.93 The c-KIT activation mutation is the most common mutation detected in the GIST, accounting for 75–80% of cases, followed by the platelet-derived growth factor receptor alpha (PDGFRA) mutation, which represents nearly 10–15%.94–96 Mutations in the KIT gene were mainly identified in the intracellular juxtamembrane domain at exon 11 (67%), followed by the extracellular dimerization domain at exon 9 (10%), the ATP-binding domain at exons 13 and 14 (1%), and the activation loop at exon 17 (1%), which leads to the expression of a truncated c-KIT/CD117 protein. Mutations in the PDGFRA gene were detected in exons 18 (6% of all GISTs), 12 (0.7% of GISTs), and 14 (0.1% of GISTs).95,97–99 Activation of RTKs results in increased cellular proliferation, differentiation, and migration, and consequently carcinogenesis through activating downstream signaling pathways such as AKT, STAT1, STAT3, MAPK, S6K, PI3K/mTOR, and ETV1.100,101 Therefore, efforts have been made over the past two decades to control GIST by blocking c-KIT signaling through small molecule inhibitors or monoclonal antibodies. Accordingly, GIST is a successful classical model for mutational targeted therapy, where molecular genotyping has become an essential step for managing GIST patients.102
Imatinib mesylate is a derivative of 2-phenyaminopyrimidine that acts as a small molecule inhibitor targeting RTKs. It is considered the first-line treatment for GIST patients with c-KIT or PDGFRA mutations. It inhibits the autophosphorylation and activation of those RTKs through binding to the ATP binding sites on CD117 and PDGFRA, resulting in signal transduction suppression.1,103 It was approved for the treatment of GIST patients in 2001, where it could be used in the preoperative and in adjuvant setting.104,105 Moreover, it was found to improve the progression-free survival (PFS) after 5 years of continuous imatinib treatment in 30% of metastatic GIST patients, and after 10 years in 7–9% of the patients.106,107 Unfortunately, although it can achieve a remarkable clinical outcome in most GIST patients, 70–90% of these patients still develop imatinib resistance and disease relapse within 20–24 months.102,106,108 Many studies have demonstrated that the sensitivity of GIST cells to imatinib is primarily dependent upon the underlying genotype of the tumor cell population, as patients with c-KIT exon 11 mutations are more clinically responsive to imatinib therapy compared to those with c-KIT exon 9 mutations.109,110 Other studies proposed that the failure of imatinib therapy is mostly due to acquiring secondary mutations, resulting in the reactivation of c-KIT or PDGFRA downstream signaling pathways and, consequently, preventing optimal binding of imatinib. Such mutations usually develop in c-KIT exons 13 and 14, which code for the ATP-binding pocket, and in exons 17 and 18, which code for the activation loop of the kinase domain.96,111 The secondary mutations that have evolved in PDGFRA are detected in exons 13, 14, and 15, which encode the ATP-binding pocket.112 On the other hand, it is not clear whether these secondary mutations are originally present in the tumor cell population and become clinically obvious as a result of positive selection caused by imatinib therapy, or are de novo mutations acquired during treatment.102
Two other TKIs that have been approved for imatinib-resistant cases are sunitinib and regorafenib. They are considered the second and the third lines, respectively, for the treatment of patients with advanced GIST, where they have a broad activity against secondary c-kit mutations.102,113 Sunitinib is more effective for patients with exon 13/14 secondary c-kit mutations compared to those with exon 17/18 secondary c-kit mutations,108,111 while regorafenib is an effective choice for patients whose tumors harbor secondary exon 13/14 or exon 17/18 mutations.114
Another TKI that was approved by the US FDA in 2020, based on the positive results obtained from the INVICTUS trial, is ripretinib. It was recommended for advanced or metastatic GIST patients who were resistant to three or more kinase inhibitors including imatinib.115 It hinders the conformational shift from the inactive to the active state of the RTKs by blocking the two switch sites present in the juxtamembrane domain (exon 11) and in the activation loop (exons 17 and 18).116 Therefore, it keeps the c-KIT in an inactive state regardless of the type of mutations, whether primary or secondary, thus allowing for suppression of a wide range of KIT oncoproteins.76 Although it was found that ripretinib achieved the same results regarding the overall response rate (ORR) and the mean progression-free survival (mPFS) rate as the other TKIs after imatinib failure, ripretinib is considered the preferred drug when combined with the other TKIs owing to its efficiency and its acceptable toxicity profile.115
Avapritinib is a small molecule inhibitor targeting RTKs that was approved in 2020 by the FDA for the treatment of GIST patients.117 Unlike the other TKIs, avapritinib is a potent type I inhibitor as it efficiently binds to the active form of the c-KIT at subnanomolar concentrations. It specifically targets the activation loop mutants, which are encoded by exon 17.118
Leukemia
c-KIT mutation is frequently detected in 60–80% of AML patients. It has also been identified in 33.3–45% of AML with inv(16), and in 12.8–46.8% of AML M2 with t(8;21).119 Fan et al found that c-KIT mutations represented 23% of pediatric AML with core-binding factor (CBF).120 The most common type of c-KIT mutation in AML is a gain of function, which is found in the juxtamembrane domain (Val560Gly) and in the second part of the kinase domain (sp816Val).121 It is predominantly hyperactivation, formed of in-frame insertions or deletions in the extracellular domain involved in c-KIT dimerization, which is mainly present in exon 8, or missense mutations that affect the activation loop in the c-KIT TK domain, which is found in exon 17.122 Many studies have highlighted the critical role of c-KIT expression in the proliferation, differentiation, and activation of hematopoietic progenitor cells.31,123,124 Accordingly, it was found that c-KIT is associated significantly with poor survival, increased incidence of relapse, and unfavorable outcomes of AML patients, especially in those with CBF-AML.31,125,126 Moreover, it had been reported that c-KIT exon 17 mutations are associated significantly with worse outcomes in adult de novo AML patients with RUNX1-RUNX1T1.127–129
Genetic markers underlying AML carcinogenesis are considered to be useful prognostic markers, and they could be potential therapeutic targets for AML. However, most AML patients respond initially to induction therapy and then they show resistance or relapses.130 The RTKs are approved for various cancers with mutated c-KIT; however, their role in AML has not been well established.33 Imatinib is used for the inhibition of ABL, KIT, and PDGFR in the treatment of chronic myeloid leukemia, Philadelphia chromosome-positive acute lymphoblastic leukemia, and chronic eosinophilic leukemia with PDGFRα rearrangement.33 Several clinical trials have been performed to assess the utility of RTKs in c-KIT mutated AML.29–31,131,132 Advani et al reported that imatinib can improve the outcome of newly diagnosed AML patients when it is used as a maintenance therapy after the completion of post-remission therapy.29 Moreover, Kampa-Schittenhelm et al reported that dasatinib has an anti-leukemic effect on KIT-mutated CBF-AML.30 Similarly, Heo et al demonstrated that dasatinib and radotinib induce AML cell death by targeting c-KIT both in vivo and in vitro. Therefore, dasatinib and radotinib could have a potential role in anti-leukemic therapy for c-KIT-positive AML patients.31
Another line of AML therapy that has been developed is the heat shock protein-90 (HSP90) inhibitor. HSP90 is a molecular chaperone that regulates the function and folding of various client proteins, including c-KIT.133 Pimitespib (TAS-116) is a HSP90 highly selective inhibitor that was proved by Honma et al, in the phase 3 CHAPTER-GIST-301 trial, to improve the PFS of GIST patients who were resistant to imatinib, sunitinib, and regorafenib.28 Similarly, Ikebe et al proposed that pimitespib showed anti-adult T-cell leukemia/lymphoma (ATL) activity in ATL-related cell lines, in primary ATL cells ex vivo, and in tumors developed in ATL cell-xenografted mice.32 Hence, HSP90 inhibitors in combination with TKIs could be a successful line of treatment in AML.134
Melanoma
Melanoma is an aggressive type of skin cancer.39 c-KIT mutations are identified in 3–9.5% of all melanoma cases, which is mutually exclusive of B-Raf proto-oncogene serine/threonine kinase (BRAF) and neuroblastoma rat sarcoma viral oncogene homolog (NRAS) mutations.39,135,136 The most common c-KIT mutations detected are constitutive activating mutations, in nearly 70% of cases, comprising 39% of mucosal melanomas, 36% of acral melanoma, and 28% of chronically sun-damaged melanoma.1 These mutations include a lysine to-proline mutation at codon 576 (L576P) in exon 11 and a methionine-to-glutamic mutation at codon 642 (K642E) in exon 13, which lead to stimulation of the down-signaling MAPK and PI3K/AKT pathways.1,34,35 Other mutations were also found in exons 9 and 17.36 In addition, c-KIT overexpression was detected in melanoma cases by immunohistochemical analysis, especially in those with ocular melanoma (36–91%).37,38 In this context, Lukenda et al reported a significant association between c-KIT overexpression and shorter overall and disease-free survival in patients with choroidal and ciliary body melanoma.137 Although most of the c-KIT alteration melanomas showed activated mutations, other studies reported a loss of c-KIT expression with tumor progression.20,138 Accordingly, the role of c-KIT mutation in melanoma is controversial, with proper detection of the mutation type being required before starting the appropriate personalized therapy. Regarding the treatment modalities of melanomas, TKIs are one of the molecular-targeted options that provide efficacy against c-KIT mutated melanomas, especially the metastatic type. However, the emergence of resistance is common when it is used as a monotherapy for a prolonged period.139,140 This resistance could be explained by secondary mutations, leading to the expression of various oncogenic proteins, or by the development of epithelial–mesenchymal transition.141 Therefore, several clinical trials have worked on combining TKIs such as imatinib, nilotinib, dasatinib, sunitinib, sorafenib, and masitinib together with other modalities of treatment.39 Some of these clinical trials combined imatinib with anti-PD-1 (pembrolizumab) immunotherapy, which improved the outcomes of the patients.40 Other studies investigated the possibility of combining TKIs and immunotherapies targeting key protein elements involved in the oncogenic RAS/RAF/MAPK or PI3K/AKT pathways. This resulted in improved outcomes and prolonged PFS in patients with metastatic melanoma.41,42 Moreover, Delyon et al43 proposed the potential of combining JAK/STAT inhibitors and c-KIT inhibitors (nilotinib) for the treatment of melanoma, which could improve patients’ response to nilotinib. Overall, melanoma patients with c-KIT mutations could benefit from combining c-KIT inhibitors and immunotherapy, which may be a successful treatment option for these patients in the future.
Breast Cancer (BC)
The role of c-KIT mutation in BC is still a controversial issue, as some research has reported that most BC cases showed loss of c-KIT expression.22,44 This loss of c-KIT function is considered the main deriving factor for malignant transformation due to c-kit gene promoter DNA hypermethylation.45 In contrast, other types of breast cancer, such as adenoid cystic carcinoma (ACC), showed increased c-KIT expression in about 90% of cases, especially those with tubular and cribriform carcinomas of the breast.46–48 Similarly, it had been found that c-KIT was significantly overexpressed in 20–89% of triple-negative breast cancers (TNBCs), which could provide a useful molecular target for this aggressive type of BC.49–52 Moreover, López-Mejía et al53 found that c-Kit overexpression in TNBC results in increasing cell migration and metastasis through the activation of STAT3, Akt, and ERK1/2 pathways. They proposed that TNBC cells expressing functional c-Kit are more sensitive to nilotinib than the other available TKIs. Therefore, nilotinib could be considered a potent drug for TNBC treatment.53 In addition, Funkhouser et al reported a significant association between c-KIT missense mutations (p.M541L) and elevated serum levels of galectins in patients with metastatic disease.142 On the other hand, Vahdatinia et al concluded that c-KIT mutation is a rare event in BC (0.32%) and that the c-KIT-altered BC patients showed high-grade tumors with poor outcomes.143 Many TKIs are commercially available for blocking c-KIT functions, such as imatinib, sorafenib, sunitinib, nilotinib, and dasatinib; however, the only approved TKI for treating BC patients is lapatinib, especially for those with HER2-positive BC, as it also targets epidermal growth factor receptors (EGFRs).53,54
Colorectal Carcinoma (CRC)
The role of c-KIT mutations in CRC is not yet well understood. It has been reported that c-KIT hyperexpression is detected in approximately 50% of CRC tissues.144,145 Moreover, c-KIT expression is significantly associated with the most aggressive subtype, the consensus molecular subtype 4 (CMS4), which has a high rate of disease recurrence and shows poor response to treatment.55 Bellone et al found that c-KIT is highly expressed in the premalignant and malignant tissues of colonic lesions, where it is associated with unfavorable outcomes of the patients.146 In a study performed by Küçükköse et al, c-KIT expression was found to increase the aggressiveness of the colonic tumor cells through the inhibition of SMAD2, which resulted in the stimulation of transforming growth factor-β (TGFβ) expression.57 Another study, by Ma et al, demonstrated that c-KIT could promote CRC progression through the activation of the ERK1/2-ELK1 pathway, resulting in increased carcinoembryonic antigen (CEA) expression.55 Therefore, they suggested TKIs as a therapeutic option for the treatment of CRC patients with CMS4 subtype.55,58 In addition, Wang et al reported that the SCF/c-kit-JNK/AP-1 signaling pathway could maintain tumorigenesis in CRC by stimulating claudin-3 expression.147 Similarly, Li et al reported that SCF/c-KIT signaling was significantly activated in mucinous colorectal adenocarcinoma (MCA), where it induced mucus secretion by goblet cells through the activation of PKCδ-MARCKS.148 Hence, it could provide a new targeted marker for MCA treatment.148 Many published studies have confirmed the supportive role of c-KIT in colonic cancer stem cells (CSCs), as it promotes migration, invasiveness, and EMT.56,149 In this context, Ding et al proposed that bufalin has anti-stemness activity by blocking the C-Kit/Slug signaling axis, and therefore it could be a potent treatment for CRC patients.56 Although c-KIT has been reported in many recently published studies to have an important role in CRC carcinogenesis, imatinib has not yet been approved for the treatment of CRC cases.59 Accordingly, many clinical trials have been performed to assess the therapeutic effect of imatinib on CRC-CMS4. They revealed that imatinib could mitigate the aggressive behavior of CRC-CMS4, and it induces genes responsible for CMS switching, resulting in improvals in patient survival and outcomes.57–60 Therefore, c-KIT targeting therapy could be a useful strategy for CRC-CMS4 management.60
Renal Cell Carcinoma (RCC)
Several studies have reported that c-KIT protein is significantly overexpressed in renal tumor cells in comparison to its normal counterpart, especially in chromophobe RCC and renal oncocytoma subtypes, using immunohistochemical staining.61–65 It was less frequently present in renal angiomyolipomas, papillary RCC, and clear-cell RCC.61–63,65 Regarding the c-KIT mRNA expression analysis, Huo et al demonstrated that c-KIT mRNA was significantly overexpressed in chromophobe RCC and renal oncocytoma compared to the other RCC subtypes, using cDNA expression microarrays.61 Moreover, Lin et al performed mutational analyses of the c-kit gene in different subtypes of RCC. They reported that no mutation was detected in exons 9, 11, 13, or 17 in RCC subtypes, apart from papillary RCC, which showed point mutations in 94% (17/18) of the cases.73 Similarly, Zimpfer et al demonstrated that c-KIT was significantly overexpressed in chromophobe RCC and renal oncocytoma without c-kit mutations.64 Ergün et al, in a study on the possible association between RCC and truncated KIT (tr-KIT), which is an alternative variant of c-KIT protein, proposed that the tr-KIT/c-KIT expression ratio was upregulated in RCC tissues, including clear cell, papillary, and chromophobe RCC. Added to that, the increased tr-KIT/c-KIT ratio was associated significantly with a more aggressive clinical course and inferior patient outcomes.150
Regarding the treatment options for RCC, vascular endothelial growth factor receptors (VEGFRs) and the TKIs sorafenib, sunitinib, pazopanib, axitinib, cabozantinib, and lenvatinib were considered the standard of care in patients with advanced RCC. However, most patients showed disease relapse and drug resistance later.66–69 Many clinical trials have investigated the combination of TKIs and immune checkpoint inhibitors, including nivolumab, ipilimumab, and pembrolizumab, which showed longer overall survival, better clinical prognosis, and more favorable patient outcomes.151–153
Mastocytosis MC)
MC is a neoplastic proliferation of abnormal mast cells which infiltrate different organs. It can present in children as a cutaneous lesion, which undergoes spontaneous regression at puberty, whereas it commonly presents in adults as a more aggressive systemic MC that can lead to multiorgan failure.154,155 More than 90% of systemic MC in adults is associated with c-KIT missense mutations affecting exon 17, codon 816, in which aspartate is substituted by valine (KIT D816V).5 This mutation (KIT D816V) renders c-KIT 586-fold more active than the native c-KIT, which is responsible for imatinib resistance and a poor clinical prognosis.70,71 Similarly, the D816V mutation is detected in 42% of children with systemic MC.16 Other less frequent types of c-KIT mutation that occur in systemic MC and are imatinib sensitive include those occurring in exons 17 and 18 (eg D820G or N822I/K), exon 10 (eg F522C), exon 11 (eg V560G/I), exon 8 (eg deletion of codon 419), and/or exon 9 (deletion of codon p.A502_Y503dup).72 Therefore, the treatment of MC is highly individualized according to the type of mutation detected.154 Patients who have the KIT D816V mutation, which is the most common oncogenic driver for MC, usually experience primary resistance to the TKIs imatinib and masitinib.156,157 Similarly, nilotinib and dasatinib have low clinical significance for these patients.158,159 Imatinib is currently approved by the US FDA for patients with systemic MC who are negative for KIT D816V, have unknown KIT mutation status, or have the previously mentioned imatinib-sensitive KIT mutations.160 Midostaurin is a multikinase/KIT inhibitor that confers a suppressive activity against D816V mutant MC.74 It is approved by the FDA, the European Medicines Agency (EMA), and Agenzia Italiana del Farmaco (AIFA) as a targeted therapy for the treatment of patients with advanced systemic MC, indolent systemic MC, and severe MC mediator-release symptoms (MCMRS).7,75 It is also approved as a first-line treatment for patients with mast cell leukemia (MCL), or as a maintenance therapy after allogeneic stem cell transplantation (ASCT).161 Another available drug that is approved by the FDA and EMA for the treatment of advanced systemic MC is avapritinib.74 Avapritinib is a small molecule kinase inhibitor of the activation-loop mutations of c-KIT, including KIT D816V.74 Another investigational drug that is now under trial (NCT02571036) for the treatment of MC is ripretinib (DCC-2618), which is a type II switch pocket control inhibitor of c-KIT exon 17.76
Germ Cell Tumors (GCTs)
c-KIT mutations have also been detected in human GCTs. Regarding testicular germ cell tumors (TGCTs), the incidence of c-KIT mutation is ten-fold higher in seminoma (20–25%) than in non-seminoma TGCTs.162,163 The most common c-KIT alteration in seminoma is the activating c-KIT mutation, in 10–40% of cases, which present in exons 11 and 17. About two-thirds of seminoma cases have missense point mutations in exon 17, mainly at codon 816, where X is either valine [V] or histidine [H].5,164–166 In addition, McIntyre et al reported that amplification of chromosome 4q12, containing the c-KIT gene, in TGCT was associated with the progression to the seminoma subtype.164 Notably, activating c-KIT mutations have also been found in other tumors having the same histological features as testicular seminoma, including mediastinal seminomas, intracranial germinomas, and ovarian dysgerminomas.167,168 c-KIT mutations have been detected in approximately half of pure ovarian dysgerminoma cases, where all are located in exon 17.169 Moreover, Hersmus et al proposed that c-KIT mutations could have a role in disorders of sex development (DSD).169 It had been reported that c-KIT protein overexpression using immunohistochemistry was also significantly found in ovarian dysgerminoma, which represents about 87% of cases of ovarian dysgerminoma. However, there was no significant correlation between c-KIT protein overexpression and c-kit mutations in the assessed cases.170,171 In this context, Stemberger-Papic et al reported that c-KIT overexpression in patients with primary ovarian high-grade serous carcinoma was associated significantly with shorter disease-free survival and peritoneal metastasis.172 On the other hand, Gao et al assessed c-KIT expression in intracranial GCTs and reported that c-KIT protein expression was found in 59.1% of patients, whereas c-KIT mutations were found in only 5.9% of CNS germinoma cases. This mutation was present in exon 11 at codon 557–558 WK (tryptophan-lysine).77 They concluded that imatinib could be an effective treatment for CNS germinoma patients who are resistant to standard chemotherapy and exhibit c Kit mutations.77 Taken together, these studies show that c-KIT could be a potential prognostic marker and therapeutic target for patients with GCTs.
Thyroid Cancer (TC)
It had been reported that c-KIT has an important role in the differentiation and growth of the thyroid epithelium.21,173 Hence, several studies have investigated the impact of c-KIT mutations on the development of different histological types of TC.21,78–80,173,174 They proposed that c-KIT was strongly downregulated in papillary,81 follicular,82 anaplastic,78 poorly differentiated,79 as well as differentiated TC,80 in comparison to benign thyroid nodules. Moreover, Tomei et al demonstrated that assessment of c-KIT expression in thyroid fine-needle aspiration cytology (FNAC) smears could improve the diagnostic accuracy of the cytological analysis of TC.81 Therefore, several studies were performed to investigate the usefulness of TKIs as a systemic therapy for radioactive iodine-refractory TC. Such TKIs include motesanib diphosphate,83 lenvatinib,84,85 sorafenib,86 pazopanib,87 sunitinib,88 cabozantinib,89 and anlotinib,90 which are able to inhibit cell proliferation and angiogenesis by blocking VEGFR, FGFR, PDGFR, and c-Kit.90 However, there are still some limitations regarding the administration of TKIs in TC, such as secondary resistance or the escape phenomenon, which may occur after a certain period of administration.84
Lung Cancer
c-KIT is reported to be overexpressed in about 70% of small cell lung cancer (SCLC) cases, by immunohistochemical staining, without c-KIT mutation being detected.175–177 Also, it was reported to be overexpressed in 46.9% of advanced non-small cell lung cancer (NSCLC) cases with ALK fusion.178,179 However, the prognostic significance of c-KIT aberrations in lung cancer is still a debatable issue. This discrepancy could be explained by the variability in the type of the sample assessed, the tumor stage, the immunohistochemistry technique, and associated other genetic mutations.1 In this regard, Yang et al demonstrated that c-Kit activation is a good predictor for crizotinib efficacy. They also concluded that c-KIT is a useful prognostic marker, as it is associated with shorter overall survival rates in patients with advanced-stage ALK fusion NSCLC.178 Although c-KIT protein overexpression was found significantly in lung cancer patients, imatinib did not achieve valuable therapeutic efficacy in patients with SCLC.180–182 Anlotinib is a tyrosine multikinase inhibitor that has been approved by the Chinese FDA for the treatment of advanced NSCLC.91 Currently, anlotinib is being investigated as a treatment option for SCLC, CRC, and soft tissue sarcoma.183,184 In addition, Kim et al developed a combination treatment using anti-c-Kit ADC plus anti-PD-L1 therapy (4C9-DM1) that exhibits a potential therapeutic efficacy against SCLC.92
Conclusions
c-KIT has a crucial role in the pathogenesis and development of many cancers; however, its role is not well known yet, especially in certain tumors including breast cancer, melanoma, mastocytosis, thyroid cancer, germ cell tumors, and renal cell carcinoma. The role of c-KIT is still controversial as it varies according to the type of the tumor, as either gain or loss of function. Hence, TKIs are considered an important line of treatment in such c-KIT mutated tumors. However, attention should be directed toward proper detection of the underlying c-KIT mutational type and localization using advanced techniques such as next-generation sequencing. This will allow for better designing a personalized therapy according to the underlying mutations, and consequently maximize the benefit, which will be reflected in the prognosis and outcomes of the patients. Research is now being directed toward combination therapy including TKIs and other therapeutic modalities, such as immunotherapy or checkpoint inhibitors. Moreover, HSP90 is evolving for the treatment of AML patients in combination with TKIs. In summary, c-KIT is still a hot topic for more research on its diagnostic, prognostic, and therapeutic role. Therefore, more efforts should be made to achieve the proper management of cancer patients.
Disclosure
All authors declare that there are no possible conflicts of interest.
References
1. Sheikh E, Tran T, Vranic S, Levy A, Bonfil RD. Role and significance of c-KIT receptor tyrosine kinase in cancer: a review. Bosn J Basic Med Sci. 2022;22(5):683–698. doi:10.17305/bjbms.2021.7399
2. Waskow C, Paul S, Haller C, Gassmann M, Rodewald HR. Viable c-Kit(W/W) mutants reveal pivotal role for c-kit in the maintenance of lymphopoiesis. Immunity. 2002;17(3):277–288. doi:10.1016/s1074-7613(02)00386-2
3. Yavuz AS, Lipsky PE, Yavuz S, Metcalfe DD, Akin C. Evidence for the involvement of a hematopoietic progenitor cell in systemic mastocytosis from single-cell analysis of mutations in the c-kit gene. Blood. 2002;100(2):661–665. doi:10.1182/blood-2002-01-0203
4. Li E, Hristova K. Receptor tyrosine kinase transmembrane domains: function, dimer structure and dimerization energetics. Cell Adh Migr. 2010;4(2):249–254. doi:10.4161/cam.4.2.10725
5. Lennartsson J, Ronnstrand L. Stem cell factor receptor/c-Kit: from basic science to clinical implications. Physiol Rev. 2012;92(4):1619–1649. doi:10.1152/physrev.00046.2011
6. Chan PM, Ilangumaran S, La Rose J, Chakrabartty A, Rottapel R. Autoinhibition of the kit receptor tyrosine kinase by the cytosolic juxtamembrane region. Mol Cell Biol. 2003;23(9):3067–3078. doi:10.1128/mcb.23.9.3067-3078.2003
7. Gilreath JA, Tchertanov L, Deininger MW. Novel approaches to treating advanced systemic mastocytosis. Clin Pharmacol. 2019;11:77–92. doi:10.2147/CPAA.S206615
8. Liang J, Wu YL, Chen BJ, Zhang W, Tanaka Y, Sugiyama H. The C-kit receptor-mediated signal transduction and tumor-related diseases. Int J Biol Sci. 2013;9(5):435–443. doi:10.7150/ijbs.6087
9. Yasuda A, Sawai H, Takahashi H, et al. Stem cell factor/c-kit receptor signaling enhances the proliferation and invasion of colorectal cancer cells through the PI3K/Akt pathway. Dig Dis Sci. 2007;52(9):2292–2300. doi:10.1007/s10620-007-9759-7
10. Feng Z-C, Riopel M, Popell A, Wang R. A survival Kit for pancreatic beta cells: stem cell factor and c-Kit receptor tyrosine kinase. Diabetologia. 2015;58(4):654–665. doi:10.1007/s00125-015-3504-0
11. Chaix A, Lopez S, Voisset E, Gros L, Dubreuil P, De Sepulveda P. Mechanisms of STAT protein activation by oncogenic KIT mutants in neoplastic mast cells. J Biol Chem. 2011;286(8):5956–5966. doi:10.1074/jbc.m110.182642
12. Levy DE, Darnell JE. Stats: transcriptional control and biological impact. Nat Rev Mol Cell Biol. 2002;3(9):651–662. doi:10.1038/nrm909
13. Hendriks RW. Drug discovery: new Btk inhibitor holds promise. Nat Chem Biol. 2011;7(1):4–5. doi:10.1038/nchembio.502
14. Duensing A, Heinrich MC, Fletcher CD, Fletcher JA. Biology of gastrointestinal stromal tumors: KIT mutations and beyond. Cancer Invest. 2004;22(1):106–116. doi:10.1081/cnv-120027585
15. Curtin JA, Busam K, Pinkel D, Bastian BC. Somatic activation of KIT in distinct subtypes of melanoma. J Clin Oncol. 2006;24(26):4340–4346. doi:10.1200/jco.2006.06.2984
16. Bodemer C, Hermine O, Palmerini F, et al. Pediatric mastocytosis is a clonal disease asso ciated with D816V and other activating c-KIT mutations. J Invest Dermatol. 2010;130(3):804–815. doi:10.1038/jid.2009.281
17. Beghini A, Ripamonti CB, Cairoli R, et al. KIT activating mutations: incidence in adult and pedi atric acute myeloid leukemia, and identification of an internal tan dem duplication. Haematologica. 2004;89(8):920–925.
18. Sakuma Y, Sakurai S, Oguni S, Hironaka M, Saito K. Alterations of the c-kit gene in testicular germ cell tumors. Cancer Sci. 2003;94(6):486–491.
19. Hongyo T, Li T, Syaifudin M, et al. Specific c-kit mutations in sinonasal natural killer/T-cell lymphoma in China and Japan. Cancer Res. 2000;60(9):2345–2347.
20. Bello DM, Ariyan CE, Carvajal RD. Melanoma mutagenesis and aberrant cell signaling. Cancer Control. 2013;20(4):261–281. doi:10.1177/107327481302000404
21. Franceschi S, Lessi F, Panebianco F, et al. Loss of c-KIT expression in thyroid cancer cells. PLoS One. 2017;12(3):e0173913. doi:10.1371/journal.pone.0173913
22. Simon R, Panussis S, Maurer R, et al. KIT (CD117)-positive breast cancers are infrequent and lack KIT gene mutations. Clin Cancer Res. 2004;10(1 Pt 1):178–183. doi:10.1158/1078-0432.ccr-0597-3
23. Kantarjian H, Sawyers C, Hochhaus A, et al. Hematologic and cytogenetic responses to imatinib mesylate in chronic myelogenous leukemia. N Engl J Med. 2002;346(9):645–652. doi:10.1056/nejmoa011573
24. Kelly CM, Gutierrez Sainz L, Chi P. The management of metastatic GIST: current standard and investigational therapeutics. J Hematol Oncol. 2021;14(1):2. doi:10.1186/s13045-020-01026-6
25. Pathania S, Pentikäinen OT, Singh PK. A holistic view on c-Kit in cancer: structure, signaling, pathophysiology and its inhibitors. Biochim Biophys Acta Rev Cancer. 2021;1876(2):188631. doi:10.1016/j.bbcan.2021.188631
26. Shi X, Sousa LP, Mandel-Bausch EM, et al. Distinct cellular properties of oncogenic KIT receptor tyrosine kinase mutants enable alternative courses of cancer cell inhibition. Proc Natl Acad Sci USA. 2016;113(33):E4784–E4793. doi:10.1073/pnas.1610179113
27. Hafeez U, Parakh S, Gan HK, Scott AM. Antibody-Drug Conjugates for Cancer Therapy. Molecules. 2020;25(20):4764. doi:10.3390/molecules25204764
28. Honma Y, Kurokawa Y, Sawaki A, et al. Randomized, double-blind, placebo (PL)-controlled, Phase III trial of pimitespib (Tas-116), an oral inhibitor of heat shock protein 90 (HSP90), in patients (pts) with advanced gastrointestinal stromal tumor (GIST) refractory to imatinib (IM), sunitinib (SU) and regorafenib (REG). J Clin Oncol. 2021;39(Suppl 15):11524.
29. Advani AS, Tse W, Li H, et al. A Phase II Trial of Imatinib Mesylate as Maintenance Therapy for Patients with Newly Diagnosed C-kit-positive Acute Myeloid Leukemia. Clin Lymphoma Myeloma Leuk. 2021;21(2):113–118. doi:10.1016/j.clml.2020.11.018
30. Kampa-Schittenhelm KM, Vogel W, Bonzheim I, et al. Dasatinib overrides the differentiation blockage in a patient with mutant-KIT D816V positive CBFβ-MYH11 leukemia. Oncotarget. 2018;9(14):11876–11882. doi:10.18632/oncotarget.24376
31. Heo SK, Noh EK, Kim JY, et al. Targeting c-KIT (CD117) by dasatinib and radotinib promotes acute myeloid leukemia cell death. Sci Rep. 2017;7(1):15278. doi:10.1038/s41598-017-15492-5
32. Ikebe E, Shimosaki S, Hasegawa H, et al. Tas-116 (pimitespib), a heat shock protein 90 inhibitor, shows efficacy in preclinical models of adult T-cell leukemia. Cancer Sci. 2022;113(2):684–696. doi:10.1111/cas.15204
33. Katagiri S, Chi S, Minami Y, et al. Mutated KIT tyrosine kinase as a novel molecular target in acute myeloid leukemia. Int J Mol Sci. 2022;23(9):4694. doi:10.3390/ijms23094694
34. Pham DDM, Guhan S, Tsao H. KIT and melanoma: biological insights and clinical implications. Yonsei Med J. 2020;61(7):562–571. doi:10.3349/ymj.2020.61.7.562
35. Carvajal RD, Lawrence DP, Weber JS, et al. Phase II study of nilotinib in melanoma harboring KIT alterations following progression to prior KIT inhibition. Clin Cancer Res. 2015;21(10):2289–2296. doi:10.1158/1078-0432.CCR-14-1630
36. Lee SJ, Kim TM, Kim YJ, et al. Phase II trial of nilotinib in patients with metastatic malignant melanoma harboring KIT gene aberration: a multicenter trial of Korean cancer study group (UN10-06). Oncologist. 2015;20(11):1312–1319. doi:10.1634/theoncologist.2015-0161
37. Beadling C, Jacobson-Dunlop E, Hodi FS, et al. KIT gene mutations and copy number in melanoma subtypes. Clin Cancer Res. 2008;14(21):6821–6828. doi:10.1158/1078-0432.ccr-08-0575
38. Hofmann UB, Kauczok-Vetter CS, Houben R, Becker JC. Overexpression of the KIT/SCF in uveal melanoma does not translate into clinical efficacy of imatinib mesylate. Clin Cancer Res. 2009;15(1):324–329. doi:10.1158/1078-0432.ccr-08-2243
39. Rager T, Eckburg A, Patel M, et al. Treatment of metastatic melanoma with a combination of immunotherapies and molecularly targeted therapies. Cancers. 2022;14(15):3779. doi:10.3390/cancers14153779
40. Abdou Y, Kapoor A, Hamad L, Ernstoff MS. Combination of pembrolizumab and imatinib in a patient with double KIT mutant melanoma: a case report. Medicine. 2019;98(44):e17769. doi:10.1097/MD.0000000000017769
41. Namikawa K, Yamazaki N. Targeted therapy and immunotherapy for melanoma in Japan. Curr Treat Options Oncol. 2019;20(1):7. doi:10.1007/s11864-019-0607-8
42. Luke JJ, Flaherty KT, Ribas A, Long GV. Targeted agents and immunotherapies: optimizing outcomes in melanoma. Nat Rev Clin Oncol. 2017;14(8):463–482. doi:10.1038/nrclinonc.2017.43
43. Delyon J, Chevret S, Jouary T, et al. STAT3 mediates nilotinib response in KIT-altered melanoma: a phase ii multicenter trial of the French skin cancer network. J Invest Dermatol. 2018;138(1):58–67. doi:10.1016/j.jid.2017.07.839
44. Kondi-Pafiti A, Arkadopoulos N, Gennatas C, Michalaki V, Frangou-Plegmenou M, Chatzipantelis P. Expression of c-kit in common benign and malignant breast lesions. Tumori. 2010;96(6):978–984.
45. Janostiak R, Vyas M, Cicek AF, Wajapeyee N, Harigopal M. Loss of c-KIT expression in breast cancer correlates with malignant transformation of breast epithelium and is mediated by KIT gene promoter DNA hypermethylation. Exp Mol Pathol. 2018;105(1):41–49. doi:10.1016/j.yexmp.2018.05.011
46. Crisi GM, Marconi SA, Makari-Judson G, Goulart RA. Expression of c-kit in adenoid cystic carcinoma of the breast. Am J Clin Pathol. 2005;124(5):733–739. doi:10.1309/61MV-ENEK-5EJ7-JKGF
47. Mastropasqua MG, Maiorano E, Pruneri G, et al. Immunoreactivity for c-kit and p63 as an adjunct in the diagnosis of adenoid cystic carcinoma of the breast. Mod Pathol. 2005;18(10):1277–1282. doi:10.1038/modpathol.3800423
48. Vranic S, Bender R, Palazzo J, Gatalica Z. A review of adenoid cystic carcinoma of the breast with emphasis on its molecular and genetic characteristics. Hum Pathol. 2013;44(3):301–309. doi:10.1016/j.humpath.2012.01.002
49. Zhu Y, Wang Y, Guan B, et al. C-kit and PDGFRA gene mutations in triple negative breast cancer. Int J Clin Exp Pathol. 2014;7(7):4280–4285.
50. Millis SZ, Gatalica Z, Winkler J, et al. Predictive biomarker profiling of > 6000 breast cancer patients shows heterogeneity in TNBC, with treatment implications. Clin Breast Cancer. 2015;15(6):473–481.e3. doi:10.1016/j.clbc.2015.04.008
51. Ahmed R, Ud Din H, Akhtar F, Afzal S, Muhammad I, Hashmi SN. Immunohistochemical expression of epidermal growth factor receptor and c-Kit in triple negative breast cancer. J Coll Physicians Surg Pak. 2016;26(7):570–572.
52. Gromova I, Espinoza JA, Grauslund M, et al. Functional proteomic profiling of triple-negative breast cancer. Cells. 2021;10(10):2768. doi:10.3390/cells10102768
53. López-Mejía JA, Tallabs-Utrilla LF, Salazar-Sojo P, Mantilla-Ollarves JC, Sánchez-Carballido MA, Rocha-Zavaleta L. c-Kit induces migration of triple-negative breast cancer cells and is a promising target for tyrosine kinase inhibitor treatment. Int J Mol Sci. 2022;23(15):8702. doi:10.3390/ijms23158702
54. Voigtlaender M, Schneider-Merck T, Trepel M. Lapatinib. Recent Results Cancer Res. 2018;211:19–44. doi:10.1007/978-3-319-91442-8_2
55. Ma J, Liu X, Chen H, et al. c-KIT-ERK1/2 signaling activated ELK1 and upregulated carcinoembryonic antigen expression to promote colorectal cancer progression. Cancer Sci. 2021;112(2):655–667. doi:10.1111/cas.14750
56. Ding L, Yang Y, Lu Q, et al. Bufalin inhibits tumorigenesis, stemness, and epithelial-mesenchymal transition in colorectal cancer through a C-Kit/Slug Signaling axis. Int J Mol Sci. 2022;23(21):13354. doi:10.3390/ijms232113354
57. Küçükköse E, Peters NA, Ubink I, et al. KIT promotes tumor stroma formation and counteracts tumor-suppressive TGFβ signaling in colorectal cancer. Cell Death Dis. 2022;13(7):617. doi:10.1038/s41419-022-05078-z
58. Ubink I, Bloemendal HJ, Elias SG, et al. Imatinib treatment of poor prognosis mesenchymal-type primary colon cancer: a proof-of-concept study in the preoperative window period (ImPACCT). BMC Cancer. 2017;17(1):282. doi:10.1186/s12885-017-3264-y
59. Peters NA, Constantinides A, Ubink I, et al. Consensus molecular subtype 4 (CMS4)-targeted therapy in primary colon cancer: a proof-of-concept study. Front Oncol. 2022;12:969855. doi:10.3389/fonc.2022.969855
60. Nalli M, Puxeddu M, La Regina G, Gianni S, Silvestri R. Emerging therapeutic agents for colorectal cancer. Molecules. 2021;26(24):7463. doi:10.3390/molecules26247463
61. Huo L, Sugimura J, Tretiakova MS, et al. C-kit expression in renal oncocytomas and chromophobe renal cell carcinomas. Hum Pathol. 2005;36(3):262–268. doi:10.1016/j.humpath.2005.01.011
62. Kruger S, Sotlar K, Kausch I, Horny HP. Expression of KIT (CD117) in renal cell carcinoma and renal oncocytoma. Oncology. 2005;68(2–3):269–275. doi:10.1159/000086783
63. Skenderi F, Ulamec M, Vranic S, et al. Cystic renal oncocytoma and tubulocystic renal cell carcinoma: morphologic and immunohistochemical comparative study. Appl Immunohistochem Mol Morphol. 2016;24(2):112–119. doi:10.1097/pai.0000000000000156
64. Zimpfer A, Janke S, Huhns M, et al. C-kit overexpression is not associated with KIT gene muta tions in chromophobe renal cell carcinoma or renal oncocytoma. Pathol Res Pract. 2014;210(8):521–525. doi:10.1016/j.prp.2014.04.013
65. Norouzinia F, Abbasi F, Dindarian S, et al. Immunohistochemical study of C-kit expression in subtypes of renal cell carcinoma. Turk J Urol. 2018;44(1):31–35. doi:10.5152/tud.2018.91455
66. Motzer RJ, Hutson TE, Tomczak P, et al. Sunitinib versus interferon alfa in metastatic renal-cell carcinoma. N Engl J Med. 2007;356:115–124. doi:10.1056/nejmoa065044
67. Motzer RJ, Hutson TE, Cella D, et al. Pazopanib versus sunitinib in metastatic renal-cell carcinoma. N Engl J Med. 2013;369:722–731. doi:10.1056/nejmoa1303989
68. Choueiri TK, Motzer RJ. Systemic therapy for metastatic renal-cell carcino ma. N Engl J Med. 2017;376:354–366.
69. Medina López RA, Rivero Belenchon I, Mazuecos-Quirós J, Congregado-Ruíz CB, Couñago F. Update on the treatment of metastatic renal cell carcinoma. World J Clin Oncol. 2022;13(1):1–8. doi:10.5306/wjco.v13.i1.1
70. De Beauchene IC, Allain A, Panel N, et al. Hotspot mutations in KIT receptor differentially modulate its allosterically coupled conformational dynamics: impact on activation and drug sensitivity. PLoS Comput Biol. 2014;10(7):e1003749.
71. Naumann N, Lubke J, Baumann S, et al. Adverse prognostic impact of the KIT D816V transcriptional activity in advanced systemic mastocytosis. Int J Mol Sci. 2021;22(5):2562.
72. Arock M, Sotlar K, Akin C, et al. KIT mutation analysis in mast cell neoplasms: recommendations of the European Competence Network on Mastocytosis. Leukemia. 2015;29(6):1223–1232. doi:10.1038/leu.2015.24
73. Lin ZH, Han EM, Lee ES, et al. A distinct expression pattern and point mutation of c-kit in papillary renal cell carcinomas. Mod Pathol. 2004;17(6):611–616. doi:10.1038/modpathol.3800108
74. Papayannidis C, Federico V, Fianchi L, et al. Treatment of advanced systemic mastocytosis with midostaurin: practical guidance for optimal therapy and management. Mediterr J Hematol Infect Dis. 2022;14(1):e2022073. doi:10.4084/MJHID.2022.073
75. van Anrooij B, Oude Elberink JNG, Span LFR, et al. Midostaurin in patients with indolent systemic mastocytosis: an open-label Phase 2 trial. J Allergy Clin Immunol. 2018;142(3):1006–1008.e7. doi:10.1016/j.jaci.2018.06.003
76. Smith BD, Kaufman MD, Lu WP, et al. Ripretinib (DCC-2618) is a switch control kinase inhibitor of a broad spectrum of oncogenic and drug-resistant KIT and PDGFRA variants. Cancer Cell. 2019;35(5):738–751.e9. doi:10.1016/j.ccell.2019.04.006
77. Gao YP, Jiang JY, Liu Q. Expression and mutation of c-Kit in intracranial germ cell tumors: a single-centre retrospective study of 30 cases in China. Oncol Lett. 2016;11(5):2971–2976. doi:10.3892/ol.2016.4373
78. Broecker-Preuss M, Sheu SY, Worm K, et al. Expression and mutation analysis of the tyrosine kinase c-kit in poorly differentiated and anaplastic thyroid carcinoma. Horm Metab Res. 2008;40(10):685–691. doi:10.1055/s-2008-1080895
79. Murakawa T, Tsuda H, Tanimoto T, Tanabe T, Kitahara S, Matsubara O. Expression of KIT, EGFR, HER-2 and tyrosine phosphorylation in undifferentiated thyroid carcinoma: implication for a new therapeutic approach. Pathol Int. 2005;55(12):757–765. doi:10.1111/j.1440-1827.2005.01902.x
80. Tanaka T, Umeki K, Yamamoto I, et al. c-Kit proto-oncogene is more likely to lose expression in differentiated thyroid carcinoma than three thyroid-specific genes: thyroid peroxidase, thyroglobulin, and thyroid stimulating hormone receptor. Endocr J. 1995;42(5):723–728. doi:10.1507/endocrj.42.723
81. Tomei S, Mazzanti C, Marchetti I, et al. c-KIT receptor expression is strictly associated with the biological behaviour of thyroid nodules. J Transl Med. 2012;10:7. doi:10.1186/1479-5876-10-7
82. Makhlouf AM, Chitikova Z, Pusztaszeri M, et al. Identification of CHEK1, SLC26A4, c-KIT, TPO and TG as new biomarkers for human follicular thyroid carcinoma. Oncotarget. 2016;7(29):45776–45788. doi:10.18632/oncotarget.10166
83. Sherman SI, Wirth LJ, Droz JP, et al. Motesanib diphosphate in progressive differentiated thyroid cancer. N Engl J Med. 2008;359(1):31–42. doi:10.1056/NEJMoa075853
84. Lorusso L, Pieruzzi L, Biagini A, et al. Lenvatinib and other tyrosine kinase inhibitors for the treatment of radioiodine refractory, advanced, and progressive thyroid cancer. Onco Targets Ther. 2016;9:6467–6477. doi:10.2147/OTT.S84625
85. Takahashi S, Kiyota N, Yamazaki T, et al. A phase II study of the safety and efficacy of lenvatinib in patients with advanced thyroid cancer. Future Oncol. 2019;15:717–726.
86. Ito Y, Onoda N, Ito KI, et al. Sorafenib in Japanese patients with locally advanced or metastatic medullary thyroid carci noma and anaplastic thyroid carcinoma. Thyroid. 2017;27:1142–1148.
87. Bible KC, Suman VJ, Molina JR, et al. A multicenter phase 2 trial of pazopanib in metastatic and progressive medullary thyroid carcinoma: MC057H. J Clin Endocrinol Metab. 2014;99:1687–1693.
88. Ferrari SM, Centanni M, Virili C, et al. Sunitinib in the treatment of thyroid cancer. Curr Med Chem. 2019;26(6):963–972. doi:10.2174/0929867324666171006165942
89. Schlumberger M, Elisei R, Muller S, et al. Overall survival analysis of EXAM, a phase III trial of cabozantinib in patients with radiographically progressive medullary thyroid carcinoma. Ann Oncol. 2017;28:2813–2819.
90. Li D, Chi Y, Chen X, et al. Anlotinib in locally advanced or metastatic medullary thyroid carcinoma: a randomized, double-blind phase IIB trial. Clin Cancer Res. 2021;27(13):3567–3575. doi:10.1158/1078-0432.CCR-20-2950
91. Han B, Li K, Wang Q, et al. Effect of anlotinib as a third-line or further treatment on overall survival of patients with advanced non-small cell lung cancer: the ALTER 0303 phase 3 randomized clinical trial. JAMA Oncol. 2018;4(11):1569–1575. doi:10.1001/jamaoncol.2018.3039
92. Kim KH, Kim JO, Park JY, Seo MD, Park SG. Antibody-drug conjugate targeting c-Kit for the treatment of small cell lung cancer. Int J Mol Sci. 2022;23(4):2264. doi:10.3390/ijms23042264
93. Kindblom LG, Remotti HE, Aldenborg F, Meis-Kindblom JM. Gastrointestinal pacemaker cell tumor (GIPACT): gastrointestinal stromal tumors show phenotypic characteristics of the interstitial cells of Cajal. Am J Pathol. 1998;152(5):1259–1269.
94. Heinrich MC, Corless CL, Duensing A, et al. PDGFRA activating mutations in gastrointestinal stromal tumors. Science. 2003;299(5607):708–710. doi:10.1126/science.1079666
95. Hirota S, Isozaki K, Moriyama Y, et al. Gain-of-function mutations of c-kit in human gastrointestinal stromal tumors. Science. 1998;279(5350):577–580. doi:10.1126/science.279.5350.577
96. Serrano C, George S. Gastrointestinal stromal tumor: challenges and opportunities for a new decade. Clin Cancer Res. 2020;26(19):5078–5085. doi:10.1158/1078-0432.CCR-20-1706
97. Wang WJ, Li HT, Yu JP, et al. Identification of key genes and associated pathways in KIT/PDGFRA wild‑type gastrointestinal stromal tumors through bioinformatics analysis. Mol Med Rep. 2018;18(5):4499–4515. doi:10.3892/mmr.2018.9457
98. Li K, Cheng H, Li Z, et al. Genetic progression in gastrointestinal stromal tumors: mechanisms and molecular interventions. Oncotarget. 2017;8(36):60589–60604. doi:10.18632/oncotarget.16014
99. Niinuma T, Suzuki H, Sugai T. Molecular characterization and pathogenesis of gastrointestinal stromal tumor. Transl Gastroenterol Hepatol. 2018;3:2.
100. Duensing A, Medeiros F, McConarty B, et al. Mechanisms of oncogenic KIT signal transduction in primary gastrointestinal stromal tumors (GISTs). Oncogene. 2004;23(22):3999–4006. doi:10.1038/sj.onc.1207525
101. Ran L, Sirota I, Cao Z, et al. Combined inhibition of MAP kinase and KIT signaling synergistically destabilizes ETV1 and suppresses GIST tumor growth. Cancer Discov. 2015;5(3):304–315. doi:10.1158/2159-8290.CD-14-0985
102. Serrano C, George S, Valverde C, et al. Novel insights into the treatment of imatinib-resistant gastrointestinal stromal tumors. Target Oncol. 2017;12(3):277–288. doi:10.1007/s11523-017-0490-9
103. Din OS, Woll PJ. Treatment of gastrointestinal stromal tumor: focus on imatinib mesylate. Ther Clin Risk Manag. 2008;4(1):149–162. doi:10.2147/tcrm.s1526
104. Joensuu H, Roberts PJ, Sarlomo-Rikala M, et al. Effect of the tyrosine kinase inhibitor STI571 in a patient with a metastatic gastrointestinal stromal tumor. N Engl J Med. 2001;344(14):1052–1056. doi:10.1056/NEJM200104053441404
105. von Mehren M, Joensuu H. Gastrointestinal Stromal Tumors. J Clin Oncol. 2018;36(2):136–143. doi:10.1200/JCO.2017.74.9705
106. Casali PG, Zalcberg J, Le Cesne A, et al. Ten-year progression-free and overall survival in patients with unresectable or metastatic GI stromal tumors: long-term analysis of the European organisation for research and treatment of cancer, Italian sarcoma group, and Australasian gastrointestinal trials group intergroup phase III randomized trial on imatinib at two dose levels. J Clin Oncol. 2017;35(15):1713–1720. doi:10.1200/JCO.2016.71.0228
107. Heinrich MC, Rankin C, Blanke CD, et al. Correlation of Long-term Results of Imatinib in Advanced Gastrointestinal Stromal Tumors With Next-Generation Sequencing Results: analysis of Phase 3 SWOG Intergroup Trial S0033. JAMA Oncol. 2017;3(7):944–952. doi:10.1001/jamaoncol.2016.6728
108. Blanke CD, Demetri GD, von Mehren M, et al. Long-term results from a randomized phase II trial of standard- versus higher-dose imatinib mesylate for patients with unresectable or metastatic gastrointestinal stromal tumors expressing KIT. J Clin Oncol. 2008;26(4):620–625. doi:10.1200/jco.2007.13.4403
109. Heinrich MC, Corless CL, Blanke CD, et al. Molecular correlates of imatinib resistance in gastrointestinal stromal tumors. J Clin Oncol. 2006;24(29):4764–4774. doi:10.1200/JCO.2006.06.2265
110. Tuveson DA, Willis NA, Jacks T, et al. STI571 inactivation of the gastrointestinal stromal tumor c-KIT oncoprotein: biological and clinical implications. Oncogene. 2001;20(36):5054–5058. doi:10.1038/sj.onc.1204704
111. Heinrich MC, Maki RG, Corless CL, et al. Primary and secondary kinase genotypes correlate with the biological and clinical activity of sunitinib in imatinib-resistant gastrointestinal stromal tumor. J Clin Oncol. 2008;26(33):5352–5359. doi:10.1200/jco.2007.15.7461
112. Grunewald S, Klug LR, Mühlenberg T, et al. Resistance to Avapritinib in PDGFRA-driven GIST is caused by secondary mutations in the PDGFRA Kinase domain. Cancer Discov. 2021;11(1):108–125. doi:10.1158/2159-8290.CD-20-0487
113. Demetri GD, Reichardt P, Kang YK, et al. Efcacy and safety of regorafenib for advanced gastrointestinal stromal tumours after failure of imatinib and sunitinib (GRID): an international, multicentre, randomised, placebo-controlled, phase 3 trial. Lancet. 2013;381(9863):295–302. doi:10.1016/S0140-6736(12)61857-1
114. Jeffers M, Kappeler C, Kuss I, et al. Broad spectrum of regorafenib activity on mutant KIT and absence of clonal selection in gastrointestinal stromal tumor (GIST): correlative analysis from the GRID trial. Gastric Cancer. 2022;25(3):598–608. doi:10.1007/s10120-021-01274-6
115. Serrano C, Bauer S. New tyrosine kinase inhibitors for the treatment of gastrointestinal stromal tumors. Curr Oncol Rep. 2022;24(2):151–159. doi:10.1007/s11912-021-01165-0
116. Mol CD, Dougan DR, Schneider TR, et al. Structural basis for the autoinhibition and STI-571 inhibition of c-Kit tyrosine kinase. J Biol Chem. 2004;279(30):31655–31663. doi:10.1074/jbc.M403319200
117. Heinrich MC, Jones RL, von Mehren M, et al. Avapritinib in advanced PDGFRA D842V-mutant gastrointestinal stromal tumour (NAVIGATOR): a multicentre, open-label, Phase 1 trial. Lancet Oncol. 2020;21(7):935–946. doi:10.1016/S1470-2045(20)30269-2
118. Evans EK, Gardino AK, Kim JL, et al. A precision therapy against cancers driven by KIT/PDGFRA mutations. Sci Transl Med. 2017;9(414):eaao1690. doi:10.1126/scitranslmed.aao1690
119. Cioccio J, Claxton D. Therapy of acute myeloid leukemia: therapeutic targeting of tyrosine kinases. Expert Opin Investig Drugs. 2019;28(4):337–349. doi:10.1080/13543784.2019.1584610
120. Fan J, Gao L, Chen J, Hu S. Influence of KIT mutations on prognosis of pediatric patients with core-binding factor acute myeloid leukemia: a systematic review and meta-analysis. Transl Pediatr. 2020;9(6):726–733. doi:10.21037/tp-20-102
121. Lennartsson J, Jelacic T, Linnekin D, Shivakrupa R. Normal and oncogenic forms of the receptor tyrosine kinase kit. Stem Cells. 2005;23(1):16–43. doi:10.1634/stemcells.2004-0117
122. Allen C, Hills RK, Lamb K, et al. The importance of relative mutant level for evaluating impact on outcome of KIT, FLT3 and CBL mutations in core-binding factor acute myeloid leukemia. Leukemia. 2013;27(9):1891–1901. doi:10.1038/leu.2013.186
123. Ashman LK, Griffith R. Therapeutic targeting of c-KIT in cancer. Expert Opin Investig Drugs. 2013;22(1):103–115. doi:10.1517/13543784.2013.740010
124. Marcucci G, Haferlach T, Döhner H. Molecular genetics of adult acute myeloid leukemia: prognostic and therapeutic implications. J Clin Oncol. 2011;29(5):475–486. doi:10.1200/JCO.2010.30.2554
125. Nabil R, Hassan NM, Abdellateif MS, Gawdat RM, Elshazly SS. The prognostic role of C-KIT, TET1 and TET2 gene expression in acute myeloid leukemia. Mol Biol Rep. 2023;50(1):641–653. doi:10.1007/s11033-022-08000-0
126. Ayatollahi H, Shajiei A, Sadeghian MH, et al. Prognostic importance of C-KIT mutations in core binding factor acute myeloid leukemia: a systematic review. Hematol Oncol Stem Cell Ther. 2017;10(1):1–7. doi:10.1016/j.hemonc.2016.08.005
127. Krauth MT, Eder C, Alpermann T, et al. High number of additional genetic lesions in acute myeloid leukemia with t(8;21)/RUNX1-RUNX1T1: frequency and impact on clinical outcome. Leukemia. 2014;28(7):1449–1458. doi:10.1038/leu.2014.4
128. Kim HJ, Ahn HK, Jung CW, et al. KIT D816 mutation associates with adverse outcomes in core binding factor acute myeloid leukemia, especially in the subgroup with RUNX1/RUNX1T1 rearrangement. Ann Hematol. 2013;92(2):163–171. doi:10.1007/s00277-012-1580-5
129. Ishikawa Y, Kawashima N, Atsuta Y, et al. Prospective evaluation of prognostic impact of KIT mutations on acute myeloid leukemia with RUNX1-RUNX1T1 and CBFB-MYH11. Blood Adv. 2020;4(1):66–75. doi:10.1182/bloodadvances.2019000709
130. Padmakumar D, Chandraprabha VR, Gopinath P, et al. A concise review on the molecular genetics of acute myeloid leukemia. Leuk Res. 2021;111:106727. doi:10.1016/j.leukres.2021.106727
131. Carter JL, Hege K, Yang J, et al. Targeting multiple signaling pathways: the new approach to acute myeloid leukemia therapy. Signal Transduct Target Ther. 2020;5(1):288. doi:10.1038/s41392-020-00361-x
132. Klug LR, Corless CL, Heinrich MC. Inhibition of KIT tyrosine kinase activity: two decades after the first approval. J Clin Oncol. 2021;39(15):1674–1686. doi:10.1200/JCO.20.03245
133. Woodford MR, Sager RA, Marris E, et al. Tumor suppressor Tsc1 is a new Hsp90 co-chaperone that facilitates folding of kinase and non-kinase clients. EMBO J. 2017;36(24):3650–3665. doi:10.15252/embj.201796700
134. Chi SG, Minami Y. Emerging targeted therapy for specific genomic abnormalities in acute myeloid leukemia. Int J Mol Sci. 2022;23(4):2362. doi:10.3390/ijms23042362
135. Yaman B, Akalin T, Kandiloğlu G. Clinicopathological characteristics and mutation profiling in primary cutaneous melanoma. Am J Dermatopathol. 2015;37(5):389–397. doi:10.1097/DAD.0000000000000241
136. Carlino MS, Haydu LE, Kakavand H, et al. Correlation of BRAF and NRAS mutation status with outcome, site of distant metastasis and response to chemotherapy in metastatic melanoma. Br J Cancer. 2014;111(2):292–299. doi:10.1038/bjc.2014.287
137. Lukenda A, Dotlic S, Vukojevic N, Saric B, Vranic S, Zarkovic K. Expression and prognostic value of putative cancer stem cell markers CD117 and CD15 in choroidal and ciliary body melanoma. J Clin Pathol. 2016;69(3):234–239. doi:10.1136/jclinpath-2015-203130
138. Montone KT, van Belle P, Elenitsas R, Elder DE. Proto-oncogene c-kit expression in malignant melanoma: protein loss with tumor progression. Mod Pathol. 1997;10(9):939–944.
139. Montor WR, Salas AROSE, Melo FHM. Receptor tyrosine kinases and downstream pathways as druggable targets for cancer treatment: the current arsenal of inhibitors. Mol Cancer. 2018;17(1):55. doi:10.1186/s12943-018-0792-2
140. Czarnecka AM, Bartnik E, Fiedorowicz M, Rutkowski P. Targeted Therapy in Melanoma and Mechanisms of Resistance. Int J Mol Sci. 2020;21(13):4576. doi:10.3390/ijms21134576
141. Iderzorig T, Kellen J, Osude C, et al. Comparison of EMT mediated tyrosine kinase inhibitor resistance in NSCLC. Biochem Biophys Res Commun. 2018;496(2):770–777. doi:10.1016/j.bbrc.2018.01.069
142. Funkhouser AT, Strigenz AM, Blair BB, et al. KIT mutations correlate with higher galectin levels and brain metastasis in breast and non-small cell lung cancer. Cancers. 2022;14(11):2781. doi:10.3390/cancers14112781
143. Vahdatinia M, Derakhshan F, Da Cruz Paula A, et al. KIT genetic alterations in breast cancer. J Clin Pathol. 2022. doi:10.1136/jcp-2022-208611
144. Chen EC, Karl TA, Kalisky T, et al. KIT signaling promotes growth of colon xenograft tumors in mice and is up-regulated in a subset of human colon cancers. Gastroenterology. 2015;149(3):705–17.e2. doi:10.1053/j.gastro.2015.05.042
145. Zhang B, Wang J, Wang X, et al. Proteogenomic characterization of human colon and rectal cancer. Nature. 2014;513(7518):382–387. doi:10.1038/nature13438
146. Bellone G, Smirne C, Carbone A, et al. KIT/stem cell factor expression in premalignant and malignant lesions of the colon mucosa in relationship to disease progression and outcomes. Int J Oncol. 2006;29(4):851–859. doi:10.3892/ijo.29.4.851
147. Wang Y, Sun T, Sun H, Yang S, Li D, Zhou D. SCF/C-Kit/JNK/AP-1 signaling pathway promotes claudin-3 expression in colonic epithelium and colorectal carcinoma. Int J Mol Sci. 2017;18(4):765. doi:10.3390/ijms18040765
148. Li G, Yang S, Shen P, et al. SCF/c-KIT signaling promotes mucus secretion of colonic goblet cells and development of mucinous colorectal adenocarcinoma. Am J Cancer Res. 2018;8(6):1064–1073.
149. Rothenberg ME, Nusse Y, Kalisky T, et al. Identification of a cKit(+) colonic crypt base secretory cell that supports Lgr5(+) stem cells in mice. Gastroenterology. 2012;142(5):1195–1205.e6. doi:10.1053/j.gastro.2012.02.006
150. Ergün S, Altay DU, Güneş S, et al. Tr-KIT/c-KIT ratio in renal cell carcinoma. Mol Biol Rep. 2019;46(5):5287–5294. doi:10.1007/s11033-019-04985-3
151. Albiges L, Tannir NM, Burotto M, et al. Nivolumab plus ipilimumab versus sunitinib for first-line treatment of advanced renal cell carcinoma: extended 4-year follow-up of the phase III CheckMate 214 trial. Esmo Open. 2020;5:e001079. doi:10.1136/esmoopen-2020-001079
152. Capitanio U, Fallara G, Raggi D, et al. Pembrolizumab in advanced renal cell carcinoma: a meta-analysis providing level 1a evidence. Curr Probl Cancer. 2022;46(4):100875. doi:10.1016/j.currproblcancer.2022.100875
153. Motzer R, Alekseev B, Rha SY, et al. Lenvatinib plus pembrolizumab or everolimus for advanced renal cell carcinoma. N Engl J Med. 2021;384(14):1289–1300. doi:10.1056/NEJMoa2035716
154. Pardanani A. Systemic mastocytosis in adults: 2021 Update on diagnosis, risk stratification and management. Am J Hematol. 2021;96(4):508–525. doi:10.1002/ajh.26118
155. Lim KH, Tefferi A, Lasho TL, et al. Systemic mastocytosis in 342 consecutive adults: survival studies and prognostic factors. Blood. 2009;113:5727–5736.
156. Growney JD, Clark JJ, Adelsperger J, et al. Activation mutations of human c-KIT resistant to imatinib mesylate are sensitive to the tyrosine kinase inhibitor PKC412. Blood. 2005;106(2):721–724.
157. Dubreuil P, Letard S, Ciufolini M, et al. Masitinib (AB1010), a potent and selective tyrosine kinase inhibitor targeting KIT. PLoS One. 2009;4(9):e7258.
158. Hochhaus A, Baccarani M, Giles FJ, et al. Nilotinib in patients with systemic mastocytosis: analysis of the phase 2, open-label, single-arm nilotinib registration study. J Cancer Res Clin Oncol. 2015;141(11):2047–2060.
159. Verstovsek S, Tefferi A, Cortes J, et al. Phase II study of dasatinib in Philadelphia chromosome-negative acute and chronic myeloid diseases, including systemic mastocytosis. Clin Cancer Res. 2008;14(12):3906–3915.
160. Alvarez-Twose I, Matito A, Morgado JM, et al. Imatinib in systemic mastocytosis: a Phase IV clinical trial in patients lacking exon 17 KIT mutations and review of the literature. Oncotarget. 2016;8(40):68950–68963.
161. Ustun C, Courville EL. Resolution of osteosclerosis after alloHCT in systemic mastocytosis. Blood. 2016;127(14):1836. doi:10.1182/blood-2016-01-690669
162. Oosterhuis JW, Looijenga LH. Testicular germ-cell tumours in a broader perspective. Nat Rev Cancer. 2005;5(3):210–222. doi:10.1038/nrc1568
163. Coffey J, Linger R, Pugh J, et al. Somatic KIT mutations occur predominantly in seminoma germ cell tumors and are not predictive of bilateral disease: report of 220 tumors and review of literature. Genes Chromosomes Cancer. 2008;47(1):34–42. doi:10.1002/gcc.20503
164. McIntyre A, Summersgill B, Grygalewicz B, et al. Amplification and overexpression of the KIT gene is associated with progression in the seminoma subtype of testicular germ cell tumors of adolescents and adults. Cancer Res. 2005;65(18):8085–8089. doi:10.1158/0008-5472.CAN-05-0471
165. Nakai Y, Nonomura N, Oka D, et al. KIT (c-kit oncogene product) pathway is constitutively activated in human testicular germ cell tumors. Biochem Biophys Res Commun. 2005;337(1):289–296. doi:10.1016/j.bbrc.2005.09.042
166. Willmore-Payne C, Holden JA, Chadwick BE, Layfield LJ. Detection of c-kit exons 11- and 17-activating mutations in testicular seminomas by high-resolution melting amplicon analysis. Mod Pathol. 2006;19(9):1164–1169. doi:10.1038/modpathol.3800623
167. Przygodzki RM, Hubbs AE, Zhao FQ, O’Leary TJ. Primary mediastinal seminomas: evidence of single and multiple KIT mutations. Lab Invest. 2002;82(10):1369–1375. doi:10.1097/01.lab.0000032410.46986.7b
168. Sakuma Y, Sakurai S, Oguni S, Satoh M, Hironaka M, Saito K. c-kit gene mutations in intracranial germinomas. Cancer Sci. 2004;95(9):716–720. doi:10.1111/j.1349-7006.2004.tb03251.x
169. Hersmus R, Stoop H, van de Geijn GJ, et al. Prevalence of c-KIT mutations in gonadoblastoma and dysgerminomas of patients with disorders of sex development (DSD) and ovarian dysgerminomas. PLoS One. 2012;7(8):e43952. doi:10.1371/journal.pone.0043952
170. Cheng L, Roth LM, Zhang S, et al. KIT gene mutation and amplification in dysgerminoma of the ovary. Cancer. 2011;117(10):2096–2103. doi:10.1002/cncr.25794
171. Sever M, Jones TD, Roth LM, et al. Expression of CD117 (c-kit) receptor in dysgerminoma of the ovary: diagnostic and therapeutic implications. Mod Pathol. 2005;18(11):1411–1416.
172. Stemberger-Papić S, Vrdoljak-Mozetic D, Ostojić DV, et al. Expression of CD133 and CD117 in 64 serous ovarian cancer cases. Coll Antropol. 2015;39(3):745–753.
173. Natali PG, Berlingieri MT, Nicotra MR, et al. Transformation of thyroid epithelium is associated with loss of c-kit receptor. Cancer Res. 1995;55(8):1787–1791.
174. Pusztaszeri MP, Sadow PM, Faquin WC. CD117: a novel ancillary marker for papillary thyroid carcinoma in fine-needle aspiration biopsies. Cancer Cytopathol. 2014;122(8):596–603. doi:10.1002/cncy.21437
175. Hida T, Ueda R, Sekido Y, et al. Ectopic expression of c-kit in small-cell lung cancer. Int J Cancer Suppl. 1994;8:108–109. doi:10.1002/ijc.2910570723
176. Yokouchi H, Nishihara H, Harada T, et al. Immunohistochemical profiling of receptor tyrosine kinases, MED12, and TGF-betaRII of surgically resected small cell lung cancer, and the potential of c-kit as a prognostic marker. Oncotarget. 2017;8(24):39711–39726. doi:10.18632/oncotarget.14410
177. Lu HY, Zhang G, Cheng QY, et al. Expression and mutation of the c-kit gene and correlation with prognosis of small cell lung cancer. Oncol Lett. 2012;4(1):89–93. doi:10.3892/ol.2012.679
178. Yang H, Wang F, Deng Q, et al. Predictive and prognostic value of phosphorylated c-KIT and PDGFRA in advanced non-small cell lung cancer harboring ALK fusion. Oncol Lett. 2019;17(3):3071–3076. doi:10.3892/ol.2019.9972
179. Zhang L, Jiang T, Li X, et al. Clinical features of Bim deletion polymorphism and its relation with crizotinib primary resistance in Chinese patients with ALK/ROS1 fusion-positive non-small cell lung cancer. Cancer. 2017;123(15):2927–2935. doi:10.1002/cncr.30677
180. Wang WL, Healy ME, Sattler M, et al. Growth inhibition and modulation of kinase pathways of small cell lung cancer cell lines by the novel tyrosine kinase inhibitor STI 571. Oncogene. 2000;19(31):3521–3528. doi:10.1038/sj.onc.120369831
181. Soria JC, Johnson BE, Chevalier TL. Imatinib in small cell lung cancer. Lung Cancer. 2003;41 Suppl 1:S49–S53. doi:10.1016/s0169-5002(03)00142-9
182. Johnson BE, Fischer T, Fischer B, et al. Phase II study of imatinib in patients with small cell lung cancer. Clin Cancer Res. 2003;9(16 Pt 1):5880–5887.
183. Shen G, Zheng F, Ren D, et al. Anlotinib: a novel multi-targeting tyrosine kinase inhibitor in clinical development. J Hematol Oncol. 2018;11(1):120. doi:10.1186/s13045-018-0664-7
184. Cheng Y, Wang Q, Li K, et al. OA13.03 anlotinib as thirdline or further-line treatment in relapsed SCLC: a multicentre, randomized, double-blind phase 2 trial. J Thorac Oncol. 2018;13:S351–2.
 © 2023 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2023 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.