Back to Journals » OncoTargets and Therapy » Volume 10
Bystander signaling via oxidative metabolism
Authors Sawal HA ![]() , Asghar K, Bureik M, Jalal N
, Asghar K, Bureik M, Jalal N
Received 3 March 2017
Accepted for publication 3 June 2017
Published 4 August 2017 Volume 2017:10 Pages 3925—3940
DOI https://doi.org/10.2147/OTT.S136076
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Samir Farghaly
Humaira Aziz Sawal,1 Kashif Asghar,2 Matthias Bureik,3 Nasir Jalal4
1Healthcare Biotechnology Department, Atta-ur-Rahman School of Applied Biosciences, National University of Sciences and Technology, Islamabad, 2Basic Sciences Research, Shaukat Khanum Memorial Cancer Hospital and Research Centre, Lahore, Pakistan; 3Health Science Platform, School of Pharmaceutical Science and Technology, Tianjin University, Tianjin, China; 4Health Science Platform, Department of Molecular and Cellular Pharmacology, Tianjin University, Tianjin, China
Abstract: The radiation-induced bystander effect (RIBE) is the initiation of biological end points in cells (bystander cells) that are not directly traversed by an incident-radiation track, but are in close proximity to cells that are receiving the radiation. RIBE has been indicted of causing DNA damage via oxidative stress, besides causing direct damage, inducing tumorigenesis, producing micronuclei, and causing apoptosis. RIBE is regulated by signaling proteins that are either endogenous or secreted by cells as a means of communication between cells, and can activate intracellular or intercellular oxidative metabolism that can further trigger signaling pathways of inflammation. Bystander signals can pass through gap junctions in attached cell lines, while the suspended cell lines transmit these signals via hormones and soluble proteins. This review provides the background information on how reactive oxygen species (ROS) act as bystander signals. Although ROS have a very short half-life and have a nanometer-scale sphere of influence, the wide variety of ROS produced via various sources can exert a cumulative effect, not only in forming DNA adducts but also setting up signaling pathways of inflammation, apoptosis, cell-cycle arrest, aging, and even tumorigenesis. This review outlines the sources of the bystander effect linked to ROS in a cell, and provides methods of investigation for researchers who would like to pursue this field of science.
Keywords: radiation-induced bystander effect (RIBE), reactive oxygen species (ROS), oxidative stress, bystander signaling, tumorigenesis
Introduction
Since the discovery of radiation-induced bystander effect (RIBE)-mediated mutations,1 the associated gene expressions2 and carcinogenesis3 have been studied extensively. Several well-established signaling pathways of the RIBE have been established that include but are not limited to TGF-β1,4 TNFα,5 IL8,6 nitric oxide (NO),7,8 COX2,9 and carbon monoxide (CO).10 On the basis of these factors, a unified model was proposed in 2008.11 Recent research has shown that the regulation of the RIBE depends upon the secretion of certain important proteins that allow communication between cells, and could be as diverse as the reactive oxygen species (ROS) produced during oxidative metabolism that lead to stress.12 Oxidative stress is mainly caused by increased levels of ROS in the environment of a cell. The generation of ROS is dependent on endogenous sources, such as by-products of cellular metabolism, or exogenous sources, such as exposure to ionizing radiation, ultraviolet radiation, or redox-cycling drugs, carcinogenic compounds, and anticancer alkylating agents.13 Prolonged exposure to ROS leads to chronic inflammation, which contributes to tumor development and progression, and is hence recognized as a critical hallmark of cancer.14 Some of the commonly occurring ROS species in radiation-targeted cells are shown in Figure 1.
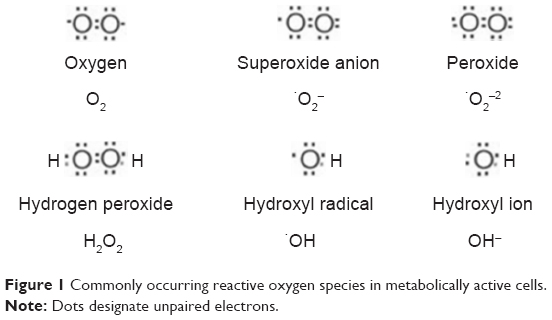 | Figure 1 Commonly occurring reactive oxygen species in metabolically active cells. |
Several biological abnormalities occur in both naïve and bystander cells. These abnormalities can be mutations and alterations in protein expression at transcription, posttranscription, translation, and even posttranslation levels.15–17 These signaling factors can affect bystander cells through the culture medium and/or the gap junction between directly communicating cells, but the source of this signal has been widely debated.8,18 Evidence suggests that nuclear DNA could not be a direct target in the induction of the RIBE, but that the incident-radiation track must pass through the cytoplasm and membranous organelles before it reaches the nucleus, and hence any of the cytoplasmic organelles in the path of a radiation track could be the cause. It has been suggested that microbeam-targeted cytoplasmic irradiation can also induce bystander responses, where cell-membrane rafts may also be involved.19 In addition, as key cell organelles in the cytoplasm, mitochondria have been shown to be sensors of the RIBE,20 and the bystander response cannot be induced in cells without mitochondria.21–23 The involvement of induced DNA strand breaks or their repair pathways contributing to bystander mutagenesis has also been argued.24 Considering the new advances in the RIBE, this review focuses on the signaling pathways that are involved in bystander signaling due to exogenous or endogenous sources of oxidative stress.
Oxidative stress-related pathways of RIBE
In one of the earliest reported studies on the RIBE, it was shown that when glioblastoma cells were treated with helium ions, the frequency of micronuclei formation in the surrounding cells increased.1 In normal cultures of fibroblast cells, it has been demonstrated that gap junctions, ROS, and TGF-β1 are involved in bystander signaling.2–4 NO has been considered to contribute towards the initiation of damage to DNA in the medium.5,6 Lyng et al122 concluded that the RIBE led to the production of micronuclei and apoptosis7,8 and that radiotherapy-induced stress can cause TGF-β1 release (Figure 2).
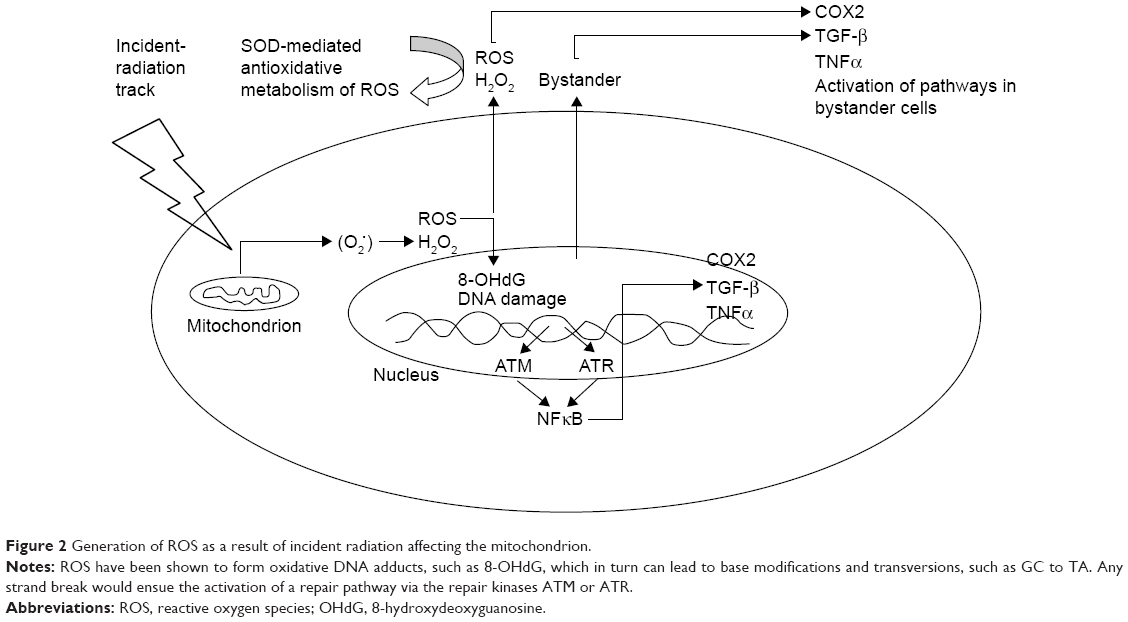 | Figure 2 Generation of ROS as a result of incident radiation affecting the mitochondrion. |
DNA damage-induced activation of ATM-p53 and ATM leads to the activation of NFκB-signaling pathways, followed by the stimulation of NFκB-dependent gene expression, including those for IL8, TNFα, COX2, iNOS, and the production of prostaglandin E2 (PGE2), ROS, and NO.23,25 The stimulation of bystander cells due to the paracrine mechanism using cytokine or growth-factor interactions with the corresponding receptors results in an induction of cell-signaling pathways and specific gene expression in bystander cells,9,23 which in turn lead to the translocation of ROS and NO from directly irradiated to bystander cells through gap junctions with the secondary damaging effects on mitochondrial and genomic DNA in bystander cells.
Genetic defects caused by radiation and ensuing signaling pathways
In similar experiments, it has been reported that after bombarding cells in vitro with radiation, DNA is damaged and genetic or chromosomal rearrangements occur. In mammalian cell lines, most genetic alterations occur in cells when they are exposed to a low frequency of α-particles.11–14 When radiation hits the target cell, because of its high-energy deposition (due to linear energy transfer), the chemical bonds in the target are cleaved and nucleotide bases damaged (Figure 3). Most lethal damage is due to the production of lesions, such as double-strand breaks, which occur because DNA in the nucleus becomes directly damaged. DNA damage can be repaired by the cells using DNA-repair machinery; however, if complete repair does not occur, there can be two possible outcomes: chromosomal abnormalities with depletion of genetic material that lead to cell-cycle arrest or the overwhelming extent of unrepaired damage could trigger apoptosis. The double-strand breaks thus caused can either be repaired efficiently via homologous recombination or with major defects via nonhomologous end joining and produce a bystander effect.11,17,19
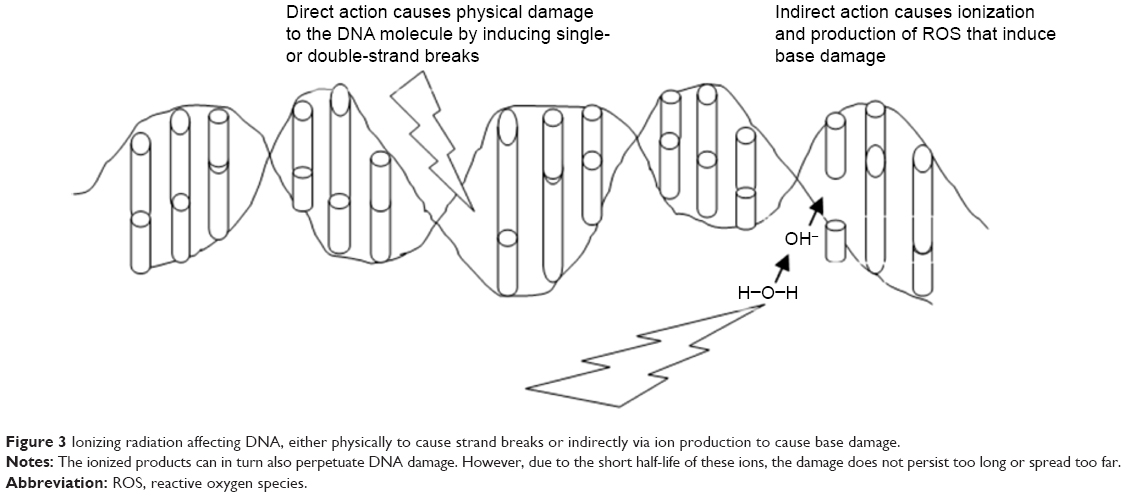 | Figure 3 Ionizing radiation affecting DNA, either physically to cause strand breaks or indirectly via ion production to cause base damage. |
Accumulation of major damage over several generations can lead to tumorigenic progeny or ultimately to cell death.15 The RIBE generally has two modes of action to proliferate bystander effects. For cells that are in direct contact with each other, bystander signaling uses gap junctions. These tiny pores can transport small molecules between cells that are involved in signaling.16 An important group of proteins called connexins combine with one another to form gap junctions. Such junctions can allow the transport of signaling molecules in the size range of 1–1.5 kDa. Important molecules that pass through these are small proteins, secondary messengers, and nucleotides. Attached cell lines, on the other hand, transfer signals through gap junctions, while the suspended cell lines transmit signals through released cytokines and other soluble molecules or even ROS.17,18
Microbeam low-dose irradiation has been used to target helium ions (3He2+) to individual cells in a population of radioresistant glioma cells, either cultured alone or in coculture with primary human fibroblasts. The study indicated that even when a single cell within the glioma population had one 3He2+ ion pass through the cytoplasm, bystander responses were induced in the neighboring unirradiated glioma or fibroblasts. This resulted in a micronuclei yield increased by 36% for the glioma population and 78% for the bystander-fibroblast population.19 Direct low linear energy-transfer radiation induces point mutations in mitochondrial DNA. Some of the genes that undergo deletion include MTATP6, MTCO3, MTND3, MTND4L, MTND4, MTND5, and five tRNAs. As a stress response, the number of mitochondria increases in cells traversed with low-dose radiation. Such mitochondria may proliferate more rapidly than in untargeted cells.20
Oxidative stress and Keap1–Nrf2 stress-response pathway
Upon targeting with ionizing radiation or oxidative xenobiotics, cells are usually confronted with oxidative stress, and hence they must quickly augment their antioxidant capacity to deal with increased ROS production and maintain homeostasis. Nrf2 is a transcription factor regulating the redox-homeostatic gene-regulatory network (Figure 4). The Nrf2-signaling pathway is activated under oxidative stress to increase the expression of a number of antioxidants and drug-detoxification phase II enzymes, such as UGTs, that can jointly work to restore redox homeostasis. Keap1, a cysteine-rich protein, while being anchored to actin in the cytosol, interacts with Nrf2 to work as an adaptor protein for the Cul3 ubiquitin-ligase complex. Under normal conditions, Keap1 promotes ubiquitination and eventual degradation of Nrf2. This is a very quick event, because Nrf2 has a relatively short half-life of 13–21 minutes.26,27 The Keap1–Nrf2 pathway plays a pivotal role in providing protection against oxidative and xenobiotic stresses. The Nrf2 transcription factor activates the transcription of various cytoprotective genes that have been indicted in cancer and neurodegenerative diseases.28 In fact, this pathway is now recognized as an important anticancer target.29 While the Keap1–Nrf2 pathway enables the cell to counter oxidative and xenobiotic stresses, its disruption does quite the opposite, making the cell susceptible to carcinogens and chronic inflammation that eventually modify the phenotype of a cell from normal to tumorigenic.30,31 The importance of Nrf2 has been highlighted in the gene regulation of several phase II drug-metabolism enzymes. Homeostatic regulatory control of the expression of phase II enzymes in a cell is Nrf2-dependent.32
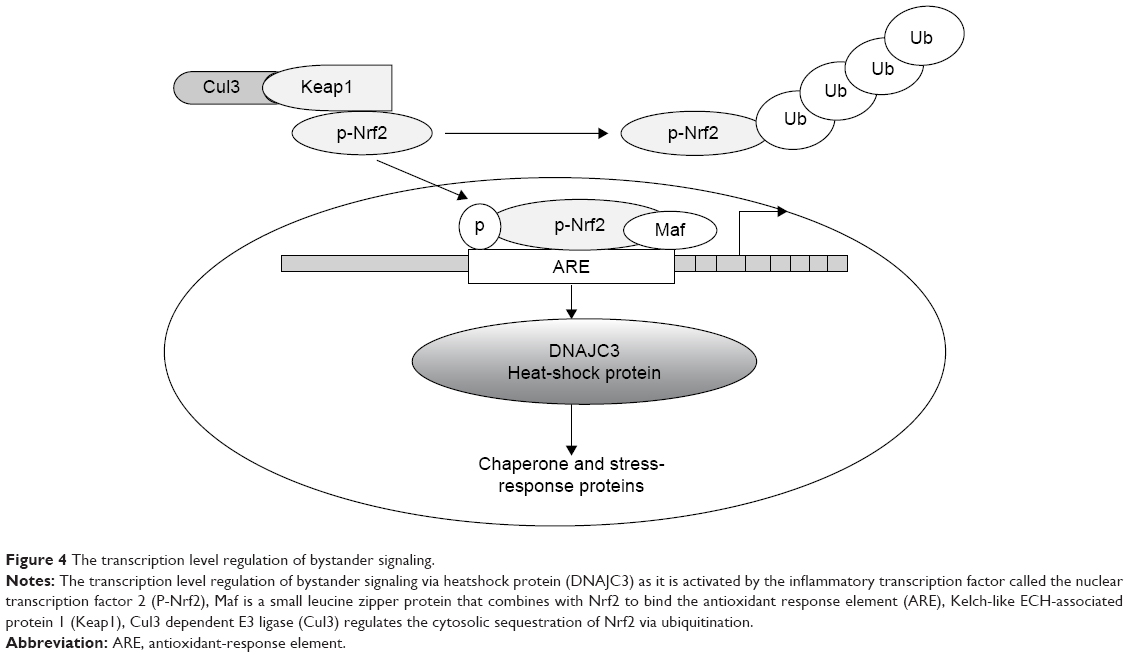 | Figure 4 The transcription level regulation of bystander signaling. |
At genetic and transcription levels, Nrf2 activation mediates bystander signaling via oxidative metabolism. The expression level of DNAJC3, sometimes known as protein kinase-inhibitor p58, was shown to increase by 1.6-fold in the untargeted liver of irradiated rats. DNAJC3 is known to be involved in the endoplasmic reticulum stress response. It is also a target of Nrf2, a critical transcription factor for mediating amplification of the mammalian defense system against various environmental stresses. The Nrf2 protein is a key regulator of response to oxidative stress. It acts as a transcription factor that regulates the expression and induction of a range of defensive genes encoding detoxifying enzymes and antioxidant proteins.36
Bystander signaling at translation level
When simulated space radiation using 48Ti ions at 50 cGy (1,100 MeV/nucleon) is targeted to the head of male Sprague Dawley rats, it can modify a number of proteins in the untargeted liver. Altered protein structure would eventually have modified function or cause complete loss of function. Although the list of cellular proteins that are modified or can possibly be modified is not exhaustive, one radiation-induced bystander-mutagenesis experiment reported 25 proteins to suffer that fate (Table 1).36
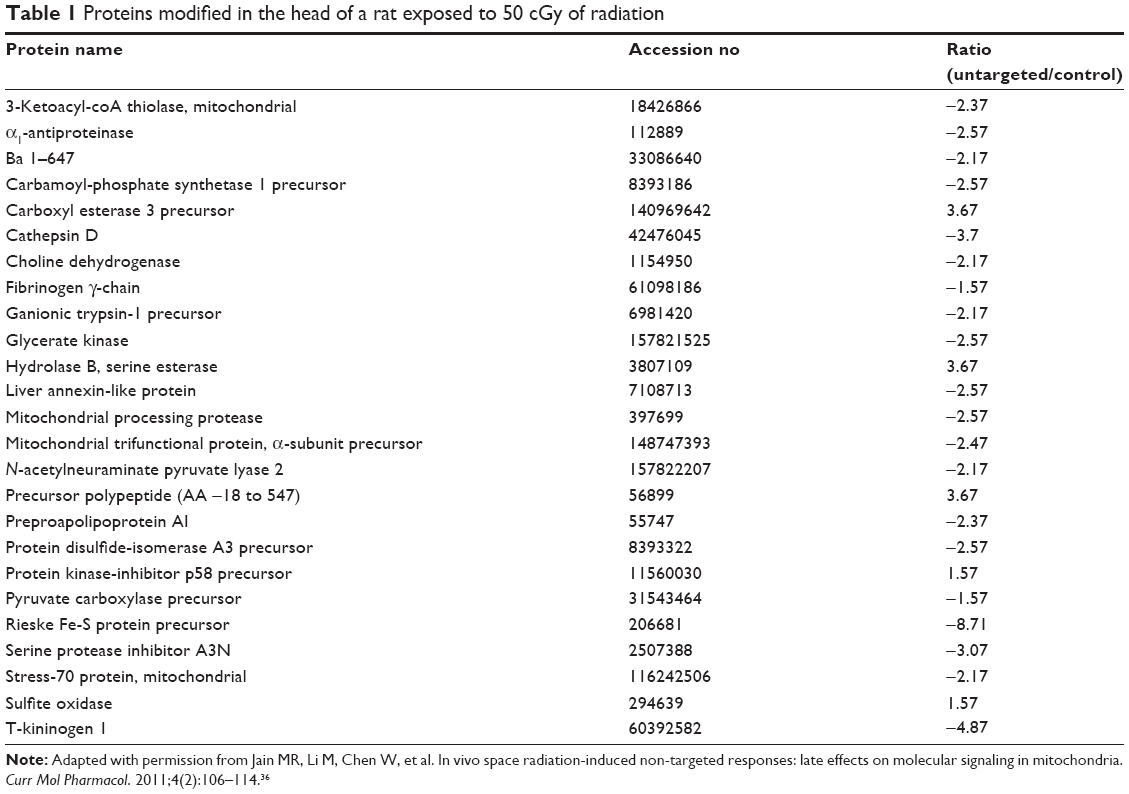 | Table 1 Proteins modified in the head of a rat exposed to 50 cGy of radiation |
Some other ways in which signaling molecules could be modified are NO-dependent. NO-mediated posttranslational modification of macromolecules is a new area that we have just now started to explore. Peroxynitrite, a strongly oxidizing intermediate of NO metabolism, has been shown to modify the structure of macromolecules to induce apoptosis in a p38-mediated manner.33 Although the process is not yet fully known, it was clear that oxidative damage was not the primary cause,34 but Akt and MLK/MAPK pathways are believed to be involved.35 It is also logical to assume that the modified proteins (identified in Table 1) could have been due to the involvement of peroxynitrite.
ROS-generating enzymes in cells
Several cellular organelles, such as mitochondria (as part of the electron-transport chain), the endoplasmic reticulum (especially during ER stress), and peroxisomes (in the metabolism of long-chain fatty acids),37 regulate metabolic reactions that lead to ROS generation. Most of the enzymes involved in such metabolic reactions are either lipoxygenases, xanthine oxidases, cyclooxygenases, cytochrome P450 monooxygenases, NO synthases (NOSs), and NADPH oxidases.38 Three types of such enzymes – lipoxygenases, cyclooxygenases and epoxygenases/monooxygenases (eg, cytochrome P450) – can metabolize arachidonic acid to biologically active eicosanoids. Various lipid peroxides and bioactive lipids produced via this metabolism can affect cell proliferation, apoptosis, differentiation, and senescence, consequently leading to carcinogenesis.39,40 An imbalance in the generation and removal of ROS can lead to a pathological condition associated with oxidative stress.41 Prime candidates of oxidative stress are not only limited to the free radicals ·OH, RO2·, NO· and O2·−, but also include the nonradicals HOCl, 1O2, ONOO–, O3, and H2O2.42
Lipoxygenase
Lipoxygenase (Lox)-catalyzed metabolism of arachidonic acid into various eicosanoids has been widely studied, and the enzyme expression varies throughout the phases of cancer initiation and progression, and hence regulates aspects of tumor development.43 Lox comprises a family of nonheme iron dioxygenases that insert molecular oxygen into free and esterified polyunsaturated fatty acids and based on regional specificity may be classified as 5-, 8-, 12-, and 15-Lox.44 The Lox enzymes metabolize arachidonic acid to the biologically active metabolite hydroperoxyeicosatetraenoic acids (HPETE), which can be reduced to hydroxyeicosatetraenoic acid (HETE). The products of Lox metabolism can either produce intermediary metabolites, such as HPETE, or they can be catalyzed into secondary products, such as lipoxins, hepoxilins, and HETE,45 which can act as signaling molecules independently or give rise to ROS. The enzyme 15-Lox1 is expressed mainly in reticulocytes, eosinophils, epithelial cells of air passages, and macrophages of atherosclerotic lesions.46 This enzyme has been shown to play a role in cell differentiation and maturation, inflammation, asthma, carcinogenesis, and atherogenesis.47 While many investigations indicate that 5- and 12-Lox metabolites promote angiogenesis and carcinogenesis, in contrast 15-Lox may play an inhibitory role in tumor angiogenesis and thus may slow down carcinogenesis.48,49 In a study that explored the contribution of photodynamic stress toward bystander signaling in WTK1 cells, lipid peroxidation was identified as playing a key role in the perpetuation of this signal.50 The involvement of Lox enzymes in carcinogenesis and angiogenesis is a clear indication of the involvement of ROS, which could logically contribute to the overall bystander burden in an environment of oxidative stress.
Cyclooxygenase
The role of Lox and COX is widely recognized in cancers.39,51,52 Tumor-cell functions at primary and secondary sites are controlled by many different factors, including growth factors and their receptors, nuclear receptors, intercellular interactions, cell–matrix interactions, and various chemokines, as well as oxygenated metabolites of arachidonic acid.51,52 In 1971, Vane first demonstrated that aspirin and indomethacin inhibited PG production by blocking the enzymatic activity of COX.53 Since then, the direct correlation between COX and anti-inflammatory drugs, such as nonsteroidal anti-inflammatory drugs, has been explained. These drugs either covalently modify the enzyme or selectively inhibit (eg, COX2 inhibited by APHS, found in aspirin) by competing for the active site.54 Aspirin blocks COX enzymatic activity via the acetylation of Ser530 in COX1 and Ser516 in COX2.55 The COX1 and COX2 enzymes catalyze rate-limiting steps in the production of PGs and thromboxane from arachidonic acid. Both COX enzymes convert arachidonic acid to endoperoxide PGH2, and downstream-selective isomerases convert PGH2 to prostacyclin, PGD2, PGE2, PGF2α, or thromboxane A2.56 COX2 expression has been shown to increase adenomas in mouse models of adenoma and implicated in human colorectal cancer, followed by the observation that COX2 also plays a role in colorectal metastasis into liver56 and angiogenesis.57 Also, the chemical inhibition of COX2 has demonstrated immense therapeutic significance for halting tumor growth and progression.58 The enzymatic contribution of Lox- or COX-mediated ROS toward bystander mutagenesis has not yet been investigated, and thus is a good direction to pursue.
Cytochrome P450 enzymes
Cytochrome P450 enzymes (CYPs or P450s) are a large superfamily of monooxygenases present in all biological kingdoms.59 Within the context of this review, they are of special interest, because their activity can cause oxidative stress not only via ROS production but also by the generation of reactive oxygenated metabolites (ROMs).62–64 We summarize the current knowledge for both of these aspects.
P450s are B-type hemoproteins that are named after the unusual spectral properties of the complex between reduced CYP and carbon monoxide, which is characterized by a pronounced peak at 450 nm.60 This spectral feature is caused by a cysteine-thiolate group that forms the fifth ligand of the heme iron. More than 200,000 distinct CYP genes have been identified to date61 and P450 systems can be found throughout all life forms, although there are organisms (such as Escherichia coli) that lack them.62 In general, CYPs are the terminal oxidase enzymes in short electron-transfer chains that encompass a CYP enzyme together with one or two electron-transfer proteins and constitute a P450 system.63 P450 systems can metabolize a huge variety of small and large molecules, and are best known for catalyzing hydroxylations, but some of them can also perform N-, O-, and S-dealkylations, sulfoxidations, epoxidations, deaminations, and even other reaction types.64–66
In mammals, CYP content is highest in the liver, but they are also present in kidneys, small intestine, lungs, adrenal cortex, skin, brain, testes, placenta, and other tissues. Humans have 57 functional CYP genes, and the enzymes encoded by them are membrane-bound proteins that are either located on the cytoplasmic side of the endoplasmic reticulum or on the matrix side of the inner mitochondrial membrane. In addition, the nuclear envelope and plasma membranes contain low amounts of CYP.67,68 Many human CYP systems are involved in the phase I metabolism of xenobiotics, while others have their predominant role in the biosynthesis of physiologically important compounds, such as steroids and fatty acids; in addition, around half a dozen members are still considered “orphans”, because their main function remains unknown.69 Importantly, the two groups of drug-metabolizing CYPs on one hand and those CYPs that catalyze vital endogenous reactions on the other are not strictly separated from each other, eg, some steroid hydroxylases can also metabolize xenobiotics with steroidal structures.70–74
Despite the diversity of P450 chemistries, all cytochrome P450-enzyme reactions follow a common catalytic cycle, in which an iron(IV)–oxo species with an additional oxidizing equivalent delocalized over the porphyrin, and thiolate ligands (P450 compound I) are the principal intermediate.75 According to current understanding, the individual steps in the P450 catalytic cycle are: 1) substrate binding, 2) first single-electron reduction, 3) oxygen binding, 4) second single-electron reduction, 5) protonation of the distal oxygen coordinating with iron, 6) formation of the reactive heme iron–oxygen species (compound I), 7) hydrogen-atom abstraction, 8) oxygen rebound with the radical intermediate, and 9) product release.76 While the different steps of the cycle are believed to be very similar for most CYP enzymes, the rates of each step can differ widely between different CYPs and sometimes even for different reactions catalyzed by the same enzyme. For instance, the second electron transfer is the rate-limiting step for most bacterial P450 enzymes, which can reach turnover numbers of over 1,000/secons. By comparison, eukaryotic steroid hydroxylases are typically much slower, and display turnover numbers in the range of 5–250/minute.
Importantly, the process of electron transfer during the activity of a P450 system may be either “coupled” or “leaky”: in a coupled P450 system, all electrons from NADPH are utilized in the biotransformation. However, if some electrons are transferred to other acceptors, then this process is referred to as “uncoupling” of electron transfer from biotransformation, as “leaky” electron transfer, or as “futile” oxidation. Uncoupling can occur during electron transfer from NADPH to the CYP enzyme and also during the P450-reaction cycle during the so-called shunt reactions that lead to production of water or ROS instead of substrate turnover.77 These include the autoxidation shunt, which results in the formation of superoxide anion (·O2−) and the peroxide shunt that generates hydrogen peroxide (H2O2).
Therefore, ROS are always generated during NADPH-dependent drug metabolism in liver microsomes, as well as in isolated hepatocytes, and CYP systems are a significant source of ROS in biological systems, especially in tissues like the liver, where P450 is present in high amounts.78 It is important to remember that ROS can have important physiological effects on gene signaling and gene activation, so it is probably inappropriate to attribute only adverse effects to ROS generation by CYP systems.79 Still, the toxicity of many reagents is at least partially due to the increased ROS production that occurs when they are metabolized by CYPs; some examples are acetaminophen, benzene, CCl4 and other halogenated hydrocarbons, and nitrosamines. In addition, some xenobiotics are able to enhance their own metabolism by inducing the expression of CYPs that metabolize them, eg, polycyclic aromatic hydrocarbons and barbiturates, such as phenobarbital, increase CYP1A2 levels, while glucocorticoid drugs, such as dexamethasone, induce CYP3A4.80
ROS generation depends on several factors, including the specific form of CYP, entry of the second electron into the P450 cycle, and the presence and nature of the substrate.81–84 For instance, one study determined a rank order for ROS-generating activity by microsomes from lymphoblasts expressing human CYPs to be CYP3A4 > CYP1A1 > CYP1A2 = CYP2B6.85 CYP2E1 was repeatedly shown to be highly uncoupled,86,87 and appears to be the most significant generator of ROS in liver. Its induction by chronic ethanol exposure is thought to contribute significantly to ethanol-induced oxidative stress and hepatotoxicity.80,88–91 Significant evidence for this notion also comes from the observation that ROS production in E47 cells (ie, HepG2 cells that constitutively express recombinant human CYP2E1) is about 50% higher than in normal hepatocytes and associated with roughly 30% lower ATP levels, which is probably due to ROS damage to complex I of the respiratory chain.92 ROS production by CYP2E1 also causes higher rates of microsomal lipid peroxidation within microsomes and liposomes, and this effect can be suppressed by inhibition of the enzyme’s activity with anti-CYP2E1 antibodies.93,94
In general, microsomal CYPs obtain the electrons necessary for their activity from cytochrome P450 reductase (CPR or POR), with cytochrome B5 sometimes acting as an allosteric factor to facilitate CPR–CYP interaction. After being reduced by cytochrome B5 reductase, cytochrome B5 may even act as an unusual donor for the second (but not the first) electron in the P450 cycle and has also been shown to be a source of ROS formation, albeit at lower levels than the CYPs themselves.90 It is important to note that CPR protein levels in the ER membrane are typically much lower than those of all CYPs together, eg, the total CYP:CPR ratio is about 40:1 in rat hepatocytes.95 Therefore, multiple P450 species compete for CPR interaction, which adds another dimension to the complexity of these processes.
As far as is known today, the seven human mitochondrial CYPs (CYP11A1, CYP11B1, CYP11B2, CYP24A1, CYP27A1, CYP27B1, and CYP27C1) are generally involved in adrenal steroidogenesis, bile-acid formation, and the metabolism of vitamins A and D.96–99 Among these, CYP11A1, which catalyzes the conversion of cholesterol to pregnenolone (the rate-limiting step in adrenal steroid biosynthesis), is the best-studied enzyme in terms of uncoupling and ROS production;100 however, uncoupling in CYP11B enzymes might even be higher.101 Many years ago, it was demonstrated in a seminal study that ROS are efficiently produced in vitro by CYP11A1 in the presence of NADPH and adrenodoxin reductase, but in the absence of its substrate cholesterol.102 We later showed that transient overexpression of CYP11A1 decreased cell survival in some cancer cell lines but not in others; moreover, we showed that in those cases where loss of cell viability occurred, it depended on ROS formation.103 In mammalian mitochondria, electrons are transferred from NADPH via adrenodoxin reductase (AdR or FdxR) to adrenodoxin (Adx or Fdx1), which in turn reduces mitochondrial CYP enzymes.63,104–106 Therefore, as in the microsomal P450 system, the mitochondrial electron-transfer proteins are not specific for individual enzymes, but serve as electron donors for different CYPs in different tissues. AdR and Adx are expressed in all human tissues, and their highest levels of expression are observed in steroidogenic cells, especially in the adrenal cortex and ovarian corpus luteum.107,108 However, with respect to their capability for ROS production, the properties of the two proteins differ remarkably: in vitro data show that while electrons can be transferred from NADPH via AdR and Adx to O2, AdR alone is not a major source of electron leakage in the presence of NADPH but without Adx.102,109,110 Accordingly, AdR overexpression failed to exert an effect on cell proliferation in any cell lines tested, although it does sensitize cells to oxidative stress-induced apoptosis.103,111,112 Therefore, in the absence of a mitochondrial CYP but in the presence of NADPH, Adx is the major source of ROS, so much so that it has been labeled an “electron gun” that is constantly being loaded and fired until electron supply via NADPH is exhausted.102 Consistently with the results observed in reconstituted systems, transient overexpression of either human or bovine Adx led to reduced cell viability in all eleven tumor-derived and nontumorigenic cell lines tested.103
Formation of mitochondrial ROS is one of the major internal triggers for the initiation of apoptotic cell death.113 An early step in this process is the release of cytochrome C from within the intermembrane space of the mitochondria, which then binds to Apaf1 to form apoptosomes. The apoptosomes bind to and activate caspase 9, which in turn activates downstream effector caspases, spreading a cascade of proteolytic activity that leads to breakdown of the mitochondrial membrane and eventually phagocytosis of the cell. Consistent with the process of apoptosis, we showed that transient overexpression of human Adx in HCT116 cells caused a disruption of the mitochondrial transmembrane potential, to cytochrome C release from the mitochondria, and to increased caspase activity in the cell; by contrast, overexpression of an Apo mutant of adrenodoxin (C46S) that cannot bind the [2Fe-2S] cluster essential for electron transfer did not cause apoptosis.103 Interestingly, recombinant expression of Adx does not cause apoptosis in the microbial model system Schizosaccharomyces pombe, in spite of a strong increase in ROS, which is indeed much higher than that observed in mammalian cells.114 An explanation for this observation might well be connected to the very recent finding that S. pombe, in contrast to baker’s yeast (Saccharomyces cerevisiae), does not age.115
As mentioned, CYP systems are not only a source of ROS but can also cause ROM-mediated oxidative stress to either the CYP enzymes themselves or the cells that express them. The transition intermediates formed in the CYP catalytic cycle are sometimes highly reactive and bind directly to the enzyme. Mechanism-based inhibition (also known as time-dependent inhibition) then occurs, and results in partial or total irreversible inactivation of the enzyme. Mechanism-based inactivation can potentiate the risk of drug–drug interactions and toxicity from coadministered drugs by increasing their steady-state levels. If the reactive species formed in the CYP are able to escape the active site and bind covalently to other proteins or nucleic acids, this can culminate in adverse drug reactions and toxicity.116 Indeed, in the majority of cases, toxicity is caused by the actions of such reactive metabolites, rather than by the parent chemicals.116 Generation of such ROMs is also a major reason for the failure of many drugs in pre- and postclinical trials.69
Although CYPs are quite well conserved, it is important to note that they are not identical, as polymorphic variants exist, sometimes at high frequencies. Even small differences in amino-acid sequence may have a significant impact on substrate affinity and turnover. This is exemplified by the considerable variation in the efficiency and amount of CYP2D6 produced between individuals, which cause drugs that are mainly metabolized by CYP2D6 to be eliminated by some individuals much more rapidly than by others. These variations give rise to four different drug-metabolizing phenotypes: poor metabolizers, which are characterized by a complete lack of enzyme activity due to two defective alleles; intermediate metabolizers, which are carriers of either one defective allele or two partially functional alleles; extensive metabolizers, which have two functional alleles; and ultrarapid metabolizers, which have either multiple functional alleles or alleles that code for activated enzymes. Therefore, the dose of drugs that are mainly metabolized by CYP2D6 should be adjusted to take into account the speed at which they are metabolized by the individual patient. Increasing knowledge in this field has led to a transition from population-based dosing and prescriptions to patient individualization in both drug development and clinical practice, or in other words to personalized medicine.117,118
To complicate matters further, CYP-dependent drug metabolism is often followed by conjugation reactions catalyzed by phase II enzymes, such as UGTs or SULTs. In the context of safety testing, availability of all phase I and phase II metabolites is thus an essential prerequisite for the assessment of drug-caused side effects or toxicity.119,120 Finally, differences in the activity of drug-metabolic enzymes in humans and animals are known to cause species differences in metabolism-related toxicity. An important example is the CYP-dependent metabolism of the breast cancer prodrug tamoxifen, which leads to the formation of desired active metabolites, such as 4-hydroxytamoxifen, but also to the undesired and DNA-reactive α-hydroxytamoxifen. In human females, this latter metabolite is efficiently detoxified by UGTs, yielding a product that is rapidly eliminated from the body; rodent females lack sufficient UGT activity to derivatize all the α-hydroxytamoxifen, and this reactive metabolite can then cause DNA damage and liver cancer.121
Measurement of ROS production in bystander cells
The oxidative metabolism of γ-radiation-targeted cells can be investigated through the use of antioxidants, such as L-lactate and L-deprenyl, or functionally related drugs that can inhibit the collapse of mitochondrial membrane potential, resulting in the inhibition of cytotoxic effects on bystander cells when transferred from irradiated cultures. Chemical inhibition of the bystander signal with antioxidants signifies it to be ROS-based, while drugs that collapse mitochondrial membrane potential indicate the involvement of calcium-ion signaling or components of apoptotic signaling as possible candidates of bystander signal.122–124
Other in vivo sources of free radicals
The redox state of a cell is generally linked to an iron/copper–redox couple and maintained within a strict physiological range.125 This tight iron regulation ensures that there is no free intracellular iron; however, in vivo under stress, superoxide may sometimes release free iron. This phenomenon was demonstrated for the [4Fe-4S] cluster-containing enzymes of the dehydratase-lyase family.126 The Fe(II) thus released can take part in the Fenton reaction, producing highly reactive hydroxyl radical (Fe[II] + H2O2 → Fe[III] + ·OH + OH−). Therefore, under stress conditions the O2·- acts as an oxidant of the [4Fe-4S] cluster-containing enzymes and facilitates ·OH production from H2O2 by providing Fe(II) for the Fenton reaction.127 This hydroxyl radical has a half life of <1 nanosecond, and thus in vivo it reacts very close to the source.128 Production of ·OH close to the DNA can lead to its reaction with DNA bases or the deoxyribosyl backbone of DNA to produce damaged DNA strands. Another commonly occurring radical in cellular environments is the peroxyl radical (ROO·). This radical is a high-energy species with a reduction potential varying between 0.77 and 1.44 V, depending on the functional group.129 The role of peroxyl radicals in DNA cleavage and disease is also well documented.130–132
Free radicals as part of the immune defense mechanism
Oxygen-derived free radicals are purposely produced to neutralize invading viruses and bacteria. Macrophage- and neutrophil-mediated phagocytosis stimulates certain cellular mechanisms, including the “respiratory burst” that enhances oxygen uptake to produce potent oxidant bactericidal substances, such as hypochlorous acid and hydroxyl radicals. Simultaneously, NO, a gaseous radical produced by macrophages, reacts with superoxide to form peroxynitrite, to work as an additional bactericidal agent.133
While oxidants are continually being produced as by-products of normal metabolism, they are also rendered harmless by a range of antioxidant enzymes, such as glutathione peroxidase, glutathione reductase, catalase, and SOD, as well as nonenzymatic antioxidants, including thiol antioxidants, melatonin, coenzyme Q, and metal-chelating proteins, which are efficient enough to fight against excessive free radicals. Nutrient antioxidants, such as the most abundant fat-soluble vitamin E and the most abundant water-soluble vitamin C, also efficiently and effectively remove oxidants from the living system.134
This “two-faced” character of ROS has recently been characterized as a secondary messenger that induces and maintains the tumorigenic phenotype of cancer cells. DNA mutations are a critical step in cancer induction and progression, with enhanced levels of oxidative DNA lesions (8–OH–G) reported in various tumors.135
SODs are a class of metalloenzymes that are ubiquitously distributed among all oxygen-using organisms, and work by providing protection against the oxidative damage of high concentrations of superoxide radical anion (O2·−).136 The redox activity of SOD can be used137 as the primary cellular defense against oxidative damage by simply reducing superoxide (O2·−) to oxygen (O2) and hydrogen peroxide (H2O2). Overexpression of SOD has been demonstrated to have antiproliferative and antitumor effects in vitro. Oxidative stress as a source of ROS that leads to bystander signaling was shown to be inhibited with SOD treatment in vitro.138 The ROS-induced mutation frequency in DNA that directly damaged and indirectly damaged (bystander) cells was able to be significantly reduced.
One of the most toxic and common species of ROS found in vitro is H2O2. It plays an important part in cell-cycle regulation and mitogenic stimulation. Measurement of H2O2 in vitro can be facilitated by the use of homovannillic acid or Amplex red, which dimerizes upon oxidation by H2O2 via horseradish-peroxidase catalysis. The monomer of both compounds is nonfluorescent, but becomes fluorescent upon dimerization with an emission wavelength of 425 nm and a peak excitation wavelength of 315 nm.139 Colorimetric analysis of the sample using an enzyme-linked immunosorbent-assay reader can provide an optical density that can be quantified and plotted to measure ROS in the sample.
Superoxide radicals can be measured via an interaction between superoxide and another compound to produce quantifiable data. This can be achieved by the reduction of ferricytochrome C to ferrocytochrome C. The transformation of citrate to isocitrate is catalyzed by the aconitase enzyme. This enzyme can be inactivated in vitro by superoxide through the oxidization of the Fe moiety from its cubane cluster. Subsequently, the concentrations of superoxide in culture can be estimated by the extent of enzyme inactivation. Activation of p53 and the Ras–Raf pathway was also implicated in oxidatively stressed conditions.139,140 The ROS generated in cells usually have a very short half-life (nanosecond scale), and can travel only a short distance (micrometer scale) before being neutralized. Therefore, the readout obtained using certain techniques is not a very accurate measure, although new fluorescent probes are being developed to get a high-throughput analysis via live-cell imaging.149
Reactive nitrogen species
Another oxidative species produced as a result of radiation targeting and hence usually studied in bystander signaling is NO. It is another commonly found reactive species, specifically referred to as reactive nitrogen species (RNS). RNS are a group of compounds with different properties and reactivity. Some RNS are highly reactive, and their interaction with target macromolecules can lead to permanent modification of the target, suggesting that RNS might not be cell-signal transducers; however, our understanding of RNS as oxidizers of macromolecules has evolved over the years.141 Nitrated proteins are removed by degradation142 and other mechanisms that are not yet fully understood.143 NO is produced from L-arginine by three main isoforms of NOS: epithelial (eNOS), neuronal (nNOS), and inducible (iNOS). The isoforms eNOS and nNOS are tightly regulated by calcium in a calmodulin-dependent manner.144 On the other hand, the inducible isoform iNOS is present in hepatocytes, macrophages, and fibroblasts, and can produce large amounts of NO in response to inflammatory stimuli, such as ionizing radiation and oxidative xenobiotics, and provides host defense through its oxidative toxicity. A standard way to determine NO is to measure composition products, such as nitrate (NO3) and nitrite (NO2), colorimetrically. This reaction is catalyzed by nitrate reductase as NO3 is first reduced to NO2.139,140 iNOS is activated in response to a variety of cytokine and endotoxin signals that can lead to fast production of huge fluxes of NO. iNOS expression is also tightly regulated by MAPK and JNK–STAT pathways.145
End points of bystander effect produced by targeted cells
Cell proliferation has been widely used as an end point of bystander signaling. Oxidative stress causes the proliferation process to slow down dramatically. This has been shown through several in vitro and in vivo experiments. When directly targeted (exposed to radiation directly), epithelial cells of rat liver were mixed with naïve bystander cells (not exposed to radiation directly) and incubated together for 24 hours. The naïve cells demonstrated a rate of cell proliferation that was higher by 14%–17% than the directly targeted cells.146
The bystander effect has been checked by using the membrane-localizing deuteroporphyrin (dimethyl ester) dye and separating the direct and bystander cells in a migration transwell chamber. The extranuclear localization of the photosensitizer used suggested that primary DNA damage is not the trigger for bystander response in naïve cells. The end points measured included elevated oxidative stress, DNA damage (micronucleus formation), mutagenesis, and decreased clonogenic survival. Furthermore, the antioxidant effect of vitamin E in targeted cells was shown to be responsible for preventing oxidative stress.50
A range of end points can be measured in direct and naïve bystander cells to quantify overall bystander effects, and the list below is only a few of them:
- genetic or epigenetic16
- small- or large-scale mutations or loss of heterozygosity17
- protein-signaling pathways11,15
- DNA damage-sensor proteins, such as γ-H2AX148
- proliferation and apoptosis in direct or naïve cells122,149
- sister-chromatid exchanges in direct or naïve cells1
- micronucleation in direct or naïve cells2,150
- neoplastic transformation in naïve cells.17
Cell models used for RIBE
In many of the in vitro studies mentioned in this article, lymphoblasts were commonly used as cell models, primarily in conditioned-culture studies. This is an asset, because lymphoblasts are suspended cell lines and can be spun down easily without having to trypsinize the cells. Conditioned medium can then be decanted and applied to naïve cells for bystander analysis. Some cell lines used as models for the RIBE include TK6 (p53 wild), WTK1 (p53 mutant), and NH32 (p53 null). These cell lines also provide an added layer of analytical advantage, due to their p53 status.164
For bystander effects on human development, the use of stem cells is inevitable. Human stem cells are responsible for many important biological developmental processes, including growth and differentiation of embryos, environment maintenance, and aging. Regarding the RIBE in human stem cells, not much research has been conducted, but it was reported in 2010 that these cells are not prone to bystander signaling in comparison to other somatic cells in humans.151 Fish cell lines have also been used for qualitative bystander-effect analysis. Different types of bystander effects produced in different target cell lines have been identified, but already-established fish cell lines do not generate death signals due to bystander effect.152
In 2009, the role of p53 was reported by Zhao et al.121 Three liver cancer cell lines were used in this study: HepG2 (p53 wild type), PLC/PRF/5 (p53 mutated), and Hep3B (p53 null). In order to elucidate the RIBE, all of them were exposed to γ-radiation and then cultured with normal Chang hepatocytes having p53 wild type. Only HepG2 cells were able to initiate bystander signaling in surrounding normal Chang hepatocytes. When inhibitors of p53 and cytochrome C were used with HepG2 cells, the bystander effect was able to be downregulated. Radiation targeting in these cell lines activated cytochrome C only in HepG2 cells because of p53. Conclusively, it can be said that cytochrome C release is p53-dependent in the RIBE.153
A group has also investigated the production of bystander signaling in primary tissue culture under in vitro conditions. Bladder samples were taken from mice and then treated with radiation, and irradiated tissue-conditioned medium taken from directly exposed cultures was applied to naïve tissue. The subsequent bystander signal produced was of high intensity compared to single cells, and so was the nuclear fragmentation that results in micronucleation. It was concluded that the extent of the bystander effect induced in 3D geometric tissue was higher when compared to single cells.154
Chemical signals from targeted cells activate/stimulate various signaling pathways in naïve cells
It has been discovered that in bystander signaling between irradiated and unirradiated cells, cytokines play an important role.155,156 A group of scientists have reported the inflammatory and proinflammatory action of cytokines by using the culture medium in which they treated human glioblastoma cells with different dosage of radiation. IL6 is an important cytokine with multiple functions, eg, in cancer, it causes malignancy and helps evasion of apoptosis.157,158 IL8 levels have also been shown to increase in human gliomas, and it has an important role in tumor progression.159 Generally, the immune system is involved in the production of different cytokines against disease. In one study, a heavy-ion beam was used to target T cells, in order to check the production of IL6 and TNFα. It was determined that cytokine generation decreased in the directly targeted cells after exposure to radiation, and might have been induced in the indirect cells through the bystander effect. In the cells that are directly exposed to radiation, the expressions of genes of various cytokines that are dependent on NFκB produced some other cytokines that are involved in autocrine and paracrine signals, which further initiated signaling processes involving the expression of NFκB in bystander cells. After radiation therapy, the changes produced in DNA because of heredity can be transmitted to many generations, and the naïve bystander cells have the same abnormalities in the genome as in the directly targeted cells.160
Genetic abnormalities and apoptosis are initiated because of radiation, which ultimately leads to DNA damage. When bone marrow in mice is exposed to radiation, it leads to cell death and DNA damage in bone marrow cells not directly targeted by radiation. Signaling in this scenario shows the involvement of various cytokines.161
Some bioinformatic tools have also been utilized to analyze gene modulation and signaling at early stages after the exposure of radiation in IMR90 fibroblasts. Real-time quantitative polymerase chain reaction and whole-human-genome microarrays were the techniques use to authenticate an in silico analysis. For signaling studies specifically, semiquantitative image analysis and immunoblotting revealed the involvement of NFκB and Akt–GSK3β pathways.162
In 2013, Guo et al reported on the RIBE between germ cells and somatic cells in Caenorhabditis elegans. Radiation given to posterior pharynx bulbs and tails of C. elegans increased germ-cell death, elevated DNA damage, and introduced genomic inconsistencies in the F1 generation, which demonstrated a risk of tumorigenic phenotype in the next generation. RIBE-induced germ-cell apoptosis also showed a complicated collaboration among many signaling pathways in somatic and germ cells.163
Conclusion
The RIBE can play a critical role in initiating secondary tumorigenesis via oxidative stress. The ROS produced as a result of radiation exposure can act as secondary messengers to cause bystander effects in naïve cells. Although the ROS have a very short half-life (nanoseconds) and travel only short distances (micrometers), their cumulative effect in cells directly targeted with radiation can trigger the activation of bystander-signaling pathways. Some sources of generation of ROS in a cell include direct ionization of molecules targeted with ionizing radiation or metabolic enzymes, such as COX, Lox, and monooxygenases. These ROS or ROM can set up intracellular and intercellular communication. Gap-junction-dependent signaling proteins in attached cell lines or soluble signaling proteins in suspended cell lines can communicate with neighboring cells via hormones and cytokines, which can activate several signaling pathways, including NFκB, TGF-β1, TNFα, and COX2, either in the directly targeted cells or naïve bystander cells. Another recently discovered stress-response pathway is mediated by Nrf2, which is a transcription factor regulating the redox-homeostatic gene-regulatory network. The Nrf2-signaling pathway is activated under oxidative stress to increase the expression of a number of antioxidant and drug-detoxification phase II enzymes, such as UGTs, which contribute to restoring redox homeostasis.
DNA damage, genetic changes, and double-strand breaks are the other consequences of radiation targeting. Because of radiation, the rate of division in bystander cells (indirect cells) is comparatively higher compared to exposed cells (direct cells). The antioxidative properties of vitamin E reduce oxidative stress. In developmental analyses, the germ-cell death rate shows that complex signaling is involved between somatic and germ cells. If somatic cells are under radiation exposure, there is a risk of cancer initiation and progression in the next generation. Human stem cells are not responsive to bystander signaling, and untargeted effects are produced if only cytoplasm is exposed to radiation but not the nucleus. Therefore, the nucleus cannot be the primary cause of a DNA damage-induced bystander effect. Because of radiation, genetic changes introduced in DNA can be transmitted to the next generation if not repaired.
Disclosure
The authors report no conflicts of interest in this work.
References
Nagasawa H, Little JB. Induction of sister chromatid exchanges by extremely low doses of α-particles. Cancer Res. 1992;52(22):6394–6396. | ||
Azzam EI, de Toledo SM, Spitz DR, Little JB. Oxidative metabolism modulates signal transduction and micronucleus formation in bystander cells from α-particle-irradiated normal human fibroblast cultures. Cancer Res. 2002;62(19):5436–5442. | ||
Mancuso M, Pasquali E, Leonardi S, et al. Oncogenic bystander radiation effects in Patched heterozygous mouse cerebellum. Proc Natl Acad Sci U S A. 2008;105(34):12445–12450. | ||
Iyer R, Lehnert BE. Effects of ionizing radiation in targeted and nontargeted cells. Arch Biochem Biophys. 2000;376(1):14–25. | ||
Emerit I. Reactive oxygen species, chromosome mutation, and cancer: possible role of clastogenic factors in carcinogenesis. Free Radic Biol Med. 1994;16(1):99–109. | ||
Narayanan PK, LaRue KE, Goodwin EH, Lehnert BE. Alpha particles induce the production of interleukin-8 by human cells. Radiat Res. 1999;152(1):57–63. | ||
Matsumoto H, Hayashi S, Hatashita M, et al. Induction of radioresistance by a nitric oxide-mediated bystander effect. Radiat Res. 2001;155(3):387–396. | ||
Shao C, Furusawa Y, Aoki M, Ando K. Role of gap junctional intercellular communication in radiation-induced bystander effects in human fibroblasts. Radiat Res. 2003;160(3):318–323. | ||
Zhou H, Ivanov VN, Gillespie J, et al. Mechanism of radiation-induced bystander effect: role of the cyclooxygenase-2 signaling pathway. Proc Natl Acad Sci U S A. 2005;102(41):14641–14646. | ||
Han W, Wu L, Chen S, Yu KN. Exogenous carbon monoxide protects the bystander Chinese hamster ovary cells in mixed coculture system after alpha-particle irradiation. Carcinogenesis. 2010;31(2):275–280. | ||
Hei TK, Zhou H, Ivanov VN, et al. Mechanism of radiation-induced bystander effects: a unifying model. J Pharm Pharmacol. 2008;60(8):943–950. | ||
Azzam EI, de Toledo SM, Little JB. Stress signaling from irradiated to non-irradiated cells. Curr Cancer Drug Targets. 2004;4(1):53–64. | ||
Halliwell B, Gutteridge JM. Oxygen free radicals and iron in relation to biology and medicine: some problems and concepts. Arch Biochem Biophys. 1986;246(2):501–514. | ||
Colotta F, Allavena P, Sica A, Garlanda C, Mantovani A. Cancer-related inflammation, the seventh hallmark of cancer: links to genetic instability. Carcinogenesis. 2009;30(7):1073–1081. | ||
Pardali K, Moustakas A. Actions of TGF-β as tumor suppressor and pro-metastatic factor in human cancer. Biochim Biophys Acta. 2007;1775(1):21–62. | ||
Hagan M, Yacoub A, Dent P. Ionizing radiation causes a dose-dependent release of transforming growth factor α in vitro from irradiated xenografts and during palliative treatment of hormone-refractory prostate carcinoma. Clin Cancer Res. 2004;10(17):5724–5731. | ||
Seymour CB, Mothersill C, Alper T. High yields of lethal mutations in somatic mammalian cells that survive ionizing radiation. Int J Radiat Biol Relat Stud Phys Chem Med. 1986;50(1):167–179. | ||
Mitchell SA, Randers-Pehrson G, Brenner DJ, Hall EJ. The bystander response in C3H 10T1/2 cells: the influence of cell-to-cell contact. Radiat Res. 2004;161(4):397–401. | ||
Shao C, Folkard M, Michael BD, Prise KM. Targeted cytoplasmic irradiation induces bystander responses. Proc Natl Acad Sci U S A. 2004;101(37):13495–13500. | ||
Murphy JE, Nugent S, Seymour C, Mothersill C. Mitochondrial DNA point mutations and a novel deletion induced by direct low-LET radiation and by medium from irradiated cells. Mutat Res. 2005;585(1–2):127–136. | ||
Tartier L, Gilchrist S, Burdak-Rothkamm S, Folkard M, Prise KM. Cytoplasmic irradiation induces mitochondrial-dependent 53BP1 protein relocalization in irradiated and bystander cells. Cancer Res. 2007;67(12):5872–5879. | ||
Chen HH, Jia RF, Yu L, Zhao MJ, Shao CL, Cheng WY. Bystander effects induced by continuous low-dose-rate 125I seeds potentiate the killing action of irradiation on human lung cancer cells in vitro. Int J Radiat Oncol Biol Phys. 2008;72(5):1560–1566. | ||
Zhou H, Ivanov VN, Lien YC, Davidson M, Hei TK. Mitochondrial function and nuclear factor-κB-mediated signaling in radiation-induced bystander effects. Cancer Res. 2008;68(7):2233–2240. | ||
Jalal N, Haq S, Anwar N, Nazeer S, Saeed U. Radiation induced bystander effect and DNA damage. J Cancer Res Ther. 2014;10(4):819–833. | ||
Shiloh Y. The ATM-mediated DNA-damage response: taking shape. Trends Biochem Sci. 2006;31(7):402–410. | ||
Hong F, Sekhar KR, Freeman ML, Liebler DC. Specific patterns of electrophile adduction trigger Keap1 ubiquitination and Nrf2 activation. J Biol Chem. 2005;280(36):31768–31775. | ||
Kobayashi M, Yamamoto M. Nrf2-Keap1 regulation of cellular defense mechanisms against electrophiles and reactive oxygen species. Adv Enzyme Regul. 2006;46(1):113–140. | ||
Deshmukh P, Unni S, Krishnappa G, Padmanabhan B. The Keap1-Nrf2 pathway: promising therapeutic target to counteract ROS-mediated damage in cancers and neurodegenerative diseases. Biophys Rev. 2017;9(1):41–56. | ||
Bryan HK, Olayanju A, Goldring CE, Park BK. The Nrf2 cell defence pathway: Keap1-dependent and -independent mechanisms of regulation. Biochem Pharmacol. 2013;85(6):705–717. | ||
Slocum SL, Kensler TW. Nrf2: control of sensitivity to carcinogens. Arch Toxicol. 2011;85(4):273–284. | ||
Takahashi H, Jin C, Rajabi H, et al. MUC1-C activates the TAK1 inflammatory pathway in colon cancer. Oncogene. 2015;34(40):5187–5197. | ||
Jaramillo MC, Zhang DD. The emerging role of the Nrf2-Keap1 signaling pathway in cancer. Genes Dev. 2013;27(20):2179–2191. | ||
Al-Shobaili HA, Rasheed Z. Mitochondrial DNA acquires immunogenicity on exposure to nitrosative stress in patients with vitiligo. Hum Immunol. 2014;75(10):1053–1061. | ||
Abdelmegeed MA, Song BJ. Functional roles of protein nitration in acute and chronic liver diseases. Oxid Med Cell Longev. 2014;2014:149627. | ||
Quijano C, Alvarez B, Gatti RM, Augusto O, Radi R. Pathways of peroxynitrite oxidation of thiol groups. Biochem J. 1997;322(Pt 1):167–173. | ||
Jain MR, Li M, Chen W, et al. In vivo space radiation-induced non-targeted responses: late effects on molecular signaling in mitochondria. Curr Mol Pharmacol. 2011;4(2):106–114. | ||
Shu L, Hollenberg PF. Identification of the cytochrome P450 isozymes involved in the metabolism of N-nitrosodipropyl-, N-nitrosodibutyl- and N-nitroso-n-butyl-n-propylamine. Carcinogenesis. 1996;17(4):839–848. | ||
Holmström KM, Finkel T. Cellular mechanisms and physiological consequences of redox-dependent signalling. Nat Rev Mol Cell Biol. 2014;15(6):411–421. | ||
Furstenberger G, Krieg P, Müller-Decker K, Habenicht AJ. What are cyclooxygenases and lipoxygenases doing in the driver’s seat of carcinogenesis? Int J Cancer. 2006;119(10):2247–2254. | ||
Krysan K, Reckamp KL, Sharma S, Dubinett SM. The potential and rationale for COX-2 inhibitors in lung cancer. Anticancer Agents Med Chem. 2006;6(3):209–220. | ||
Rani V, Yadav UC. Free Radicals in Human Health and Disease. New Delhi: Springer; 2015. | ||
Aruoma OI. Nutrition and health aspects of free radicals and antioxidants. Food Chem Toxicol. 1994;32(7):671–683. | ||
Pidgeon GP, Lysaght J, Krishnamoorthy S, et al. Lipoxygenase metabolism: roles in tumor progression and survival. Cancer Metastasis Rev. 2007;26(3–4):503–524. | ||
Funk CD. Molecular biology in the eicosanoid field. Prog Nucleic Acid Res Mol Biol. 1993;45:67–98. | ||
Norel X, Brink C. The quest for new cysteinyl-leukotriene and lipoxin receptors: recent clues. Pharmacol Ther. 2004;103(1):81–94. | ||
Conrad DJ. The arachidonate 12/15 lipoxygenases: a review of tissue expression and biologic function. Clin Rev Allergy Immunol. 1999;17(1–2):71–89. | ||
Kuhn H, Walther M, Kuban RJ. Mammalian arachidonate 15-lipoxygenases structure, function, and biological implications. Prostaglandins Other Lipid Mediat. 2002;68–69:263–290. | ||
Shureiqi I, Lippman SM. Lipoxygenase modulation to reverse carcinogenesis. Cancer Res. 2001;61(17):6307–6312. | ||
van Leyen K, Duvoisin RM, Engelhardt H, Wiedmann M. A function for lipoxygenase in programmed organelle degradation. Nature. 1998;395(6700):392–395. | ||
Chakraborty A, Held KD, Prise KM, Liber HL, Redmond RW. Bystander effects induced by diffusing mediators after photodynamic stress. Radiat Res. 2009;172(1):74–81. | ||
Nie D. Cyclooxygenases and lipoxygenases in prostate and breast cancers. Front Biosci. 2007;12:1574–1585. | ||
Schneider C, Pozzi A. Cyclooxygenases and lipoxygenases in cancer. Cancer Metastasis Rev. 2011;30(3–4):277–294. | ||
Vane JR. Inhibition of prostaglandin synthesis as a mechanism of action for aspirin-like drugs. Nat New Biol. 1971;231(25):232–235. | ||
Gabriel SE, Jaakkimainen L, Bombardier C. Risk for serious gastrointestinal complications related to use of nonsteroidal anti-inflammatory drugs: a meta-analysis. Ann Intern Med. 1991;115(10):787–796. | ||
Wennogle LP, Liang H, Quintavalla JC, et al. Comparison of recombinant cyclooxygenase-2 to native isoforms: aspirin labeling of the active site. FEBS Lett. 1995;371(3):315–320. | ||
Chen WS, Wei SJ, Liu JM, Hsiao M, Kou-Lin J, Yang WK. Tumor invasiveness and liver metastasis of colon cancer cells correlated with cyclooxygenase-2 (COX-2) expression and inhibited by a COX-2-selective inhibitor, etodolac. Int J Cancer. 2001;91(6):894–899. | ||
Gately S, Li WW. Multiple roles of COX-2 in tumor angiogenesis: a target for antiangiogenic therapy. Semin Oncol. 2004;31(2 Suppl 7):2–11. | ||
Ghosh N, Chaki R, Mandal V, Mandal SC. COX-2 as a target for cancer chemotherapy. Pharmacol Rep. 2010;62(2):233–244. | ||
Bernhardt R. Cytochromes P-450. In: Lennarz WJ, Lane MD, Modrich P, et al, editors. Encyclopedia of Biological Chemistry. Cambridge (MA): Academic Press; 2004:544–549. | ||
Omura T, Sato R. The carbon monoxide-binding pigment of liver microsomes – II: solubilization, purification, and properties. J Biol Chem. 1964;239(7):2379–2385. | ||
Nelson D. The cytochrom P450 homepage. Human Genomics 4, 59–65. Available from: http://drnelson.uthsc.edu/CytochromeP450.html. Accessed July 25, 2017. | ||
Omura T. Contribution of cytochrome P450 to the diversification of eukaryotic organisms. Biotechnol Appl Biochem. 2013;60(1):4–8. | ||
Hannemann F, Bichet A, Ewen KM, Bernhardt R. Cytochrome P450 systems: biological variations of electron transport chains. Biochim Biophys Acta. 2007;1770(3):330–344. | ||
Bernhardt R, Urlacher VB. Cytochromes P450 as promising catalysts for biotechnological application: chances and limitations. Appl Microbiol Biotechnol. 2014;98(14):6185–6203. | ||
Guengerich FP. Common and uncommon cytochrome P450 reactions related to metabolism and chemical toxicity. Chem Res Toxicol. 2001;14(6):611–650. | ||
Guengerich FP. Uncommon P450-catalyzed reactions. Curr Drug Metab. 2001;2(2):93–115. | ||
Romano M, Facchinetti T, Salmona M. Is there a role for nuclei in the metabolism of xenobiotica? A review. Drug Metab Rev. 1983;14(4):803–829. | ||
Loeper J, Descatoire V, Maurice M, et al. Cytochromes P-450 in human hepatocyte plasma membrane: recognition by several autoantibodies. Gastroenterology. 1993;104(1):203–216. | ||
Guengerich FP. Intersection of the roles of cytochrome P450 enzymes with xenobiotic and endogenous substrates: relevance to toxicity and drug interactions. Chem Res Toxicol. 2017;30(1):2–12. | ||
Neunzig J, Milhim M, Schiffer L, et al. The steroid metabolite 16β-OH-androstenedione generated by CYP21A2 serves as a substrate for CYP19A1. J Steroid Biochem Mol Biol. 2017;167:182–191. | ||
Parr MK, Zöllner A, Fusshöller G, et al. Unexpected contribution of cytochrome P450 enzymes CYP11B2 and CYP21, as well as CYP3A4 in xenobiotic androgen elimination: Insights from metandienone metabolism. Toxicol Lett. 2012;213(3):381–391. | ||
Schiffer L, Brixius-Anderko S, Hannemann F, et al. Metabolism of oral turinabol by human steroid hormone-synthesizing cytochrome P450 enzymes. Drug Metab Dispos. 2016;44(2):227–237. | ||
Schiffer L, Müller AR, Hobler A, et al. Biotransformation of the mineralocorticoid receptor antagonists spironolactone and canrenone by human CYP11B1 and CYP11B2: characterization of the products and their influence on mineralocorticoid receptor transactivation. J Steroid Biochem Mol Biol. 2016;163:68–76. | ||
Zöllner A, Parr MK, Drăgan CA, et al. CYP21-catalyzed production of the long-term urinary metandienone metabolite 17β-hydroxymethyl-17α-methyl-18-norandrosta-1,4,13-trien-3-one: a contribution to the fight against doping. Biol Chem. 2010;391(1):119–127. | ||
Rittle J, Green MT. Cytochrome P450 compound I: capture, characterization, and C-H bond activation kinetics. Science. 2010;330(6006):933–937. | ||
Yoshimoto FK, Auchus RJ. Rapid kinetic methods to dissect steroidogenic cytochrome P450 reaction mechanisms. J Steroid Biochem Mol Biol. 2016;161:13–23. | ||
Lewis DF, Ito Y. Cytochrome P450 structure and function: an evolutionary perspective. In: Ioannides C, editor. Cytochromes P450: Role in the Metabolism and Toxicity of Drugs and Other Xenobiotics. Cambridge: RSC Publishing; 2008. | ||
Rifkind AB. CYP1A in TCDD toxicity and in physiology: with particular reference to CYP dependent arachidonic acid metabolism and other endogenous substrates. Drug Metab Rev. 2006;38(1–2):291–335. | ||
Moorthy B. The CYP1A subfamily. In: Ioannides C, editor. Cytochromes P450: Role in the Metabolism and Toxicity of Drugs and Other Xenobiotics. Cambridge: RSC Publishing; 2008:97–135. | ||
Cederbaum AI. Molecular mechanisms of the microsomal mixed function oxidases and biological and pathological implications. Redox Biol. 2015;4:60–73. | ||
Kuthan H, Ullrich V. Oxidase and oxygenase function of the microsomal cytochrome P450 monooxygenase system. Eur J Biochem. 1982;126(3):583–588. | ||
Blanck J, Ristau O, Zhukov AA, Archakov AI, Rein H, Ruckpaul K. Cytochrome P-450 spin state and leakiness of the monooxygenase pathway. Xenobiotica. 1991;21(1):121–135. | ||
White RE, Coon MJ. Oxygen activation by cytochrome P-450. Annu Rev Biochem. 1980;49:315–356. | ||
Pompon D. Rabbit liver cytochrome P-450 LM2: roles of substrates, inhibitors, and cytochrome B5 in modulating the partition between productive and abortive mechanisms. Biochemistry. 1987;26(20):6429–6435. | ||
Puntarulo S, Cederbaum AI. Production of reactive oxygen species by microsomes enriched in specific human cytochrome P450 enzymes. Free Radic Biol Med. 1998;24(7–8):1324–1330. | ||
Gorsky LD, Koop DR, Coon MJ. On the stoichiometry of the oxidase and monooxygenase reactions catalyzed by liver microsomal cytochrome P-450: products of oxygen reduction. J Biol Chem. 1984;259(11):6812–6817. | ||
Ekström G, Ingelman-Sundberg M. Rat liver microsomal NADPH-supported oxidase activity and lipid peroxidation dependent on ethanol-inducible cytochrome P-450 (P-450IIE1). Biochem Pharmacol. 1989;38(8):1313–1319. | ||
Rashba-Step J, Turro NJ, Cederbaum AI. ESR studies on the production of reactive oxygen intermediates by rat liver microsomes in the presence of NADPH or NADH. Arch Biochem Biophys. 1993;300(1):391–400. | ||
Ding XX, Pernecky SJ, Coon MJ. Purification and characterization of cytochrome P450 2E2 from hepatic microsomes of neonatal rabbits. Arch Biochem Biophys. 1991;291(2):270–276. | ||
Puntarulo S, Cederbaum AI. Role of cytochrome P-450 in the stimulation of microsomal production of reactive oxygen species by ferritin. Biochim Biophys Acta. 1996;1289(2):238–246. | ||
Dai Y, Rashba-Step J, Cederbaum AI. Stable expression of human cytochrome P4502E1 in HepG2 cells: characterization of catalytic activities and production of reactive oxygen intermediates. Biochemistry. 1993;32(27):6928–6937. | ||
Mari M, Cederbaum AI. CYP2E1 overexpression in HepG2 cells induces glutathione synthesis by transcriptional activation of γ-glutamylcysteine synthetase. J Biol Chem. 2000;275(20):15563–15571. | ||
Krikun G, Cederbaum AI. Effect of chronic ethanol consumption on microsomal lipid peroxidation: role of iron and comparison between controls. FEBS Lett. 1986;208(2):292–296. | ||
Ekström G, von Bahr C, Ingelman-Sundberg M. Human liver microsomal cytochrome P-450IIE1: immunological evaluation of its contribution to microsomal ethanol oxidation, carbon tetrachloride reduction and NADPH oxidase activity. Biochem Pharmacol. 1989;38(4):689–693. | ||
Watanabe J, Asaka Y, Fujimoto S, Kanamura S. Densities of NADPH-ferrihemoprotein reductase and cytochrome P-450 molecules in the endoplasmic reticulum membrane of rat hepatocytes. J Histochem Cytochem. 1993;41(1):43–49. | ||
Nebert DW, Wikvall K, Miller WL. Human cytochromes P450 in health and disease. Philos Trans R Soc Lond B Biol Sci. 2013;368(1612):20120431. | ||
Enright JM, Toomey MB, Sato SY, et al. Cyp27c1 red-shifts the spectral sensitivity of photoreceptors by converting vitamin A1 into A2. Curr Biol. 2015;25(23):3048–3057. | ||
Schiffer L, Anderko S, Hannemann F, Eiden-Plach A, Bernhardt R. The CYP11B subfamily. J Steroid Biochem Mol Biol. 2015;151:38–51. | ||
Bird IM, Conley AJ. Steroid biosynthesis: enzymology, integration and control. In: Mason JI, editor. Genetics of Steroid Biosynthesis and Function. London: Taylor & Francis; 2002:1–35. | ||
Hanukoglu I. Antioxidant protective mechanisms against reactive oxygen species (ROS) generated by mitochondrial P450 systems in steroidogenic cells. Drug Metab Rev. 2006;38(1–2):171–196. | ||
Rapoport R, Sklan D, Hanukoglu I. Electron leakage from the adrenal cortex mitochondrial P450scc and P450c11 systems: NADPH and steroid dependence. Arch Biochem Biophys. 1995;317(2):412–416. | ||
Hanukoglu I, Rapoport R, Weiner L, Sklan D. Electron leakage from the mitochondrial NADPH-adrenodoxin reductase-adrenodoxin-P450scc (cholesterol side chain cleavage) system. Arch Biochem Biophys. 1993;305(2):489–498. | ||
Derouet-Hümbert E, Römer K, Bureik M. Adrenodoxin (Adx) and CYP11A1 (P450scc) induce apoptosis by the generation of reactive oxygen species in mitochondria. Biol Chem. 2005;386(5):453–461. | ||
Sheftel AD, Stehling O, Pierik AJ, et al. Humans possess two mitochondrial ferredoxins, Fdx1 and Fdx2, with distinct roles in steroidogenesis, heme, and Fe/S cluster biosynthesis. Proc Natl Acad Sci U S A. 2010;107(26):11775–11780. | ||
Midzak A, Papadopoulos V. Adrenal mitochondria and steroidogenesis: from individual proteins to functional protein assemblies. Front Endocrinol (Lausanne). 2016;7:106. | ||
Bernhardt R. The role of adrenodoxin in adrenal steroidogenesis. Curr Opin Endocrin Diabetes. 2000;7(3):109–115. | ||
Brentano ST, Black SM, Lin D, Miller WL. cAMP post-transcriptionally diminishes the abundance of adrenodoxin reductase mRNA. Proc Natl Acad Sci U S A. 1992;89(9):4099–4103. | ||
Hanukoglu I, Hanukoglu Z. Stoichiometry of mitochondrial cytochromes P-450, adrenodoxin and adrenodoxin reductase in adrenal cortex and corpus luteum: implications for membrane organization and gene regulation. Eur J Biochem. 1986;157(1):27–31. | ||
Chu JW, Kimura T. Studies on adrenal steroid hydroxylases: complex formation of the hydroxylase components. J Biol Chem. 1973;248(14):5183–5187. | ||
Mitani F, Ichiyama A. Enzymic studies on adrenocortical deoxycorticosterone 11β-hydroxylase system. J Biol Chem. 1975;250(20):8010–8015. | ||
Hwang PM, Bunz F, Yu J, et al. Ferredoxin reductase affects p53-dependent, 5-fluorouracil-induced apoptosis in colorectal cancer cells. Nat Med. 2001;7(10):1111–1117. | ||
Liu G, Chen X. The ferredoxin reductase gene is regulated by the p53 family and sensitizes cells to oxidative stress-induced apoptosis. Oncogene. 2002;21(47):7195–7204. | ||
Bras M, Queenan B, Susin SA. Programmed cell death via mitochondria: different modes of dying. Biochemistry (Mosc). 2005;70(2):231–239. | ||
Derouet-Hümbert E, Dragan CA, Hakki T, Bureik M. ROS production by adrenodoxin does not cause apoptosis in fission yeast. Apoptosis. 2007;12(12):2135–2142. | ||
Spivey EC, Jones SK, Rybarski JR, Saifuddin FA, Finkelstein IJ. An aging-independent replicative lifespan in a symmetrically dividing eukaryote. Elife. 2017;6:e20340. | ||
Davis CD, Hanumegowda UM. The role of drug metabolism in toxicity. In: Nassar AF, Hollenberg PF, Scatina J, editors. Drug Metabolism Handbook: Concepts and Applications. Hoboken (NJ): John Wiley & Sons; 2009:561–628. | ||
Stingl J, Viviani R. Polymorphism in CYP2D6 and CYP2C19, members of the cytochrome P450 mixed-function oxidase system, in the metabolism of psychotropic drugs. J Intern Med. 2015;277(2):167–177. | ||
Ingelman-Sundberg M. Personalized medicine into the next generation. J Intern Med. 2015;277(2):152–154. | ||
Zöllner A, Buchheit D, Meyer MR, Maurer HH, Peters FT, Bureik M. Production of human phase 1 and 2 metabolites by whole-cell biotransformation with recombinant microbes. Bioanalysis. 2010;2(7):1277–1290. | ||
Hvastkovs EG, Rusling JF. State-of-the-art metabolic toxicity screening and pathway evaluation. Anal Chem. 2016;88(9):4584–4599. | ||
Zhao L, Krishnan S, Zhang Y, Schenkman JB, Rusling JF. Differences in metabolite-mediated toxicity of tamoxifen in rodents versus humans elucidated with DNA/microsome electro-optical arrays and nanoreactors. Chem Res Toxicol. 2009;22(2):341–347. | ||
Lyng FM, Seymour CB, Mothersill C. Production of a signal by irradiated cells which leads to a response in unirradiated cells characteristic of initiation of apoptosis. Br J Cancer. 2000;83(9):1223–1230. | ||
Lyng FM, Maguire P, McClean B, Seymour C, Mothersill C. The involvement of calcium and MAP kinase signaling pathways in the production of radiation-induced bystander effects. Radiat Res. 2006;165(4):400–409. | ||
Mothersill C, Stamato TD, Perez ML, Cummins R, Mooney R, Seymour CB. Involvement of energy metabolism in the production of ‘bystander effects’ by radiation. Br J Cancer. 2000;82(10):1740–1746. | ||
Valko M, Morris H, Cronin MT. Metals, toxicity and oxidative stress. Curr Med Chem. 2005;12(10):1161–1208. | ||
Liochev SI, Fridovich I. The role of O2·− in the production of HO·: in vitro and in vivo. Free Radic Biol Med. 1994;16(1):29–33. | ||
Liochev SI, Fridovich I. The Haber-Weiss cycle – 70 years later: an alternative view. Redox Rep. 2002;7(1):55–57. | ||
Pastor N, Weinstein H, Jamison E, Brenowitz M. A detailed interpretation of OH radical footprints in a TBP-DNA complex reveals the role of dynamics in the mechanism of sequence-specific binding. J Mol Biol. 2000;304(1):55–68. | ||
Burcham PC. Genotoxic lipid peroxidation products: their DNA damaging properties and role in formation of endogenous DNA adducts. Mutagenesis. 1998;13(3):287–305. | ||
Cadenas E, Sies H. The lag phase. Free Radic Res. 1998;28(6):601–609. | ||
Pham-Huy LA, He H, Pham-Huy C. Free radicals, antioxidants in disease and health. Int J Biomed Sci. 2008;4(2):89–96. | ||
Gupta RK, Patel AK, Shah N, et al. Oxidative stress and antioxidants in disease and cancer: a review. Asian Pac J Cancer Prev. 2014;15(11):4405–4409. | ||
Knight JA. Review: free radicals, antioxidants, and the immune system. Ann Clin Lab Sci. 2000;30(2):145–158. | ||
Aslani BA, Ghobadi S. Studies on oxidants and antioxidants with a brief glance at their relevance to the immune system. Life Sci. 2016;146:163–173. | ||
Valko M, Rhodes CJ, Moncol J, Izakovic M, Mazur M. Free radicals, metals and antioxidants in oxidative stress-induced cancer. Chem Biol Interact. 2006;160(1):1–40. | ||
Moscone D, Miscini M. Determination of superoxide dismutase activity with an electrochemical oxygen probe. Anal Chim Acta. 1988;211:195–204. | ||
Davis CA, Hearn AS, Fletcher B, et al. Potent anti-tumor effects of an active site mutant of human manganese-superoxide dismutase: evolutionary conservation of product inhibition. J Biol Chem. 2004;279(13):12769–12776. | ||
Jalal N. DNA strand break associated bystander effect (DSB-ABE) is linked to gene mutations in naïve cells and indicates the involvement of a MAPK pathway.2012. Available from: https://dspace.library.colostate.edu/handle/10217/68182. Accessed June 27, 2017. | ||
Passos JF, Miwa S, von Zglinicki T. Measuring reactive oxygen species in senescent cells. Methods Mol Biol. 2013;965:253–263. | ||
Ho JH, Chen YF, Ma WH, Tseng TC, Chen MH, Lee OK. Cell contact accelerates replicative senescence of human mesenchymal stem cells independent of telomere shortening and p53 activation: roles of Ras and oxidative stress. Cell Transplant. 2011;20(8):1209–1220. | ||
Adams L, Franco MC, Estevez AG. Reactive nitrogen species in cellular signaling. Exp Biol Med. 2015;240(6):711–717. | ||
Gow AJ, Duran D, Malcolm S, Ischiropoulos H. Effects of peroxinitrite-induced protein modifications on tyrosine phosphorylation and degradation. FEBS Lett. 1996;385(1–2):63–66. | ||
Deeb RS, Nuriel T, Cheung C, et al. Characterization of a cellular denitrase activity that reverses nitration of cyclooxygenase. Am J Physiol Heart Circ Physiol. 2013;305(5):H687–H698. | ||
Murad F. Signal transduction using nitric oxide and cyclic guanosine monophosphate. JAMA. 1996;276(14):1189–1192. | ||
Jung CH, Jung H, Shin YC, et al. Eleutherococcus senticosus extract attenuates LPS-induced iNOS expression through the inhibition of Akt and JNK pathways in murine macrophage. J Ethnopharmacol. 2007;113(1):183–187. | ||
Gerashchenko BI, Howell RW. Flow cytometry as a strategy to study radiation-induced bystander effects in co-culture systems. Cytometry A. 2003;54(1):1–7. | ||
Hancock JT, Desikan R, Neil SJ. Role of reactive oxygen species in cell signalling pathways. Biochem Soc Trans. 2001;29(2):345–350. | ||
Grosovsky AJ, Walter BN, Giver CR. DNA-sequence specificity of mutations at the human thymidine kinase locus. Mutat Res. 1993;289(2):231–243. | ||
O’Reilly MS, Holmgren L, Shing Y, et al. Angiostatin: a novel angiogenesis inhibitor that mediates the suppression of metastases by a Lewis lung carcinoma. Cell. 1994;79(2):315–328. | ||
Prise KM. Ionizing radiation therapy. In: Schwab M, editor. Encyclopedia of Cancer. 3rd ed. Heidelberg: Springer; 2009:1907–1909. | ||
Sokolov MV, Neumann RD. Radiation-induced bystander effects in cultured human stem cells. PLoS One. 2010;5(12):e14195. | ||
O’Neill-Mehlenbacher A, Kilemade M, Elliott A, Mothersill C, Seymour C. Comparison of direct and bystander effects induced by ionizing radiation in eight fish cell lines. Int J Radiat Biol. 2007;83(9):593–602. | ||
He M, Zhao M, Shen B, Prise KM, Shao C. Radiation-induced intercellular signaling mediated by cytochrome-C via a p53-dependent pathway in hepatoma cells. Oncogene. 2011;30(16):1947–1955. | ||
Vines AM, Lyng FM, McClean B, Seymour C, Mothersill CE. Bystander effect induced changes in apoptosis related proteins and terminal differentiation in in vitro murine bladder cultures. Int J Radiat Biol. 2009;85(1):48–56. | ||
Facoetti A, Mariotti L, Ballarini F, et al. Experimental and theoretical analysis of cytokine release for the study of radiation-induced bystander effect. Int J Radiat Biol. 2009;85(8):690–699. | ||
McBride WH, Chiang CS, Olson JL, et al. A sense of danger from radiation. Radiat Res. 2004;162(1):1–19. | ||
Van Meir E, Sawamura Y, Diserens AC, Hamou MF, de Tribolet N. Human glioblastoma cells release interleukin 6 in vivo and in vitro. Cancer Res. 1990;50(20):6683–6688. | ||
Weissenberger J, Loeffler S, Kappeler A, et al. IL-6 is required for glioma development in a mouse model. Oncogene. 2004;23(19):3308–3316. | ||
Brat DJ, Bellail AC, Van Meir EG. The role of interleukin-8 and its receptors in gliomagenesis and tumoral angiogenesis. Neuro Oncol. 2005;7(2):122–133. | ||
Hamada N, Maeda M, Otsuka K, Tomita M. Signaling pathways underpinning the manifestations of ionizing radiation-induced bystander effects. Curr Mol Pharmacol. 2011;4(2):79–95. | ||
Burr K, Robinson J, Rastogi S, et al. Radiation-induced delayed bystander-type effects mediated by hemopoietic cells. Radiat Res. 2010;173(6):760–768. | ||
Ghandhi SA, Ming L, Ivanov VN, Hei TK, Amundson SA. Regulation of early signaling and gene expression in the α-particle and bystander response of IMR-90 human fibroblasts. BMC Med Genomics. 2010;3:31. | ||
Guo X, Sun J, Bian P, et al. Radiation-induced bystander signaling from somatic cells to germ cells in Caenorhabditis elegans. Radiat Res. 2013;180(3):268–275. | ||
Honma M, Hayashi M, Sofuni T. Cytotoxic and mutagenic responses to X-rays and chemical mutagens in normal and p53-mutated human lymphoblastoid cells. Mutat Res. 1997;374(1):89–98. |
 © 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.