")
Back to Journals » International Journal of Nanomedicine » Volume 10 » Issue 1
Bypassing the EPR effect with a nanomedicine harboring a sustained-release function allows better tumor control
Authors Shen YA, Shyu IL, Lu M, He CL, Hsu YM, Liang HF, Liu CP, Liu RS, Shen BJ, Wei YH, Chuang CM
Received 28 November 2014
Accepted for publication 9 February 2015
Published 30 March 2015 Volume 2015:10(1) Pages 2485—2502
DOI https://doi.org/10.2147/IJN.S78321
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Lei Yang
Yao An Shen,1,* Ing Luen Shyu,2,* Maggie Lu,3 Chun Lin He,4 Yen Mei Hsu,2 Hsiang Fa Liang,3 Chih Peng Liu,3 Ren Shyan Liu,5,6 Biing Jiun Shen,7 Yau Huei Wei,1 Chi Mu Chuang2,4
1Institute of Biochemistry and Molecular Biology, School of Life Sciences, 2Institute of Clinical Medicine, School of Medicine, National Yang-Ming University, Taipei, 3Biomedical Technology and Device Research Laboratories, Industrial Technology Research Institute, Hsinchu, 4Division of Gynecologic Oncology, Department of Obstetrics and Gynecology, Taipei Veterans General Hospital, 5Department of Biomedical Imaging and Radiological Sciences, National Yang-Ming University, 6National PET/Cyclotron Center, Taipei Veterans General Hospital, Taipei, Taiwan; 7Division of Psychology, Nanyang Technological University, Singapore
*These authors contributed equally to the work
Abstract: The current enhanced permeability and retention (EPR)-based approved nanomedicines have had little impact in terms of prolongation of overall survival in patients with cancer. For example, the two Phase III trials comparing Doxil®, the first nanomedicine approved by the US Food and Drug Administration, with free doxorubicin did not find an actual translation of the EPR effect into a statistically significant increase in overall survival but did show less cardiotoxicity. In the current work, we used a two-factor factorial experimental design with intraperitoneal versus intravenous delivery and nanomedicine versus free drug as factors to test our hypothesis that regional (intraperitoneal) delivery of nanomedicine may better increase survival when compared with systemic delivery. In this study, we demonstrate that bypassing, rather than exploiting, the EPR effect via intraperitoneal delivery of nanomedicine harboring a sustained-release function demonstrates dual pharmacokinetic advantages, producing more efficient tumor control and suppressing the expression of stemness markers, epithelial-mesenchymal transition, angiogenesis signals, and multidrug resistance in the tumor microenvironment. Metastases to vital organs (eg, lung, liver, and lymphatic system) are also better controlled by intraperitoneal delivery of nanomedicine than by standard systemic delivery of the corresponding free drug. Moreover, the intraperitoneal delivery of nanomedicine has the potential to replace hyperthermic intraperitoneal chemotherapy because it shows equal efficacy and lower toxicity. In terms of efficacy, exploiting the EPR effect may not be the best approach for developing a nanomedicine. Because intraperitoneal chemotherapy is a type of regional chemotherapy, the pharmaceutical industry might consider the regional delivery of nanomedicine as a valid alternative pathway to develop their nanomedicine(s) with the goal of better tumor control in the future.
Keywords: enhanced permeability and retention effect, liposome, paclitaxel, ovarian cancer
Corrigendum for this paper has been published
Introduction
The enhanced permeability and retention (EPR) effect is becoming the “gold standard” for the development of nanomedicines. All nanoparticle-based drugs use the EPR effect as a guiding principle.1 In theory, the use of nanoparticles should have a pharmacokinetic advantage by prolonging the half-life of drugs in the systemic circulation.2 However, the EPR effect provides only relatively modest specificity, offering 20%–30% increases in delivery of payload compared with critical normal organs.3 Nonetheless, despite this pharmacokinetic advantage and numerous publications and patents regarding nanotechnology, only a few nanomedicines have demonstrated success in practice.4
As an example, although increased tumor accumulation has been achieved with PEGylated liposomal doxorubicin (Doxil®, Johnson & Johnson Pharmaceutical Research and Development, LLC, Raritan, NJ, USA; Caelyx®, Schering-Plough Corporation, Kenilworth, NJ, USA), the first nanomedicine approved by the US Food and Drug Administration, the two Phase III trials that compared Doxil with free doxorubicin in the treatment of metastatic breast cancer did not show an actual translation of the EPR effect into a statistically significant improvement in tumor response rate and overall survival.5,6 Another approved nanomedicine, Abraxane® (Celgene, Summit, NJ, USA) an EPR-based albumin-binding paclitaxel, necessitated a 50% higher dose than free paclitaxel to achieve a better tumor response rate and progression-free survival, but an increase in overall survival was still not achieved for metastatic breast cancer (65.0 weeks versus 55.7 weeks, P=0.374).7 Moreover, in the pivotal Phase III study leading to the approval of Abraxane for the treatment of metastatic non-small lung cancer, the median overall survival with Abraxane was only 12.1 months compared with the 11.2 months obtained with conventional paclitaxel,8 resulting in only a 0.9-month survival benefit (P=0.271). Of note, overall survival remains the “gold standard” for evaluating the outcome of an investigational drug in the current era, but exploiting the EPR effect has rarely translated into real antitumor efficacy for most currently approved nanomedicines.9
Regional chemotherapy, such as intraperitoneal chemotherapy, has the pharmacokinetic advantage of an increased ratio of the peritoneal-to-plasma area under the curve (AUC) to the tumor-containing peritoneal cavity.10,11 Despite this pharmacokinetic advantage, the clinical use of intraperitoneal therapy has been challenged by the premature clearance of a small molecular weight drug from the peritoneal cavity, a lack of target specificity, and poor drug penetration into the target tissues.12
The combination of nanotechnology and regional chemotherapy, ie, regional delivery of nanomedicine, may compensate for each other’s limitations. This combination may potentially present several advantages. First, the regional delivery of a nanomedicine may have dual pharmacokinetic advantages, as discussed previously. Second, the application of hydrophobic, poorly water-soluble chemotherapeutic agents for regional delivery is associated with serious problems of poor absorption and low bioavailability. The advent of nanotechnology can improve the aqueous solubility of poorly soluble drugs and thus may introduce more candidate drugs for the application of regional chemotherapy.13 Third, the anticancer activity of some conventional drugs, such as 5-fluorouracil, gemcitabine, paclitaxel, and camptothecin, is primarily cell cycle-dependent, resulting in the requirement of prolonged exposure times.14 The sustained-release function of nanomedicines may overcome this inherent limitation.
To accelerate the progress of development of nanomedicines, we aimed to investigate whether bypassing the EPR effect may improve their therapeutic efficacy. In this study, we tested several approved or developing nanomedicines. Two-factor factorial experiments were conducted to test the effect of the delivery method (factor 1: intraperitoneal delivery versus intravenous delivery), the drug formulation (factor 2: nanomedicine versus free drug), and their interactions on tumor control. For each factorial design, the active drug was administered on an equal dose basis.
Materials and methods
Animal models and cell lines
Tumor generation involved a single intraperitoneal injection of the human ovarian cancer cell line ES-2-luc (kindly provided by T-C Wu, Cervical Cancer Research Laboratory, Johns Hopkins University) at day 0 into 5-week-old SCID mice (CB.17 SCID/SCID) and was consistently first evident after 3 weeks via abdominal distension secondary to malignant ascites. The animals were purchased from the National Laboratory Animal Breeding and Research Center of Taipei and maintained in the oncology animal facility of Taipei Veterans General Hospital, Taipei, Taiwan. The animals were used in compliance with the institutional animal health care regulations, and all of the animal experimental procedures were approved by the Institutional Animal Care and Use Committee. The tumor-bearing mice were euthanized at the point when there were signs of distress, including fur ruffling, rapid respiratory rate, hunched posture, reduced activity, and progressive formation of ascites. Groups of mice were treated with different formulations using different delivery methods. Several approved or developing nanomedicines, including liposomal paclitaxel (Nano-taxol, Industrial Technology Research Institute, Hsinchu, Taiwan), cisplatin-incorporating polymeric micelles (Nano-platin, NanoCarrier Co Ltd, Kashiwa, Japan), polymeric micelle topotecan (Nano-topotecan, Taiwan Liposomal Company, Taipei, Taiwan), PEGylated liposomal doxorubicin (Nano-doxorubicin, Doxil, Caelyx, Merck & Co., Whitehouse Station, NJ, USA), and albumin-bound paclitaxel (Abraxane) were tested. The dosing schedule for each of the paired drugs (free drug and nanomedicine) was based on the maximum tolerated dose according to preliminary studies. Lung metastases were induced by tail vein injection, whereas liver metastases were induced by splenic injection, and retroperitoneal lymphatic metastases were induced by intrauterine injection of tumor cells.
In vitro release kinetics of paclitaxel by high-performance liquid chromatography
The release of paclitaxel from liposomes was assayed using the dialysis method at room temperature and was compared with that of Taxol® (Bristol-Myers Squibb, New York, NY, USA). After reconstituting the freeze-dried liposomes in phosphate-buffered saline (pH 7.4) to yield 1.5 mg/mL paclitaxel, an aliquot of each liposomal dispersion (0.1 mL) was placed in a dialysis tube (molecular weight cutoff 6,000–8,000DA, Gene Bio-Application Ltd, Kfar HaNagid, Israel), which was then tightly sealed. The tube was then immersed in 200 mL of release medium, ie, phosphate-buffered saline (pH 7.4) containing 0.1% (v/v) Tween 80, to maintain the sink conditions. While stirring medium using a magnetic stirrer at 300 rpm, samples (0.5 mL) were collected at predetermined time intervals from the release medium over a period of 24 hours, and the displaced medium was replaced with the same volume of fresh medium. The concentration of paclitaxel was determined by high-performance liquid chromatography after appropriate dilution with acetonitrile without further treatment.
Paclitaxel assay in tumor tissue
The paclitaxel concentration was analyzed using an Agilent 1100 series chromatographic system (Agilent Technologies, Palo Alto, CA, USA). Chromatographic separation was achieved with a C18 Altima™ column (4.6 mm ID, 150 mm; 5 μm) purchased from Grace (Columbia, MD, USA) and protected by a SecurityGuard™ in-line filter unit (Phenomenex, Torrance, CA, USA). The mobile phase consisted of water, methanol, and acetonitrile (25:35:40, v/v). The mobile phase was filtered through a 0.45 μm filter (Millipore, Bedford, MA, USA) and degassed before analysis. The high-performance liquid chromatography system was run at ambient temperature with a flow rate of 1.0 mL per minute, and quantitative analysis of the peak AUC was performed at a wavelength of 230 nm, with a total chromatographic run time of 8 minutes. The calibration curve for the quantification of paclitaxel was linear over the standard paclitaxel concentration range of 50–50,000 ng/mL with a correlation coefficient of R2=0.999. The limit of detection was 50 ng/mL.
Determining synergy of intraperitoneal delivery of nanomedicine by the Chou–Talalay method
Six dose levels were used to evaluate the synergy effects. To calculate the combined drug effects, we used the combination index (CI)-isobologram method developed by Chou–Talalay.15 Intraperitoneal delivery of Nano-taxol was viewed as a combined effect, whereas systemic delivery of Nano-taxol or intraperitoneal delivery of free Taxol was viewed as a single effect. The Chou–Talalay method involves the plotting of dose-effect curves for each agent and combinations thereof in multiple diluted concentrations using the median-effect equation to calculate how much the experimental effect differs from the effect expected assuming additive effects. CI =1, <1, and >1 indicate additive, synergistic, and antagonistic effects, respectively. By definition, synergy is an effect that is more than additive, whereas an antagonistic effect is an effect that is less than additive. The CI values can be determined at different effect levels and different dose levels, and the isobolograms can be automatically generated using Compusyn software.
Statistical analysis
The results are given as the mean ± standard deviation of at least three experiments. Statistical comparisons were performed using the Student’s t-test. Overall survival was defined as the time from the date of tumor implantation to the date of death and is presented as the hazard ratio (with 95% confidence interval). Kaplan–Meier survival curves were plotted to assess the difference between the hyperthermic intraperitoneal chemotherapy (HIPEC) group and the group that received intraperitoneal delivery of Nano-taxol. A two-factor factorial analysis was used to test the effect of the delivery method (intraperitoneal versus intravenous), the drug formulation (nanomedicine versus free drug), and their interactions on tumor control. The efficacy of tumor control was evaluated by IVIS® luminescence (PerkinElmer, Waltham, MA, USA). The luminescence data obtained for each mouse were averaged for statistical purposes. All of the statistical tests were two-sided with a significance level (P-value) of 0.05 unless otherwise specified. The statistical calculations were performed using Stata version 12.0 (Statacorp, College Station, TX, USA).
For other details, please refer to the materials and methods in the Supplementary materials section.
Results
Pharmacokinetic characteristics and mathematical proof of the synergistic tumor-killing effect obtained by intraperitoneal delivery of sustained-release nanomedicine
In the current work, we used Nano-taxol, which harbors a sustained-release function, as the major test drug. Transmission electron microscopy (upper panel, Figure 1A) and dynamic light scattering (lower panel, Figure 1A) analyses of this drug are presented. The particle size distribution obtained by dynamic light scattering corresponded to a 100 nm-sized vesicular formulation. The review published by Nagayasu et al reports that size is a key property in the optimization of liposomes directed for blood circulation, tumor accumulation, and in vivo drug release, and concludes that 100 nm appears to be the most suitable size for obtaining an effective antitumor preparation.16 Applying kinetic equation models, the in vitro release process was calculated by treating the release profile mathematically using zero-order, first-order, and Higuchi equations.17 Higher correlation coefficients were obtained with the Higuchi equation, indicating that the release of the free drug was sustained and driven mainly by a diffusion-controlled mechanism because the plots showed high linearity with a correlation coefficient (R2) value of 0.9864 (Figure 1B), as observed with other liposomal products.18
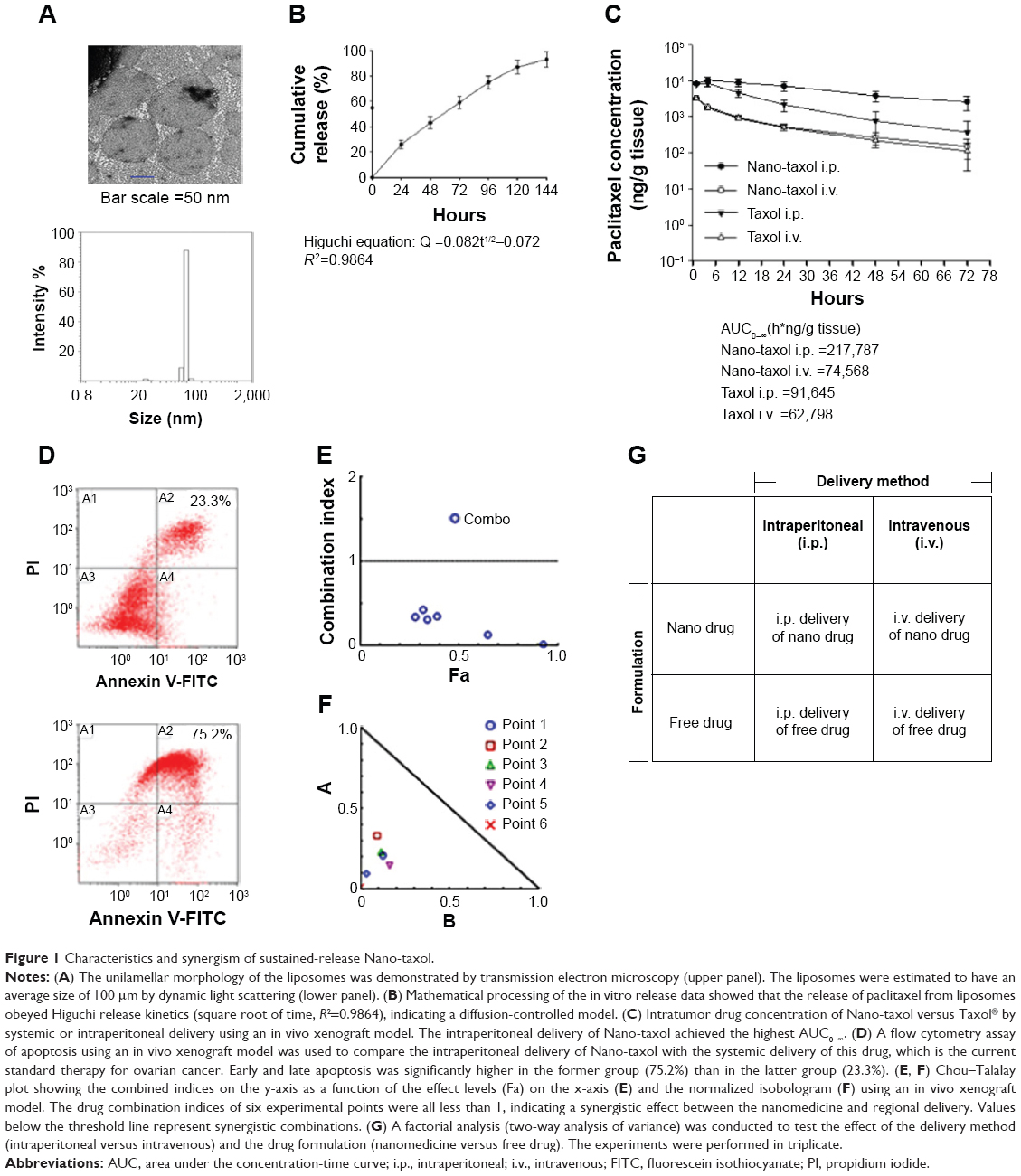 | Figure 1 Characteristics and synergism of sustained-release Nano-taxol. |
We then determined the pharmacokinetics and pharmacodynamics using the AUC and apoptosis, respectively, as end points in an in vivo xenograft model. Increased AUC of the intratumor drug concentration (Figure 1C) and apoptosis (Figure 1D) were also observed in the group to which Nano-taxol was intraperitoneally delivered. AUC measurements have been used to estimate the total drug exposure because they often correlate with pharmacodynamic effects.19 In the present study, the intraperitoneal delivery of Nano-taxol demonstrated an approximately threefold higher AUC compared with that obtained by systemic delivery of Taxol, which is currently a standard regimen for the treatment of ovarian cancer. With respect to apoptosis, intraperitoneal delivery of Nano-taxol also resulted in a significantly higher proportion of combined early and late apoptosis than that induced by systemic delivery of Taxol (75.2% versus 23.3%), even though late apoptotic cells may contain some necrotic cells.
We further evaluated the synergistic tumor-killing effects of intraperitoneal delivery of Nano-taxol using the Chou–Talalay method.20 In this experiment, we used an in vivo xenograft model for the calculation. A total of six dose levels were used to evaluate the synergistic effects. The Fa-CI plot demonstrates that the drug combination indices were all less than 1, indicating that intraperitoneal delivery of Nano-taxol can produce synergistic tumor killing when compared with either systemic delivery of Nano-taxol or intraperitoneal delivery of free Taxol (Figure 1E). Furthermore, the normalized isobologram for combinations at different ratios further corroborate this synergistic tumor-killing effect (Figure 1F).
The factorial design used to test the therapeutic effect of the delivery method (intraperitoneal versus intravenous) and drug formulation (nanomedicine versus free drug) is presented in Figure 1G. All of the tested paired drugs followed this factorial design.
Intraperitoneal delivery of Nano-taxol has a better tumor-killing effect than systemic delivery
We next asked whether the observed synergy could be translated into real tumor killing. Under the treatment scheme presented in Figure 2A, intraperitoneal delivery of Nano-taxol showed a markedly better tumor-killing ability than any other type of treatment (Figure 2B). Intraperitoneal delivery of Nano-topotecan also showed positive therapeutic effects (Figure S1). In contrast, intraperitoneal delivery of Nano-platin (Figure S2), Nano-doxorubicin (Figure S3), and Abraxane (Figure S4) failed to show this therapeutic benefit.
 | Figure 2 Therapeutic effects of intraperitoneal delivery of Nano-taxol. |
Further, intraperitoneal delivery of free drug, such as Taxol (Figure 2B), topotecan (Figure S1), cisplatin (Figure S2), and doxorubicin (Figure S3), did show a modest therapeutic benefit, which paralleled the results obtained in each of the corresponding human trials.21–24 A summary of quantification of bioluminescence signals for each presented figure is given in Figure S5.
A significant interaction between two factors obtained in a factorial analysis indicates that the effect of one variable depends on the level of the second variable.25 All of the tested drugs showed a significant interaction effect, indicating that the therapeutic effects of a drug formulation are dependent on the delivery method used (Figure 2C and Figures S1–S4).
The hazard ratio for overall survival demonstrates that intraperitoneal delivery of Nano-taxol conferred the longest survival (Figure 2D), whereas white cell counts were not significantly different between the different treatment groups (Figure 2E). The clinical implication is that intraperitoneal delivery of Nano-taxol can result in longer survival than that achieved by the current standard therapy without risking more side effects.
In summary, our work demonstrates that systemic delivery of the tested nanomedicines (ie, exploiting the EPR effect) all failed to show better therapeutic efficacy compared with each corresponding free drug, indicating that exploiting the EPR effect can hardly translate into a real therapeutic effect; in addition, intraperitoneal delivery of nanomedicines (ie, bypassing the EPR effect) shows controversial results when compared with free drugs (better for Nano-taxol and Nano-topotecan but worse for Nano-doxorubicin and Abraxane), suggesting that the structures of Nano-doxorubicin and Abraxane are presumably too stable to release the encapsulated drug in a timely manner to control intraperitoneal tumors. The clinical implication of these results is that the pharmaceutical industry should attempt to explore the therapeutic efficacy obtained by regional (intraperitoneal) delivery of a nanomedicine in development when the nanomedicine fails to show therapeutic efficacy by systemic delivery.
Intraperitoneal delivery of sustained-release nanomedicine suppresses vital organ metastases
Although direct intraperitoneal dissemination is considered the most common, ovarian cancer may also metastasize through the lymphatic channels and the hematogenous route. The effects of the consequent vital organ metastases (eg, liver or lung) are often devastating, with a dismal prognosis, despite the current standard treatment of systemic delivery of Taxol.26
Extensive liver parenchymal tumor metastases often lead to liver failure, which is speculated to be caused by extensive hepatocellular necrosis resulting from pressure atrophy and interference in the vascular supply.27 In this work, mice were induced to develop liver metastases by splenic injection of ES-2-luc cells. The results show that intraperitoneal delivery of Nano-taxol significantly suppresses tumor metastases to the liver compared with the systemic delivery of Taxol (upper panel, Figure 3A). The extirpation of liver tissue for hematoxylin and eosin staining further confirmed that intraperitoneal delivery of Nano-taxol shows the best response in terms of prevention of liver metastases.
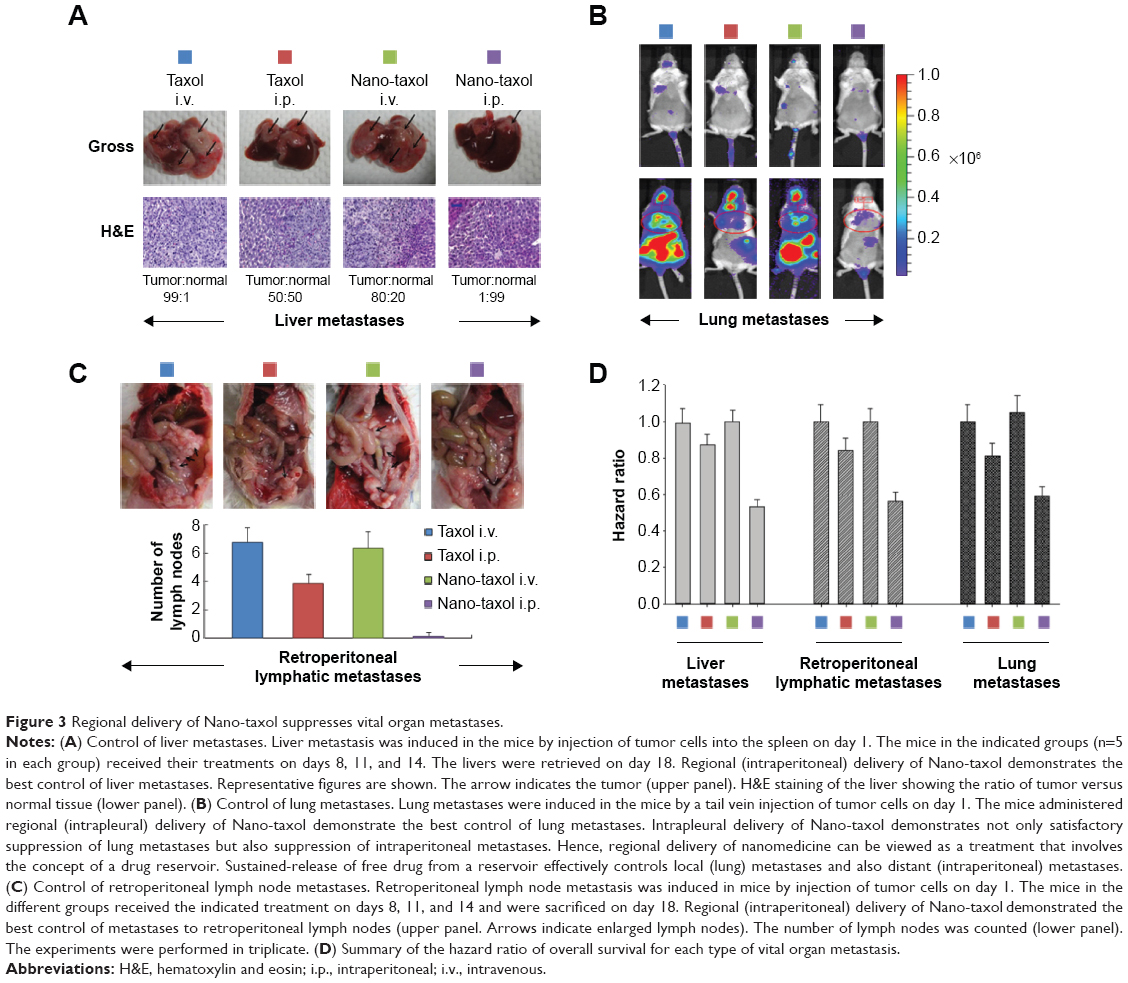 | Figure 3 Regional delivery of Nano-taxol suppresses vital organ metastases. |
Intrapleural chemotherapy (a type of regional chemotherapy) has been used as an alternative therapy in the treatment of lung cancer.28 Based on the observed prominent tumor-killing ability of peritoneal tumor spreading obtained by intraperitoneal delivery of Nano-taxol, we then asked whether this finding could also be obtained in the treatment of lung metastases. Mice were induced to develop lung metastases by tail vein injection of ES-2-luc cells. Intrapleural delivery of Nano-taxol showed the best control of lung metastases (Figure 3B). A summary of quantification of bioluminescence signals is given in Figure S5. In contrast, the current standard therapy of systemic delivery of Taxol shows unsatisfactory tumor control, whereas the intrapleural delivery of Taxol did show some therapeutic efficacy, which parallels that observed in a Phase II trial.29
To date, studies focusing on the intrapleural delivery of nanomedicine have been very rare, but the results have shown favorable control of lung cancer.30 The results of the current study may consolidate these published findings and could trigger more research on the intrapleural delivery of nanomedicine for the control of primary or secondary lung cancer.
The pathways of lymphatic drainage in ovarian cancer have been recognized. The presence of lymph node metastases in patients with advanced ovarian cancer often indicates a poor prognosis. Moreover, metastatic cells lodged in the lymph nodes are indicators, in a statistical sense, of the metastatic cell load, risk of later clinical metastases, and death from disease.31 With the exception of conventional systemic delivery of Taxol, there exists no novel therapy for the treatment of lymphatic spread.32 In this study, we demonstrate that intraperitoneal delivery of Nano-taxol outperforms the systemic delivery of Taxol in terms of control of lymph node metastases (Figure 3C and D).
A hazard ratio analysis of overall survival further demonstrated the significant therapeutic effects of intraperitoneal delivery of Nano-taxol for controlling metastases to these vital organs (Figure 3D).
Intraperitoneal delivery of Nano-taxol leads to more efficient killing of cancer stem cells
Tumor spheres grew in serum-free medium, and a side population identified by Hoechst 33342 staining through flow cytometry proved their efficiency in enriching and maintaining cancer stem cells (CSCs) from several cell lines.33 In the current experiment, intraperitoneal delivery of Nano-taxol presented the best control of tumor sphere formation (Figure 4A and B) and the side population (Figure 4C). In contrast, systemic or intraperitoneal delivery of Taxol and systemic delivery of Nano-taxol increased tumor sphere formation and the side population when compared with the control group; this phenomenon paralleled the findings obtained in other studies, indicating that increased CSCs are commonly observed in the tumor microenvironment after conventional chemotherapy.33 Because CSCs are often linked with recurrence of cancer, suppression of tumor sphere formation and the side population by intraperitoneal delivery of Nano-taxol may decrease the chance of cancer recurrence.
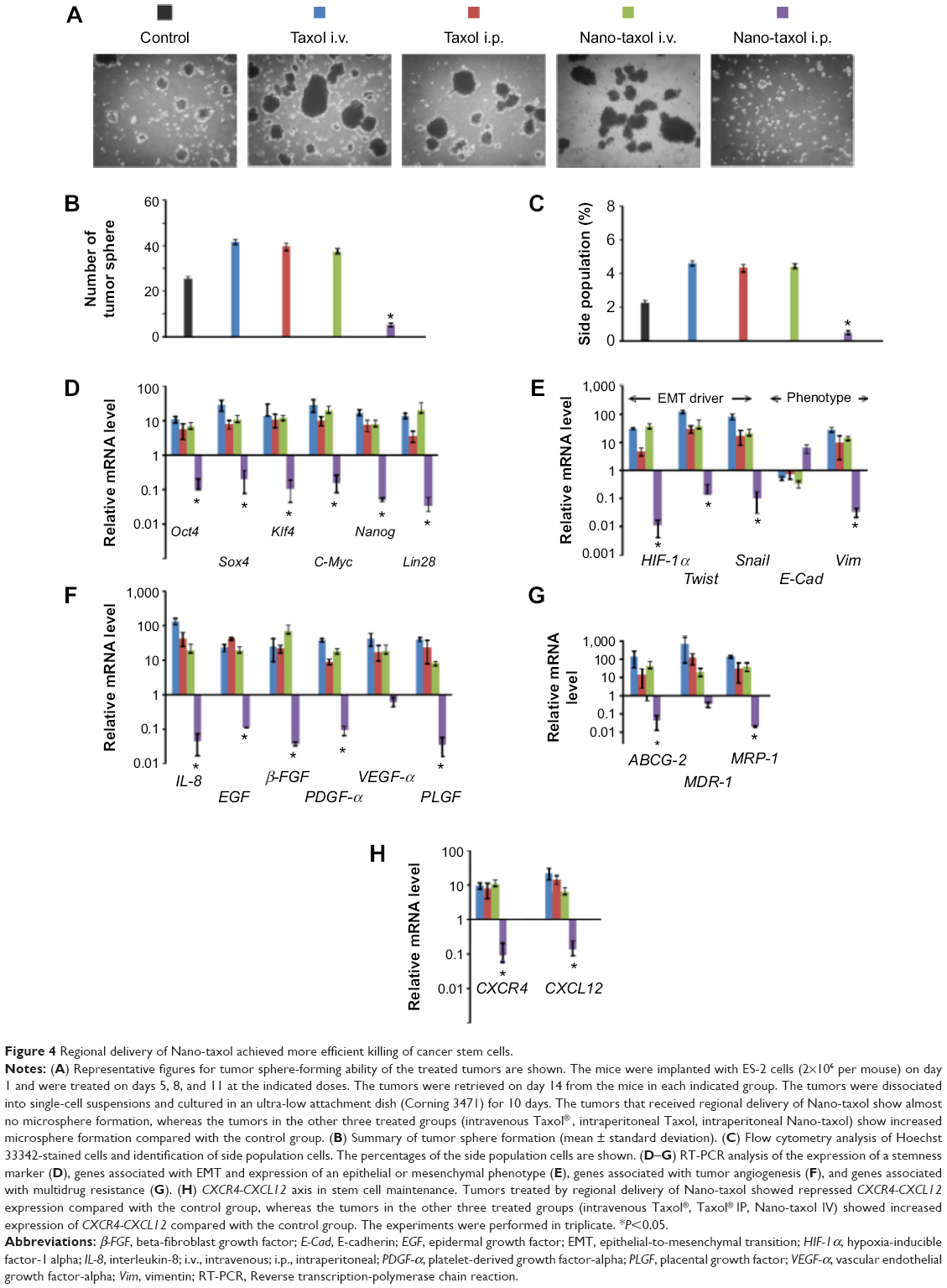 | Figure 4 Regional delivery of Nano-taxol achieved more efficient killing of cancer stem cells. |
Moreover, in this work, intraperitoneal delivery of Nano-taxol overwhelmingly suppressed the expression of a stemness marker (Figure 4D), an epithelial-to-mesenchymal transition (EMT) driver and phenotype (Figure 4E), an angiogenesis signal (Figure 4F), a multidrug resistance signal (Figure 4G), and the chemokine CXCR4-CXCL12 axis (Figure 4H) in the tumor microenvironment. Please refer to Table 1 for primer sequence for each molecule presented. To summarize, intraperitoneal delivery of Nano-taxol suppresses the expression of CSCs, indicating better tumor control.
 | Table 1 Primer sequences |
Intraperitoneal delivery of sustained-release nanomedicine shows comparable efficacy to HIPEC
Peritoneal carcinomatosis is one of the main indications for HIPEC in ovarian cancer.34 To determine whether intraperitoneal delivery of Nano-taxol may replace HIPEC (because the former is less time-consuming and less labor-intensive), we developed a recurrent ovarian cancer model to simulate peritoneal carcinomatosis. Flow cytometry analysis by Hoechst 33342 staining showed a higher percentage of the side population of recurrent tumor cells than that of primary tumor cells (Figure 5A), indicating the more chemoresistant nature of the recurrent tumor. The HIPEC procedure is depicted in Figure 5B. The results demonstrate that intraperitoneal delivery of Nano-taxol and HIPEC have comparable therapeutic efficacy (Figure 5C), with the former showing less toxicity (Figure 5D). A summary of the quantification of bioluminescence signals is given in Figure S5.
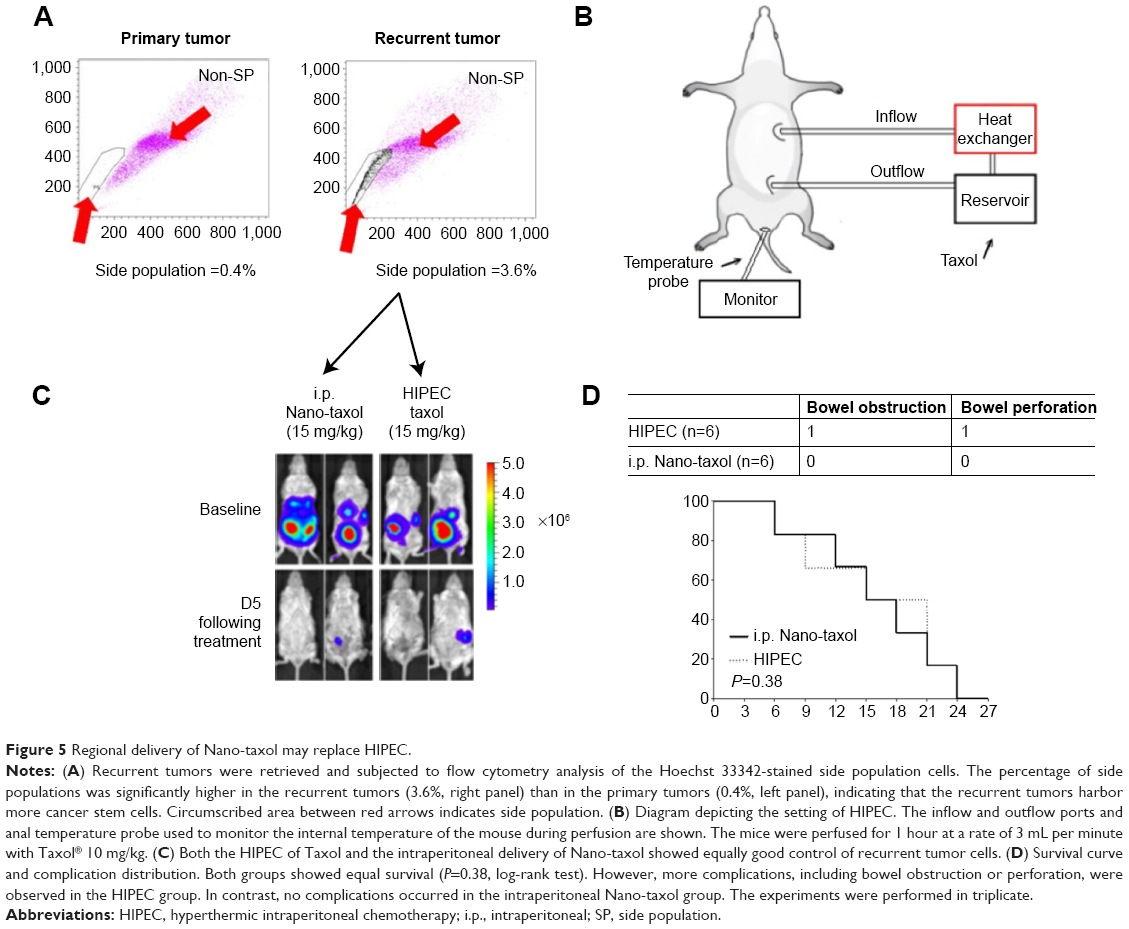 | Figure 5 Regional delivery of Nano-taxol may replace HIPEC. |
In summary, intraperitoneal delivery of Nano-taxol may replace HIPEC because it has comparable efficacy but comes with fewer complications and is less labor-intensive.
Bypassing the EPR effect by regional delivery of sustained-release nanomedicine achieves better tumor control
The results of the current work show that exploiting the EPR effect by systemic delivery of nanomedicine has limited therapeutic efficacy. Although the EPR effect is a well-established phenomenon in the tumor microenvironment, exploiting this effect rarely produces therapeutic efficacy compared with its corresponding free drug on an equal dose basis. In contrast, bypassing the EPR effect by regional (eg, intraperitoneal) delivery of nanomedicine harboring a sustained-release function yields markedly better results in terms of tumor control than those obtained by systemic delivery.
We recommend that the pharmaceutical industry aim to reduce toxicity but not the tumor-killing effect by exploiting the EPR effect (the classical pathway) when developing a nanomedicine. However, if the tumor-killing effect is the major goal, the industry might consider bypassing the EPR effect as an alternative pathway for development of nanomedicines. Intraperitoneal delivery is one type of regional delivery. Therefore, several types of regional nanomedicine delivery, such as intrathecal, intrapleural, and intravesical delivery, can be explored (Figure 6A).
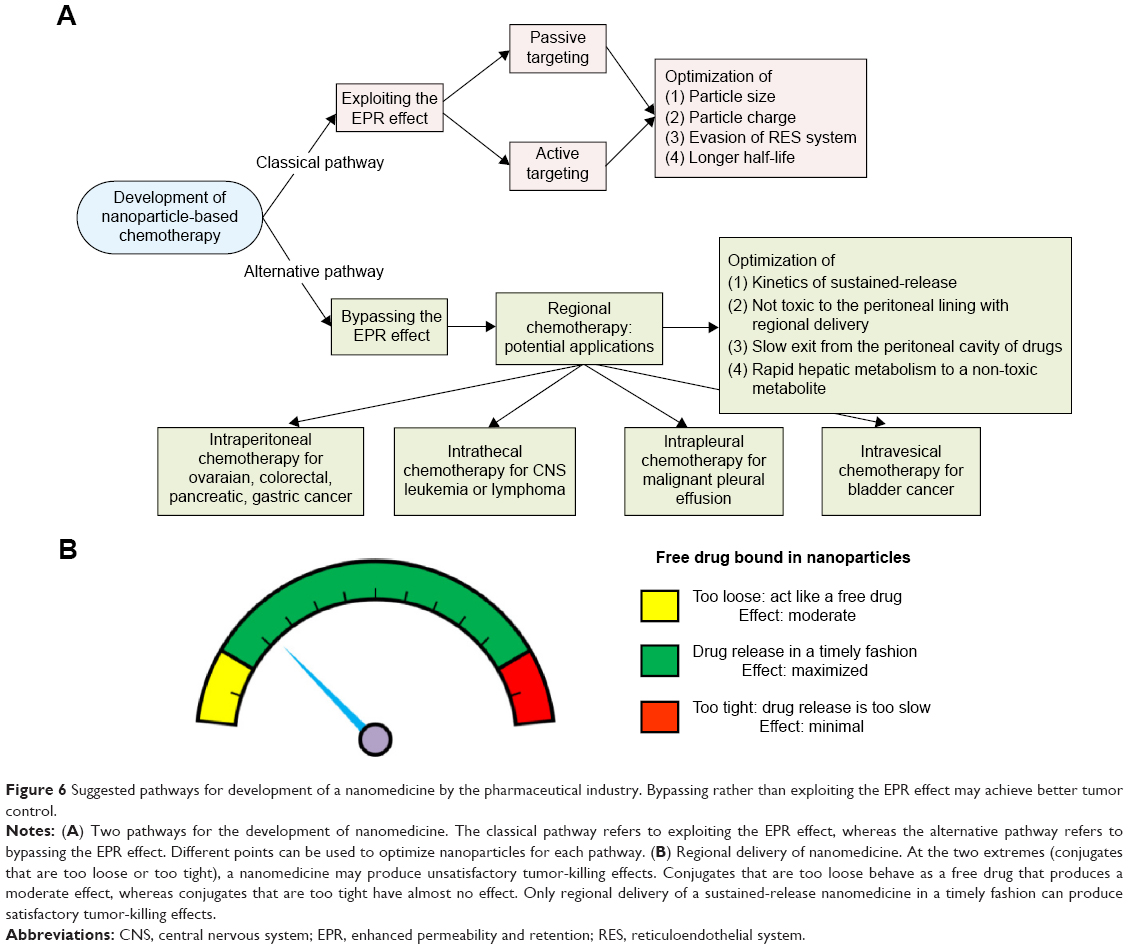 | Figure 6 Suggested pathways for development of a nanomedicine by the pharmaceutical industry. Bypassing rather than exploiting the EPR effect may achieve better tumor control. |
Of note, not all nanomedicines are suitable for intraperitoneal delivery. We propose that some nanomedicines fail to demonstrate better tumor killing by regional delivery (eg, Nano-platin, Nano-doxorubicin, and Abraxane, as demonstrated in the current work) because the covalent or non-covalent conjugates of free drug and nanoparticles are too stable to release free drug in a timely fashion. The current work demonstrates that, at one extreme, when the nanomedicine releases free drug too fast, it behaves as a free drug, and at the other extreme, when the nanomedicine releases free drug too slowly, the velocity of tumor growth may topple the effect of the released free drug (Figure 6B).
Discussion
The results of the current study suggest that exploiting the EPR effect may not be the best way to develop nanomedicine in terms of therapeutic efficacy. Bypassing the EPR effect may be a valid alternative for development of nanomedicines. Investigation of regional delivery of nanomedicine can be traced back decades.35,36 However, compared with the many studies of nanomedicines that explore the effect of systemic delivery (exploiting the EPR effect), studies of regional delivery of nanomedicines (bypassing the EPR effect) are limited.
The other novel findings of the current work include the following:
- Vital organ (liver, lung, and lymphatic) metastases can be better controlled by regional delivery of a nanomedicine compared with current standard therapy.
- Intrapleural delivery of Nano-taxol achieves better control of intraperitoneal metastases (Figure 3B). Hence, regional delivery of nanomedicine may be viewed as delivery of nanomedicine into a reservoir. Once a primary reservoir fails, eg, due to severe adhesion induced by carcinomatosis in the peritoneal cavity, the pleural cavity can replace the peritoneal cavity as a substitute reservoir for delivery of nanomedicine to treat tumors lodged in the primary reservoir.
- Better killing of CSCs can be achieved by regional delivery of nanomedicine.
- Comparable therapeutic efficacy can be seen with regional delivery of nanomedicine and HIPEC.
It is important to analyze the kinetics of the release of nanoparticles. The impact of the release profile of a nanomedicine has already been discussed by other investigators, who comment that “If the formulations lose their associated drug rapidly following administration, the drug accumulation will be poor. At the other extreme, if the formulations never release their associated drug, the level of drug accumulation will be great, but the effects of the drug may be limited unless there is a functional feature that facilitates drug release or internalization”.37 All these observations warrant further in-depth studies.
CSCs are currently viewed as key tumor-initiating cells that may have an integral role in recurrence. Their unique molecular machinery enables these cells to become resistant to most chemotherapeutic agents. Depletion of CSCs or inhibition of the molecular signature of CSCs may reduce the risk of recurrence and improve clinical outcomes.38 Cancer cells may also undergo adaptive changes following therapy that exacerbate drug resistance. In epithelial cancers, these adaptive changes may involve, at least in part, the EMT. Of interest, the EMT can trigger reversion of cancer cells to the CSC-like phenotype, indicating an association between EMT, CSCs, and drug resistance.39 Moreover, activation of the chemokine CXCR4-CXCL12 axis may be indicative of a metastatic CSC population. The CXCR4-CXCL12 axis plays a central role in proliferation, invasion, and dissemination of cancer cells in the majority of malignant diseases.40 There exist some strategies for the killing of CSCs, but most of these remain in the investigative phase.41–43
Besides ovarian cancer, peritoneal carcinomatosis is also the most commonly seen type of recurrence (15%-40%) in patients with gastric cancer and the second most commonly seen type of recurrence in patients with colon cancer.44 HIPEC has been shown to increase progression-free and overall survival in different models of these tumors.45–47 However, HIPEC is time-consuming, requiring more effort on the part of nursing and medical staff, and is associated with high rates of potentially life-threatening complications. The prevalent complications in most series are digestive fistulae, either in the form of an anastomotic leak or bowel perforation away from the anastomotic lines.48
The results of the current work can be echoed by the results obtained by intrathecal delivery of liposomal cytarabine (DepoCyt®), a nanomedicine approved for the treatment of leptomeningeal metastases of lymphoma or leukemia. In a randomized controlled trial, DepoCyt was delivered intrathecally at lower doses compared with free cytarabine. Nonetheless, DepoCyt achieved an increased tumor response rate (71% versus 15%).49
The sustained-release function of nanomedicines is similar to the concept of metronomic chemotherapy, ie, frequent, repetitive administration of chemotherapeutic drugs at relatively low doses and without a prolonged drug-free break, and is an emerging strategy in the fight against cancer.50 Its mechanism of action is regarded as primarily antiangiogenic in nature, affecting both the endothelial cells of tumor-supplying blood vessels and circulating endothelial progenitor cells.51 Our results are in parallel with the effects of metronomic chemotherapy, showing a reduction in the signal for tumor angiogenesis (Figure 4F).
In conclusion, bypassing rather than exploiting the EPR effect via regional delivery of nanomedicine (exploiting the EPR effect) may enable better tumor control and killing of CSCs than intravenous delivery of nanomedicine (bypassing the EPR effect). The traditional concept of exploiting the EPR effect as the only “gold standard” should be questioned. However, not all nanomedicines are suitable for regional delivery. Only a nanomedicine that harbors a sustained-release function, ie, that releases the encapsulated drug in a timely fashion between the two extremes (too-slow or too-fast release kinetics), can efficiently kill tumor cells. In the future, to develop nanomedicines for use in the peritoneal cavity, further research should be undertaken to optimize the release profile, biocompatibility, biodistribution, and formulation stability in the peritoneal cavity.
Acknowledgments
This work was supported in part by a grant from the National Science Council of Taiwan (NSC 101-2623-E-010-001-NU). The authors thank the Genomics Research Center, National Yang Ming University, for providing facilities and support for tumor imaging and flow cytometric analysis.
Disclosure
The authors report no conflicts of interest in this work.
References
Prabhakar U, Maeda H, Jain RK, et al. Challenges and key considerations of the enhanced permeability and retention effect for nanomedicine drug delivery in oncology. Cancer Res. 2013;73:2412–2417. | ||
Danhier F, Feron O, Preat V. To exploit the tumor microenvironment: passive and active tumor targeting of nanocarriers for anti-cancer drug delivery. J Control Release. 2010;148:135–146. | ||
Kobayashi H, Watanabe R, Choyke PL. Improving conventional enhanced permeability and retention (EPR) effects; what is the appropriate target? Theranostics. 2013;4:81–89. | ||
Venditto VJ, Szoka FC Jr. Cancer nanomedicines: so many papers and so few drugs! Adv Drug Deliv Rev. 2013;65:80–88. | ||
O’Brien ME, Wigler N, Inbar M, et al. Reduced cardiotoxicity and comparable efficacy in a phase III trial of pegylated liposomal doxorubicin HCl (Caelyx/Doxil) versus conventional doxorubicin for first-line treatment of metastatic breast cancer. Ann Oncol. 2004;15:440–449. | ||
Harris L, Batist G, Belt R, et al. Liposome-encapsulated doxorubicin compared with conventional doxorubicin in a randomized multicenter trial as first-line therapy of metastatic breast carcinoma. Cancer. 2002;94:25–36. | ||
Gradishar WJ, Tjulandin S, Davidson N, et al. Phase III trial of nanoparticle albumin-bound paclitaxel compared with polyethylated castor oil-based paclitaxel in women with breast cancer. J Clin Oncol. 2005;23:7794–7803. | ||
Socinski MA, Bondarenko I, Karaseva NA, et al. Weekly nab-paclitaxel in combination with carboplatin versus solvent-based paclitaxel plus carboplatin as first-line therapy in patients with advanced non-small-cell lung cancer: final results of a phase III trial. J Clin Oncol. 2012;30:2055–2062. | ||
Driscoll JJ, Rixe O. Overall survival: still the gold standard. Why overall survival remains the definitive end point in cancer clinical trials. Cancer J. 2009;15:401–405. | ||
Markman M. Intraperitoneal antineoplastic drug delivery: rationale and results. Lancet Oncol. 2003;4:277–283. | ||
Ceelen WP, Flessner MF. Intraperitoneal therapy for peritoneal tumors: biophysics and clinical evidence. Nat Rev Clin Oncol. 2010;7:108–115. | ||
Minchinton AI, Tannock IF. Drug penetration in solid tumours. Nat Rev Cancer. 2006;6:583–592. | ||
Bharali DJ, Mousa SA. Emerging nanomedicines for early cancer detection and improved treatment: current perspective and future promise. Pharmacol Ther. 2010;128:324–335. | ||
Lapenna S, Giordano A. Cell cycle kinases as therapeutic targets for cancer. Nat Rev Drug Discov. 2009;8:547–566. | ||
Chou TC. Drug combination studies and their synergy quantification using the Chou-Talalay method. Cancer Res. 2010;70:440–446. | ||
Nagayasu A, Uchiyama K, Kiwada H. The size of liposomes: a factor which affects their targeting efficiency to tumors and therapeutic activity of liposomal antitumor drugs. Adv Drug Deliv Rev. 1999;40:75–87. | ||
Higuchi T. Rate of release of medicaments from ointment bases containing drugs in suspension. J Pharm Sci. 1961;50:874–875. | ||
Nounou MM, El-Khordagui LK, Khalafallah NA, Khalil SA. In vitro release of hydrophilic and hydrophobic drugs from liposomal dispersions and gels. Acta Pharm. 2006;56:311–324. | ||
Lindholm A, Kahan BD. Influence of cyclosporine pharmacokinetics, trough concentrations, and AUC monitoring on outcome after kidney transplantation. Clin Pharmacol Ther. 1993;54:205–218. | ||
Chou TC. Theoretical basis, experimental design, and computerized simulation of synergism and antagonism in drug combination studies. Pharmacol Rev. 2006;58:621–681. | ||
Alberts DS, Liu PY, Hannigan EV, et al. Intraperitoneal cisplatin plus intravenous cyclophosphamide versus intravenous cisplatin plus intravenous cyclophosphamide for stage III ovarian cancer. N Engl J Med. 1996;335:1950–1955. | ||
Armstrong DK, Bundy B, Wenzel L, et al. Intraperitoneal cisplatin and paclitaxel in ovarian cancer. N Engl J Med. 2006;354:34–43. | ||
Muntz HG, Malpass TW, McGonigle KF, Robertson MD, Weiden PL. Phase 2 study of intraperitoneal topotecan as consolidation chemotherapy in ovarian and primary peritoneal carcinoma. Cancer. 2008;113:490–496. | ||
Delgado G, Potkul RK, Treat JA, et al. A phase I/II study of intraperitoneally administered doxorubicin entrapped in cardiolipin liposomes in patients with ovarian cancer. Am J Obstet Gynecol. 1989;160:812–817. | ||
Triola MF. Elementary Statistics. 12th ed. Upper Saddle River, NJ, USA: Pearson Education Limited; 2014. | ||
Lengyel E. Ovarian cancer development and metastasis. Am J Pathol. 2010;177:1053–1064. | ||
Allison KH, Fligner CL, Parks WT. Radiographically occult, diffuse intrasinusoidal hepatic metastases from primary breast carcinomas: a clinicopathologic study of 3 autopsy cases. Arch Pathol Lab Med. 2004;128:1418–1423. | ||
Baba T, Uramoto H, Kuwata T, et al. Intrapleural chemotherapy improves the survival of non-small cell lung cancer patients with positive pleural lavage cytology. Surg Today. 2013;43:648–653. | ||
Lombardi G, Nicoletto MO, Gusella M, et al. Intrapleural paclitaxel for malignant pleural effusion from ovarian and breast cancer: a phase II study with pharmacokinetic analysis. Cancer Chemother Pharmacol. 2012;69:781–787. | ||
Liu J, Meisner D, Kwong E, Wu XY, Johnston MR. Translymphatic chemotherapy by intrapleural placement of gelatin sponge containing biodegradable paclitaxel colloids controls lymphatic metastasis in lung cancer. Cancer Res. 2009;69:1174–1181. | ||
Gervasoni JE Jr, Taneja C, Chung MA, Cady B. Biologic and clinical significance of lymphadenectomy. Surg Clin North Am. 2000;80:1631–1673. | ||
Halkia E, Spiliotis J, Sugarbaker P. Diagnosis and management of peritoneal metastases from ovarian cancer. Gastroenterol Res Pract. 2012;2012:541842. | ||
Kondo T. Stem cell-like cancer cells in cancer cell lines. Cancer Biomark. 2007;3:245–250. | ||
Harless WW. Cancer treatments transform residual cancer cell phenotype. Cancer Cell Int. 2011;11:1. | ||
Helm CW. Current status and future directions of cytoreductive surgery and hyperthermic intraperitoneal chemotherapy in the treatment of ovarian cancer. Surg Oncol Clin N Am. 2012;21:645–663. | ||
Parker RJ, Hartman KD, Sieber SM. Lymphatic absorption and tissue disposition of liposome-entrapped [14C]adriamycin following intraperitoneal administration to rats. Cancer Res. 1981;41:1311–1317. | ||
Patel KR, Baldeschwieler JD. Mouse Lewis lung carcinoma and hepatoma ascites treatment by combination of liposome chemotherapy and non-specific immunotherapy. Int J Cancer. 1984;34:717–723. | ||
Leung AW, Kalra J, Santos ND, Bally MB, Anglesio MS. Harnessing the potential of lipid-based nanomedicines for type-specific ovarian cancer treatments. Nanomedicine (Lond). 2014;9:501–522. | ||
Nguyen LV, Vanner R, Dirks P, Eaves CJ. Cancer stem cells: an evolving concept. Nat Rev Cancer. 2012;12:133–143. | ||
Polyak K, Weinberg RA. Transitions between epithelial and mesenchymal states: acquisition of malignant and stem cell traits. Nat Rev Cancer. 2009;9:265–273. | ||
Cojoc M, Peitzsch C, Trautmann F, et al. Emerging targets in cancer management: role of the CXCL12/CXCR4 axis. Onco Targets Ther. 2013;6:1347–1361. | ||
Singh A, Settleman J. EMT, cancer stem cells and drug resistance: an emerging axis of evil in the war on cancer. Oncogene. 2010;29:4741–4751. | ||
Zhao Y, Alakhova DY, Kabanov AV. Can nanomedicines kill cancer stem cells? Adv Drug Deliv Rev. 2013;65:1763–1783. | ||
Elias D, Quenet F, Goere D. Current status and future directions in the treatment of peritoneal dissemination from colorectal carcinoma. Surg Oncol Clin N Am. 2012;21:611–623. | ||
Jacquet P, Averbach A, Stuart OA, Chang D, Sugarbaker PH. Hyperthermic intraperitoneal doxorubicin: pharmacokinetics, metabolism, and tissue distribution in a rat model. Cancer Chemother Pharmacol. 1998;41:147–154. | ||
Yan TD, Black D, Sugarbaker PH, et al. A systematic review and meta-analysis of the randomized controlled trials on adjuvant intraperitoneal chemotherapy for resectable gastric cancer. Ann Surg Oncol. 2007;14:2702–2713. | ||
Yan TD, Black D, Savady R, Sugarbaker PH. A systematic review on the efficacy of cytoreductive surgery and perioperative intraperitoneal chemotherapy for pseudomyxoma peritonei. Ann Surg Oncol. 2007;14:484–492. | ||
Di Miceli D, Alfieri S, Caprino P, et al. Complications related to hyperthermia during hypertermic intraoperative intraperitoneal chemotherapy (HIPEC) treatment. Do they exist? Eur Rev Med Pharmacol Sci. 2012;16:737–742. | ||
Glantz MJ, LaFollette S, Jaeckle KA, et al. Randomized trial of a slow-release versus a standard formulation of cytarabine for the intrathecal treatment of lymphomatous meningitis. J Clin Oncol. 1999;17:3110–3116. | ||
Pasquier E, Kieran MW, Sterba J, et al. Moving forward with metronomic chemotherapy: meeting report of the 2nd International Workshop on Metronomic and Anti-Angiogenic Chemotherapy in Paediatric Oncology. Transl Oncol. 2011;4:203–211. | ||
Pasquier E, Kavallaris M, Andre N. Metronomic chemotherapy: new rationale for new directions. Nat Rev Clin Oncol. 2010;7:455–465. |
Supplementary materials
Side population assay
The cell density was adjusted to 106 cells/mL with Dulbecco’s Modified Eagle’s Medium containing 2% fetal bovine serum and 5 μM Hoechst 33342 (Sigma-Aldrich, St Louis, MO, USA) and incubated for 90 minutes at 37°C. The negative control groups were treated with fumitremorgin C (10μM, Sigma-Aldrich) for 30 minutes at 37°C prior to and during Hoechst 33342 staining. After two washes with phosphate-buffered saline, propidium iodide (2 μg/mL, Sigma-Aldrich) staining was used to exclude dead cells. The cells were kept at 4°C in the dark and then isolated using a BD FACSAria™ flow cytometer (BD Biosciences, San Jose, CA, USA).
Microsphere formation activity
The treated tumors were washed and subjected to enzymatic dissociation. The tumor cells were then resuspended in tumor sphere medium consisting of human recombinant epidermal growth factor (20 ng/mL; Sigma-Aldrich), β-fibroblast growth factor (20 ng/mL; Upstate Biotechnology Inc, Lake Placid, NY, USA), leukemia inhibitory factor (10 ng/mL; Chemicon International Inc, Temecula, CA, USA), neuronal survival factor (1×; Clonetics, Lonza, Walkersville, MD, USA), and N-acetylcysteine (60 μg/mL; Sigma-Aldrich) and plated at a density of 3×106 live cells/60 mm plate. Red blood cells were removed using Lympholyte-M® (Cedarlane Laboratories, Burlington, ON, Canada). The cells were grown under these conditions for 10 days, and the nonadherent spheres that formed were counted.
RT-PCR for stemness marker
The total RNA from the cells was isolated using TRIzol reagent (Invitrogen, Carlsbad, CA, USA) according to the manufacturer’s instructions. The RNA samples were treated with deoxyribonuclease I (Invitrogen). Reverse transcription-polymerase chain reaction (RT-PCR) was performed using the Super-Script First-Strand Synthesis System (Invitrogen), and real-time PCR was performed using the Step One-Plus and SYBR-green system (Applied Biosystems, Warrington, UK). The internal control was 18s RNA. The primer sequences are listed in Table 1.
Annexin V/propidium iodide apoptosis assays
An in vivo xenograft model was used to detect apoptotic cells. The mice were intraperitoneally injected with ES-2 cells on day 0 (2×105/each mouse, n=3). At days 9 and 11, the mice were treated by either intraperitoneal delivery of Nano-taxol or systemic delivery of Taxol®. Six hours after the last treatment, the mice were sacrificed, and the tumors were harvested and made into single-cell suspensions. A total of 106 harvested tumor cells were then stained with 5 μL of Annexin V-fluorescein isothiocyanate and 5 μL of propidium iodide (PI) (5 μg/mL, BD Bioscience) in 1× binding buffer (10 mM HEPES, pH 7.4, 140 mM NaOH, 2.5 mM CaCl2) for 15 minutes at room temperature, and apoptosis was determined using a FACSCalibur flow cytometer. Both early apoptotic (Annexin V+/PI−) and late apoptotic (Annexin V+/PI+) cells were regarded as cell death.
Cytometric detection and quantification of cells undergoing apoptosis, cells dying by necrosis, and cells remaining viable were performed as determined through preliminary studies. The definition of necrotic cells was predetermined using a protocol proposed by Reed and Mixter.1 In brief, Jurkat cells treated with ethanol to induce necrosis provide these control populations.
Hyperthermic intraperitoneal chemotherapy
The open hyperthermic intraperitoneal chemotherapy (HIPEC) method was used because this technique is believed to provide optimal thermal homogeneity and spatial diffusion, and 250 mL of heated saline containing 10 mg/kg Taxol was administered to each animal in the HIPEC group. An inflow catheter was inserted into the upper abdomen between the hepatic and diaphragmatic surface, and an outflow catheter was placed at the pelvic floor. The perfusion solution was heated to 42.0°C±0.5°C and infused into the peritoneal cavity at a rate of 3 mL per minute through the inflow tube introduced from the automatic perfusion pump. The perfusion in the peritoneal cavity was stirred manually to obtain an equal spatial distribution. The temperature of the perfusion solution in the peritoneal space was maintained at 42.0°C±0.5°C and monitored using a thermometer. The total HIPEC time was 60 minutes, and the perfusion solution in the abdominal cavity was then removed.
Bioluminescence imaging
The animals were injected intraperitoneally with D-luciferin (300 mg/kg [0.05 mL/10 g of body weight]) and anesthetized 5 minutes before the peak luciferin uptake time (determined by a preliminary in vitro bioluminescence imaging experiment) with 2%–3% isoflurane. Isoflurane was reduced to 2% after transferring the animals to the imaging chamber. Dorsal and ventral bioluminescence imaging (maximum of five animals) was performed (IVIS®, PerkinElmer Inc, Waltham, MA, USA) at the optimal imaging time.
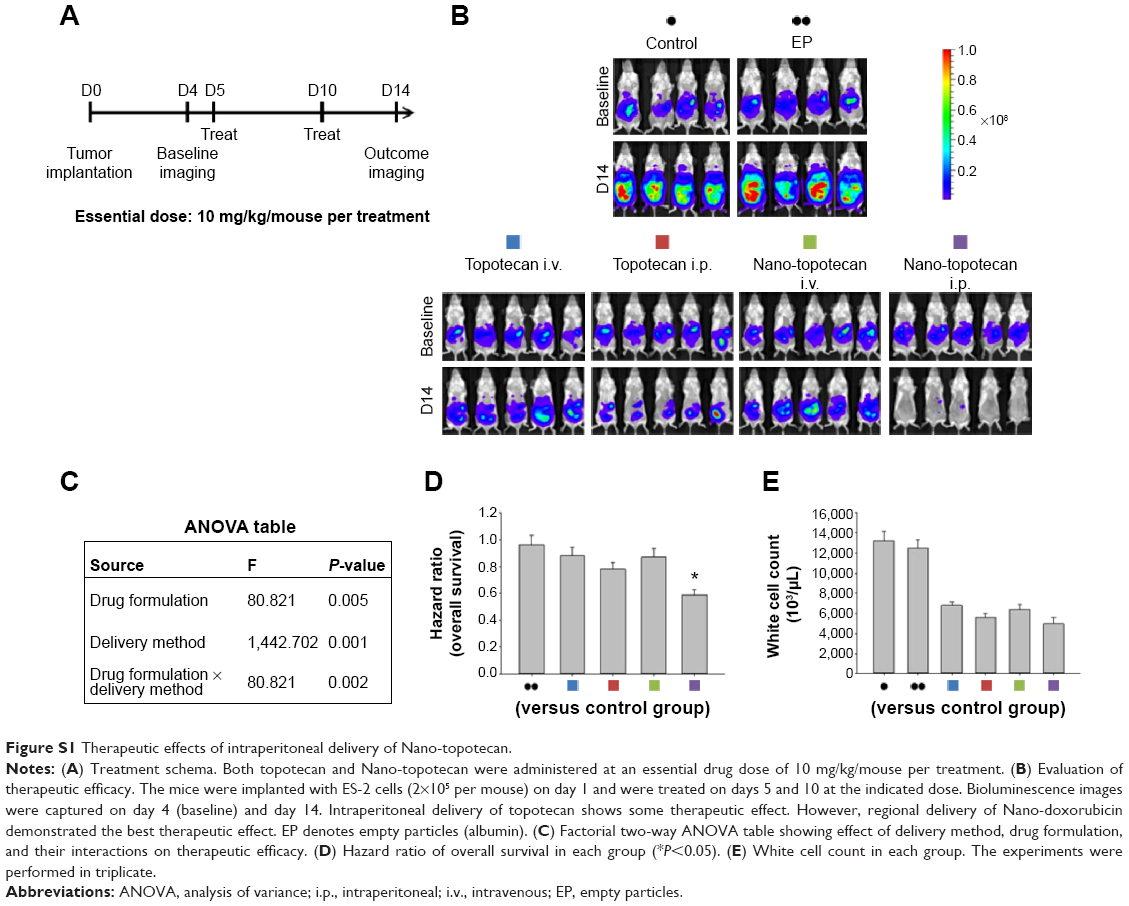 | Figure S1 Therapeutic effects of intraperitoneal delivery of Nano-topotecan. |
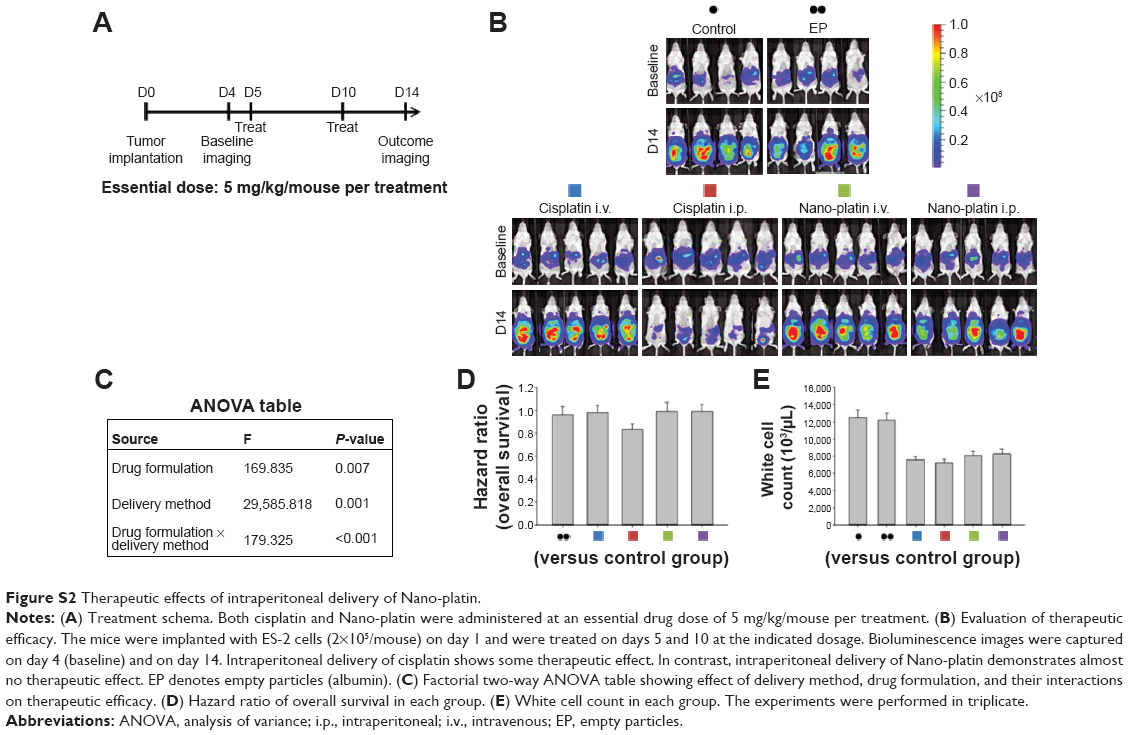 | Figure S2 Therapeutic effects of intraperitoneal delivery of Nano-platin. |
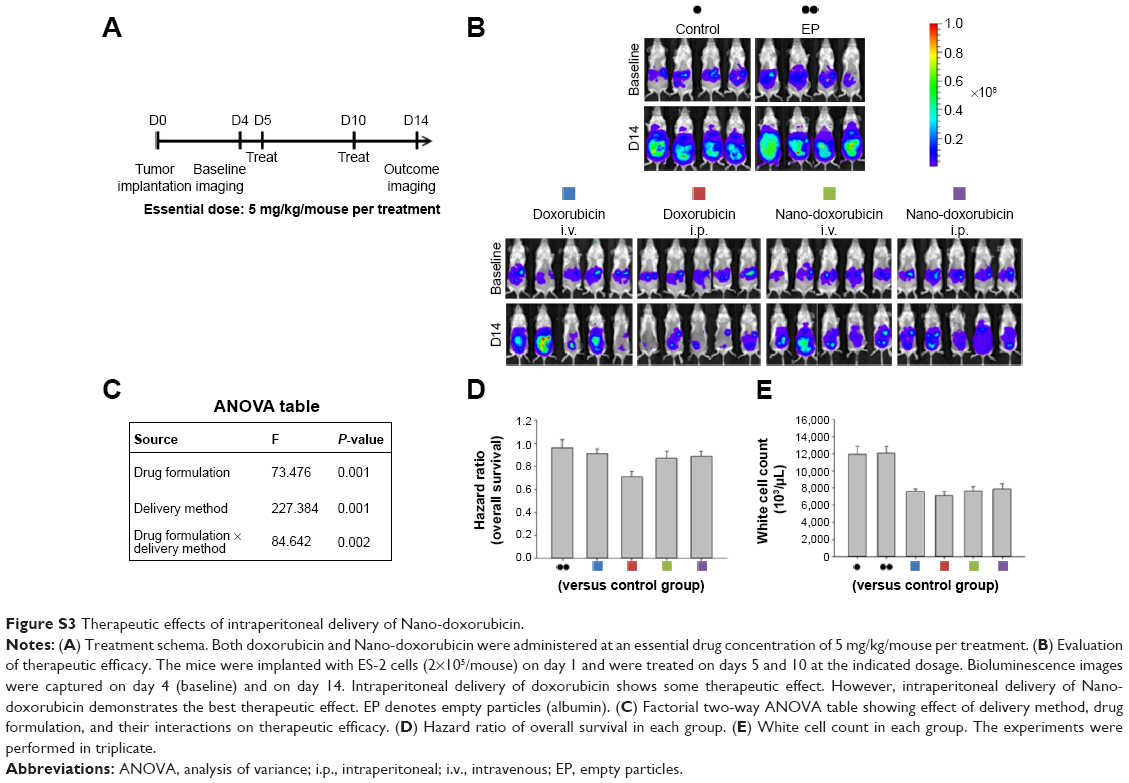 | Figure S3 Therapeutic effects of intraperitoneal delivery of Nano-doxorubicin. |
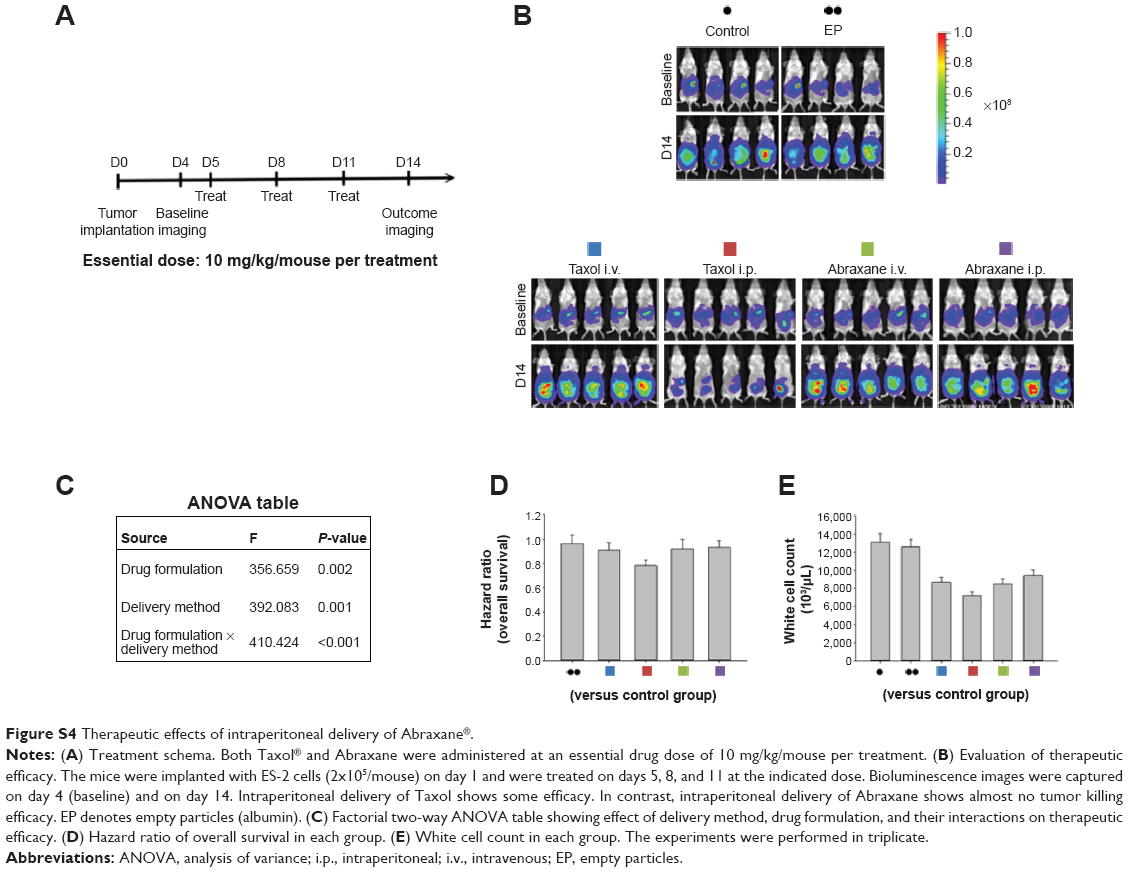 | Figure S4 Therapeutic effects of intraperitoneal delivery of Abraxane®. |
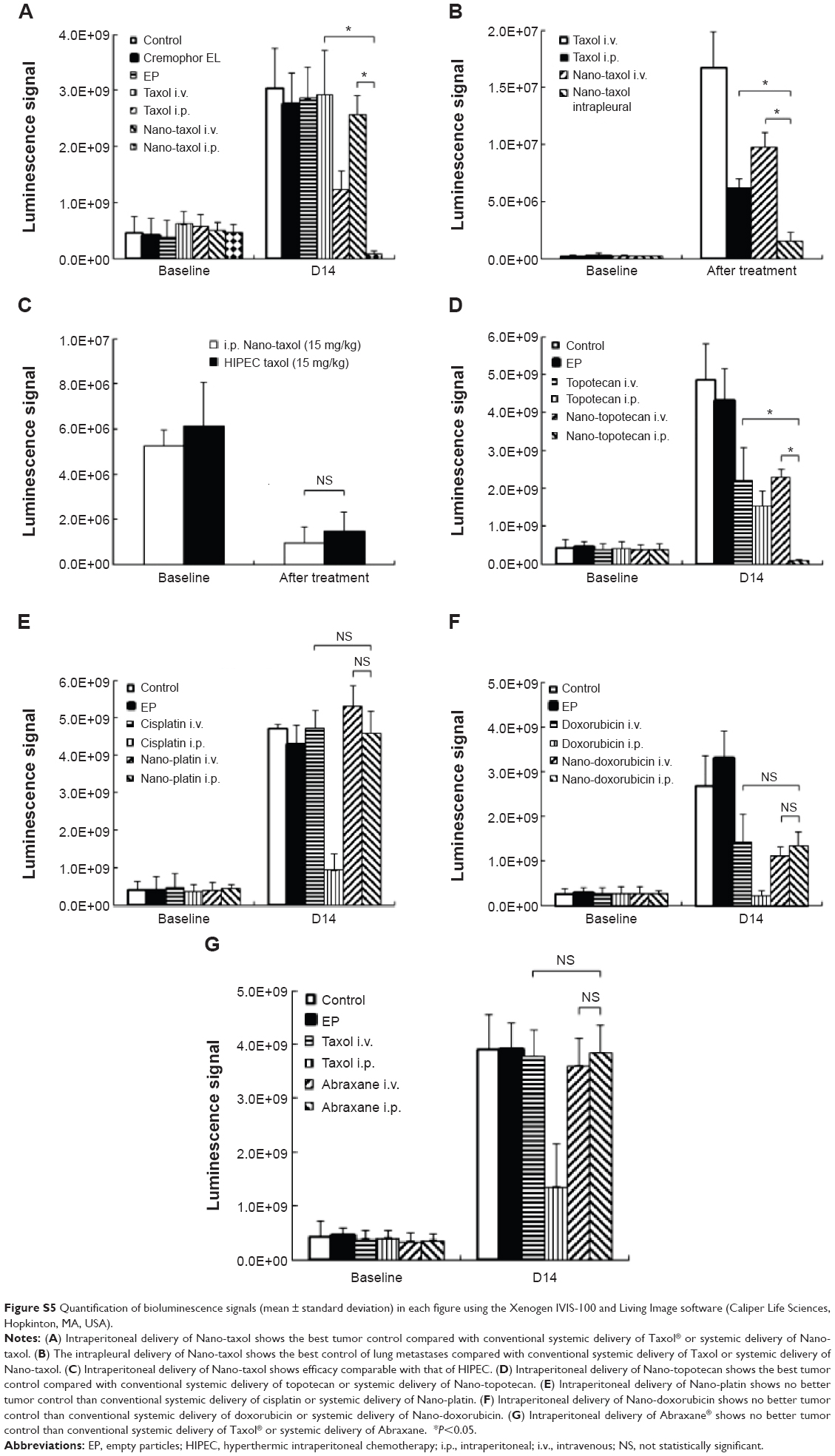 | Figure S5 Quantification of bioluminescence signals (mean ± standard deviation) in each figure using the Xenogen IVIS-100 and Living Image software (Caliper Life Sciences, Hopkinton, MA, USA). |
Reference
Reed CL, Mixter PF. Control samples for Annexin V-FITC vs propidium iodide quantitation of apoptosis and necrosis. Purdue Cytometry CDROM volume 4. Available from http://www.cyto.purdue.edu/cdroms/cyto4/15_apop/data/reed.htm |
 © 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms.php
and incorporate the Creative Commons Attribution
- Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms.php
and incorporate the Creative Commons Attribution
- Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.