")
Back to Journals » OncoTargets and Therapy » Volume 11
Butein activates p53 in hepatocellular carcinoma cells via blocking MDM2-mediated ubiquitination
Authors Zhou Y, Wang K, Zhou N, Huang T, Zhu J, Li J
Received 17 December 2017
Accepted for publication 9 February 2018
Published 6 April 2018 Volume 2018:11 Pages 2007—2015
DOI https://doi.org/10.2147/OTT.S160119
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Yao Dai
Yuanfeng Zhou,1,2 Kuifeng Wang,2 Ni Zhou,2 Tingting Huang,2 Jiansheng Zhu,2 Jicheng Li1
1Institute of Cell Biology, Zhejiang University, Hangzhou, People’s Republic of China; 2Department of Infectious Diseases, Affiliated Taizhou Hospital of Wenzhou Medical University, Taizhou, People’s Republic of China
Introduction: In this study, we aimed to investigate the effect of butein on p53 in hepatocellular carcinoma (HCC) cells and the related molecular mechanisms by which p53 was activated.
Methods: MTS assay and clonogenic survival assay were used to examine the antitumor activity of butein in vitro. Reporter gene assay was adopted to evaluate p53 transcriptional activity. Flow cytometry and western blotting were performed to study apoptosis induction and protein expression respectively. Xenograft model was applied to determine the in vivo efficacy and the expression of p53 in tumor tissue was detected by immunohistochemistry.
Results: HCC cell proliferation and clonogenic survival were significantly inhibited after butein treatment. With the activation of cleaved-PARP and capsase-3, butein induced apoptosis in HCC cells in a dose-dependent manner. The transcriptional activity of p53 was substantially promoted by butein, and the expression of p53-targeted gene was increased accordingly. Mechanism studies demonstrated that the interaction between MDM2 and p53 was blocked by butein and MDM2-mediated p53 ubiquitination was substantially decreased. Short-hairpin RNA experiment results showed that the sensitivity of HCC cells to butein was substantially impaired after p53 was knocked down and butein-induced apoptosis was dramatically decreased. In vivo experiments validated substantial antitumor efficacy of butein against HepG2 xenograft growth, and the expression of p53 in butein-treated tumor tissue was significantly increased.
Conclusion: Butein demonstrated potent antitumor activities in HCC by activating p53, and butein or its analogs had therapeutic potential for HCC management.
Keywords: butein, cell apoptosis, hepatocellular carcinoma, MDM2, natural product, p53, ubiquitination
Introduction
Genomic instability, a tendency of gene alteration during the life cycle of cells, is considered to be the major driving force of tumorigenesis.1 The genes engaged in the regulation of cell cycle arrest and DNA damage repair are considered to have an important role to maintain the stability of whole genome.2 p53, a transcription factor which is often activated when cells are in various stressed conditions, has a critical role in keeping genome integrity. Owing to the importance, so far, the TP53 gene mutation has been identified in over half of the human cancers.3 According to the different effects on p53 function, the gene mutations can be divided into contact mutations and structural mutations. The contact mutations affect the binding between p53 and DNA and give rise to the loss of its transcriptional activity, while the structural mutations can cause the instability of local structure of the p53 core domain.4–6
In addition to the gene mutation, p53 activity is also mediated by multiple posttranslational modifications. The well-known mechanism to inactivate p53 is via overexpression of its negative regulators, such as MDM2. MDM2 amplification or overexpression has been identified in various cancers, including gastric cancer, breast cancer, and prostate cancer, and is closely related to p53 inactivation.7–9 MDM2 mediates p53 activity by two distinct mechanisms: directly suppressing p53 transcriptional activity via binding to the N-terminal, or promoting its ubiquitination and resulting in p53 degradation. Under normal physiological conditions, owing to the continuous degradation mediated by MDM2, the half-life of p53 is very short and it is maintained at a low level. Besides MDM2, other ubiquitin ligases such as RIPH2, COP1, and CHIP have also been identified as negative regulators of p53.10 Apart from ubiquitination, phosphorylation, acetylation, sumoylation, or methylation of p53 has been demonstrated to promote p53 stabilization and activation.11 Once p53 is activated, expressions of genes involved in cell cycle mediation, cell apoptosis or senescence induction, or tumor angiogenesis are induced by p53.12
Butein, a chalconoid isolated from Rhus verniciflua Stokes, has been used as a food additive for a long time in Southeast Asia.13 Butein has also been shown to exhibit promising therapeutic efficacy in chronic diseases, such as inflammation, glaucoma, and cardiovascular diseases.14–16 Recently, increasing evidence has demonstrated that butein could exert substantial antitumor activity against various cancers.17–20 As a multitargeted compound, butein was found to demonstrate inhibitory effects on several important signaling pathways in cancer cells, including EGFR, NF-κB, PI3K/AKT/mTOR, and STAT3 pathways.20–22
In the present study, we investigated the antitumor potency as well as the underlying mechanism of butein in hepatocellular carcinoma (HCC). Butein had shown profound efficacy against HCC cells in vitro, and p53 was significantly stabilized and activated after butein treatment. The activation of p53 by butein was mainly attributed to the blockade of the interaction between p53 and MDM2, and the suppression of MDM2-mediated p53 degradation. Further investigations revealed that butein exerted its function in a p53-dependent manner. Our study suggested that butein, or its analog, may have therapeutic potential in HCC.
Materials and methods
Cell lines and reagents
The SMMC-7721 and HepG2 cells were purchased from the Cell Bank of Chinese Academy of Sciences (Shanghai, People’s Republic of China) and the American Type Culture Collection (Manassas, VA, USA), respectively, and cultured following the protocols provided by supplier. Butein was a product of Sigma-Aldrich (St Louis, MO, USA). The primary anti-cleaved-PARP, cleaved-caspase3, ubiquitin, p53, Bax, MDM2, and Ki67 antibodies and the secondary goat anti-rabbit IgG-horseradish peroxidase (HRP) were from Cell Signaling Technology (Beverly, MA, USA). Lentivirus plasmid (pLKO.1-shp53, TRCN0000003753) was purchased from Thermo Scientific (Huntsville, AL, USA). LipofectamineTM 2000 was from Invitrogen (Carlsbad, CA, USA).
Cell proliferation assay
SMMC-7721 or HepG2 cells were seeded into 96-well plates with appropriate cell density and then treated with different concentrations of butein (0, 15, 30, and 60 μM). At different time points (24, 48, and 72 h), the cell viability was determined with the MTS assay kit (Promega, Madison, WI, USA).
Clonogenic survival assay
After treatment with butein for 24 h, SMMC-7721 or HepG2 cell suspensions (1×103 cells/well) were plated into a 6-well plate and cultured for about 1–2 weeks until visible colonies (≥50 cells) were formed. The colonies were fixed with 4% paraformaldehyde and stained with 1% crystal violet, and then the number of colonies was counted.
Flow cytometry
After treatment with butein, HepG2 cells were harvested and centrifuged at 2,000 rpm for 5 min and then the cell pellets were washed and resuspended with cold PBS solution. 5 μL Annexin V–fluorescein isothiocyanate (FITC) solution was incubated with the cell suspension for 10–15 min avoiding light, then 10 μL propidium iodide (PI) was added and the mixture was incubated for another 5 min; the stained cells were subjected to flow cytometry analysis after 1 h.
Western blot analysis
The cells were lysed with radioimmunoprecipitation assay (RIPA) buffer (Beyotime Biotechnology, Jiangsu, People’s Republic of China) on ice and the lysates were collected; then the proteins in cell lysates were quantified with the BCA Protein Assay Kit (Beyotime Biotechnology). After resolving the proteins with sodium dodecyl sulfate–polyacrylamide gel electrophoresis (SDS-PAGE), the proteins were transferred onto polyvinylidene difluoride membranes. After blocking with 5% nonfat milk, the membranes were incubated with corresponding primary antibodies at 4°C overnight. By hybridizing with HRP-conjugated secondary antibodies, the bands on the membranes were visualized with an enhanced Western lightning plus-ECL kit (#32132; PIERCE, Rockford, IL, USA).
Immunoprecipitation
For MDM2–p53 interaction experiments, after treatment with butein the HepG2 cells in 100 mm dish were lysed with CO-IP lysis buffer (#87787; Thermo Scientific) containing protease inhibitors. 2 μg p53 antibodies and 100 μL protein A/G agarose beads (Amersham Biosciences AB, Buckinghamshire, UK) were added into the lysates and incubated at 4°C overnight, and then the beads were collected by centrifugation. The precipitated proteins on the beads were washed at least three times and mixed with the loading buffer, boiled at 100°C for 5 min, subjected to SDS-PAGE and then probed with anti-MDM2 antibody. For p53 ubiquitination analysis, the HepG2 cells in the 100 mm dish were treated with butein and lysed with RIPA lysis buffer (#89900; Thermo Scientific) containing protease inhibitors; appropriate p53 antibodies and 100 μL protein A/G agarose beads (Amersham Biosciences AB) were added into the lysates and incubated at 4°C overnight, then the beads were collected by centrifugation. The precipitated proteins on the beads were washed at least three times and mixed with the loading buffer, boiled at 100°C for 5 min, and subjected to SDS-PAGE, and p53 ubiquitination was detected with an anti-ubiquitin antibody.
Luciferase reporter assay
The p53 firefly luciferase reporter plasmid (pp53-TA-luc; Beyotime Biotechnology) and the internal control Renilla luciferase reporter plasmid (Promega) were cotransfected into HepG2 using Lipofectamine 2000 according to supplier’s directions. After transfection, the transfected cells were treated butein for 24 h and the luciferase activities in cell lysates were determined with the Dual Luciferase Reporter Assay kit (Promega).
Lentiviral infection
pLKO.1-shp53 was cotransfected with PSPAX2 and PMD2-G into 293T cells using Lipofectamine 2000 reagent. Fourty-eight hours after transfection, viral fractions in the supernatant were collected by centrifugation. Next, the virus was infected into HepG2 cells which were at 80% confluence and were growing in 10 cm dish along with 10 μg/mL polybrene. Twenty-four hours later, fresh medium with puromycin was added. After the cells with no lentiviral infection were completely dead, p53-short-hairpin RNA (shRNA) cells were used for further studies.
In vivo xenograft experiment
The animal experiments were approved by Wenzhou Medical University Animal Care and Use Committee. Balb/c nude mice were used in the in vivo experiment, and all procedures were performed based on the guidelines approved by Wenzhou Medical University Animal Care and Use Committee. The mice with HepG2 xenografts were randomly assigned to groups when the tumor volume was about 50–100 mm3; the vehicle group was dosed with 0.2 mL 0.5% carboxymethylcellulose (the vehicle), and the treatment group was administered 5 mg/kg butein (butein was dissolved with 0.5% carboxymethylcellulose and 10% polyethylene glycol 400) three times per week by intraperitoneal injection. The tumor volume and mice body weight were recorded twice per week.
Immunohistochemical staining
Tumor tissues embedded in paraffin were dewaxed in xylene and hydrated in ethanol. By placing the slides in boiling sodium citrate buffer (10 mmol/L, pH 6.0) for 5 min, the antigen was retrieved. The endogenous peroxidase activity in tumor tissues was blocked with 3% H2O2. After incubation with goat serum to block the unspecific binding sites, the slides were incubated with anti-p53 or -Ki67 antibodies at 4°C overnight. Biotinylated goat anti-rabbit IgG antibodies were added at a 1:100 dilution and incubated. Finally, the slides were incubated with HRP-conjugated streptavidin and then developed with DAB solution. After counterstaining with Harris’ hematoxylin, the sections were dehydrated and mounted. The expression of p53 or Ki67 in tumor tissue was analyzed using Image-Pro Plus software (version 6.2; Media Cybernetics, Rockville, MD, USA).
Statistical analysis
Data are presented as mean ± SD in this study, and the statistical difference was analyzed using the Student’s t-test or one-way analysis of variance (SPSS software, version 13.0; SPSS, Chicago, IL, USA). p<0.05 indicated a significant difference.
Results
Butein suppressed HCC cell proliferation and clonogenic survival
First, the antitumor activity of butein against HCC cell proliferation was examined by MTS assay, and the results demonstrated that HCC proliferation was inhibited significantly by butein in a time- and dose-dependent manner. At 60 μM, the inhibition rate reached 43.8% and 54.7% in SMMC-7721 and HepG2, respectively (Figure 1A). In addition to proliferation inhibition, we also investigated the effect of butein on HCC cells by clonogenic survival assay, which is regarded as the “gold standard” cellular sensitivity assay. After butein treatment, the number of colonies was substantially decreased. At 60 μM, few colonies were observed in contrast with the control (Figure 1B).
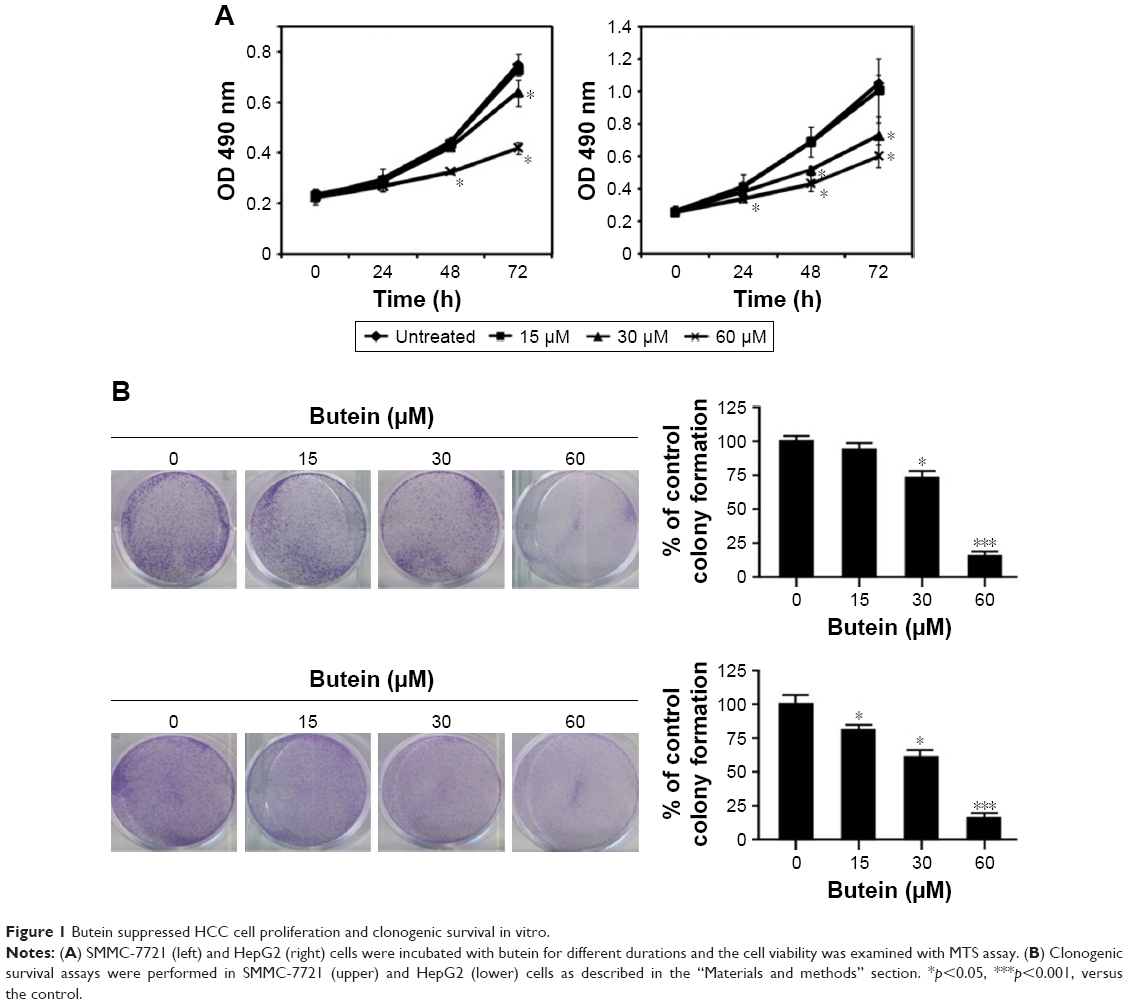 | Figure 1 Butein suppressed HCC cell proliferation and clonogenic survival in vitro. |
Butein induced apoptosis in HepG2 cells
To confirm cell apoptosis induction by butein, after the treatment with butein for 24 h, HepG2 cells were stained with Annexin V–FITC and PI to detect apoptotic status. As shown in Figure 2A, butein dramatically induced apoptosis in HepG2 cells in a dose-dependent manner (>20% of HepG2 cells undergone apoptosis at 60 μM). Moreover, the effect of butein on protein involved in apoptosis induction was also examined. Western blotting results showed that cleaved PAPR and caspase-3, which are two important biomarkers to indicate apoptosis, were substantially increased (Figure 2B). This further demonstrated that HCC cells were subjected to apoptosis after butein treatment.
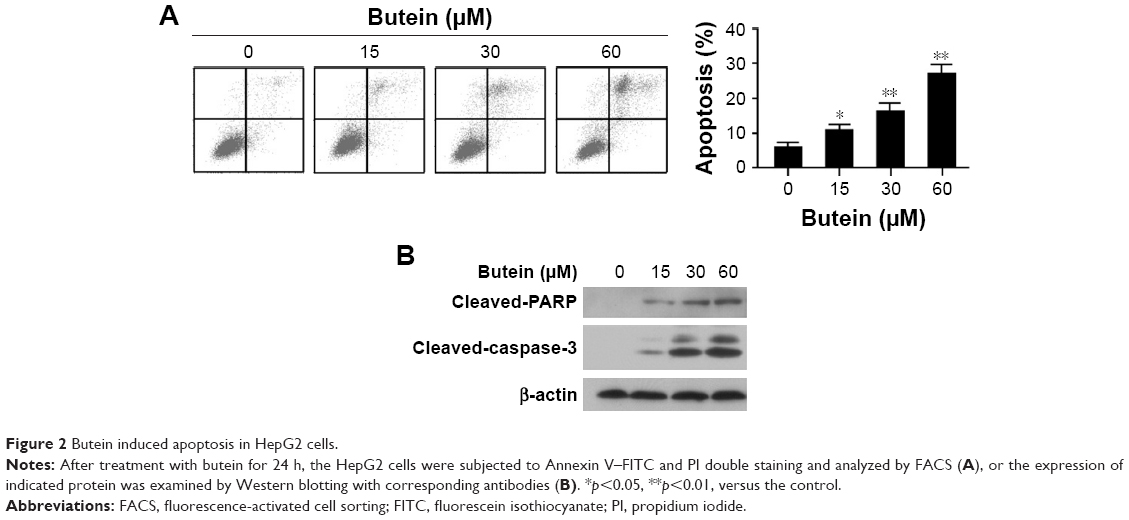 | Figure 2 Butein induced apoptosis in HepG2 cells. |
Butein activated p53 transcriptional activity
To investigate the effect of butein on p53, firefly luciferase pp53-TA-luc reporter plasmid was transfected into HepG2 cells. Given luciferase activities were tied closely to p53 transcriptional activity, the luciferase activities were used to examine the effect of butein on p53 transcriptional activity. As demonstrated in Figure 3A, the luciferase activities were substantially increased in a dose-dependent fashion and the activities were promoted to nearly 6-fold after 30 μM butein treatment, suggesting that the transcriptional activity of p53 was significantly increased. Moreover, we assessed the expression of Bax, which is an important target gene of p53 for apoptosis mediation. After butein treatment, the expressions of p53 and Bax were significantly increased, confirming that p53 transcriptional activity was significantly promoted by butein (Figure 3B).
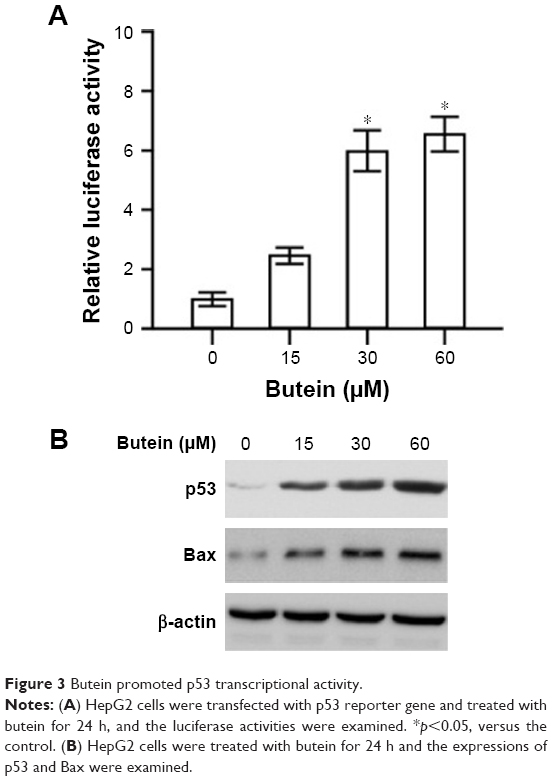 | Figure 3 Butein promoted p53 transcriptional activity. |
Butein suppressed MDM2-mediated p53 ubiquitination
MDM2-mediated ubiquitination is an important posttranslational modification of p53, and could result in p53 degradation. Our results showed that p53 was increased after butein treatment; so we further investigated whether butein had an effect on MDM2-mediated p53 ubiquitination. As shown in Figure 4A, butein blocked the interaction between MDM2 and p53. After butein treatment, MDM2 binding to p53 was significantly decreased. Due to the blockade of interaction between MDM2 and p53, p53 ubiquitination was decreased in a dose-dependent manner (Figure 4B), which led to p53 activation.
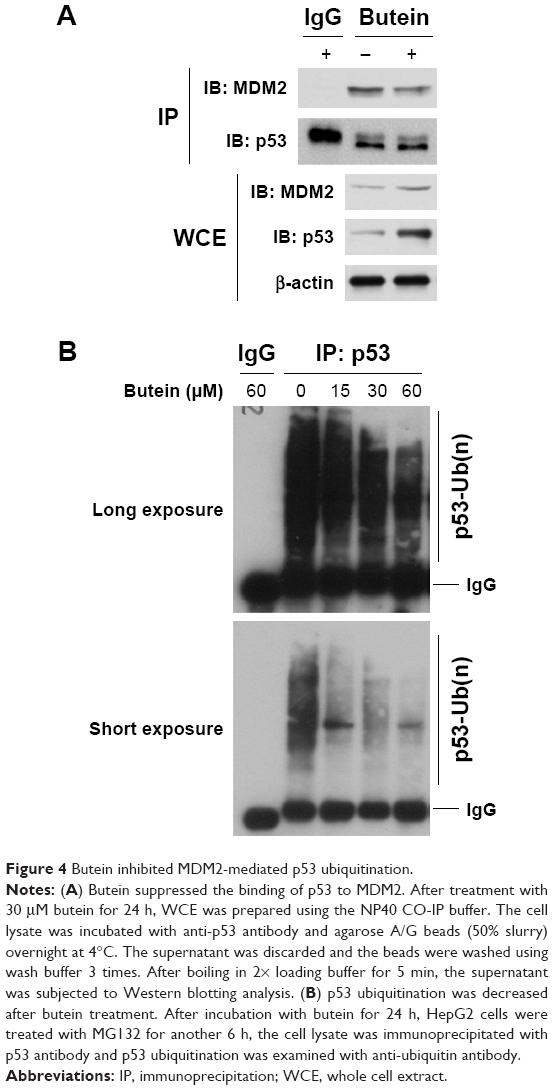 | Figure 4 Butein inhibited MDM2-mediated p53 ubiquitination. |
p53 knockdown impaired the sensitivity to butein
To confirm the importance of p53 in butein-mediated antitumor activity, shRNA was utilized to silence p53 expression in HepG2 cells. As shown in Figure 5B, in p53-knockdown cells, butein-induced apoptosis was dramatically attenuated. In the shGFP group, more than 20% of the cells were subjected to apoptosis by butein; however, cell apoptosis was significantly decreased in p53-knockdown cells. Western blotting results further validated that after p53 was silenced the increase in the level of Bax, as well as that of cleaved-PARP and caspase-3, was substantially impaired (Figure 5A).
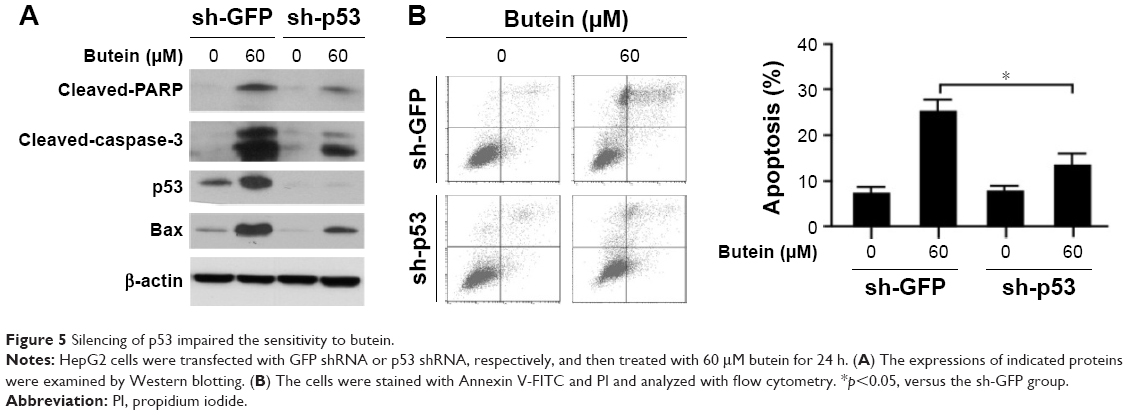 | Figure 5 Silencing of p53 impaired the sensitivity to butein. |
Butein suppressed HepG2 xenograft growth
Finally, the antitumor potency of butein was determined in HepG2 xenograft model in vivo, and the results demonstrated that butein substantially suppressed tumor growth (Figure 6A–C). The volume of tumor in the vehicle group reached about 700 mm3, but in the butein-treated group, the average tumor volume was reduced to 300 mm3. Moreover, the average tumor weights between these two groups were significantly different (0.68 g vs 0.29 g, p<0.05). Also, no obvious toxicity was detected in the butein-treated group; the average body weights in both vehicle- and butein-treated group were equal (Figure 6D). We further detected p53 expression in tumor tissue. Immunohistochemical staining demonstrated that p53 expression was remarkably increased in butein-treated tissue (Figure 6E). Meanwhile, the level of Ki67, a proliferation maker, was significantly decreased, indicating that with p53 activation, the proliferation potential of HepG2 cells was weakened.
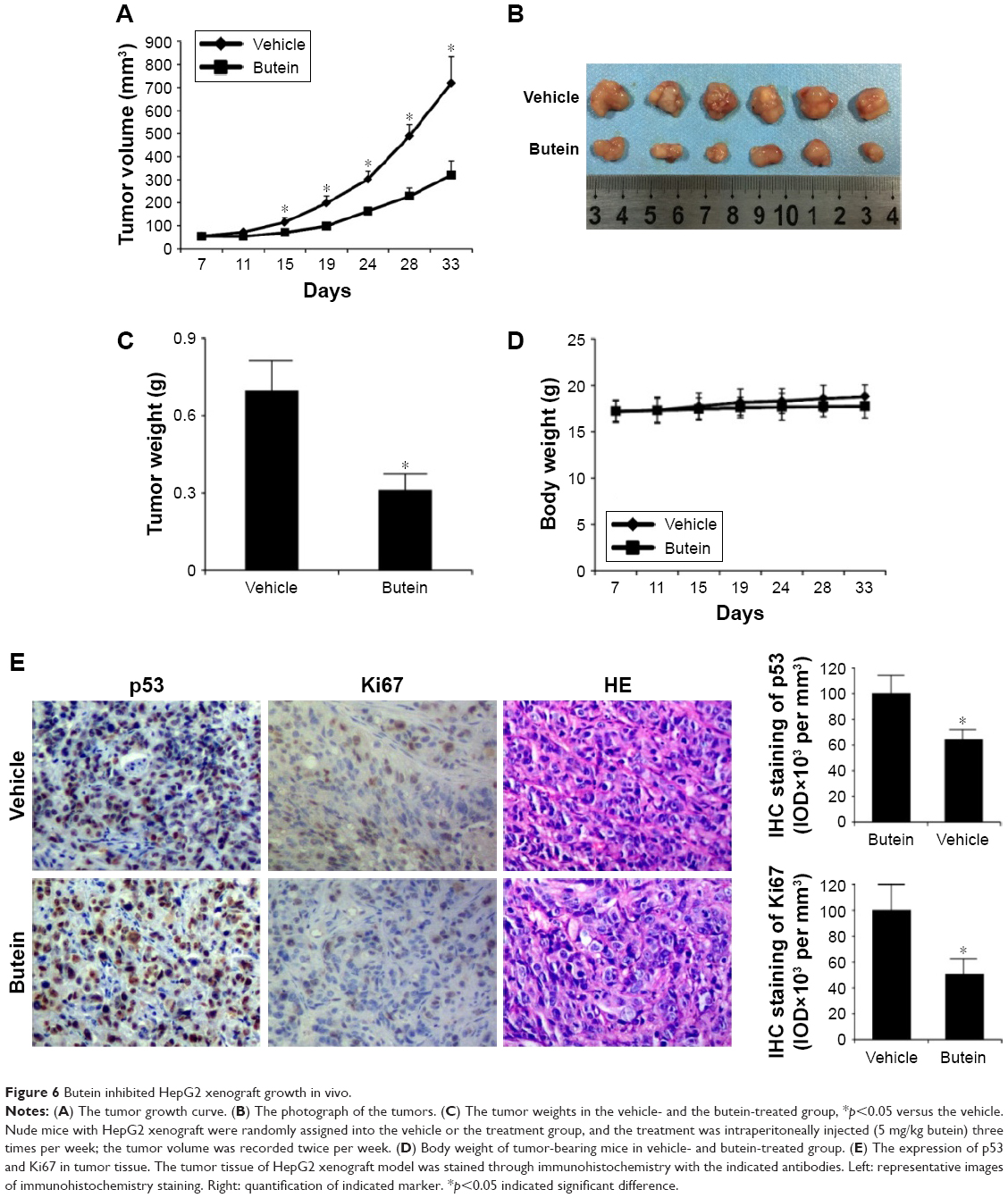 | Figure 6 Butein inhibited HepG2 xenograft growth in vivo. |
Discussion
HCC is the cancer that ranks second in terms of mortality rate and is a great threat to public health.23 Due to the increasing prevalence of this disease, the development of novel therapeutics is in urgent demand. Leaving aside the chemical entities obtained from rational design, natural compounds have also demonstrated great potential in cancer prevention and therapy.24 Through the analysis of the failure of several phase III trials in HCC, drug safety was identified to be an important factor.25 In terms of drug safety, owing to butein being used as food additive, it may have an advantage. The in vivo experimental results also confirmed that no obvious toxicity was observed in butein-treated mice (Figure 6D).
It has been demonstrated previously that butein administration could induce cell cycle arrest at G2/M phase, trigger reactive oxygen species generation, and increase mitochondrial permeability.26–28 All these stress signals would result in the activation of p53. As demonstrated in Figures 3 and 6E, p53 expression was significantly increased after butein treatment. Meanwhile, the transcriptional activity of p53 was substantially enhanced. With the improvement in p53 activity, Bax, which is the main downstream target of p53, was also significantly increased. As an important protein involved in cell apoptosis regulation, Bax elevation led to mitochondrial depolarization, cytochrome C release, and induction of cell apoptosis. Further investigations validated that butein-induced apoptosis was closely correlated with the p53 activation, as evidenced by the significantly impaired apoptosis in p53-knockdown HCC cells (Figure 5). In chronic myeloid leukemia, Woo et al29 also observed that butein had shown more potent antitumor activity in p53 wild-type cell then in p53-null cell.
Abnormalities of p53–MDM2 pathway, such as TP53 gene mutations and MDM2 overexpression, are common in HCC, and the restoration of p53 was proven to be effective to trigger tumor clearance.30,31 Different strategies, including disruption of the MDM2–p53 interaction, stabilization of active p53 conformation, and delivery of exogenous therapeutic wild-type p53 were applied to restore p53 function.32 Nutlin, which has been proven to possess antitumor activity against different types of cancer cells, was discovered as the first MDM2 inhibitor that can displace MDM2–p53 binding, causing the stabilization and accumulation of p53.33 So far, except Nutlin and its derivative RG7112, other MDM2 antagonists such as MK-8242 and RO5503781, are also in clinical studies.34 p53 expression was substantially increased after butein treatment (Figures 3 and 4A), owing to the fact that p53 can bind to the specific response elements of MDM2 promoter region (the autoregulatory feedback loop), and thus the increase of p53 would enhance MDM2 transcription. In our study, the increase of MDM2 expression was observed after butein treatment (Figure 4A). However, the increase of MDM2 did not give rise to elevation of p53 ubiquitination; on the contrary, p53 ubiquitination was decreased in a dose-dependent manner by butein, proving that the interaction between MDM2 and p53 was blocked by butein. Therefore, we proposed that the activation of p53 was mainly attributed to the suppression of the interaction between p53 and MDM2. In addition to ubiquitination, other modifications such as phosphorylation and acetylation are also involved in the regulation of p53 stability. It has been reported previously that phosphorylation of p53 at a specific site could free p53 from MDM2.11 The multiple effects of butein in cancer cells would result in stress signals that lead to the induction of p53 posttranslational modifications. Therefore, further studies are needed to elaborate whether butein blocked the interaction between p53 and MDM2 via directly binding to MDM2 or p53, or in an indirect way by inducing other modifications.
Conclusion
Our study demonstrated that butein could display significant antitumor activity against HCC cells by stabilizing and activating p53 function. Investigations of the mechanism revealed that activation of p53 was mainly attributed to the blockade of p53 binding to MDM2 and the suppression of MDM2-mediated p53 ubiquitination. Our study disclosed a novel mechanism by which butein displayed its antitumor activity, and hence we suggest that butein or its analogs have therapeutic potential in HCC.
Acknowledgment
This work was funded by the Zhejiang Province Major Science and Technology Programs (No. 2012C13018-3) and Taizhou Postdoctoral Programs (No. 201602).
Disclosure
The authors report no conflicts of interest in this work.
References
Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144:646–674. | ||
Beishline K, Azizkhan-Clifford J. Interplay between the cell cycle and double-strand break response in mammalian cells. Methods Mol Biol. 2014;1170:41–59. | ||
Muller PA, Vousden KH. p53 mutations in cancer. Nat Cell Biol. 2013;15:2–8. | ||
Bullock AN, Fersht AR. Rescuing the function of mutant p53. Nat Rev Cancer. 2001;1:68–76. | ||
Eldar A, Rozenberg H, Diskin-Posner Y, Rohs R, Shakked Z. Structural studies of p53 inactivation by DNA-contact mutations and its rescue by suppressor mutations via alternative protein-DNA interactions. Nucleic Acids Res. 2013;41:8748–8759. | ||
Rivlin N, Brosh R, Oren M, Rotter V. Mutations in the p53 tumor suppressor gene: important milestones at the various steps of tumorigenesis. Genes Cancer. 2011;2:466–474. | ||
Ye Y, Li X, Yang J, et al. MDM2 is a useful prognostic biomarker for resectable gastric cancer. Cancer Sci. 2013;104:590–598. | ||
Hori M, Shimazaki J, Inagawa S, Itabashi M, Hori M. Overexpression of MDM2 oncoprotein correlates with possession of estrogen receptor alpha and lack of MDM2 mRNA splice variants in human breast cancer. Breast Cancer Res Treat. 2002;71:77–83. | ||
Khor LY, Bae K, Paulus R, et al. MDM2 and Ki-67 predict for distant metastasis and mortality in men treated with radiotherapy and androgen deprivation for prostate cancer: RTOG 92-02. J Clin Oncol. 2009;27:3177–3184. | ||
Hock AK, Vousden KH. The role of ubiquitin modification in the regulation of p53. Biochim Biophys Acta. 2014;1843:137–149. | ||
Gu B, Zhu WG. Surf the post-translational modification network of p53 regulation. Int J Biol Sci. 2012;8:672–684. | ||
Briest F, Grabowski P. The p53 network as therapeutic target in gastroenteropancreatic neuroendocrine neoplasms. Cancer Treat Rev. 2015;41:423–430. | ||
Song NJ, Yoon HJ, Kim KH, et al. Butein is a novel anti-adipogenic compound. J Lipid Res. 2013;54:1385–1396. | ||
Yadav VR, Prasad S, Sung B, Aggarwal BB. The role of chalcones in suppression of NF-kappaB-mediated inflammation and cancer. Int Immunopharmacol. 2011;11:295–309. | ||
Choi SJ, Lee MY, Jo H, Lim SS, Jung SH. Preparative isolation and purification of neuroprotective compounds from Rhus verniciflua by high speed counter-current chromatography. Biol Pharm Bull. 2012;35:559–567. | ||
Lee JH, Kim M, Chang KH, et al. Antiplatelet effects of Rhus verniciflua stokes heartwood and its active constituents – fisetin, butein, and sulfuretin – in rats. J Med Food. 2015;18:21–30. | ||
Cho SG, Woo SM, Ko SG. Butein suppresses breast cancer growth by reducing a production of intracellular reactive oxygen species. J Exp Clin Cancer Res. 2014;33:51. | ||
Huang YT, Lin CI, Chien PH, Tang TT, Lin J, Chao JI. The depletion of securin enhances butein-induced apoptosis and tumor inhibition in human colorectal cancer. Chem Biol Interact. 2014;220:41–50. | ||
Pandey MK, Sung B, Ahn KS, Aggarwal BB. Butein suppresses constitutive and inducible signal transducer and activator of transcription (STAT) 3 activation and STAT3-regulated gene products through the induction of a protein tyrosine phosphatase SHP-1. Mol Pharmacol. 2009;75:525–533. | ||
Khan N, Adhami VM, Afaq F, Mukhtar H. Butein induces apoptosis and inhibits prostate tumor growth in vitro and in vivo. Antioxid Redox Signal. 2012;16:1195–1204. | ||
Jung SK, Lee MH, Lim DY, et al. Butein, a novel dual inhibitor of MET and EGFR, overcomes gefitinib-resistant lung cancer growth. Mol Carcinog. 2015;54:322–331. | ||
Rajendran P, Ong TH, Chen L, et al. Suppression of signal transducer and activator of transcription 3 activation by butein inhibits growth of human hepatocellular carcinoma in vivo. Clin Cancer Res. 2011;17:1425–1439. | ||
Torre LA, Bray F, Siegel RL, Ferlay J, Lortet-Tieulent J, Jemal A. Global cancer statistics, 2012. CA Cancer J Clin. 2015;65:87–108. | ||
Kaulmann A, Bohn T. Carotenoids, inflammation, and oxidative stress – implications of cellular signaling pathways and relation to chronic disease prevention. Nutr Res. 2014;34:907–929. | ||
Llovet JM, Hernandez-Gea V. Hepatocellular carcinoma: reasons for Phase III failure and novel perspectives on trial design. Clin Cancer Res. 2014;20:2072–2079. | ||
Moon DO, Kim MO, Choi YH, Hyun JW, Chang WY, Kim GY. Butein induces G(2)/M phase arrest and apoptosis in human hepatoma cancer cells through ROS generation. Cancer Lett. 2010;288:204–213. | ||
Yang LH, Ho YJ, Lin JF, Yeh CW, Kao SH, Hsu LS. Butein inhibits the proliferation of breast cancer cells through generation of reactive oxygen species and modulation of ERK and p38 activities. Mol Med Rep. 2012;6:1126–1132. | ||
Cui Z, Song E, Hu DN, Chen M, Rosen R, McCormick SA. Butein induces apoptosis in human uveal melanoma cells through mitochondrial apoptosis pathway. Curr Eye Res. 2012;37:730–739. | ||
Woo SM, Choi YK, Kim AJ, Cho SG, Ko SG. p53 causes butein-mediated apoptosis of chronic myeloid leukemia cells. Mol Med Rep. 2016;13:1091–1096. | ||
Nishida N, Kudo M. Recent advancements in comprehensive genetic analyses for human hepatocellular carcinoma. Oncology. 2013;84 (Suppl 1):93–97. | ||
Xue W, Zender L, Miething C, et al. Senescence and tumour clearance is triggered by p53 restoration in murine liver carcinomas. Nature. 2007;445:656–660. | ||
Xiao X, He Q, Huang K. Novel amino-modified silica nanoparticles as efficient vector for hepatocellular carcinoma gene therapy. Med Oncol. 2010;27:1200–1207. | ||
Vassilev LT, Vu BT, Graves B, et al. In vivo activation of the p53 pathway by small-molecule antagonists of MDM2. Science. 2004;303:844–848. | ||
Zhang Q, Zeng SX, Lu H. Targeting p53-MDM2-MDMX loop for cancer therapy. Subcell Biochem. 2014;85:281–319. |
 © 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.