Back to Journals » Drug Design, Development and Therapy » Volume 8
Buccal acetaminophen provides fast analgesia: two randomized clinical trials in healthy volunteers
Authors Pickering G, Macian N, Libert F, Cardot J, Coissard S, Perovitch P, Maury M, Dubray C
Received 4 March 2014
Accepted for publication 9 April 2014
Published 26 September 2014 Volume 2014:8 Pages 1621—1627
DOI https://doi.org/10.2147/DDDT.S63476
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Gisèle Pickering,1–3 Nicolas Macian,1 Frédéric Libert,2,4 J Michel Cardot,1 Séverine Coissard,1 Philippe Perovitch,5 Marc Maury,5 Claude Dubray1–3
1CHU Clermont-Ferrand, Centre de Pharmacologie Clinique, Clermont-Ferrand, France; 2Inserm, Clermont-Ferrand, France; 3Clermont Université, Laboratoire de Pharmacologie, Faculté de Médecine, Clermont-Ferrand, France; 4Laboratoire de Pharmacologie, CHU Clermont-Ferrand, France; 5Unither Pharamceuticals, Paris, France
Background: Acetaminophen (APAP) by oral or intravenous (iv) routes is used for mild to moderate pain but may take time to be effective. When fast relief is required and/or oral or iv routes are not available because of the patient's condition, the transmucosal route may be an alternative.
Methodology: A new transmucosal/buccal (b) pharmaceutical form of APAP dissolved in 50% wt alcohol is compared with other routes of administration. Two consecutive randomized, crossover, double-blind clinical trials (CT1: NCT00982215 and CT2: NCT01206985) included 16 healthy volunteers. CT1 compared the pharmacology of 250 mg bAPAP with 1 g iv APAP. CT2 compared the pharmacodynamics of 125 mg bAPAP with 1 g iv and 125 mg sublingual (s) APAP. Mechanical pain thresholds are recorded in response to mechanical stimuli applied on the forearm several times during 120 minutes. The objective is to compare the time of onset of antinociception and the antinociception (area under the curve) between the routes of administration with analysis of variance (significance P<0.05).
Results: bAPAP has a faster time of antinociception onset (15 minutes, P<0.01) and greater antinociception at 50 minutes (P<0.01, CT1) and 30 minutes (P<0.01, CT2) than ivAPAP and sAPAP. All routes are similar after 50 minutes.
Conclusion: bAPAP has a faster antinociceptive action in healthy volunteers. This attractive alternative to other routes would be useful in situations where oral or iv routes are not available. This finding must now be confirmed in patients suffering from acute pain of mild and moderate intensity.
Keywords: transmucosal delivery, pain, healthy volunteer
Introduction
Acetaminophen (acetyl-p-aminophenol; APAP; paracetamol) is an effective analgesic for mild and moderate pain.1 Plasma concentrations of APAP required to provide analgesia are not well defined.2 An antipyretic effect is obtained with 10–20 μg/mL, and 10 μg/mL produces an analgesic effect,3 but higher and lower concentrations have also been described as effective.4,5 When prompt analgesia is required, intravenous (iv) APAP is considered a more effective alternative to the rectal route, which has a slow onset time and a wide variation in bioavailability, and to the oral route when iv administration is not appropriate.6 Different noninvasive technologies have been developed to provide faster pain relief using the oral mucosa route of administration (sublingual [s] or buccal [b]), and a number of rapid-onset fentanyl formulations for the management of severe cancer breakthrough pain have recently emerged.7,8 In postoperative conditions for acute pain of mild to moderate intensity, the quickest reported time to onset of analgesia with APAP is 8 minutes9 for the iv route and 37 minutes6 for the oral route. So far, there are no noninvasive and rapid-onset APAP formulations that would be very useful to rapidly alleviate moderate pain in emergency situations before switching to other routes if necessary. A pharmaceutical form of APAP for transmucosal/buccal delivery has recently been designed and has been trialed in healthy volunteers with induced mechanical pain and recording of antinociception. Our study reports two successive randomized clinical trials that evaluate its pharmacokinetics and antinociceptive properties by comparing 250 mg and then 125 mg bAPAP with ivAPAP and sAPAP in healthy human volunteers.
Materials and methods
Study
We report on two successive clinical trials, CT1 and CT2. CT1 is a prospective, randomized, double-blind, crossover, controlled study with two arms. CT2 is a similar study with three arms and was designed to complete the results of CT1 and explore whether the 125 mg dose was antinociceptive. Both studies took place in the Clinical Investigation Centre, University Hospital of Clermont-Ferrand, France. All subjects were followed by the same medical and technical teams. The studies received the agreement of the Regional Ethics Committee and the French Drug Agency, were declared on clinicaltrials.gov (CT1: NCT00982215 and CT2: NCT01206985), and followed standardized ethical and safety Good Clinical Practice Guidelines.
Volunteers
In CT1, 20 healthy Caucasian male subjects were selected after signing the informed consent, and 16 were randomized. In CT2, 20 healthy Caucasian male subjects, different from those included in CT1, were selected after signing informed consent, and 16 were randomized. Exclusion criteria included a known hypersensitivity to APAP or alcohol, lack of concentration and unsatisfactory results in the psychophysics tests, concomitant pathologies, consumption of tobacco, habitual excessive consumption of tea or coffee, and consumption of, and addiction to, street drugs. Smoking, eating, and drinking were forbidden during the period of the study. Other exclusion criteria were any consumption of drugs on the day of and during the 2 weeks preceding the date of the selection.
Study design
In CT1, volunteers came twice at 1-week intervals to the Clinical Pharmacology Centre and were randomized. This was all carried out in blinded setup so none of the staff knew what was dosed. For each of the treatment periods, pain was assessed using a pressure instrument, the electronic Von Frey (Bioseb, Chaville, France). The subjects determined their pain threshold (PT) when they began to feel pain in response to increasing intensity of the stimulus. In each of the treatment periods (Figure 1), after clinical examination, baseline blood sampling, and pain test recording (T-1h), subjects were injected at T0 over a 15-minute period with APAP (1 g) or placebo (0.9% saline), and concomitantly 250 mg of APAP dissolved in 1 mL of a hydroalcoholic solution (HAS) or placebo (HAS only) was applied in the left mucogingival sulcus. The timer was started at the start of iv injection concomitant with the first drop on the buccal mucosa. Volunteers were asked not to close their mouth or to swallow for 1.5 minutes. This double-blind clinical trial had a double-arm design, with volunteers being administered ivAPAP + bHAS or iv saline + bAPAP. Pain tests were then carried out at T0+3 minutes, T0+7 minutes, T0+15 minutes, T0+30 minutes, T0+50 minutes, and T0+90 minutes. Blood samples for pharmacokinetic measurements were taken at T0+1 minutes, T0+5 minutes, T0+10 minutes, T0+20 minutes, T0+40 minutes, T0+60 minutes, and T0+120 minutes.
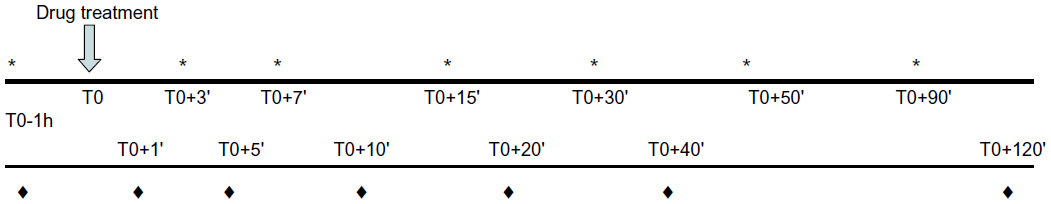 | Figure 1 Chronology of clinical trials 1 and 2, indicating assessment of pain thresholds (*), elapsed times, drug administration, and blood samplings (♦), and blood samplings only for clinical trial 2. |
After completion of CT1, the data analysis showed encouraging results, and a second trial at half the APAP dose used in CT1 (125 mg) was carried out focusing only on pharmacodynamic evaluation.
CT2 followed the same methodology as CT1 but volunteers came three times at 1-week intervals. In each of the treatment periods (Figure 1), after a clinical examination and the baseline pain test recording (T-1h), subjects were injected over a 15-minute period with APAP (1 g) or placebo (0.9% saline). Concomitantly, in randomized fashion, 125 mg sAPAP was applied sublingually under the frenulum of the tongue or on the left mucogingival sulcus (bAPAP), or 1 mL HAS was applied in the mucogingival sulcus. Pain tests were then carried out at the same times as in CT1. There was no blood sampling. This double-dummy clinical trial had a three-arm design, with volunteers being administered ivAPAP + bHAS or iv saline + bAPAP or iv saline + sAPAP.
APAP pharmaceutical form
The patented form of APAP (Patent Europe N07871968.9) consists of paracetamol in a stable and complete dissolution state in a hydroalcoholic solution containing 50 wt% of alcohol for rapid absorption through the mucosa of the oral cavity and/or the oropharynx. Glass vials contained 250 mg/2 mL and 125 mg/1 mL in CT1 and CT2, respectively. The drug was applied locally by a nurse with a pipette.
Pain induction for pharmacodynamic assessment
Pain induction was carried out with a mechanical test commonly used to explore antinociception in experimental settings, the electronic Von Frey test (Bioseb), which consists of applying pressure on the medial aspect of the dominant arm via the tip of a plastic cone. The strain gauge was connected to a plastic sterile cone (Eppendorf, Hamburg, Germany). The tip was applied in a perpendicular way on the studied skin area. The pressure was gradually increased with a constant slope under visual control of the pressure value up to the detection of mechanical PT. Threshold was defined as the lowest pressure that produced a sensation of pain. Three threshold values were averaged to determine the PT (in grams). The study was performed in a quiet and distraction-free room. All the sessions took place at the same time of day to minimize possible circadian influences on pain ratings. The subjects had their arm on a stable surface in a sitting position. Each volunteer had two training sessions before being included in the protocol. Twenty volunteers were selected and the 16 who had the best reproducibility in the tests were kept for randomization. The value measured at T-1h was considered the baseline. The main parameter under consideration was the mechanical PT difference between T+n and T-1h (ΔPT in grams). The difference was chosen in order to correct for control/placebo effects. Derived parameters were calculated: maximum of action observed (Emax) and area under the curve (AUC) calculated using linear trapezoidal rule on ΔPT data (in g · minute [min]). Acceptability of the drug was assessed by a scale (very bad, bad, satisfactory, good, very good) at the end of each session.
Pharmacokinetics
APAP plasma levels were determined using a specific high-performance liquid chromatographic method. Blood samples of 5 mL were collected from all patients. Samples were centrifuged and plasma stored at −20°C until analysis. At first, 50 μL of internal standard (3-acetamidolol 40 μg/ml) were added to 1 mL of plasma samples. The sample was extracted with 5 mL of ethyl acetate by shaking for 10 minutes and centrifuging for 10 minutes. The organic layer was evaporated to dryness by blowing a slow stream of nitrogen into the tubes. The residue was dissolved in 200 μL of a mixture of acetonitrile/water (80/20) and injected into a liquid chromatograph. Standard curves were made with spiked serum at the following concentrations: 0 – 0.1 – 1 – 5 – 10 – 30 – 50 μg/mL. All calibration samples were taken through the extraction protocol described.
Separation of analytes was obtained with Symmetry C8 250×4.6 mm (Waters Ltd, Hertfordshire, UK). The mobile phase consisted of a gradient of phosphate buffer, 50 mM, pH 3.6 acetonitrile, increasing linearity from 13% to 35% acetonitrile over 8 minutes (flow rate: 1 mL/min) and to 80% for the next 4 minutes (flow rate gradient: from 1 mL/min to 1.5 mL/min). The system was then stabilized for an additional 3 minutes with initial conditions (flow rate: 1 mL/min).
Detection was carried out with a diode array detector (Waters Alliance 996) set at 200 nm, and spectral data were analyzed in order to assess the purity of the APAP peak.
A linear relationship (r>0.999) was found for the APAP calibration curve, and its limit of quantitation was 0.05 μg/mL.
Statistical analysis
Due to the exploratory nature of the study and the absence of previous data with such types of administration, the sample size was computed based on data in our department with electronic Von Frey. In this study the mean value on healthy skin was around 105 g. Intersubject variability was estimated in the previous study to be 30%. The difference was set at 20%, α at 5%, and β at 20%. Based on these assumptions, a final sample size of 16 patients was calculated. Statistics were performed on SAS Version 9.1 (SAS Institute Inc., Cary, NC, USA) for Windows XP. Values are given as means ± standard deviations (SD) in Figures 2 and 3 and means, SDs, coefficient of variation, and medians in Tables 1 and 2.
 | Figure 2 Pharmacokinetic profile of 1 g intravenous (iv) and 250 mg buccal acetaminophen. |
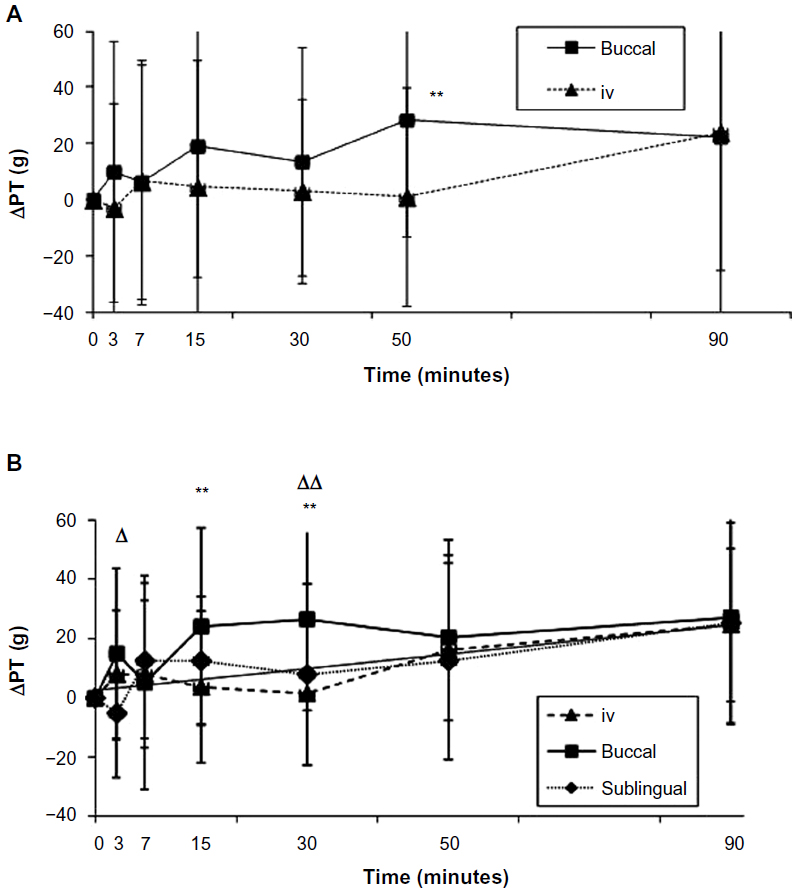 | Figure 3 Antinociceptive profiles obtained during (A) clinical trial 1 for 1 g intravenous (iv) and 250 mg buccal (b) acetaminophen (APAP) and (B) during clinical trial 2 for 1 g iv APAP, 125 mg bAPAP, and 125 mg sublingual (s)APAP. |
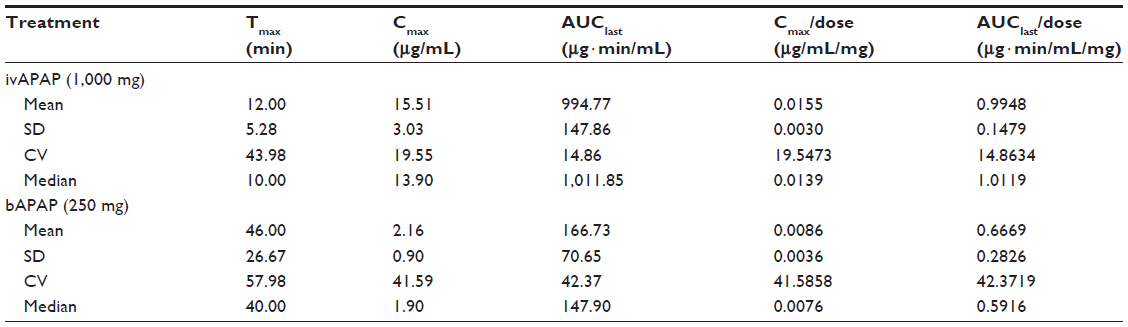 | Table 1 Clinical trial 1: descriptive statistics on pharmacokinetic parameters |
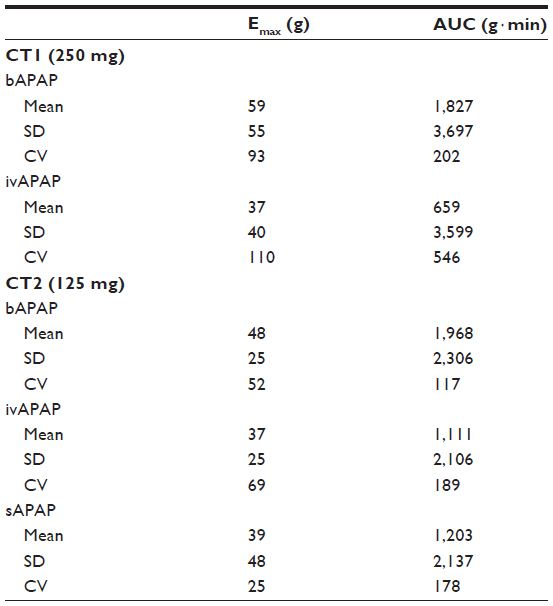 | Table 2 Clinical trials 1 (CT1) and 2 (CT2): descriptive statistics on pharmacodynamic-derived parameters (pain thresholds) |
The bioavailability analysis was carried out by analysis of variance (ANOVA) on log transformed parameters (AUC and peak concentration [Cmax]) using sequence subject within sequence, period, and treatment as effects. The residual error was extracted and then a 90% confidence interval (CI) calculated on the difference between test and reference. The lower and higher limits of the CI were back-calculated using exponential function and compared with the limits of 0.8 and 1.25. For pharmacodynamics raw data, significant differences between treatments were tested by ANOVA with repeated-measures analysis. For the analysis of pharmacodynamics-derived parameters (Emax and AUC), ANOVA was used including sequence subject within sequence, period, and treatment as effects. A probability level of P<0.05 was considered to be statistically different.
Results
For CT1 and CT2, respectively, data of 16 male subjects (age 26±2 years and 25±2 years) were analyzed, and iv perfusion took 14±3 minutes and 14±2 minutes to be administered (14 minutes is the recommended time).
Pharmacokinetic data
For CT1 (Table 1, Figure 2), repeated-measures ANOVA shows a statistical difference between treatments for both dose-corrected Cmax and AUC (P<0.0001 and P=0.0009, respectively) associated with intrasubject variability of 30.8% and 27.4% for AUC/dosage and Cmax/dosage. Ratios are 0.619 and 0.528 for AUC/dosage and Cmax/dosage, respectively, associated with 90% CI of 0.507–0.756 and 0.442–0.630, indicating a nonequivalence and a lower bioavailability for buccal administration.
Pharmacodynamic data
For CT1 (Table 2, Figure 3A), repeated-measures ANOVA shows a statistical difference at 50 minutes (P=0.0077) between 250 mg bAPAP and ivAPAP. The variability observed in the ANOVA (residual error) is large, with 58% and 106% for Emax and AUC, respectively. A statistical difference could be observed for Emax and AUC (P=0.0489 and P=0.0246, respectively), with a mean variation of +46% and +97%, respectively. Time to exhibit a statistical significance in PT compared with baseline is 50 minutes for bAPAP (P=0.0098) and 90 minutes for ivAPAP (P=0.0274).
For CT2 (Figure 3B), repeated-measures ANOVA shows a statistical difference at 3 minutes between buccal and sublingual (P=0.04), at 15 minutes between buccal and iv (P=0.0249), and 30 minutes between buccal and iv (P=0.006) and buccal and sublingual (P=0.0386) routes. No statistical difference was observed for Emax and AUC. Time to exhibit a statistical significance in PT compared with baseline is 15 minutes for bAPAP (P=0.0083), 50 minutes for ivAPAP (P=0.0073), and 90 minutes for sAPAP (P=0.0058).
Acceptability is satisfactory, despite a bitter taste noticed by the volunteers at all sessions for the placebo and active drug.
Discussion
The pharmacological characteristics of a new buccal noninvasive pharmaceutical form of APAP have been studied in two successive randomized clinical trials at dosages of 250 mg then 125 mg, compared with 1 g ivAPAP in healthy volunteers. bAPAP has a larger analgesic efficacy (CT1) and a faster time to reach analgesia (CT1 and CT2) when compared with iv and sublingual routes. Time to significant analgesia occurs from 15 minutes after bAPAP administration and 40 minutes earlier than with the other forms in both trials. These findings challenge a dose–effect relationship in the analgesia of APAP,10 as analgesia is achieved here earlier and despite lower APAP blood concentrations obtained with the buccal route. Although APAP has been widely used for the last century as an analgesic and antipyretic agent, its mechanism of action is still unclear, but its central action has been well demonstrated in humans11–13 and in animals,14–18 and its effective dose is known to be dependent on the species and on the route of administration.16
The present findings may be related on the one hand to the physicochemical properties of the pharmaceutical form itself, and on the other hand to the physiology of the buccal mucosa.
Physicochemical properties of the pharmaceutical form may play a major role, as diffusion of APAP, a lipophilic drug with a small molecular weight, through the bilipidic layer of the thin cell membrane (500 μm) of the buccal mucosa19–21 will be enhanced by alcohol.22 Alcohol also has a detersive effect on the main components of the intercellular lipids (ceramids, cholesterol, and glycolipids). It also increases vascular dilatation in the richly vascularized oral mucosa, especially in the area of the lower first canine, where a vascular–nerve bunch is anatomically located (facial artery, vein, and nerve). Compared with sublingual administration, we noticed that administration on the buccal mucosa triggers less salivation and hence less dilution of the active principle and deglutition reflex. The observed analgesic effect with the buccal delivery is probably linked to the rapid absorption of bolus APAP, but enzymatic and molecular mechanisms also need to be considered.
APAP metabolism takes place predominantly in the liver because the majority of enzyme genes are expressed abundantly there, but some of these enzymes, like cytochromes, are also reported in extrahepatic tissues, including the oral mucosa.23,24 APAP metabolites have for a long time been considered for their toxic effects, and several of them do, in fact, participate in the analgesic effect of APAP. N-acetyl-p-benzoquinoneimine (NAPQI), a Phase I metabolite of APAP via cytochrome P450 (CYP450) is known for inducing liver failure in APAP overdose.25,26 A recent paper16 elegantly demonstrates the antinociceptive property of intrathecal NAPQI in mice and discloses the activation of spinal transient receptor potential ankyrin 1 channel, a peripheral and central sensor of noxious stimuli. Likewise, previous studies27 demonstrate the antinociceptive property of another APAP metabolite, AM404 (N-[4-hydroxyphenyl]-5Z,8Z,11Z,14Z-eicosatetraenamide). AM404 is obtained centrally via fatty acid amide hydrolase from p-aminophenol via N-deacetylase from APAP,15,27 although it is also suggested that NAPQI, rather than APAP, may be the actual substrate for N-deacetylase.28,29 The role of enzymes located in the buccal or in the lung mucosa/circulation where the drug will be transported after oral diffusion has not been demonstrated so far for the in situ production of paracetamol antinociceptive metabolites. N-deacetylases, present in extrahepatic tissues,30 and CYP450 isozymes, more specifically those involved in APAP metabolism (CYP1A2, CYP2A6, CYP2D6, CYP2E1, and CYP3A4),21 are all expressed in buccal epithelium,30–32 but their precise roles in the oral mucosa are poorly described. A dynamic histological study of the buccal mucosa using APAP would help to evaluate the possible in situ production of APAP metabolites that could rapidly reach central target receptors while bypassing hepatic metabolism in the early minutes of APAP administration.
These findings of a rapid analgesia (15 minutes) in healthy volunteers with a noninvasive APAP administration must now be confirmed in patients with pain. A randomized clinical trial (NCT01586143) in patients admitted to the accident and emergency department for minor trauma (ankle sprains) and requiring APAP for alleviation of pain of mild or moderate intensity is now starting. The confirmation in patients could lead to the use of bAPAP (one-eighth of the iv dosing) as an alternative to the systematic ivAPAP injection routinely done in patients for minor trauma generating moderate pain, and could be relayed later by other dosages and routes of administration. Such a pharmaceutical form could also be assessed in breakthrough pain episodes and in vulnerable populations such as older persons, when swallowing is difficult.
Acknowledgment
Financial support: Unither Laboratories.
Disclosure
The authors report no conflicts of interest in this report.
References
Ventafridda V, Tamburini M, Caraceni A, De Conno F, Naldi F. A validation study of the WHO method for cancer pain relief. Cancer. 1987;59(4):850–856. | |
Rumack B. Aspirin versus acetaminophen: a comparative view. Paediatrics. 1978;62:943–946. | |
Anderson B, Holford N, Woollard GA, Kanagasundaram S, Mahadevan M. Perioperative pharmacodynamics of acetaminophen analgesia in children. Anaesthesiology. 1999;90:411–421. | |
Holmer-Pettersson P, Owall A, Jakobsson J. Early bioavailability of paracetamol after oral or intravenous administration. Acta Anaesthesiol Scand. 2004;48:867–870. | |
Anderson B, Holford N. Rectal paracetamol dosing regimens: determination by computer simulation. Paediatr Anaesth. 1997;7:451–455. | |
Oscier M, Bosley B, Milner S. Paracetamol: a review of three routes of administration. Update in Anesthesia. 2007;23:112–114. | |
Portenoy RK, Payne R, Coluzzi P, et al. Oral transmucosal fentanyl citrate (OTFC) for the treatment of breakthrough pain in cancer patients: a controlled dose titration study. Pain. 1999;79(2–3):303–312. | |
Mercadante S. The use of rapid onset opioids for breakthrough cancer pain: the challenge of its dosing. Crit Rev Oncol Hematol. 2011;80(3):460–465. | |
Moller PL, Juhl GI, Payen-Champenois C, Skoglund LA. Intravenous acetaminophen (paracetamol): comparable analgesic efficacy, but better local safety than its prodrug, propacetamol, for postoperative pain after third molar surgery. Anesth Analg. 2005;101(1):90–96. | |
Piguet V, Desmeules J, Dayer P. Lack of acetaminophen ceiling effect on R-III nociceptive flexion reflex. Eur J Clin Pharmacol. 1998;53(5):321–324. | |
Pickering G, Loriot MA, Libert F, Eschalier A, Beaune P, Dubray C. Analgesic effect of acetaminophen in humans: first evidence of a central serotonergic mechanism. Clin Pharmacol Ther. 2006;79:371–378. | |
Pickering G, Estève V, Loriot MA, Eschalier A, Dubray C. Acetaminophen reinforces descending inhibitory pain pathways. Clin Pharm Ther. 2008;84(1):47. | |
Pickering G, Moustafa F, Desbrandes S, Cardot JM, Roux D, Dubray C. Paracetamol and opioid pathways: a pilot randomised clinical trial. Fund Clin Pharmacol. 2013;27(3):339–345. | |
Pini LA, Sandrini M, Vitale G. The antinociceptive action of paracetamol is associated with changes in the serotonergic system in the rat brain. Eur J Pharmacol. 1996;308:31–40. | |
Mallet C, Barrière DA, Ermund A, et al. TRPV(1) in brain is involved in acetaminophen-induced antinociception. PLoS One. Epub September 17, 2010. | |
Andersson DA, Gentry C, Alenmyr L, et al. TRPA1 mediates spinal antinociception induced by acetaminophen and the cannabinoid Δ(9)-tetrahydrocannabiorcol. Nat Commun. 2011;2:551. | |
Alloui A, Pelissier T, Dubray C, Lavarenne J, Eschalier A. Tropisetron inhibits the antinociceptive effect of intrathecally administered paracetamol and serotonin. Fundam Clin Pharmacol. 1996;10:406–407. | |
Raffa RB, Stone DJ Jr, Tallarida RJ. Discovery of “self-synergistic” spinal/supraspinal antinociception produced by acetaminophen (paracetamol). J Pharmacol Exp Ther. 2004;295:291–294. | |
Campisi G, Paderni C, Saccone R, Di Fede O, Wolff A, Giannola LI. Human buccal mucosa as an innovative site of drug delivery. Curr Pharm Des. 2010;6(6):641–652. | |
Senel S, Hincal AA. Drug permeation enhancement via buccal route: possibilities and limitations. J Control Release. 2001;72:133–144. | |
Squier CA, Cox P, Wertz PW. Lipid content and water permeability of skin and oral mucosa. J Invest Dermatol. 1991;96:123–126. | |
Oliveira G, Beezer AE, Hadgraft J, Lane ME. Alcohol enhanced permeation in model membranes. Part I: thermodynamic and kinetic analyses of membrane permeation. Int J Pharm. 2010;393(1–2):61–67. | |
Koskela S, Hakkola J, Hukkanen J, et al. Expression of CYP2A genes in human liver and extrahepatic tissues. Biochem Pharmacol. 1999;57(12):1407–1413. | |
Pons F, Calvet JH, Haag M, Raeppel V, Keravis T, Frossard N. Altered expression of lung cytochrome P450 3A1 in rat after exposure to sulfur mustard. Pharmacol Toxicol. 2001;88(1):40–44. | |
Pickering G, Schneider E, Papet I, et al. Acetaminophen metabolism after major surgery: a bigger challenge when aging. Clin Pharm Ther. 2011;90(5):707–711. | |
Zhao L, Pickering G. Paracetamol metabolism and related genetic differences. Drug Metab Rev. 2011;43(1):41–52. | |
Högestätt ED, Jönsson BA, Ermund A, et al. Conversion of acetaminophen to the bioactive N-acylphenolamine AM404 via fatty acid amide hydrolase-dependent arachidonic acid conjugation in the nervous system. J Biol Chem. 2005;280:31405–31412. | |
Bessems JGM, Vermeulen NPE. Paracetamol (acetaminophen)-induced toxicity: molecular and biochemical mechanisms, analogues and protective approaches. Crit Rev Toxicol. 2001;31(1):55–138. | |
Fischer V, West PR, Nelson SD, Harvison PJ, Mason RP. Formation of 4-aminophenoxyl free radical from the acetaminophen metabolite N-acetyl-p-benzoquinone imine. J Biol Chem. 1985;260(21):11446–11450. | |
Vondracek M, Xi Z, Larsson P, et al. Cytochrome P450 expression and related metabolism in human buccal mucosa. Carcinogenesis. 2011;22(3):481–488. | |
Sarikaya D, Bilgen C, Kamataki T, Topcu Z. Comparative cytochrome P450-1A1, -2A6, -2B6, -2C, -2D6, -2E1, -3A5 and -4B1 expressions in human larynx tissue analysed at mRNA level. Biopharm Drug Dispos. 2006;27:353–359. | |
Sarikaya D, Chiba I, Bilgen C, Kamatakii T, Topcu Z. RT-PCR-based cytochrome P450 expression profile of oral tissue samples. J Clin Pharm Ther. 2007;32(5):445–448. |
 © 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.