Back to Journals » Journal of Inflammation Research » Volume 14
BT2 Suppresses Human Monocytic-Endothelial Cell Adhesion, Bone Erosion and Inflammation
Authors Yeh MC, Wu BJ, Li Y, Elahy M, Prado-Lourenco L, Sockler J, Lau H, Day RO ![]() , Khachigian LM
, Khachigian LM ![]()
Received 10 December 2020
Accepted for publication 28 January 2021
Published 23 March 2021 Volume 2021:14 Pages 1019—1028
DOI https://doi.org/10.2147/JIR.S296676
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Ning Quan
Mei-Chun Yeh,1 Ben J Wu,1 Yue Li,1 Mina Elahy,1 Leonel Prado-Lourenco,1 Jim Sockler,2 Herman Lau,3 Ric O Day,4 Levon M Khachigian1
1Vascular Biology and Translational Research, School of Medical Sciences, Faculty of Medicine and Health, University of New South Wales, Sydney, NSW, 2052, Australia; 2Statistical Operations & Programming, Datapharm Australia Pty Ltd, Drummoyne, NSW, 2047, Australia; 3BJC Health, Chatswood, NSW, 2067, Australia; 4Department of Clinical Pharmacology & Toxicology, Therapeutics Centre, St Vincent’s Hospital, UNSW Medicine and Health, Darlinghurst, NSW, 2010, Australia
Correspondence: Levon M Khachigian
Vascular Biology and Translational Research, School of Medical Sciences, Faculty of Medicine and Health, University of New South Wales, Sydney, NSW, 2052, Australia
Tel +61 2 9385 2537
Email [email protected]
Introduction: Inflammation and bone erosion are processes key to the pathogenesis of rheumatoid arthritis, a systemic autoimmune disease causing progressive disability and pain, impacting around 1.3 million people in the United States alone. However, many patients do not respond sufficiently to existing therapies or benefit is not sustained and alternate therapeutic approaches are lacking. We recently identified the dibenzoxazepinone BT2, which inhibits ERK phosphorylation, from a high-throughput chemical screen and identified its ability to inhibit angiogenesis and vascular leakiness.
Methods: Here we evaluated BT2 for potential anti-inflammatory activity in in vitro models of human monocytic-endothelial cell adhesion, monocytic cell extravasation and collagen antibody-induced arthritis in mice.
Results: BT2 inhibits human monocytic cell adhesion to IL-1ß-treated human endothelial cells and inhibits monocytic transendothelial migration toward MCP-1. In mice rendered arthritic, single systemic administration of BT2 prevented footpad swelling, bone destruction and TRAP+ cells in the joints. BT2 suppressed inducible circulating levels of IL-1ß, IL-2 and IL-6 to normal levels without affecting levels of IL-4 or IL-10 among other cytokines. BT2 also inhibited the expression of pro-inflammatory adhesion molecules ICAM-1 and VCAM-1 in arthritic joints. There was no evidence of toxicity following intraperitoneal, gavage or intraarticular administration of BT2.
Conclusion: BT2 is a novel small molecule inhibitor of joint inflammation, bone erosion, pro-inflammatory cytokine and adhesion molecule expression. This suggests the potential clinical utility of BT2 as a new anti-inflammatory agent.
Keywords: inflammation, pathology, bone erosion, rheumatoid arthritis, dibenzoxazepinone
Plain Language Summary
BT2 is a novel small molecule that prevents joint inflammation, bone erosion, pro-inflammatory cytokine and adhesion molecule expression. There was no evidence of toxicity after intraperitoneal, gavage or intraarticular administration of BT2. This suggests the potential clinical utility of BT2 as a new anti-inflammatory agent.
Introduction
Rheumatoid arthritis (RA) is an inflammatory, autoimmune disorder that causes progressive disability1 characterized by synovial inflammation, cartilage and bone damage, and joint destruction.2 Cytokine-mediated pathways play a crucial role in the pathogenesis of RA.3 RA impacts around 1.3 million people in the US alone.4 There has been significant improvement in the management of RA over the last decade. However, many patients do not achieve clinical remission with current therapeutic options and are at risk of progressive joint destruction and functional disability.5 For example, anti-TNF agents such as adalimumab (human monoclonal antibody) and soluble TNF receptor (etanercept) achieve a clinical remission rate (DAS <2.6) of 43%6 and 50%7 at 12 months, respectively. Other agents such as anti-IL-6 therapy (tocilizumab), CTLA4-Ig fusion protein (abatacept) and Janus kinase (JAK) inhibitor (tofacitinib) achieve remission rates of 34%,8 41%9 and 19–30%,10 respectively. Several such agents are biosimilar drugs (e.g. IL-6R blockade is also achieved with sarilumab11) while tofacitinib is one of a number of JAK inhibitors.12 High cost also limits the broader use of anti-cytokine therapies.13 Given the world’s ageing population,14,15 the significant unmet clinical need for RA and the socioeconomic burden that this chronic disease represents,16 alternative safe and cost-effective therapeutic approaches are needed.
We recently identified a dibenzoxazepinone BT2 from a high-throughput chemical screen17 that inhibits ERK phosphorylation (pERK), and suppresses the inducible expression of the basic region-leucine zipper proteins FosB/∆FosB and c-Fos, both members of the activator protein-1 (AP-1) family of dimeric transcription factors.18 AP-1 regulates gene expression in response to a range of pathologic stimuli including cytokines, growth factors, various stresses and viral and bacterial infection18 and has been implicated in the pathogenesis of cancer, cardiovascular disease and a range of inflammatory diseases.19 For example, AP-1 DNA binding activity has been observed in human rheumatoid synovium and is associated with disease activity.20 Currently there are no clinically used AP-1 inhibitors.19 Here we report that BT2 is a small molecule inhibitor that prevents joint inflammation and bone erosion, pro-inflammatory cytokine and adhesion molecule expression.
Materials and Methods
Synthesis and Supply of Compounds
BT2 (10-ethyl-11-oxo-10,11-dihydro-dibenzo[b,f][1,4]oxazepin-2-yl)-carbamic acid ethyl ester) was prepared by reacting commercially available 2-amino-10-ethyldibenzo[b,f][1,4] oxazepin-11 (10H)-one (BT3) with diethyl pyrocarbonate as described17 by Advanced Molecular Technologies, Scoresby (Vic).
Cell Culture
Human microvascular endothelial cells (HMEC-1) were purchased from ATCC (Rockville, MD) and grown in MCDB131 medium (Invitrogen, MD), pH 7.4 supplemented with 10% FBS, hydrocortisone (1 µg/mL), epidermal growth factor (10 ng/mL), L-glutamine (2mM) and penicillin/streptomycin. Human monocytic leukemia cells (THP-1) (Sigma) were maintained in RPMI 1640 media (Gibco) supplemented with 10% heat inactivated FBS, L-glutamine (2mM) and penicillin/streptomycin (5U/mL, Sigma). Cells were routinely passaged after detachment with 0.05% trypsin/5 mM EDTA and maintained in a humidified atmosphere of 5% CO2 at 37°C.
Flow Cytometry
HMEC-1 cells (at 80–90% confluency) were growth arrested in serum-free MCDB131 medium without EGF and hydrocortisone for 40h, treated with 30 μM BT2 or BT3 for 4 h, and exposed to 20 ng/mL IL-1ß (cat. SRP3083, Sigma) in serum-free medium with BT2 or BT3 for a further 4 h. The cells were washed with PBS then Accutase was used to detach the cells. The cells were centrifuged at 300 g for 5 min and resuspended at 5x106 cells/mL in stain buffer. The cells were incubated with BV421-conjugated mouse anti-human CD54 (ICAM-1) (cat. 566,262, BD) or BV421-conjugated mouse IgG1 (cat. 562,438, BD) for 45 min at 22°C. The cells were washed with Stain Buffer and the pellet was resuspended in 0.5 mL of 1% paraformaldehyde prior to flow cytometry BD FACSCanto II.
Western Blot Analysis
HMEC-1 cells (80–90% confluency) were rendered growth-quiescent in serum-free MCDB131 medium (Invitrogen, MD) for 44h. Cells were then treated as indicated. Total protein was harvested as previously described using RIPA buffer with protease inhibitors. Proteins were resolved on 4–20% (w/v) SDS-polyacrylamide gradient gels and transferred to Immobilon-P PVDF membranes. Membranes were blocked with 5% skim milk and incubated with rabbit monoclonal VCAM-1 (cat. 13662S, 1:1000, Cell Signaling, USA), mouse monoclonal ICAM-1 (cat. sc-8439, 1:500, Santa Cruz, USA) at 4°C for overnight. Mouse monoclonal β-actin antibodies (cat. A5316, 1:10,000, Sigma-Aldrich) were used to visualize loading. Membranes were then incubated with horseradish peroxidase conjugated secondary goat anti-rabbit (cat. P0448, 1:1000, DAKO Cytomation, Denmark) or goat anti-mouse (cat. P0447, 1:1000, DAKO Cytomation, Denmark) antibodies for 1h. Chemiluminescence was detected using the Western Lightning Chemiluminescence system and ImageQuant™ LAS 4000 biomolecular imager. Band intensity in images generated with the LAS 4000 using the same settings were quantified by NIH Image J.
Endothelial-Monocytic Cell Adhesion Assay
HMEC-1 cells (80–90% confluency) in 96-well plates were deprived of serum for 24h and treated with compound at indicated concentrations for 1h then incubated with IL-1ß (20 ng/mL) for 4h. Meanwhile THP-1 cells were labeled with 5µM calcein (5x106 cells/mL, BD Bioscience) for 30 min at 37°C followed by washing 3 times with PBS. THP-1 (2.5x105 cells/well) were then added for 30 min, unbound cells were washed off 3 times in PBS. Adhesion of calcein-labeled THP-1 to the endothelium layer was determined in a fluorescent plate reader at excitation 485 nm and emission 530 nm.
Monocyte-Transendothelial Migration Assay
Millicell 8 µm polycarbonated culture plate inserts (Millipore) were coated with 0.1% porcine gelatin type A (Sigma) and then placed into 24-well plates. HMEC-1 cells (5x104 cells/well) were seeded onto the insert and allowed to adhere overnight. Cells were then serum deprived for 24 h and treated with various compound treatments for 1 h. IL-1ß (20ng/mL) was added to stimulate the cells for 4 h and 500 µL of serum-free medium was added to the bottom of the 24-well plate along with the compound. THP-1 cells (5x105 cells in 100µL) were added into the insert and 100 ng/mL MCP-1 (Sigma) was added to the lower well. After 24 h, the number of cells that had migrated though the endothelial layer was assessed by counting 100µL of the suspension in the lower chamber using a Coulter cell counter (Beckman Coulter).
Collagen Antibody-Induced Arthritis
Arthritis was induced in female Balb/c mice (6–8 week-old) as previously described with a commercially obtained cocktail of 5 monoclonal antibodies to type II collagen at 2 mg/mouse (Chondrex, Inc. Redmond, WA) followed by LPS (50µg/mouse) with or without BT2 (3 or 30 mg/kg mouse) in DMSO vehicle administered i.p. on Day 3. Hind footpad thickness was measured on Day 9 using digital calipers. Mice were sacrificed on Day 14 and microCT scanning of hind limbs was performed.
Micro-CT Scanning and Analysis
Formalin-ethanol fixed hind limbs were micro-CT scanned prior to histology processing using a Siemens Inveon micro-CT scanner (Victoria, Australia). Data was acquired with the Inveon Acquisition Workplace at 16.84µm pixel size, 360 projections, 4100ms integration time, 80keV photon energy and 140µA current. 3D models were visualized and snap shots of the limbs acquired with the Inveon Research Workplace software. Data was quantified by binary score where 0=no bone destruction and 1=destruction was given to each individual limb.
Haematoxylin Eosin (H&E) Staining
Deparaffinized sections were re-hydrated and stained in Harris hematoxylin solution, and after differentiation and bluing, sections were counterstained in eosin-phloxine solution, and then sections were dehydrated and mounted with mounting medium. Slides were scanned using an Aperio ScanScope XT slide scanner (Leica Biosystems, Mt Waverley, Vic, Australia) and images were captured using ImageScope software (Leica).
Tartrate-Resistant Acid Phosphatase (TRAP) Staining
Osteoclasts were stained using TRAP kit (Cosmo Bio, Japan, cat. PMC-AK04F-COS). Sections were heated at 65°C for 1 h prior dewaxing. Tissue sections were deparaffinised with 100% xylene and rehydrated with 100, 70 and 30% ethanol and rinsed with distilled water for 5 min. Sections were covered with TRAP staining solution containing 3 mg tartaric acid per 50 mL tartaric acid buffer. The sections were incubated at 37°C for 1h, then rinsed in distilled water 3 times to halt the reaction. Sections were counterstained with hematoxylin for 5s then washed in running water until clear then dried. Sections were dehydrated with xylene and air-dried then mounted with aqueous permanent mounting medium. Within the synovium on the medial aspect of each animal joint, 6 random areas photographed under 20x objective were selected in the blinded fashion. Numbers of osteoclasts were counted using NIH Image J. Alternatively TRAP staining was quantitated using integrated optical density (IOD) (Image-Pro Plus).
Immunohistochemical Staining of Hind Limbs for VCAM-1 and ICAM-1 and Analysis
Formalin-fixed, paraffin embedded of hind limbs were sectioned (5 µm). Dako EnVision Rabbit Kit (cat. K4011, Dako) was used for immunohistochemical staining for VCAM-1 and ICAM-1. Briefly, sections were blocked with peroxidase for 30 min and then immunostained with rabbit monoclonal VCAM-1 (cat. ab134047, 1:100, Abcam), or rabbit polyclonal ICAM-1 (cat. ab124759, 1:100, Abcam) at 4°C overnight. Staining was visualized using labeled polymer-Horse Radish Peroxidase (HRP) (anti-rabbit) and Diaminobenzidine (DAB) system followed by counter staining with haematoxylin and Scott blue. Immunostained slides were scanned using an Aperio ScanScope XT slide scanner (Leica Biosystems, Mt Waverley, Vic, Australia) and images were captured using ImageScope software (Leica Biosystems). IOD of positive staining in ankle joint (tibia and talus) articular cartilage was assessed for VCAM-1 and ICAM-1 using Image-Pro Plus software (Cybernetics, Bethesda, MD, USA). Area (µm2) of ankle joint articular cartilage was measured using Image-Pro Plus software. Total cell number and positive staining cell number in ankle joint articular cartilage were counted manually using Image-Pro Plus software. Data was represented as IOD/µm2 and percentage of positive staining cell per 20x objective view.
Mouse Cytokine Measurement
Whole blood was collected from mice and spun at 1200 rpm for 15 min at 4°C. Plasma was then collected from the top layer and stored at −80°C. Cytokine level present in mouse plasma was measured using mouse cytokine 10-plex panel system (Invitrogen) according to the manufacturer’s protocol. Cytokine levels in mouse plasma were measured alongside a standard curve. Where plasma levels were determined by the system to be out of range below the standards, a value of 0 was assigned. Values beyond the standard range were extrapolated by the system.
Toxicology
Female Balb/c mice (8–9 week-old) were given 3 or 30 mg/kg BT2 (DMSO vehicle) via intraperitoneal injection (Days 0 and 5 in DMSO), oral gavage (Days 0–4 in DMSO/0.5% methylcellulose) or intraarticular injection (Day 0 in DMSO).21 Mice were euthanized after 8–11 d. Tissues were fixed in 10% formalin, processed routinely, sectioned at 4µm and stained with hematoxylin and eosin. Sections were examined histologically for signs of toxicity by a board-certified diplomate of the American College of Veterinary Pathologists.
Study Approval
Animal experiments were approved by the Animal Care and Ethics Committee at the University of New South Wales and conducted in accordance with the Australian Code of Practice for the Care and Use of Animals for Scientific Purposes (2004).
Statistics
Statistical analysis was performed as described in the legends and differences were considered significant when P<0.05. Where indicated, *P<0.05, **P<0.01, ***P<0.001, ****P< 0.0001.
Results
BT2 Inhibits Intercellular Adhesion Molecule (ICAM)-1 Expression
We recently demonstrated the capacity of the dibenzoxazepinone BT2 (10-ethyl-11-oxo-10,11-dihydro-dibenzo[b,f][1,4]oxazepin-2-yl)-carbamic acid ethyl ester) to inhibit angiogenesis and vascular permeability.17 This led us to explore the anti-inflammatory properties of BT2. IL-1ß and the pro-inflammatory cell adhesion molecule ICAM-1 have each strongly been implicated in the pathogenesis of RA.22,23 Flow cytometry revealed BT2 inhibition of IL-1ß inducible ICAM-1 expression in HMEC-1 cells (Figure 1A). In contrast, BT317 (2-amino-10-ethyldibenzo[b,f][1,4] oxazepin-11 (10H)-one), a close structural analogue of BT2 (Figure 1B) was unable to do so (Figure 1A). BT2 inhibited ICAM-1 expression in a dose-dependent manner (Figure 1C). BT2 inhibition of vascular cell adhesion molecule (VCAM)-117 served as a further control (Figure 1D).
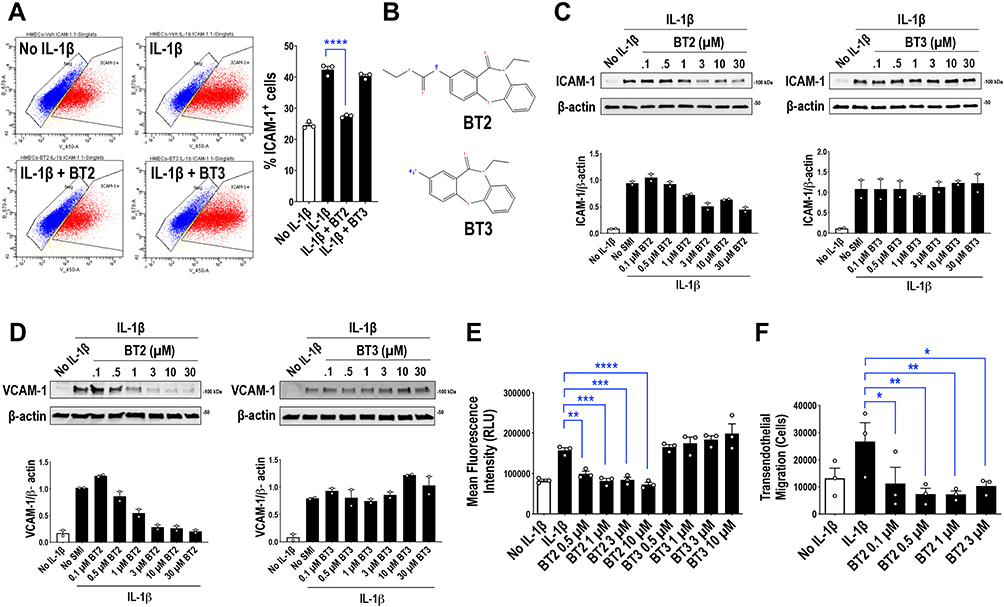 |
Figure 1 BT2 inhibits ICAM-1 and VCAM-1 expression, human monocytic cell adhesion to IL-1ß-treated endothelium in vitro and monocytic transendothelial migration toward MCP-1 in vitro. (A) Flow cytometry for ICAM-1. Serum-deprived HMEC-1 cells were treated with 30µM of BT2 or BT3 in serum free medium at 37°C for 4h and 20ng/mL IL-1ß for a further 4h. Data is representative of 3 biologically independent experiments and expressed as mean ± SEM. Statistical significance was assessed by ANOVA. (B) Structures of BT2 (C18H18N2O4) and BT3 (C15H14N2O2). (C) Western blotting for ICAM-1. Serum-deprived HMEC-1 cells were treated with 0.1–30µM of BT2 or BT3 in serum free medium at 37°C for 4h and 20ng/mL IL-1ß for a further 4h. Data is representative of 2 biologically independent experiments. (D) Western blotting for VCAM-1. Serum-deprived HMEC-1 cells were treated with 0.1–30 µM of BT2 or BT3 in serum free medium at 37°C for 4h and 20ng/mL IL-1ß for a further 4h. Data is representative of 2 biologically independent experiments. (E) THP-1 adhesion to HMEC-1 cells in vitro was assessed by first treating HMEC-1 cells with various concentrations of BT2 for 1h in 96-well plates. HMEC-1 cells were stimulated with 20ng/mL IL-1ß for 1h. Fluorescence intensity of calcein labeled THP-1 that adhered to HMEC-1 cell monolayers 30 min after adding the cells was then measured via fluorescent plate reader. RLU denotes relative light units. Data is representative of 3 experiments and expressed as mean ± SEM. Statistical significance was assessed by one-way ANOVA. (F) THP-1 transendothelial cell migration in vitro was assessed by treating HMEC-1 cells in gelatin-coated culture inserts with various concentrations of BT2 for 1h, then with 20 ng/mL IL-1ß for 1h. THP-1 cells that had undergone transendothelial migration toward MCP-1 after 24h was measured using a Coulter counter. Data is representative of 3 experiments and expressed as mean ± SEM. Statistical significance was assessed by one-way ANOVA. *P<0.05, **P<0.01, ***P<0.001, ****P< 0.0001. |
BT2 Inhibits Monocytic Cell Adhesion to IL-1ß-Treated Endothelium in vitro and Monocytic Transendothelial Migration Toward MCP-1 in vitro
ICAM-1 and VCAM-1 are well known to mediate monocyte adhesion to endothelial cells.24,25 We used in an in vitro model in which calcein-labeled THP-1 monocytic cells adhere to endothelial cells pretreated with IL-1ß. THP-1 adhesion was inhibited by BT2 in a dose-dependent manner (Figure 1E). In further experiments BT2 also perturbed THP-1 transendothelial migration from the upper to the lower chamber containing MCP-1 (Figure 1F).
Systemic Administration of BT2 Prevents Footpad Swelling and Bone Destruction in Arthritic Mice
Having established in vitro the anti-inflammatory potential of BT2, we hypothesized that BT2 may be useful in a complex pro-inflammatory setting such as collagen antibody induced arthritis.26 ERK undergoes phosphorylation in arthritic mice.27 Hind footpad thickness induced in this model was prevented by systemic administration of 30 mg/kg of BT2 (Figure 2A and B). 3D micro-CT analysis on hind limbs (Figure 2C), quantified using a binary scoring system (Figure 2D), confirmed BT2 prevention of bone destruction.
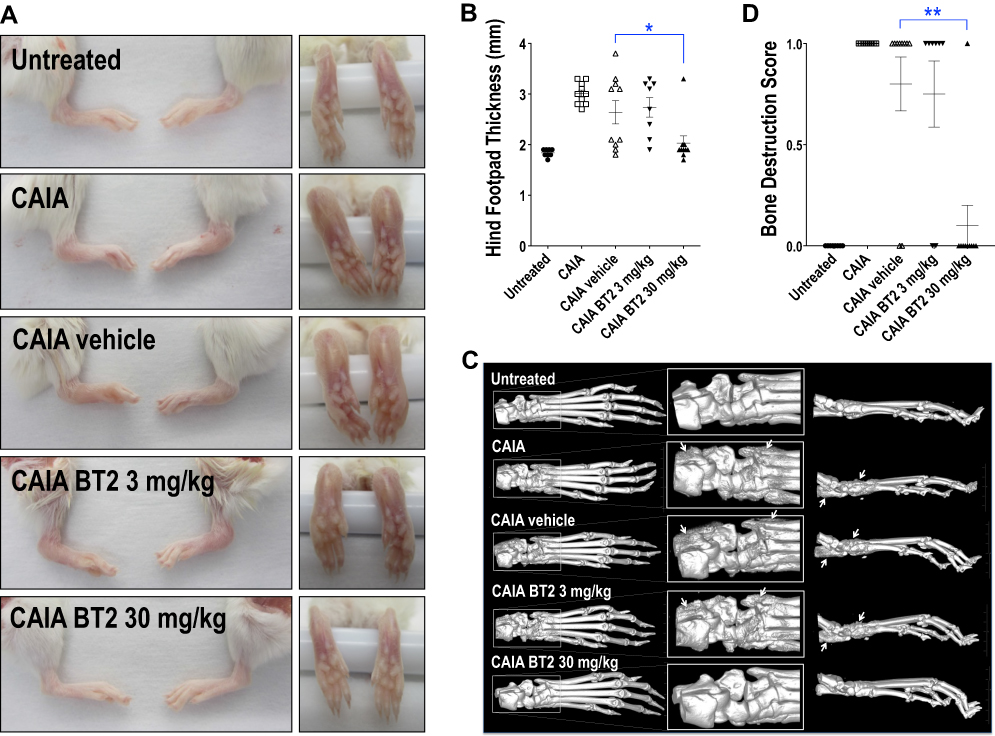 |
Figure 2 BT2 inhibits suppresses footpad swelling and bone erosion in arthritic mice. Animals were injected i.p. with antibody cocktail on Day 0 followed by LPS plus BT2 (3 or 30 mg/kg in vehicle) i.p. on Day 3. (A) Photographs of Day 14 hind feet gross specimens. (B) Hind footpad thickness was measured using digital calipers on Day 9. Data expressed as the hind footpad thickness (mm) of each limb (left and right). n=8–10 per group. Data expressed as mean ± SEM. Statistical significance was assessed by Kruskal–Wallis multiple comparisons test. (C) 3D Micro-CT images of Day 14 hind limbs. Arrows denote bone erosion and/or remodeling. (D) Micro-CT analysis of Day 14 hind limbs was quantified where a score of 0=no bone destruction and 1=destruction was given to each individual limb. Data is expressed as mean bone destruction score per hind limb (left and right) ± SEM. n=8–10 per group. Statistical significance was assessed using Firth’s penalized likelihood method test. *P<0.05, **P<0.01. |
BT2 Inhibits TRAP, ICAM-1 and VCAM-1 Staining in Arthritic Mice
H&E staining revealed extensive joint inflammation in CAIA mice (Figure 3A). We hypothesized that BT2 may influence bone-degrading osteoclast activity in these joints. BT2 reduced staining for tartrate-resistant acid phosphatase (TRAP), a histocytochemical marker of osteoclasts28 (Figure 3B). Immunohistochemical staining further revealed that BT2 inhibited ICAM-1 (Figure 3C) and VCAM-1 (Figure 3D) staining in the joints. ICAM-1 and VCAM-1 are biomarkers of inflammation in the CAIA model.29
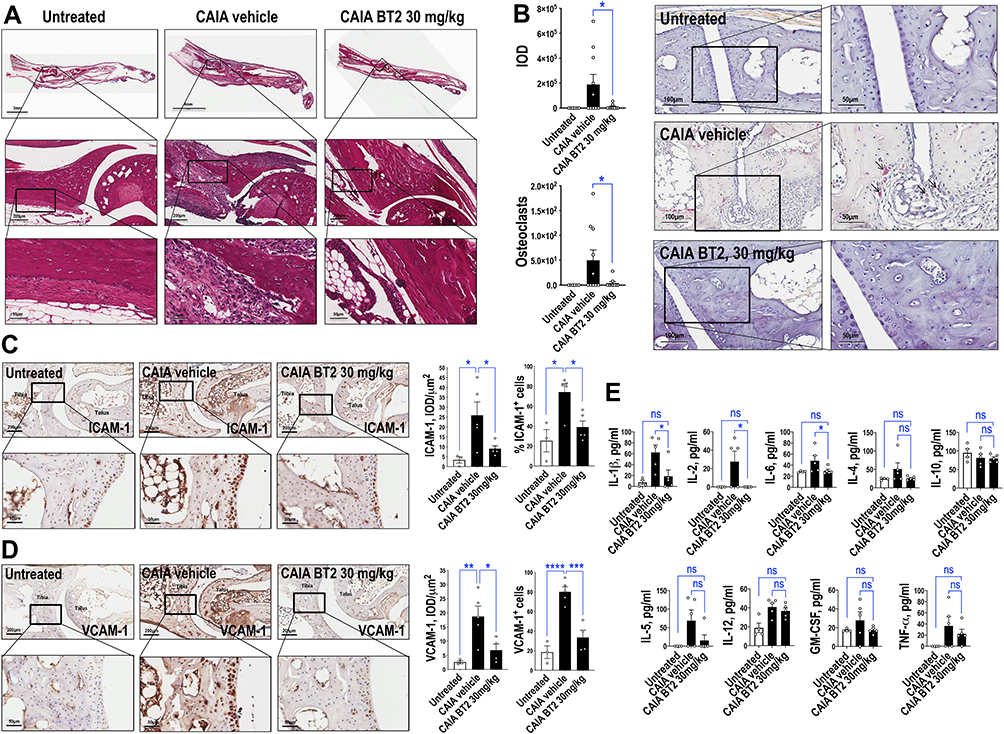 |
Figure 3 BT2 inhibits TRAP, ICAM-1 and VCAM-1 staining and circulating cytokine levels in arthritic mice. Further analysis was performed with material from mice injected i.p. with antibody cocktail on Day 0 then LPS plus BT2 (3 or 30 mg/kg in vehicle) i.p. on Day 3. (A) H&E-stained Day 14 hind limbs. The upper BT2 image was rotated for alignment as shown. (B) TRAP staining of Day 14 hind limbs. Black arrows provide examples of positive staining. Slides were photographed under 20x or 40x objectives. IOD of positive staining (red chromogen) was assessed using Image-Pro Plus software. Alternatively numbers of osteoclasts were counted using NIH Image J. Data represents the mean ± SEM of the means. Statistical significance was assessed by Wilcoxon signed-rank test. n=6–10 per group. (C) Immunohistochemical staining for ICAM-1 in Day 14 hind limbs. IOD/µm2 and percentage of positive staining cell per 20x objective view were assessed for ICAM-1 using Image-Pro Plus software. Data represents the mean ± SEM of the means. Statistical significance was assessed by one-way ANOVA and Mann Whitney test. n=3–5 per group. (D) Immunohistochemical staining for VCAM-1 in Day 14 hind limbs. IOD/µm2 and percentage of positive staining cell per 20x objective view were assessed for VCAM-1 using Image-Pro Plus software. Data represents the mean ± SEM of the means. Statistical significance was assessed by one-way ANOVA. n=3–5 per group. (E) Plasma cytokine levels in arthritic mice treated with BT2. Cytokine levels were detected using a bead-based multiplexed system according to the manufacturer’s protocol. Statistical significance was assessed by Kruskal Wallis test. n=4–5 per group. *P<0.05, **P<0.01, ***P<0.001, ****P< 0.0001. Abbreviation: ns, not significant. |
Suppression of Circulating Pro-Inflammatory Markers by BT2
Cytokines play a critical role in mediating the progression of arthritic disease development. Using a luminex measuring system, BT2 (30 mg/kg) reduced plasma levels of IL-1β, IL-2 and IL-6 to normal levels but did not change IL-4 or IL-10 (Figure 3E). We also observed reductions in levels of various other cytokines including IL-5, TNF-α and GM-CSF, which did not reach statistical significance.
No Evidence of BT2 Toxicity Following Intraarticular, Intraperitoneal or Gavage Administration
Finally, BT2 (3 or 30 mg/kg) was administered to Balb/c mice by one of 3 routes (intraarticular injection, intraperitoneal injection or oral gavage) and tissues were assessed for signs of toxicity. There was no histopathological evidence of toxic damage due to BT2 (Supplementary Table 1). Livers from most mice in all groups contained minimal to mild, infrequent inflammatory foci, occasionally associated with necrosis of individual hepatocytes or small groups of hepatocytes. This is a common, spontaneous background lesion in laboratory mice30 and not test item related. Livers from most mice in groups administered i.p. exhibited minimal to mild inflammation over the capsule, consistent with a non-specific peritoneal reaction to the injection and the effect being unrelated to the test item. Kidneys from one of 5 control mice and 4 of 30 BT2-treated mice contained infrequent inflammatory foci. Again this is a common spontaneous background lesion in laboratory mice30 and not test item related. Inflammation involving the pelvis of the kidney may have been due to ascending bacterial infection of the urinary tract. In liver and lung, there were other very infrequent, minimal changes not related to the treatment group. In summary, there was no histopathological evidence of toxicity following intraarticular, intraperitoneal or gavage administration of BT2.
Discussion
In this paper we show that BT2 prevents joint inflammation and osteoclast activity in arthritic mice. BT2 reduced inducible circulating levels of IL-1ß, IL-2 and IL-6, and suppressed ICAM-1 and VCAM-1 expression in arthritic joints. ICAM-1 and VCAM-1 mediate leukocyte recruitment including leukocyte adhesion and transendothelial migration.31,32 BT2 suppressed human monocytic cell adhesion to IL-1ß-treated endothelium and inhibited monocytic transendothelial migration toward MCP-1. There was no evidence of toxicity following intraarticular, intraperitoneal or gavage administration of BT2. Our findings suggest a potential clinical use for BT2 in inflammatory joint disease.
BT2 emerged from a high-throughput chemical screen of small molecule inhibitors of AP-1. The dibenzoxazepinone inhibits pERK and blocks IL-1β-inducible expression of c-Fos and FosB/∆FosB.17 Signaling pathways mediated by IL-1β, and other pro-inflammatory cytokines such as TNF-α and IL-6, are the subject of bDMARD antagonists in RA (e.g. anakinra as an IL-1R antagonist, tocilizumab as a IL-6R antagonist, etanercept as an TNF-α antagonist).33 pERK is increased in synovial biopsies of patients with early arthritis and in those who develop erosive RA disease suggesting that pharmacologic targeting of ERK may limit inflammation and joint destruction in the pathogenesis of RA.27,34 Focal cartilage breakdown and osteoclastogenesis are characteristic features of RA.35,36 ERK phosphorylation is found in synovial tissue of RA patients undergoing joint replacement.37 In whole joint extracts of arthritic mice, p‐ERK production increases sharply with disease severity.27 AP-1 can regulate the expression of IL-1ß, which can induce AP-1 and matrix metalloproteinase expression that mediate bone destruction.38,39 VCAM-1 and ICAM-1 promoter activity is regulated by AP-1.40,41 Serum sVCAM-1 and sICAM-1 levels are elevated in RA compared with healthy subjects and reflect clinical status in RA.42,43 Serum sVCAM-1 and sICAM-1 levels are also higher in RA compared with patients with osteoarthritis.44 Interestingly, sVCAM-1 levels decrease in RA patients as the condition is relieved.45
Cytokines play key roles in the pathogenesis and pathophysiology of RA.46 Elevated expression of IL-1 family cytokines promote inflammatory responses in RA.2 IL-1β likely mediates many pro-inflammatory pathways that relate to RA symptoms.46 IL-2 increases proliferation and expansion of T cells.47 Lower IL-2R levels coincide with joint destruction.48 In contrast, IL-4 and IL-10 are anti-inflammatory cytokines49,50 that can regulate the production of IL-1 and TNF-α in RA synovial tissue. Blocking both IL-4 and IL-10 promotes experimental arthritis.51 Administration of recombinant IL-10 results in inhibition of TNF-α and IL-1β.49,52 That BT2 reduced circulating IL-1β, IL-2 and IL-6 levels in arthritic mice to normal levels without affecting IL-4 and IL-10 levels, complements our morphologic data demonstrating BT2 inhibition of joint inflammation and bone erosion. The 10-plex panel did not capture other key cytokines, such as IL-17, which has been associated with angiogenesis and osteoclastogenesis.53
Since BT2 was administered 3 days after antibody cocktail injection before arthritis fully develops in the CAIA model53 a limitation of this study is that it demonstrates the prophylactic or preventative rather than curative activity of BT2. Nonetheless, like other studies using the CAIA model testing JAK inhibitors that also commenced therapy before paw inflammation and thickening,53 we provide in principle evidence for the potential clinical utility of BT2.
Abbreviations
AP-1, activator protein-1; BT2, (10-ethyl-11-oxo-10,11-dihydro-dibenzo[b,f][1,4]oxazepin-2-yl)-carbamic acid ethyl ester); BT3, 2-amino-10-ethyldibenzo[b,f][1,4] oxazepin-11 (10H)-one; CAIA, collagen antibody induced arthritis; HMEC-1, human microvascular endothelial cells; IL, interleukin; ICAM-1, intercellular cell adhesion molecule-1; IOD, integrated optical density; JAK, Janus kinase; pERK, ERK phosphorylation; RA, rheumatoid arthritis; THP-1, human monocytic leukemia cells; VCAM-1, vascular cell adhesion molecule-1.
Data Sharing Statement
All data generated or analysed during this study are included in this published article and its Supplementary Material.
Acknowledgments
This work was supported by grants from the National Health and Medical Research Council of Australia (NHMRC), Cancer Institute New South Wales (CINSW) and National Heart Foundation of Australia. The authors acknowledge the facilities, scientific and technical assistance of the National Imaging Facility at the Biological Resources Imaging Laboratory, Mark Wainwright Analytical Centre, UNSW) with CT scans particularly by Dr Tzong-Tyng Hung; Dr Fei Shang (Biomedical Imaging Facility, Mark Wainwright Analytical Centre, UNSW) for immunohistochemical staining; A/Professor Nicodemus Tedla for advice with TRAP staining; Professor John Mackie (University of Queensland, Australia) for pathologic assessment of sections in the toxicity study; and Emeritus Professor Ian Dawes (UNSW) for critical review of the manuscript.
Author Contributions
All authors made a significant contribution to the work reported, whether that is in the conception, study design, execution, acquisition of data, analysis and interpretation, or in all these areas; took part in drafting, revising or critically reviewing the article; gave final approval of the version to be published; have agreed on the journal to which the article has been submitted; and agree to be accountable for all aspects of the work.
Disclosure
UNSW filed a provisional patent application related to this work. The authors report no other conflicts of interest for this work.
References
1. Chen Z, Bozec A, Ramming A, Schett G. Anti-inflammatory and immune-regulatory cytokines in rheumatoid arthritis. Nat Rev Rheumatol. 2019;15(1):9–17. doi:10.1038/s41584-018-0109-2
2. McInnes IB, Schett G. The pathogenesis of rheumatoid arthritis. N Engl J Med. 2011;365(23):2205–2219. doi:10.1056/NEJMra1004965
3. McInnes IB, Schett G. Cytokines in the pathogenesis of rheumatoid arthritis. Nat Rev Immunol. 2007;7(6):429–442. doi:10.1038/nri2094
4. Clough JD. The Cleveland Clinic Guide to Arthritis. New York: Kaplan Publishing; 2009.
5. Xavier RM, Zerbini CAF, Pollak DF, et al. Burden of rheumatoid arthritis on patients’ work productivity and quality of life. Adv Rheumatol. 2019;59(1):47. doi:10.1186/s42358-019-0090-8
6. Breedveld FC, Strand V, Boklage SH, et al. The PREMIER study: a multicenter, randomized, double-blind clinical trial of combination therapy with adalimumab plus methotrexate versus methotrexate alone or adalimumab alone in patients with early, aggressive rheumatoid arthritis who had not had previous methotrexate treatment. Arthritis Rheum. 2017;69(1):26–37. doi:10.1002/art.39996
7. Emery P, Breedveld FC, Hall S, et al. Comparison of methotrexate monotherapy with a combination of methotrexate and etanercept in active, early, moderate to severe rheumatoid arthritis (COMET): a randomised, double-blind, parallel treatment trial. Lancet. 2008;372:375–382. doi:10.1016/S0140-6736(08)61000-4
8. Jones G, Sebba A, Gu J, et al. Comparison of tocilizumab monotherapy versus methotrexate monotherapy in patients with moderate to severe rheumatoid arthritis: the AMBITION study. Ann Rheum Dis. 2010;69(01):88–96. doi:10.1136/ard.2008.105197
9. Westhovens R, Robles M, Ximenes AC, et al. Clinical efficacy and safety of abatacept in methotrexate-naive patients with early rheumatoid arthritis and poor prognostic factors. Ann Rheum Dis. 2009;68:1870–1877. doi:10.1136/ard.2008.101121
10. Smolen JS, Aletaha D, Gruben D, et al. Brief report: remission rates with tofacitinib treatment in rheumatoid arthritis: a comparison of various remission criteria. Arthritis Rheum. 2017;69(4):728–734. doi:10.1002/art.39996
11. Strand V, Boklage SH, Kimura T, et al. High levels of interleukin-6 in patients with rheumatoid arthritis are associated with greater improvements in health-related quality of life for sarilumab compared with adalimumab. Arthritis Res Ther. 2020;22(1):250. doi:10.1186/s13075-020-02344-3
12. Rubbert-Roth A, Enejosa J, Pangan AL, et al. Trial of upadacitinib or abatacept in rheumatoid arthritis. N Engl J Med. 2020;383(16):1511–1521. doi:10.1056/NEJMoa2008250
13. Siebert S, Tsoukas A, Robertson J, McInnes I, Touyz RM. Cytokines as therapeutic targets in rheumatoid arthritis and other inflammatory diseases. Pharmacol Rev. 2015;67(2):280–309. doi:10.1124/pr.114.009639
14. Paulus YM, Gariano RF. Diabetic retinopathy: a growing concern in an aging population. Geriatrics. 2009;64(2):16–20.
15. van Onna M, Boonen A. The challenging interplay between rheumatoid arthritis, ageing and comorbidities. BMC Musculoskelet Disord. 2016;17(1). doi:10.1186/s12891-016-1038-3
16. Fazal SA, Khan M, Nishi SE, et al. A clinical update and global economic burden of rheumatoid arthritis. Endocr Metab Immune Disord Drug Targets. 2018;18(2):98–109. doi:10.2174/1871530317666171114122417
17. Li Y, Alhendi AMN, Yeh M-C, et al. Thermostable small-molecule inhibitor of angiogenesis and vascular permeability that suppresses a pERK-FosB/ΔFosB–VCAM-1 axis. Sci Adv. 2020;6(31):eaaz7815. doi:10.1126/sciadv.aaz7815
18. Hess J, Angel P, Schorpp-Kistner M. AP-1 subunits: quarrel and harmony among siblings. J Cell Sci. 2004;117:5965–5973. doi:10.1242/jcs.01589
19. Ye N, Ding Y, Wild C, Shen Q, Zhou J. Small molecule inhibitors targeting activator protein 1 (AP-1). J Med Chem. 2014;57(16):6930–6948. doi:10.1021/jm5004733
20. Asahara H, Fujisawa K, Kobata T, et al. Direct evidence of high DNA binding activity of transcription factor AP-1 in rheumatoid arthritis synovium. Arthritis Rheum. 1997;40(5):912–918. doi:10.1002/art.1780400520
21. Gad SC, Cassidy CD, Aubert N, Spainhour B, Robbe H. Nonclinical vehicle use in studies by multiple routes in multiple species. Int J Toxicol. 2006;25(6):499–521. doi:10.1080/10915810600961531
22. Dayer JM. The pivotal role of interleukin-1 in the clinical manifestations of rheumatoid arthritis. Rheumatology. 2003;42(Suppl 2):ii3–ii10. doi:10.1093/rheumatology/keg326
23. Okamoto H, Hoshi D, Kiire A, Yamanaka H, Kamatani N. Molecular targets of rheumatoid arthritis. Inflamm Allergy Drug Targets. 2008;7(1):53–66. doi:10.2174/187152808784165199
24. Burgazli KM, Venker CJ, Mericliler M, et al. Importance of large conductance calcium-activated potassium channels (BKCa) in interleukin-1b-induced adhesion of monocytes to endothelial cells. Eur Rev Med Pharmacol Sci. 2014;18(5):646–656.
25. Gerszten RE, Lim Y-C, Ding HT, et al. Adhesion of monocytes to vascular cell adhesion molecule-1-transduced human endothelial cells: implications for atherogenesis. Circ Res. 1998;82(8):871–878. doi:10.1161/01.RES.82.8.871
26. Khachigian LM. Collagen antibody-induced arthritis. Nat Protoc. 2006;1(5):2512–2516. doi:10.1038/nprot.2006.393
27. Thiel MJ, Schaefer CJ, Lesch ME, et al. Central role of the MEK/ERK MAP kinase pathway in a mouse model of rheumatoid arthritis: potential proinflammatory mechanisms. Arthritis Rheum. 2007;56(10):3347–3357. doi:10.1002/art.22869
28. Ballanti P, Minisola S, Pacitti MT, et al. Tartrate-resistant acid phosphate activity as osteoclastic marker: sensitivity of cytochemical assessment and serum assay in comparison with standardized osteoclast histomorphometry. Osteoporos Int. 1997;7(1):39–43. doi:10.1007/BF01623458
29. Cheng T, Wang M, Chen L, et al. Increased expression of CD40/TRAF1 and activation of nuclear factor-κκB-dependent proinflammatory gene expression in collagen-induced arthritis. Scand J Rheumatol. 2018;47(6):455–460. doi:10.1080/03009742.2018.1432684
30. Taylor. I in Background Lesions in Laboratory Animals. (Ed. E.F. McInnes). Edinburgh: Saunders Elsevier; 2012:45–75.
31. Pober JS. Endothelial activation: intracellular signaling pathways. Arthritis Res. 2002;4(Suppl 3):S109–S116. doi:10.1186/ar576
32. Schlesinger M, Bendas G. Vascular cell adhesion molecule-1 (VCAM-1)-an increasing insight into its role in tumorigenicity and metastasis. Int J Cancer. 2015;136(11):2504–2514. doi:10.1002/ijc.28927
33. Wilsdon TD, Hill CL. Managing the drug treatment of rheumatoid arthritis. Aust Prescr. 2017;40:51–58. doi:10.18773/austprescr.2017.012
34. de Launay D, van de Sande MGH, de Hair MJH, et al. Selective involvement of ERK and JNK mitogen-activated protein kinases in early rheumatoid arthritis (1987 ACR criteria compared to 2010 ACR/EULAR criteria): a prospective study aimed at identification of diagnostic and prognostic biomarkers as well as therapeutic targets. Ann Rheum Dis. 2012;71(3):415–423. doi:10.1136/ard.2010.143529
35. Karmakar S, Kay J, Gravallese EM. Bone damage in rheumatoid arthritis: mechanistic insights and approaches to prevention. Rheum Dis Clin North Am. 2010;36(2):385–404. doi:10.1016/j.rdc.2010.03.003
36. Scott DL. Radiological progression in established rheumatoid arthritis. J Rheumatol Suppl. 2004;69:55–65.
37. Schett G, Tohidast-Akrad M, Smolen JS, et al. Activation, differential localization, and regulation of the stress-activated protein kinases, extracellular signal-regulated kinase, c-JUN N-terminal kinase, and p38 mitogen-activated protein kinase, in synovial tissue and cells in rheumatoid arthritis. Arthritis Rheum. 2000;43(11):2501–2512. doi:10.1002/1529-0131(200011)43:11<2501::aid-anr18>3.0.CO;2-K
38. Shiozawa S, Tsumiyama K. Pathogenesis of rheumatoid arthritis and c-Fos/AP-1. Cell Cycle. 2009;8(10):1539–1543. doi:10.4161/cc.8.10.8411
39. Burrage PS, Mix KS, Brinckerhoff CE. Matrix metalloproteinases: role in arthritis. Front Biosci. 2006;11:529–543. doi:10.2741/1817
40. Ahmad M, Theofanidis P, Medford RM. Role of activating protein-1 in the regulation of the vascular cell adhesion molecule-1 gene expression by tumor necrosis factor-alpha. J Biol Chem. 1998;273:4616–4621. doi:10.1074/jbc.273.8.4616
41. Wang N, Verna L, Hardy S, et al. Adenovirus-mediated overexpression of c-jun and c-fos induces intercellular adhesion molecule-1 and monocyte chemoattractant protein-1 in human endothelial cells. Arterioscler Thromb Vasc Biol. 1999;19(9):2078–2084. doi:10.1161/01.ATV.19.9.2078
42. Navarro-Hernandez RE, Oregon-Romero E, Vázquez-Del Mercado M, et al. Expression of ICAM1 and VCAM1 serum levels in rheumatoid arthritis clinical activity. Association with genetic polymorphisms. Dis Markers. 2009;26(3):119–126. doi:10.1155/2009/510104
43. Littler AJ, Buckley CD, Wordsworth P, et al. A distinct profile of six soluble adhesion molecules (ICAM-1, ICAM-3, VCAM-1, E-selectin, L-selectin and P-selectin) in rheumatoid arthritis. Br J Rheumatol. 1997;36(2):164–169. doi:10.1093/rheumatology/36.2.164
44. Klimiuk PA, Sierakowski S, Latosiewicz R, et al. Soluble adhesion molecules (ICAM-1, VCAM-1, and E-selectin) and vascular endothelial growth factor (VEGF) in patients with distinct variants of rheumatoid synovitis. Ann Rheum Dis. 2002;61(9):804–809. doi:10.1136/ard.61.9.804
45. Wang L, Ding Y, Guo X, Zhao Q. Role and mechanism of vascular cell adhesion molecule-1 in the development of rheumatoid arthritis. Exp Ther Med. 2015;10(3):1229–1233. doi:10.3892/etm.2015.2635
46. McInnes IB, Buckley CD, Isaacs JD. Cytokines in rheumatoid arthritis — shaping the immunological landscape. Nat Rev Rheumatol. 2016;12(1):63–68. doi:10.1038/nrrheum.2015.171
47. Magyari L, Varszegi D, Kovesdi E, et al. Interleukins and interleukin receptors in rheumatoid arthritis: research, diagnostics and clinical implications. World J Orthop. 2014;5(4):516–536. doi:10.5312/wjo.v5.i4.516
48. Knevel R, de Rooy DPC, Zhernakova A, et al. Association of variants in IL2RA with progression of joint destruction in rheumatoid arthritis. Arthritis Rheum. 2013;65(7):1684–1693. doi:10.1002/art.37938
49. Katsikis PD, Chu CQ, Brennan FM, Maini RN, Feldmann M. Immunoregulatory role of interleukin 10 in rheumatoid arthritis. J Exp Med. 1994;179(5):1517–1527. doi:10.1084/jem.179.5.1517
50. Chatterjee P, Chiasson VL, Bounds KR, Mitchell BM. Regulation of the anti-inflammatory cytokines interleukin-4 and interleukin-10 during pregnancy. Front Immunol. 2014;5:253. doi:10.3389/fimmu.2014.00253
51. Lubberts E, van den Berg WB. Cytokines in the pathogenesis of rheumatoid arthritis and collagen-induced arthritis. Adv Exp Med Biol. 2003;520:194–202. doi:10.1007/978-1-4615-0171-8_11
52. St Clair EW. Interleukin 10 treatment for rheumatoid arthritis. Ann Rheum Dis. 1999;58(Suppl 1):I99–I102. doi:10.1136/ard.58.2008.i99
53. Stump KL, Lu LD, Dobrzanski P, et al. A highly selective, orally active inhibitor of janus kinase 2, CEP-33779, ablates disease in two mouse models of rheumatoid arthritis. Arthritis Res Ther. 2011;13(2):R68. doi:10.1186/ar3329
 © 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.