")
Back to Journals » OncoTargets and Therapy » Volume 13
Brevilin A Inhibits STAT3 Signaling and Induces ROS-Dependent Apoptosis, Mitochondrial Stress and Endoplasmic Reticulum Stress in MCF-7 Breast Cancer Cells
Authors Saleem MZ , Nisar MA , Alshwmi M, Din SRU, Gamallat Y , Khan M , Ma T
Received 26 August 2019
Accepted for publication 10 December 2019
Published 15 January 2020 Volume 2020:13 Pages 435—450
DOI https://doi.org/10.2147/OTT.S228702
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Professor Yong Teng
Muhammad Zubair Saleem, 1 Muhammad Azhar Nisar, 1 Mohammed Alshwmi, 2 Syed Riaz Ud Din, 1 Yaser Gamallat, 1 Muhammad Khan, 3 Tonghui Ma 1
1College of Basic Medical Sciences, Dalian Medical University, Dalian, Liaoning 116044, People’s Republic of China; 2Department of Clinical Laboratory, The First Affiliated Hospital of Dalian Medical University, Dalian Medical University, Dalian, Liaoning 116044, People’s Republic of China; 3Department of Zoology, University of the Punjab, Lahore, Punjab 54590, Pakistan
Correspondence: Muhammad Khan
Department of Zoology, University of the Punjab, Quaid-e-Azam Campus, Lahore, Punjab 54590, Pakistan
Tel +92-42-99231246
Email [email protected]
Tonghui Ma
College of Basic Medical Sciences, Dalian Medical University, 9 Western Section, Lvshun South Road, Dalian 116044, People’s Republic of China
Tel +86 411 8611 0278
Fax +86 411 8611 0378
Email [email protected]
Purpose: Breast cancer is the most common malignancy among women across the globe. Despite concerted efforts to improve the prevailing treatment modalities, the overall prognosis of breast cancer remains unsatisfactory. Recently, antiproliferative activity of Brevilin A (Brv-A), a sesquiterpene lactone compound of Centipeda minima, has been unveiled in various cancer types. Here, we have explored anticancer activity of Brv-A in MCF-7 breast carcinoma cells by targeting various pathways.
Materials and Methods: Cell proliferation rate was determined by CCK-8 and clonogenic assay. Cellular morphological changes were observed under phase contrast microscope while calcein-AM and PI was used for live/dead assay. Cell cycle assay was performed by flow cytometry. Apoptotic cell percentage was determined by Hoechst 33258 staining and flow cytometric analysis. ROS generation and mitochondrial membrane potential were measured using commercially available kits while protein expression was measured by Western blotting.
Results: In our study, Brv-A exerted antiproliferative effect through mitotic arrest at G 2/M phase of cell cycle and induced apoptosis in MCF-7 cells in a dose-dependent manner. Induction of apoptosis by Brv-A was found to be associated with ROS generation by targeting NOX2 and NOX3, mitochondrial dysfunction (MMP dissipation and Bcl-2 family proteins modulation), DNA fragmentation, JNK and p38 MAPK activation, endoplasmic reticulum (ER) stress by increasing Bip/GRP78, ATF4 and CHOP protein expressions and inhibition of STAT3 activation via decreased phosphorylation of JAK2 and SRC. Pretreatment of NAC, a ROS scavenger, partially reversed the aforesaid cellular events indicating ROS generation as the primary event to modulate cellular targets for induction of apoptosis. Besides, Brv-A has also been documented for inhibition of cell migration via decrease in COX-2 and MMP-2 expression.
Conclusion: Taken together, Brv-A induces G 2/M phase arrest, ROS-dependent apoptosis, ER stress, mitochondrial dysfunction and inhibits STAT3 activation in MCF-7 cells signifying it to be one of the potential anticancer therapeutics in future.
Keywords: breast cancer, Brevilin A, apoptosis, ROS, ER stress, STAT3, mitochondrial dysfunction
Introduction
Currently, cancer research has been privileged over the last century due to multiple factors. All cancer types including breast cancer have immersed the researcher’s attention to the devastating aspects in its diagnosis, care and treatment. Breast cancer, a heterogeneous disease, is diagnosed in over one million new patients every year which accounts for 23% of total new cases, 16% of all female cancers and 18% of total deaths related to cancer in both males and females.1,2 Incidence and prevalence rate of breast cancer is higher in North America, Northern and Western Europe, New Zealand and Australia compared to Eastern Asia.3 In China, however, the incidence rate of breast cancer has increased in last three decades by 20 to 30% and, according to Chinese urban cancer registries, it amounts about 3–5% each year.4,5 This rise in incidence is higher than global incidence ascends (1.5%) of breast cancer.6
On the basis of a patient’s history, characteristics and pathology of the disease, breast cancer is managed by different strategies including surgery, radiation therapy, targeted endocrine therapy, and chemotherapy. However, resistance to available therapeutics and high toxicity with severe side effects has provoked interest in antitumor drug discoveries with more accurate targets and low toxicity.7 In general, equalized state of antioxidants production is very important to maintain intracellular ROS level for normal physiological functions. Imbalance in the state brings oxidative stress that disturbs cell physiology leading to cell death.8 Role of NADPH oxidases (NOX) is well-known in immune response against microorganisms as respiratory burst, endothelial signals, cell proliferation and maintenance of renal functions. Beside respiratory chain, the main sources for ROS generation are some NOX family proteins.9–11 It is a group of transmembrane proteins involved in transportation of electrons from NADPH and reduction of oxygen to ROS superoxide anions and hydrogen peroxide.12 A lot of potential anticancer drugs are known for killing cancer cells by enhancing ROS level.13,14
Traditional Chinese Medicines (TCM) have multiple targets with low toxicity and high precision that helps to treat different diseases. Centipeda minima, a traditional Chinese herb, was initially used to treat respiratory diseases but later discovered to be effective as antibacterial, anti-allergic and antitumor agent.15 Brevilin A (Brv-A) is a natural, plant derived, bioactive sesquiterpene lactone component of Centipeda minima. It has been reported that Brv-A exerts anti-inflammatory and antiproliferative effects and exhibits anticancer activities in multiple cancer types promoting oxidative stress, mitochondrial dysfunction and more specifically STAT3 inhibition in DU145 and MDA-MB-468 cell lines.15,16 Anticancer activity of Brv-A has been shown in different types of cell lines including A549, HeLa, CT26, HepG2, A875 and U87 glioblastoma17 but precise molecular mechanism and mode of cell fatality is still unknown against breast cancer. The current study aimed to investigate cytotoxic and growth inhibitory effect of Brv-A in MCF-7 breast cancer cells along with underlying mechanism. In our study, ROS generation via modulation of NOX2, NOX3 proteins and induction of ER stress by Brv-A discloses previously unrecognized mechanisms. This multi-pathway targeting property of Brv-A augments its role as potential breast cancer therapeutics in future.
Materials and Methods
Chemicals and Reagents
Brevilin A with highest purity grade of >98% was purchased from Dalian Meilun Biotechnology, Co. Ltd. DMEM was purchased from Gibco (Thermo Fisher Scientific, Waltham, MA, USA). FBS was obtained from Gibco (Thermo Fisher). Trypsin with or without EDTA was obtained from Gibco (Thermo Fisher). Penicillin and streptomycin were purchased from Solarbio Co. Ltd. (Beijing, China). Cell counting Kit-8 (CCK-8) was purchased from Bimake (China) while dimethyl sulfoxide (DMSO), calcein-AM, propidium iodide (PI), phenylmethylsulfonyl fluoride (PMSF) and protease inhibitor cocktail were obtained from Sigma-Aldrich Co. (St Louis, MO, USA). Hoechst 33258 was purchased from Beyotime Institute of Biotechnology (Nanjing, China). Mitochondrial membrane potential assay kit (JC-1), reactive oxygen species (ROS) assay kit, annexin V-FITC apoptosis detection kit, crystal violet (CV) stain and N-Acetyl-L-cysteine (NAC) were purchased from Beyotime Biotechnology (Haimen, Jiangsu, China). Primary antibodies for cleaved PARP, cleaved caspase-9, BAX, BAK, STAT3 and p-STAT3 (Tyr 705), SRC and p-SRC, COX-2, p38 MAPK and p-p38 MAPK, JNK (1/2) and p-JNK (1/2), BiP/GRP78 and MMP-2 were purchased from Cell Signaling Technology (Beverly, MA, USA). Bcl-2, XIAP, SHP-2, NOX2, NOX3, ATF4 and GAPDH were procured from Proteintech (Wuhan, China) while CHOP was purchased from Beyotime (China). The primary antibodies for p-JAK2 and JAK2 were purchased from Abcam (Cambridge, MA, USA). Goat anti-rabbit and goat anti-mouse secondary antibodies were obtained from Sigma-Aldrich.
Cell Culture
Human MCF-7 and MDA-MB-231 breast cancer cell lines were purchased from American Type Culture Collection (Manassas, VA, USA) and were cultured in high glucose DMEM. The medium was supplemented with 10% FBS, 100 units/mL penicillin and 100 μg/mL streptomycin. Cells were kept in complete humidified atmosphere with 5% CO2 at 37°C.
Cell Viability Assay
Cell viability was measured by CCK-8 assay kit according to the manufacturer’s instructions. Briefly, MCF-7 and MDA-MB-231 cells were seeded in 96-well microtiter plates. After overnight incubation at 37°C, cells were treated with different indicated concentrations of Brv-A in presence or absence of NAC (5 mM) for 24 h. Following treatments, 10 µL of CCK-8 solution was added into each well and kept for 3 h at 37°C. Optical density was recorded at wavelength of 450 nm by using fluorescence microplate reader (Synergy neo HTS multimode microplate reader, BioTek). All experiments were performed in triplicate and cell viability was calculated using following formula.
Cell viability (%) = (A450 sample – A450 blank)/(A450 control – A450 blank) × 100
where “sample” shows optical density (OD) of drug treated cells with CCK-8 solution while “blank” represents OD value of culture medium with CCK-8 but without cells.
Microscopic Observations of Cell Morphology
MCF-7 cells were seeded and incubated in 96-well plates for 24 h at 37°C. Further, cells were treated with indicated concentrations of Brv-A in presence or absence of NAC for 24 h. The cellular morphological changes were observed under phase contrast microscope (Leica, DMIL LED, Leica Microsystems, Wetzlar, Germany) and photographed.
Live/Dead Assay
Live/dead assay was performed according to the protocol described previously.18 Briefly, cells were seeded and treated in 6-well plates with Brv-A for 24 h in presence or absence of NAC. Cells were then washed twice with and re-suspended in PBS. The samples were incubated with 2 µM calcein-AM and 4 µM PI for 20 min in dark at room temperature. Cells were washed and re-suspended in PBS and analyzed under fluorescence microscope (Leica, DMIL 4000B).
Colony Forming Assay
MCF-7 cells were treated with indicated concentrations of Brv-A for 24 h in presence or absence of NAC. After washing, cells were trypsinized and 300 cells/well were seeded and cultured into 6-well plate in DMEM. Cells were allowed to form colonies at 37°C for 10 days in humidified atmosphere. Old media was replaced after 48 h. Once colonies were formed, cells were then washed and fixed with 4% paraformaldehyde (PFA) for 15 min and stained with crystal violet. After 20 min, colonies were washed with PBS and photographed. To compute cells proliferative ratio, methanol was used to dissolve crystal violet and absorbance was measured at 595 nm by fluorescent spectrophotometer (Synergy neo HTS multimode microplate reader, BioTek).
Cell Cycle Analysis
Cell cycle was performed by flow cytometry as described previously.19 Briefly, MCF-7 cells were treated with 10 µM and 15 µM Brv-A in presence or absence of NAC for 24 h. Following treatment, cells were harvested, washed with PBS and fixed with 70% ethanol at 4°C overnight. After washing, cells were rinsed with ice-cold PBS containing 25 µL propidium iodide and 10 µL RNase for 30 min at 37°C in the dark. Cell cycle analysis was performed using BD FACS Calibur flow cytometry system. PI FlowJo 7.6 software was used to determine the proportions of cells in different cell stages of cell cycle progression (G0/G1, S, and G2/M phases).
Hoechst 33258 Staining for Nuclear Morphology
MCF-7 cells were treated with indicated concentrations of Brv-A in presence or absence of NAC for 24 h. Cells were washed, collected and fixed in 4% paraformaldehyde at room temperature for 30 min. Cells were washed twice with PBS and stained with Hoechst 33258 (20 µg/mL) for 20 min in dark at room temperature. After staining, the cells were washed with PBS to remove extra stain. Cells were re-suspended in PBS and nuclear morphology was observed under fluorescence microscope (Leica, DMI 4000B).
Apoptosis Assay
Apoptosis assay was done by apoptosis assay kit (Beyotime, China) according to the manufacturer's instructions. Briefly, cells were seeded and treated with indicated concentrations of Brv-A in presence or absence of NAC in 6-well plates for 24 h. Cells were washed with PBS and resuspended in binding buffer. The samples were incubated with 5 µL annexin V-FITC and 10 µL PI for 15 min in the dark. After filtration, the samples were analyzed by flow cytometry (BD Accuri C6) to determine percentage of apoptosis.
Measurement of Intracellular Reactive Oxygen Species (ROS) Generation
ROS generation was measured by ROS assay kit (Beyotime, China) according to manufacturer’s instructions. Briefly, MCF-7 cells were seeded in 96-well plates and treated with Brv-A in presence/absence of NAC in a time-dependent manner. Cells were then incubated with 2ʹ, 7ʹ-dichlorofluorescein diacetate (DCFH-DA) dissolved in serum-free DMEM (1:1000) in the dark for 30 min. DCF fluorescence was measured by fluorescence microplate reader (Synergy neo HTS multimode microplate reader, BioTek) with excitation wavelength of 488 nm and emission wavelength of 525 nm.
Measurement of Mitochondrial Membrane Potential (MMP)
Mitochondrial membrane potential (MMP) was measured by using MMP assay kit with JC-1 (Beyotime, China) according to manufacturer’s instructions. Briefly, MCF-7 cells were treated with 10 µM and 15 µM concentrations of Brv-A in presence/absence of NAC for 24 h. Cells were washed with serum-free DMEM and incubated in JC-I working solution for 20 min at 37°C in the dark. Cells were collected by centrifugation, washed with PBS and re-suspended in JC-1 dying buffer. After that, JC-1 monomer fluorescence distribution with excitation wavelength of 490 nm and emission wavelength of 530 nm and j-aggregates with excitation wavelength of 525 nm and emission wavelength of 590 nm was measured using fluorescent spectrophotometer (Synergy neo HTS multimode microplate reader, BioTek). MMP of control and treated groups was calculated by decrease in red/green fluorescence intensity ratio.
Wound Healing Assay
Cells were cultured in 6-well plates and kept at 37°C to attain confluence up to 80%. A monolayer of cells was scratched in a straight line using autoclaved 200 µL pipette tip. Medium was removed and cells were washed with PBS. Cells were exposed to new DMEM with indicated concentrations of Brv-A for 0, 12 and 24 h. Scratched wound was observed and photographed under microscope (Leica, DMIL 4000B).
Western Blotting
MCF-7 cells were treated with indicated concentrations of Brv-A in the presence or absence of NAC. Cells were collected and washed twice with cold PBS. After washing, cells were lysed with radio immunoprecipitation assay RIPA (Beyotime Biotechnology) supplemented with 2% sodium fluoride (NaF) in case of phosphorylated proteins and 1% phenylmethylsulfonyl fluoride (PMSF) on ice for 30 min. Cells were centrifuged at 12,000 rpm for 15 min and supernatant was collected in ice chilled tubes. BCA protein assay kit (Beyotime Biotechnology) was used to determine protein concentrations by spectrophotometer (Synergy neo HTS multimode microplate reader, BioTek). 30–40 µg proteins were loaded on 10–12% sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE) and separated. Proteins were transferred to polyvinylidene difluoride (PVDF) membranes. After transfer, PVDF membranes were incubated in 5% skim milk for one hour for blocking. After washing with Tris-buffered saline-Tween (TBST) three times, membranes were incubated with corresponding primary antibodies at 4°C overnight. After washing with TBST three times, membranes were incubated with HRP-conjugated goat anti-rabbit IgG or goat anti-mouse IgG secondary antibodies for 1 h at room temperature. Membranes were washed three times with TBST and Immobilon Western Chemi-luminescent HRP substrate (EMD Millipore, Billerica, MA, USA) was used to detect immune-reactive bands. Micro-Chemi 4.2 imaging system (DNR Bio-Imaging system) was used to capture chemi-luminescence images. In every individual blot, GAPDH was detected and measured as loading control. All blots were repeated three times and each blot is representative of three different Western blotting experiments. Variations in protein expression was determined using ImageJ software and represented in graphical scheme.
Statistical Analysis
For three different experiments, results were expressed as mean ± SD and compared statistically with control (untreated or treated group). Student’s t-test was used for comparison of only two groups. For comparison between more than two groups, one-way ANOVA was used followed by Tukey’s multiple comparison test. p <0.05 was considered to indicate significant difference.
Results
Brv-A Inhibits Proliferation and Induces Cytotoxic Effect in Breast Cancer Cells
For comparative evaluation, we measured growth inhibitory and cytotoxic effect of Brv-A in MCF-7 as well as MDA-MB-231 breast cancer cell lines. Following CCK-8 assay, Brv-A was found effective against both triple positive and triple negative breast cancer cells (Figure 1A and B). The calculated IC50 value for MCF-7 and MDA-MB-231 was approximately 17 µM and 23 µM respectively. As current study aimed to explore anticancer effect of Brv-A in triple positive breast cancer type, we selected MCF-7 cell line as a model for further mechanistic study. Among concentration gradient from 5 to 90 µM, 10 and 15 µM were found to be the most suitable concentrations to formulate effect of Brv-A in MCF-7 cells.
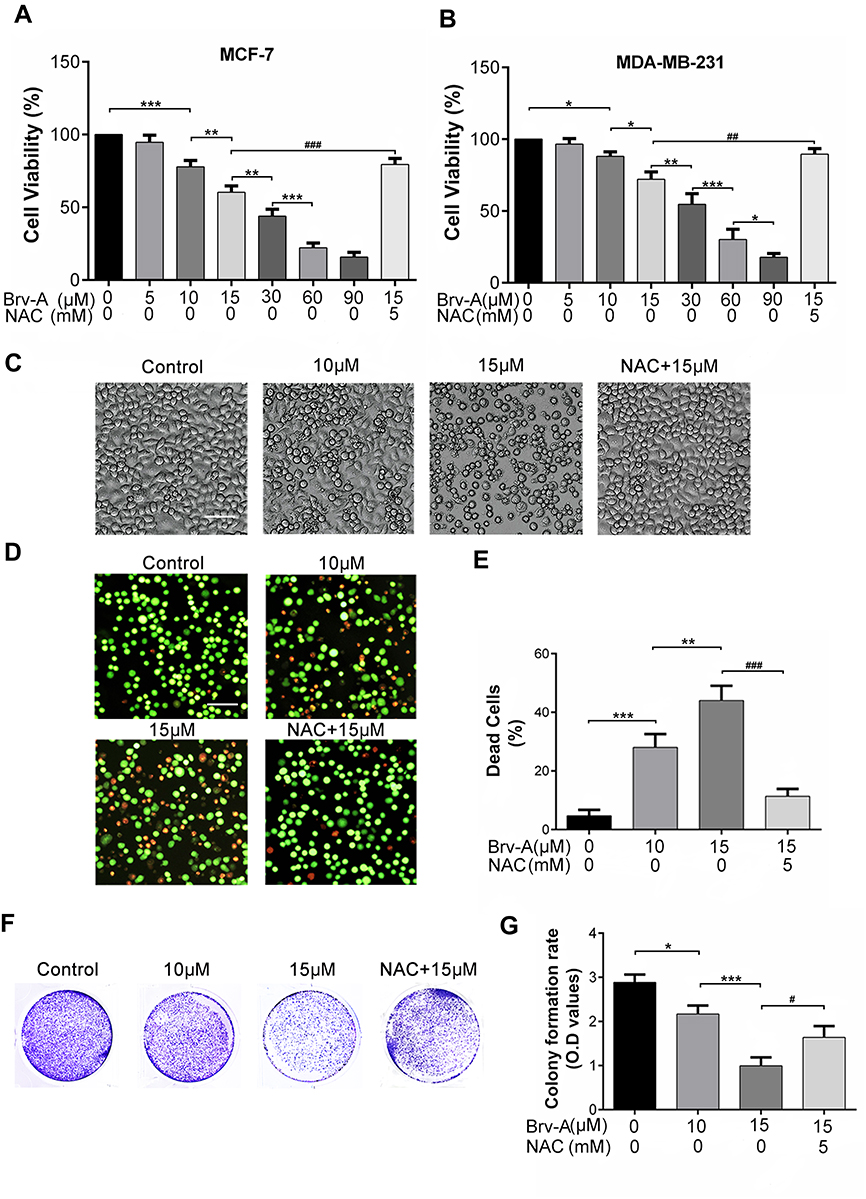 |
Figure 1 Cytotoxic and growth inhibitory effect of Brv-A in breast carcinoma cells. (A) MCF-7 and (B) MDA-MB-231 cells were treated with indicated concentrations of Brv-A in presence or absence of NAC (5 mM) for 24 h and cell viability was measured by CCK-8 kit. (C) MCF-7 cells were treated with indicated concentrations of Brv-A in presence or absence of NAC for 24 h and changes in cellular morphology were photographed by phase contrast microscope (Leica, DMIL LED). (D, E) MCF-7 cells were treated with Brv-A in dose-dependent manner in presence or absence of NAC. Cell death percentage was measured by live/dead assay using fluorescent probe calcein-AM and PI. (F) MCF-7 cells were treated with indicated concentrations of Brv-A in presence or absence of NAC for 24 h and 300 cells/well were seeded into six-well plate within DMEM. Cells were kept for 10 days to form colonies. After fixation with 4% paraformaldehyde, colonies were stained with crystal violet stain and photographed. (G) Stain picked by colonies was dissolved in methanol and optical density was measured at 595 nm. (C, D) Scale bar is 100 µm. (A, B, E, G) Data are expressed as Mean ± SD while all experiments were performed in triplicate independently. *p <0.05, **p <0.01, ***p <0.001 vs untreated group (control) while #p <0.05, ##p <0.01, ###p <0.001 vs 15 µM treated group. |
Next, we treated MCF-7 cells with 10 and 15 µM concentrations of Brv-A in presence or absence of NAC to explicate its effect on cell morphology. Under phase contrast microscope, we found that Brv-A induced several morphological changes associated with cell death in a dose-dependent manner after 24 h treatment. As shown in Figure 1C, control cells were adhesive and widened while treated cells were rounded in shape, floating in media and less in number with mislaid cellular geometry. Pretreatment of NAC partially protected cells from cytotoxic effect of Brv-A. In addition, we investigated individual effect of NAC over cell viability by CCK-8 assay and observing cell morphology. Among different concentrations, 5 mM was found most suitable for further analysis (Figure S1A and B).
Furthermore, we performed live/dead assay by using calcein-AM and PI stains to confirm Brv-A induced cell death. Data in Figure 1D and E demonstrates that Brv-A significantly induced cell death in a dose-dependent manner while NAC partially reversed the effect of Brv-A. Growth inhibitory effect of Brv-A in MCF-7 cells proliferation was also evaluated by clonogenic assay (Figure 1F). Consistent with CCK-8 and live/dead assay results, data demonstrated remarkable suppression in colony formation in MCF-7 cells. Furthermore, we quantified proliferation rate of cells by measuring optical density of uptaken crystal violet stain dissolved in methanol. Figure 1G represents significant decrease in uptake of crystal violet stain in dose-dependent fashion in MCF-7 cells. Of note, pretreatment of cells with NAC, a broad-spectrum antioxidant, significantly protected the cells from Brv-A mediated growth arrest as presented in Figure 1A–G. Collective data of CCK-8, morphological study, live/dead assay and clonogenic assay demonstrate that Brv-A exerts antiproliferative and growth inhibitory effect in MCF-7 breast carcinoma cells at least partially via ROS generation.
Brv-A Induces ROS Dependent G2/M Phase arrest in MCF-7 Cells
Cell cycle progression is one of the major regulatory mechanisms for cell growth.20 To gain further insight into mechanism underlying antiproliferative and cytotoxic effect of Brv-A, we investigated cell cycle phase profile in Brv-A treated cells in presence or absence of NAC. Flow cytometry analysis showed that Brv-A arrested MCF-7 cells in G2/M phase in dose-dependent manner. As shown in Figure 2, the percentage of G2/M phase cells was significantly increased to 37.6% and 56.4% compared to the control (28.7%) with corresponding decrease in percentage of G0/G1 and S phase cells. Pretreatment of NAC (5mM) reversed the effect of Brv-A over cell cycle progression which clearly indicates that Brv-A induces G2/M phase arrest in MCF-7 cells by promoting ROS generation.
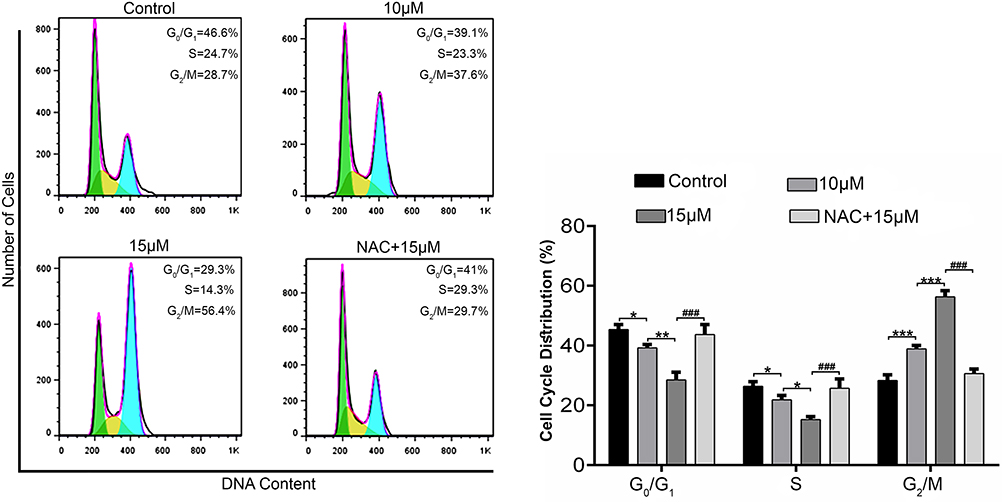 |
Figure 2 Effect of Brv-A on cell cycle distribution in MCF-7 cells. MCF-7 cells were treated with indicated concentrations of Brv-A in presence or absence of NAC for 24 h, stained with PI and analyzed by flow cytometry. Representative DNA fluorescence histograms of PI-stain cells show the cell cycle distribution. Histograms show number of cells on y-axis while DNA content on x-axis. The values display percentages of cells in indicated phases of cell cycle. Data are expressed as Mean ± SD while all experiments were performed in triplicate independently. *p <0.05, **p <0.01, ***p <0.001 vs untreated group (control) while ###p <0.001 vs 15 µM treated group. |
Brv-A Induces ROS Dependent Apoptotic Cell Death in MCF-7 Breast Cancer Cells
It has been well-known that sesquiterpene lactone compounds induce apoptosis via DNA fragmentation.21,22 We used Hoechst 33258 stain to analyze DNA fragmentation in Brv-A treated cells. Brv-A significantly increased percentage of DNA fragmentation while supplementation of NAC before drug treatment significantly protected cells from DNA fragmentation (Figure 3A and B).
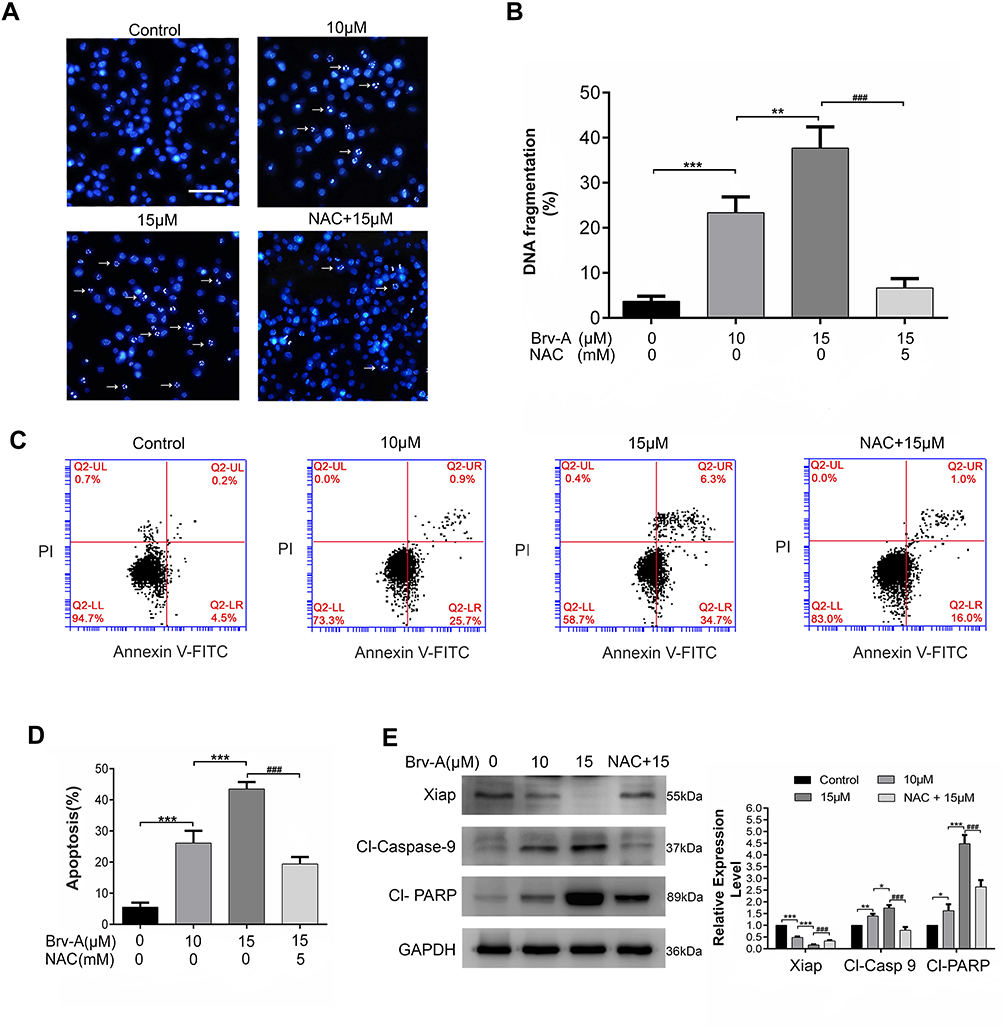 |
Figure 3 Induction of apoptosis by Brv-A in MCF-7 cells. (A) MCF-7 cells were treated with 10 µM and 15 µM concentrations of Brv-A in presence or absence of NAC for 24 h and stained with Hoechst 33258 for nuclear morphological changes. Scale bar is 100 µm. (B) In each group, among 100 cells, fragmented nuclei were counted under microscope. (C, D) Cells were treated with 10 µM and 15 µM Brv-A in presence or absence of NAC for 24 h. All samples were analyzed by flow cytometry after staining with annexin V-FITC and PI. (E) Cells were treated for 24 h with indicated concentrations of Brv-A in presence/absence of NAC. Protein extracts were prepared and analyzed by Western blotting to see expression level of apoptosis markers, ie Xiap, cleaved caspase-9 and cleaved PARP. GAPDH was used as loading control. Graphical data in (B, D, E) are expressed as Mean ± SD while all experiments were performed in triplicate independently. *p <0.05, **p <0.01, ***p <0.001, vs untreated group (control) while ###p <0.001 vs 15 µM treated group. |
To ascertain apoptotic mode of cell death, MCF-7 cells were treated with 10 and 15 µM Brv-A in presence or absence of NAC for 24 h and stained with annexin V-FITC and PI. Stained cells were analyzed by flow cytometry as shown in Figure 3C and D. Brv-A induced early apoptosis (annexin V-FITC positive), more often, gradual increase in late apoptotic/necrotic cells percentage (annexin V-FITC, PI positive) was also observed in dose-dependent manner. Pretreatment of NAC significantly reduced apoptotic cells percentage as shown in Figure 3D that further supports aforementioned data.
Brv-A induced apoptosis was further confirmed by analyzing expression of X-linked inhibitor of apoptosis (Xiap), cleavage of poly ADP ribose polymerase (PARP) and caspase-9. Figure 3E clearly shows that Brv-A decreased expression of Xiap and increased expressions of cleaved caspase-9 and cleaved PARP which are the hallmarks for apoptotic cell death. Pretreatment of NAC partially reversed the Brv-A induced change in the protein expressions. The above inclusive data shows that Brv-A induces ROS-dependent apoptotic mode of cell death in MCF-7 cell line.
Brv-A Induces ROS Production via NADPH Oxidase 2 (NOX2), NADPH Oxidase 3 (NOX3) Modulation
It is well-known that main source for intracellular ROS generation are some NOX family transmembrane proteins.10 Therefore, we hypothesized that Brv-A might exert its effect on expression of NOX family proteins. We measured NOX family proteins expression in Brv-A treated cells by Western blotting. Interestingly, we found that NOX2 and NOX3 members of NOX family transmembrane proteins were significantly over-expressed in Brv-A treated groups compared to untreated control. We also found that NAC partially reversed the Brv-A-induced expressions of NOX2 and NOX3 proteins (Figure 4A).
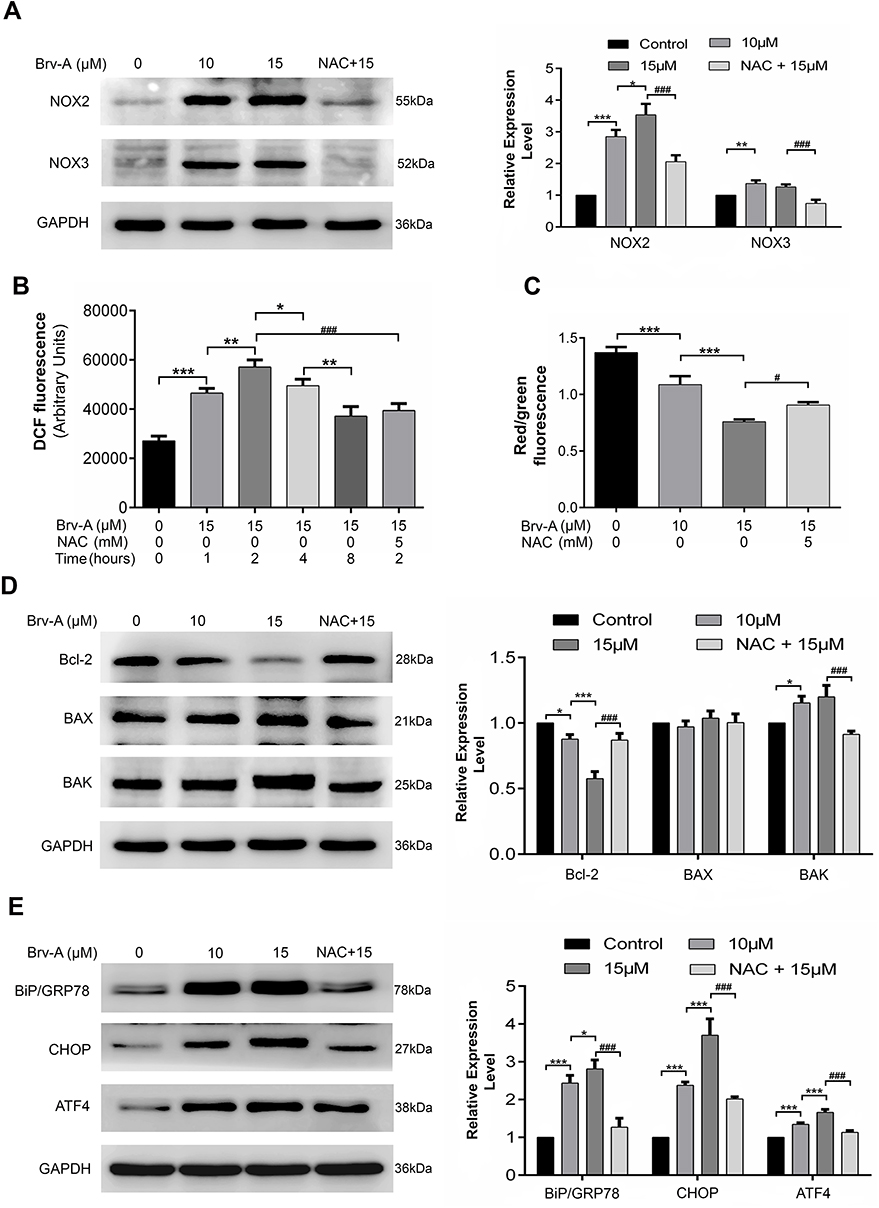 |
Figure 4 Brv-A modulates NOX family proteins, induces oxidative stress and promotes mitochondrial apoptosis and ER stress in MCF-7 cells. (A) Cells were exposed to indicated concentrations of Brv-A for 24 h in presence/absence of NAC and cell lysates were extracted by using RIPA. NOX family proteins (NOX2, NOX3) were measured by immunoblotting. (B) Cells were treated with 15 µM concentration of Brv-A for 0, 1, 2, 4 and 8 h in presence/absence of NAC and stained with DCFH-DA to determine ROS generation. (C) Cells were treated with 15 µM of Brv-A (in presence/absence of NAC) for 24 h and stained with JC-1 solution to measure MMP level. (D, E) Cells were exposed to indicated concentrations of Brv-A for 24 h in presence/absence of NAC and expression of Bcl-2 family proteins (Bcl-2, BAX and BAK) and ER-stress chaperones (BiP/GRP78, CHOP and ATF4) were measured by immunoblotting. GAPDH was used as loading control. Graphical data are expressed as Mean ± SD while all experiments were performed in triplicate independently. *p <0.05, **p <0.01, ***p <0.001, vs untreated group (control) while #p <0.05, ###p <0.001 vs 15 µM treated group. |
The role of NAC in attenuating cytotoxic effect of Brv-A is well explained in our study, therefore, we measured ROS level in a time-dependent manner. As shown in Figure 4B, Brv-A elevated ROS level in the very first hour of treatment. Moreover, highest ROS level was noticed at 2 h with gradual decrease at 4 and 8 h. After finding the highest time point of 2 h for ROS generation, we used NAC (ROS scavenger) to check reversal of Brv-A induced ROS generation. NAC with 5 mM concentration effectively reduced ROS generation up to significant level.
Brv-A Induces Mitochondrial Apoptosis and Endoplasmic Reticulum (ER) Stress via ROS Generation in MCF-7 Cells
Disruption of MMP, a distinguished feature of mitochondrial apoptosis, has been linked to ROS production. Therefore, we determined change in MMP in control and Brv-A treated cells. As indicated in Figure 4C, Brv-A induced mitochondrial dysfunction in dose-dependent manner while pretreatment of NAC restored MMP up to a significant level.
By finding dissipative change in MMP, we hypothesized that Brv-A could target and modulate Bcl-2 family proteins expression to induce mitochondrial apoptosis in MCF-7 cells. Our data demonstrates that Brv-A increased expression of pro-apoptotic protein BAK with no significant change in BAX expression and decreased the expression of anti-apoptotic Bcl-2 protein in a dose-dependent way. These modulations in Bcl-2 family proteins were attenuated under NAC pretreatment as indicated in Figure 4D. Our data demonstrate that Brv-A induces ROS-dependent mitochondrial apoptosis in MCF-7 breast cancer cells.
In earlier studies, it has been verified that homeostatic signaling of endoplasmic reticulum membrane sensors named as “unfolded protein response (UPR)” has been counter-activated by endoplasmic reticulum stress.23 In some cases, ROS production can apparently generate UPR signaling itself or become a fundamental part of it.24 Implications of NOX2 are well-known in UPR signaling that consequently lead toward oxidative shift generating ER stress and apoptosis.25,26 By going through all above mentioned results, we were able to hypothesize that Brv-A could induce ROS-dependent ER stress involving NOX family proteins modulation. Thus, we measured the expression of ER stress related proteins as shown in Figure 4E. Binding immunoglobulin protein BiP/GRP78 is activated during ER stress to degrade misfolded proteins.27 Significant increase in BiP/GRP78 expression was observed in Brv-A treated cells. We also observed significant increase in expression of activating transcription factor 4 (ATF4) and pro-death transcriptional regulator, C/EBP homologous protein (CHOP) in Brv-A treated cells which are hallmarks of ER stress-induced apoptosis. NAC treatment before drug exposure significantly reduced the Brv-A effect on expressions of BiP/GRP78, ATF4 and CHOP suggesting that Brv-A induced ER stress in MCF-7 cell line is ROS-dependent.
Brv-A Induces p38 and JNK Activation and Inhibits STAT3 Signaling
Multiple extracellular and intracellular stimuli, i.e. oxidative stress and ER stress activate mitogen-activated protein kinase (MAPK) family proteins. Among these, c-Jun N-terminal kinase (JNK) and p38 are prominent members involved in apoptosis.28,29 In previous studies, it has been demonstrated that high level of ROS generation in cells induces apoptosis via JNK pathway.8 In our study, Brv-A increased phosphorylation of JNK and p38 without affecting total JNK and p38 (Figure 5A and B). Pretreatment of NAC inhibited JNK and p38 activation which indicates that MAPK activation by Brv-A is ROS-dependent.
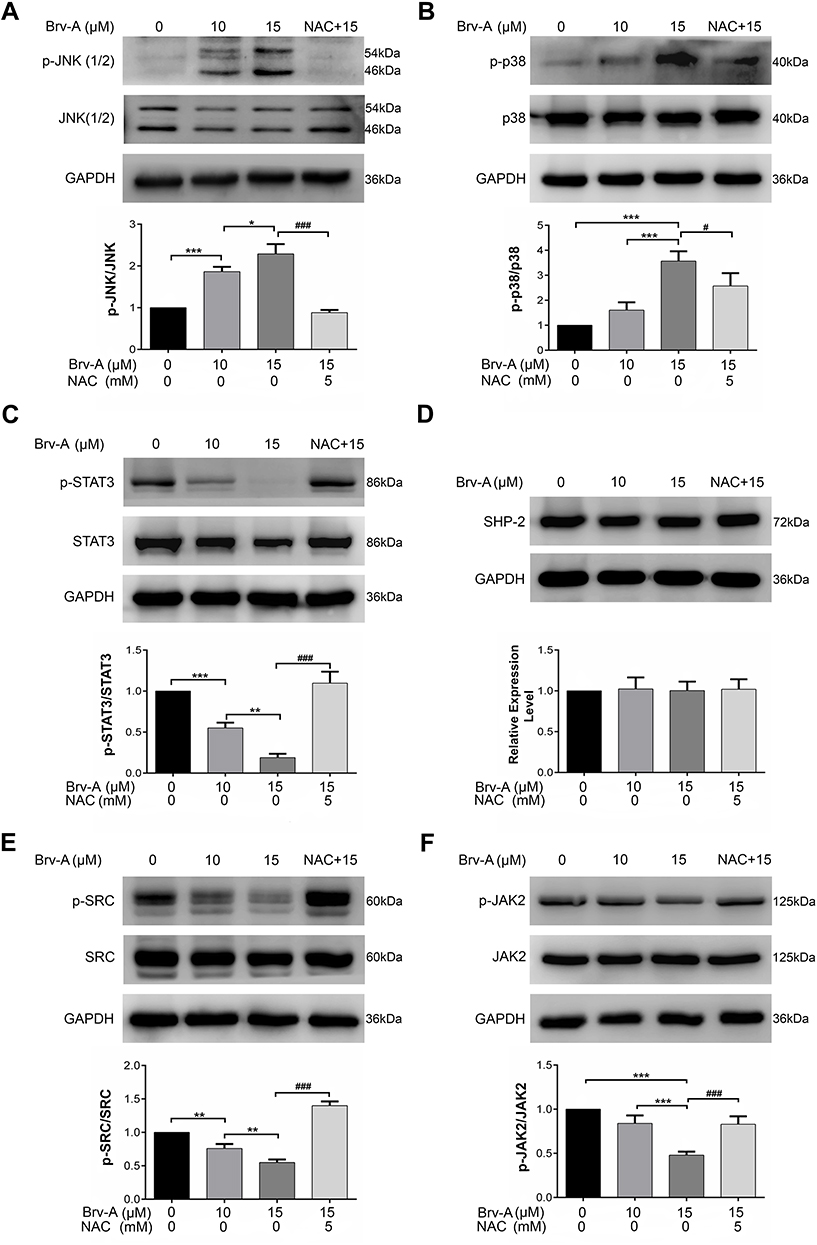 |
Figure 5 Brv-A induces JNK activation and Inhibits STAT3 signaling. (A, B) Cells were cultured in 6-well plate and treated with indicated concentrations of Brv-A for 4 h in presence/absence of NAC. Cell lysates were prepared and expression of p-JNK, JNK, p38 and p-p38 was measured by Western blotting. (C, D) Cells were exposed to10 µM and 15 µM of Brv-A in presence/absence of NAC for 24 h and cell lysates were prepared. Expression of p-STAT3, STAT3 and SHP-2 was measured by immunoblotting. (E, F) Expression of p-SRC, SRC, p-JAK2 and JAK2 was measured by Western blotting. GAPDH was used as loading control. All graphical data are expressed as Mean ± SD while all experiments were performed in triplicate independently. *p <0.05, **p <0.01, ***p <0.001 vs untreated group (control) while #p <0.05, ###p <0.001 vs 15 µM treated group. |
In various cancer types, Janus kinase (JAK)-signal transducer and activator of transcription 3 (STAT3) pathway is activated and plays an important role in drug resistance and cell survival.30 Moreover, previous studies have shown that ROS generation inhibits STAT3 activation.31 In this study, we investigated the effect of Brv-A on STAT3 activation in presence or absence of NAC. We found that Brv-A significantly inhibited STAT3 activation in a dose-dependent fashion by suppressing its phosphorylation at tyrosine 705 (Figure 5C). In the presence of NAC, Brv-A failed to inhibit STAT3 activation which highlights that Brv-A induced STAT3 inhibition is ROS-dependent. Since, STAT3 activation is regulated by various upstream signaling molecules, we measured the expressions of SHP-2 (negative regulator of STAT3) and JAK2 and SRC (positive regulators) in Brv-A treated MCF-7 cells. The expression of SHP-2 was not affected (Figure 5D); however, significant decrease in expression of phosphorylated JAK2 and SRC was observed under Brv-A treatment. In line with aforementioned data, pre-supplementation of NAC partially reversed Brv-A mediated inhibition of SRC and JAK2 (Figure 5E and F). Collective data indicate that Brv-A induced STAT3 inhibition is ROS-dependent and associated with suppression of upstream tyrosine kinases. However, further study is needed to explore the in-depth mechanism with special emphasis on immersion of ROS in STAT3 inhibition.
Brv-A Inhibits Migration in MCF-7 Breast Cancer Cells
We performed wound healing assay to monitor the inhibitory potential of Brv-A in cell migration. As shown in Figure 6A, alterations were observed in migratory abilities of MCF-7 cells after Brv-A treatment over a time period of 0, 12 and 24 h. As shown in Figure 6B, Brv-A significantly restricted migration of MCF-7 cells in 24 h time period. We measured the expression of well-known markers of cancer invasion and metastasis including cyclooxygenase-2 (COX-2) and matrix metalloproteinase 2 (MMP-2) to estimate antimetastatic activity of Brv-A up to mechanistic level. As presented in Figure 6C, Brv-A reduced the expression of COX-2 and MMP-2 in a dose-dependent manner. Collective data demonstrate that Brv-A has antimetastatic activity in MCF-7 breast cancer cell line.
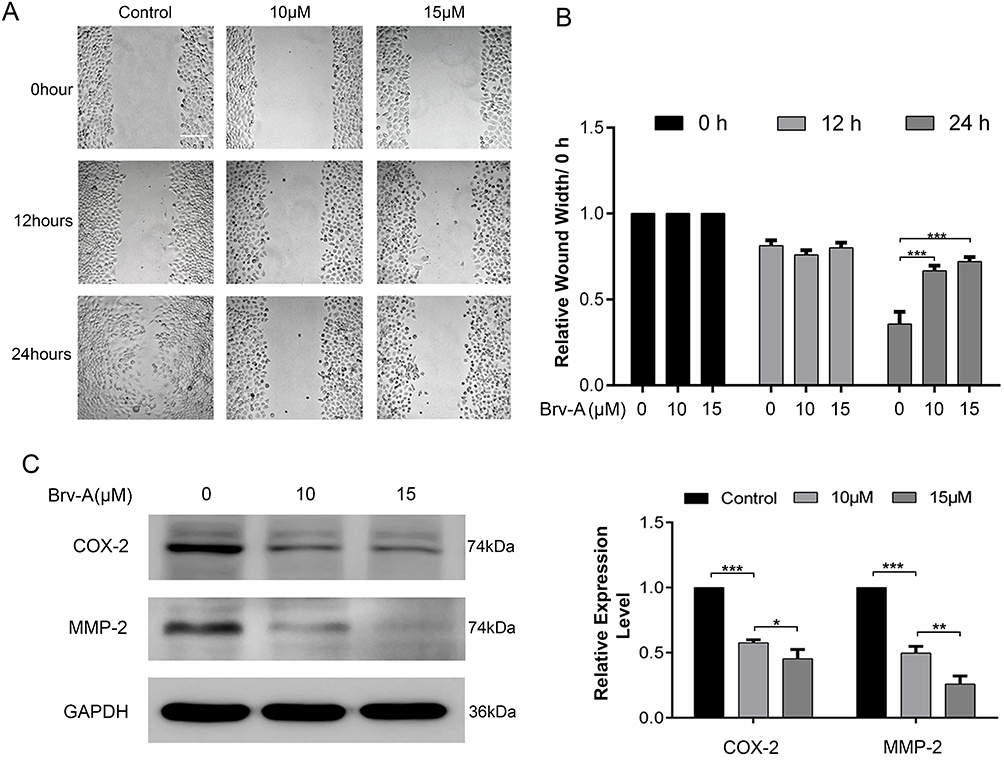 |
Figure 6 Effect of Brv-A on cell migration. (A) Cells were cultured in 6-well plate and allowed to attain 90% confluence. Scratch assay/wound healing assay was done to determine effect of Brv-A on migration in MCF-7 cells. Scratched wounds were observed and photographed in control and treated groups after 0, 12 and 24 h by using Leica, DMIL 4000B microscope. Scale bar is 100 µm. (B) Scale was used to measure width (cm) of scratch in control and treated groups. (C) MCF-7 cells were treated with Brv-A for 24 h and expression of COX-2 and MMP-2 was measured by immunoblotting. GAPDH was used as loading control. Graphical data in (B, C) are expressed as Mean ± SD while all experiments were performed in triplicate independently. *p <0.05, **p <0.01, ***p <0.001 vs untreated group (control). |
Discussion
Currently, breast cancer has become a serious concern across the whole globe.32 Surgery, chemotherapy, radiotherapy, and hormonal manipulation are the possible approaches to eradicate cancer which already has an improved five-year survival rate in breast cancer.33 Development of resistance against conventional therapies ultimately leads to cancer metastasis and recurrence. Most of the primeval antitumor drugs have limited approach to kill cancer cells, i.e. development of single drug for single target.34 Hence, the drug fails to combat survival pathways and tumor heterogeneity.35,36 Therefore, it is necessary to identify novel bioactive compounds to improve chemotherapy outcomes.17
Brv-A, a sesquiterpene lactone of Centipeda minima, is well-known for treatment of cough and asthma.15,37 Recently, Brv-A has shown antiproliferative activity in various human cancer types including CT26, A875, HeLa, HepG2 and A549 cell lines.17 Moreover, Brv-A has been reported as the inhibitor for JAK-STAT pathway in DU145 and MDA-MB-468 cell types.16 Previous study has already elaborated the role of Brv-A in generating ROS, MMP disruption, activation of caspases and inhibition of NF-κB activation in HL60 cell line.38 However, anticancer activity of Brv-A in triple positive breast cancer cells has not been investigated so far.
Cancer cells survive under higher oxidative stress than normal cells, leading to cell proliferation, cell survival, angiogenesis, and metastasis.39,40 This property of cancer cells has been therapeutically exploited for profitable purpose. Recently, ROS based drug advancement has become major attribute for cancer management irrespective of its heterogeneity and genotype.41–43 In this study, we have elucidated the functional link between ROS generation and cytocidal effect of Brv-A by using NAC. Our data demonstrated that Brv-A significantly inhibited viability of both triple positive and triple negative breast cancer cell types, however, only triple positive MCF-7 cells were used for further mechanistic study. The current study lacks cytotoxic activity of Brv-A in normal breast cell types that would probably improve its therapeutic potential. Furthermore, we performed live/dead assay, clonogenic assay and observed morphological changes associated with cell death in MCF-7 cells. In our study, Brv-A inhibited growth in a dose-dependent manner while NAC repealed cytotoxic effect of Brv-A illustrating it as ROS dependent. Our findings are parallel to the previous study where a sesquiterpene lactone, deoxyelephantopin, inhibited proliferation of HepG2 liver cancer cells by promoting ROS generation.21 Further, we were interested to find whether Brv-A could induce cell cycle arrest in MCF-7 cells. We analyzed cell cycle assay in Brv-A treated cells by flow cytometry. Brv-A induced G2/M phase arrest in MCF-7 cells in dose-dependent fashion which was consistent with previous published findings.19
Sesquiterpene lactones are well-known compounds for inducing apoptotic cell death in various cancer cell types.44 These compounds have also been confirmed to induce ROS generation.22 It is well-known that oxidative stress elicits mitochondrial apoptosis by Bcl-2 family proteins modulation. In general, Bcl-2, an anti-apoptotic protein, negatively regulates set of pro-apoptotic proteins residing in cytosol, i.e. BAX and BAK. Any apoptotic stimulus disrupts mitochondrial membrane potential by decreasing Bcl-2/BAX ratio in cancer cells and releases cytochrome c from mitochondrial inter-membrane space into cytosol. This released cytochrome c binds and triggers caspase-9 that subsequently activates caspase-3. Caspase-3 induces PARP cleavage and DNA fragmentation.45 In our study, Brv-A induced apoptotic cell death is evidenced by established parameters of mitochondrial apoptosis, i.e. DNA fragmentation, increase in BAK expression, decrease in Bcl-2 protein expression and disruption of mitochondrial membrane potential. Further, we noticed decrease in Xiap expression, and increase in cleaved caspase-9 and cleaved PARP expressions. Brv-A induced apoptosis was also confirmed by flow cytometry using annexin V-FITC and PI stains. The number of apoptotic cells were increased in Brv-A treated group in dose-dependent way. Pretreatment of NAC partially reversed the aforementioned cellular events which shows that Brv-A induces ROS-dependent mitochondrial apoptosis. Current findings are further supported by previously published studies where Brv-A has been shown to induce apoptosis in various cancer cells.15,17
ROS generation has been documented to induce ER-stress mediated apoptosis in a variety of cancer types.46 ER is chiefly involved in protein folding, post-translational modifications and export of recently formed proteins. In case of any impairment in ER homeostasis, created by harmful stimulus like ROS, unfolded proteins are mounted up in ER resulting in ER stress. If this persists for a long time without any restoration, ER stress switches unfolding protein response (UPR) from cell survival to cell death.47 In apoptotic conditions, phosphorylation of eIF2α inhibits protein translation while mRNA of ATF4 is translated which further promotes expression of pro-apoptotic CHOP. In current study, Brv-A increased expression of ATF4 and CHOP which is considered to be the hallmark for ER-stress-mediated apoptosis.48 Pretreatment of NAC partially prevented cells from Brv-A induced ER stress. Our results demonstrated that Brv-A induced ER-stress via ROS generation which has been explored for the first time in response to Brv-A treatment in any cancer type.
In various cell types, NADPH oxidases are imperative sources of ROS in UPR signaling. In previous studies, it has been demonstrated that NOX2 siRNA abolished ER-stress induced apoptosis and oxidative shift in macrophages. NOX2 is induced via CHOP-CAMKII-c-Jun N-terminal kinases (JNK) pathway to mediate apoptosis.25 In line with previously established data, we measured expression of NOX2 and NOX3 to verify source of ROS generation in ER-stress signaling pathways. Overexpression of NOX2 and NOX3 in Brv-A treated groups was reported in our data. NAC reversed the consequences of Brv-A on NOX genes, reduced ROS level and assisted cells to survive in presence of Brv-A.
MAPK family proteins, i.e. p38, JNK and ERK (1/2) are involved in several cellular processes like cell survival, proliferation and apoptosis. Activation of p38 and JNK promotes apoptosis while ERK (1/2) activation mediates cell survival. In our study, Brv-A increased phosphorylation of p38 and JNK that is consistent with previously published findings.17 It is previously reported that fate of cancer cells in case of overproduction of ROS is dependent on activation of PI3K kinase pathway for survival or ASK-1/JNK pathway leading to cell death.8 In our data, NAC partially overturned Brv-A induced phosphorylation of p38 and JNK. Hence, ROS-dependent p38 and JNK activation has been demonstrated in this study that probably led toward apoptosis.
For the most part, STAT3 contributes in many cell processes such as cell survival, metastasis, proliferation, and chemo-resistance.49 Aberrant activation of STAT3 is mediated by abnormalities in non-receptor tyrosine kinases, i.e. JAKs and SRC.50 MAPKs and PTPs also play vital role in STAT3 regulation.51 In particular reference to STAT3, investigation of JAK-STAT signal inhibitors is an efficient way to discover potential anticancer therapeutics. Brv-A has already been verified for inhibiting STAT3 in MDA-MB-468 and DU145 cells16 but the role of Brv-A induced ROS generation in STAT3 inhibition is not so far exposed in triple positive breast carcinoma. Brv-A significantly inhibited activation of STAT3 in dose-dependent way. Our findings were parallel to the previous results.16 Our data demonstrate that PTPs like SHP-2 was unaffected while Brv-A reduced phosphorylation of SRC and JAK2 in MCF-7 cells. In this study, NAC inverted Brv-A effect on JAK-STAT pathway signifying it to be ROS-dependent.
In spite of recent advancements, survival rate of patients with metastasis has not been improved proficiently.52 Finally, we measured the effect of Brv-A on cell migration using wound healing assay and evaluated its effect on biomarkers of cancer invasion and metastasis, i.e. MMP-2 and COX-2. We found that Brv-A inhibited migration of cells and decreased expression of MMP-2 and COX-2 indicating its significant antimetastatic activity in MCF-7 cells. Our data is consistent with previous published findings where alantolactone, a natural sesquiterpene lactone, inhibited MMP-2 and COX-2 in glioblastoma cells.53
Conclusion
In conclusion, we have investigated the cytotoxic, growth inhibitory effect, and underlying mechanisms of Brv-A in MCF-7 cell line. Brv-A has been identified to arrest cell cycle at G2/M phase and induce apoptosis via oxidative stress in breast carcinoma. Induction of apoptosis is partly associated with Bcl-2 family proteins modulation, MMP disruption, caspases activation, PARP cleavage, and JNK activation. However, in our study, ROS generation via modulation of NOX2, NOX3 proteins and induction of ER-stress are the new advancements in mechanistic study of Brv-A that has not been recognized previously. Furthermore, our experimental data illustrate comprehensive role of Brv-A in inhibition of constitutive STAT3 activation by suppression of upstream tyrosine kinases. Brv-A has also been remarked to reduce cell migration in MCF-7 cells. Further in vivo studies are required to explore efficacy of Brv-A for its authentication as potential therapeutic compound in future. A schematic model for molecular mechanism of Brv-A has been shown in Figure 7.
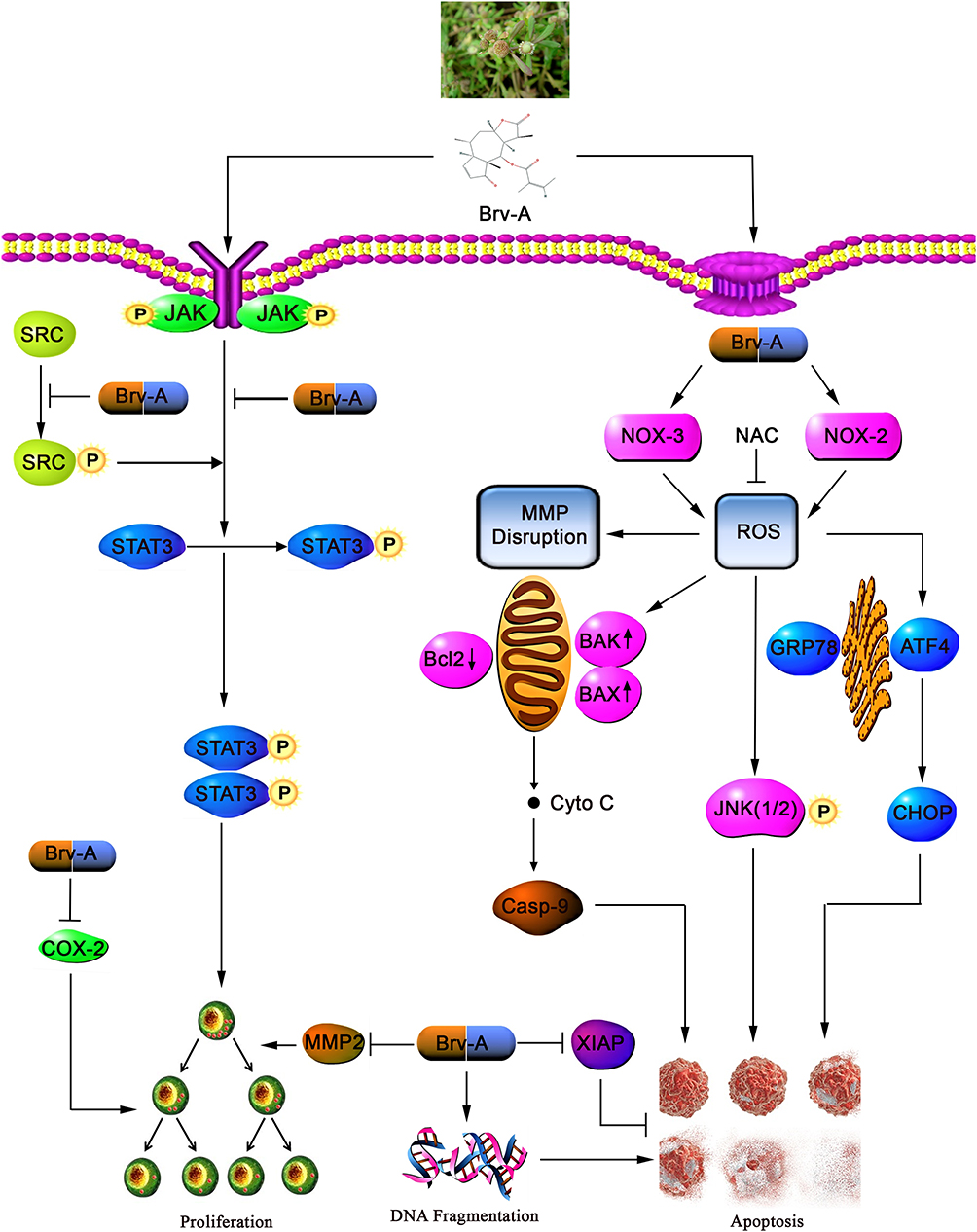 |
Figure 7 A schematic model for anticancer activity and molecular mechanism of Brv-A in MCF-7 breast carcinoma cells. |
Acknowledgment
This research work was supported by research grants from National Natural Science Foundation of China to Muhammad Khan (81650110526).
Disclosure
The authors declare no conflicts of interest.
References
1. Ballazhi L, Imeri F, Jashari A, et al. Original research paper. Hydrazinyldiene-chroman-2,4-diones in inducing growth arrest and apoptosis in breast cancer cells: synergism with doxorubicin and correlation with physicochemical properties. Acta Pharm. 2017;67(1):35–52. doi:10.1515/acph-2017-0006
2. Zhu X, Wang K, Zhang K, Zhu L, Zhou F. Ziyuglycoside II induces cell cycle arrest and apoptosis through activation of ROS/JNK pathway in human breast cancer cells. Toxicol Lett. 2014;227(1):65–73. doi:10.1016/j.toxlet.2014.03.015
3. Ferlay J, Shin HR, Bray F, Forman D, Mathers C, Parkin DM. Estimates of worldwide burden of cancer in 2008: GLOBOCAN 2008. Int J Cancer. 2010;127(12):2893–2917. doi:10.1002/ijc.25516
4. Porter P. “Westernizing” women’s risks? Breast cancer in lower-income countries. N Engl J Med. 2008;358(3):213–216. doi:10.1056/NEJMp0708307
5. Anderson BO, Yip CH, Smith RA, et al. Guideline implementation for breast healthcare in low-income and middle-income countries: overview of the breast health global initiative global summit 2007. Cancer. 2008;113(8 Suppl):2221–2243. doi:10.1002/cncr.23844
6. Li T, Mello-Thoms C, Brennan PC. Descriptive epidemiology of breast cancer in China: incidence, mortality, survival and prevalence. Breast Cancer Res Treat. 2016;159(3):395–406. doi:10.1007/s10549-016-3947-0
7. Sabzichi M, Samadi N, Mohammadian J, Hamishehkar H, Akbarzadeh M, Molavi O. Sustained release of melatonin: a novel approach in elevating efficacy of tamoxifen in breast cancer treatment. Colloids Surf B Biointerfaces. 2016;145:64–71. doi:10.1016/j.colsurfb.2016.04.042
8. Skonieczna M, Hejmo T, Poterala-Hejmo A, Cieslar-Pobuda A, Buldak RJ. NADPH oxidases: insights into selected functions and mechanisms of action in cancer and stem cells. Oxid Med Cell Longev. 2017;2017:9420539. doi:10.1155/2017/9420539
9. Angeloni C, Prata C, Dalla Sega FV, Piperno R, Hrelia S. Traumatic brain injury and NADPH oxidase: a deep relationship. Oxid Med Cell Longev. 2015;2015:370312. doi:10.1155/2015/370312
10. Loureiro ACC, Do Rêgo-Monteiro IC, Louzada RA, et al. Differential expression of NADPH oxidases depends on skeletal muscle fiber type in rats. Oxid Med Cell Longev. 2016;2016:6738701.
11. Panday A, Sahoo MK, Osorio D, Batra S. NADPH oxidases: an overview from structure to innate immunity-associated pathologies. Cell Mol Immunol. 2015;12(1):5–23. doi:10.1038/cmi.2014.89
12. Bedard K, Krause K-H. The NOX family of ROS-generating NADPH oxidases: physiology and pathophysiology. Physiol Rev. 2007;87(1):245–313. doi:10.1152/physrev.00044.2005
13. Chen L, Wang L, Shen H, Lin H, Li D. Anthelminthic drug niclosamide sensitizes the responsiveness of cervical cancer cells to paclitaxel via oxidative stress-mediated mTOR inhibition. Biochem Biophys Res Commun. 2017;484(2):416–421. doi:10.1016/j.bbrc.2017.01.140
14. Tsang WP, Chau SPY, Kong SK, Fung KP, Kwok TT. Reactive oxygen species mediate doxorubicin induced p53-independent apoptosis. Life Sci. 2003;73(16):2047–2058. doi:10.1016/S0024-3205(03)00566-6
15. You P, Wu H, Deng M, Peng J, Li F, Yang Y. Brevilin A induces apoptosis and autophagy of colon adenocarcinoma cell CT26 via mitochondrial pathway and PI3K/AKT/mTOR inactivation. Biomed Pharmacother. 2018;98:619–625. doi:10.1016/j.biopha.2017.12.057
16. Chen X, Du Y, Nan J, et al. Brevilin A, a novel natural product, inhibits janus kinase activity and blocks STAT3 signaling in cancer cells. PLoS ONE. 2013;8(5):e63697. doi:10.1371/journal.pone.0063697
17. Wang J, Li M, Cui X, et al. Brevilin A promotes oxidative stress and induces mitochondrial apoptosis in U87 glioblastoma cells. Onco Targets Ther. 2018;11:7031–7040. doi:10.2147/OTT.S179730
18. Maryam A, Mehmood T, Zhang H, Li Y, Khan M, Ma T. Alantolactone induces apoptosis, promotes STAT3 glutathionylation and enhances chemosensitivity of A549 lung adenocarcinoma cells to doxorubicin via oxidative stress. Sci Rep. 2017;7:6242. doi:10.1038/s41598-017-06535-y
19. Liu R, Qu Z, Lin Y, Lee C-S, Tai WC-S, Chen S. Brevilin A induces cell cycle arrest and apoptosis in nasopharyngeal carcinoma. Front Pharmacol. 2019;10:594. doi:10.3389/fphar.2019.00594
20. Di W, Khan D-M, Rasul A, et al. Isoalantolactone inhibits constitutive NF-κB activation and induces reactive oxygen species-mediated apoptosis in osteosarcoma U2OS cells through mitochondrial dysfunction. Oncol Rep. 2014;32.
21. Mehmood T, Maryam A, Zhang H, Li Y, Khan M, Ma T. Deoxyelephantopin induces apoptosis in HepG2 cells via oxidative stress, NF-κB inhibition and mitochondrial dysfunction. BioFactors. 2017;43(1):63–72. doi:10.1002/biof.v43.1
22. Mehmood T, Maryam A, Tian X, Khan M, Ma T. Santamarine inhibits NF-кB and STAT3 activation and induces apoptosis in HepG2 liver cancer cells via oxidative stress. J Cancer. 2017;8(18):3707–3717. doi:10.7150/jca.20239
23. Laurindo FRM, Araujo TLS, Abrahão TB. Nox NADPH oxidases and the endoplasmic reticulum. Antioxid Redox Signal. 2014;20(17):2755–2775. doi:10.1089/ars.2013.5605
24. Santos CXC, Tanaka LY, João Wosniak J, Laurindo FRM. Mechanisms and implications of reactive oxygen species generation during the unfolded protein response: roles of endoplasmic reticulum oxidoreductases, mitochondrial electron transport, and NADPH oxidase. Antioxid Redox Signal. 2009;11(10):2409–2427. doi:10.1089/ars.2009.2625
25. Li G, Scull C, Ozcan L, Tabas I. NADPH oxidase links endoplasmic reticulum stress, oxidative stress, and PKR activation to induce apoptosis. J Cell Biol. 2010;191(6):1113–1125. doi:10.1083/jcb.201006121
26. Zhang K, Kaufman RJ. From endoplasmic-reticulum stress to the inflammatory response. Nature. 2008;454(7203):455–462. doi:10.1038/nature07203
27. Cheng S-Y, Chen N-F, Kuo H-M, et al. Prodigiosin stimulates endoplasmic reticulum stress and induces autophagic cell death in glioblastoma cells. Apoptosis. 2018;23(5):314–328. doi:10.1007/s10495-018-1456-9
28. Maryam A, Mehmood T, Yan Q, Li Y, Khan M, Ma T. Proscillaridin A promotes oxidative stress and ER stress, inhibits STAT3 activation, and induces apoptosis in A549 lung adenocarcinoma cells. Oxid Med Cell Longev. 2018;2018:3853409. doi:10.1155/2018/3853409
29. Kim EK, Choi E-J. Pathological roles of MAPK signaling pathways in human diseases. Biochim Biophys Acta Mol Basis Dis. 2010;1802(4):396–405. doi:10.1016/j.bbadis.2009.12.009
30. He Y, Khan M, Yang J, Yao M, Yu S, Gao H. Proscillaridin A induces apoptosis, inhibits STAT3 activation and augments doxorubicin toxicity in prostate cancer cells. Int J Med Sci. 2018;15(8):832–839. doi:10.7150/ijms.23270
31. Jingwen Z, Seok AK, Chulwon K, et al. Nimbolide-induced oxidative stress abrogates STAT3 signaling cascade and inhibits tumor growth in transgenic adenocarcinoma of mouse prostate model. Antioxid Redox Signal. 2016;24(11):575–589. doi:10.1089/ars.2015.6418
32. Karia P, Patel KV, Rathod SSP. Breast cancer amelioration by Butea monosperma in-vitro and in-vivo. J Ethnopharmacol. 2018;217:54–62. doi:10.1016/j.jep.2017.12.026
33. Nooshinfar E, Bashash D, Safaroghli-Azar A, et al. Melatonin promotes ATO-induced apoptosis in MCF-7 cells: proposing novel therapeutic potential for breast cancer. Biomed Pharmacother. 2016;83:456–465. doi:10.1016/j.biopha.2016.07.004
34. Ji HF, Li XJ, Zhang HY. Natural products and drug discovery. Can thousands of years of ancient medical knowledge lead us to new and powerful drug combinations in the fight against cancer and dementia? EMBO Rep. 2009;10(3):194–200. doi:10.1038/embor.2009.12
35. Garraway LA, Jänne PA. Circumventing cancer drug resistance in the era of personalized medicine. Cancer Discov. 2012;2(3):214. doi:10.1158/2159-8290.CD-12-0012
36. Singh AK, Arya RK, Maheshwari S, et al. Tumor heterogeneity and cancer stem cell paradigm: updates in concept, controversies and clinical relevance. Int J Cancer. 2015;136(9):1991–2000. doi:10.1002/ijc.v136.9
37. Ren Y, Yu J, Kinghorn AD. Development of anticancer agents from plant-derived sesquiterpene lactones. Curr Med Chem. 2016;23(23):2397–2420. doi:10.2174/0929867323666160510123255
38. Changlong L, Hezhen W, Yongping H, Yanfang Y, Yanwen L, Jianwen L. 6-O-Angeloylenolin induces apoptosis through a mitochondrial/caspase and NF-kappaB pathway in human leukemia HL60 cells. Biomed Pharmacother. 2008;62(6):401–409. doi:10.1016/j.biopha.2007.10.010
39. Khan M, Maryam A, Qazi JI, Ma T. Targeting apoptosis and multiple signaling pathways with Icariside II in cancer cells. Int J Biol Sci. 2015;11(9):1100–1112. doi:10.7150/ijbs.11595
40. Khan M, Maryam A, Zhang H, Mehmood T, Ma T. Killing cancer with platycodin D through multiple mechanisms. J Cell Mol Med. 2016;20(3):389–402. doi:10.1111/jcmm.2016.20.issue-3
41. Asby DJ, Radigois MG, Wilson DC, et al. Triggering apoptosis in cancer cells with an analogue of cribrostatin 6 that elevates intracellular ROS. Org Biomol Chem. 2016;14(39):9322–9330. doi:10.1039/C6OB01591C
42. AbdulSalam SF, Thowfeik FS, Merino EJ. Excessive reactive oxygen species and exotic DNA lesions as an exploitable liability. Biochemistry. 2016;55(38):5341–5352. doi:10.1021/acs.biochem.6b00703
43. Somasundaram V, Nadhan R, Kumar Sengodan S, Srinivas P. Nitric oxide and reactive oxygen species: clues to target oxidative damage repair defective breast cancers. Crit Rev Oncol Hematol. 2016;101:184–192. doi:10.1016/j.critrevonc.2016.03.004
44. Ding Y, Wang H, Niu J, et al. Induction of ROS overload by alantolactone prompts oxidative DNA damage and apoptosis in colorectal cancer cells. Int J Mol Sci. 2016;17(4):558. doi:10.3390/ijms17040558
45. Ji YB, Qu ZY, Zou X. Juglone-induced apoptosis in human gastric cancer SGC-7901 cells via the mitochondrial pathway. Exp Toxicol Pathol. 2011;63(1–2):69–78. doi:10.1016/j.etp.2009.09.010
46. Komoike Y, Matsuoka M. Endoplasmic reticulum stress-mediated neuronal apoptosis by acrylamide exposure. Toxicol Appl Pharmacol. 2016;310:68–77. doi:10.1016/j.taap.2016.09.005
47. Flores-Martin J, Reyna L, Ridano ME, Panzetta-Dutari GM, Genti-Raimondi S. Suppression of StarD7 promotes endoplasmic reticulum stress and induces ROS production. Free Radic Biol Med. 2016;99:286–295. doi:10.1016/j.freeradbiomed.2016.08.023
48. Tsai YC, Weissman AM. The unfolded protein response, degradation from endoplasmic reticulum and cancer. Genes Cancer. 2010;1(7):764–778. doi:10.1177/1947601910383011
49. Kim SJ, Yoon S. Activated Rac1 regulates the degradation of IkappaBalpha and the nuclear translocation of STAT3-NFkappaB complexes in starved cancer cells. Exp Mol Med. 2016;48:e231. doi:10.1038/emm.2016.17
50. Garcia R, Bowman TL, Niu G, et al. Constitutive activation of Stat3 by the Src and JAK tyrosine kinases participates in growth regulation of human breast carcinoma cells. Oncogene. 2001;20(20):2499–2513. doi:10.1038/sj.onc.1204349
51. Chun J, Li RJ, Cheng MS, Kim YS. Alantolactone selectively suppresses STAT3 activation and exhibits potent anticancer activity in MDA-MB-231 cells. Cancer Lett. 2015;357(1):393–403. doi:10.1016/j.canlet.2014.11.049
52. Zibara K, Awada Z, Dib L, et al. Anti-angiogenesis therapy and gap junction inhibition reduce MDA-MB-231 breast cancer cell invasion and metastasis in vitro and in vivo. Sci Rep. 2015;5:12598. doi:10.1038/srep12598
53. Wang X, Yu Z, Wang C, et al. Alantolactone, a natural sesquiterpene lactone, has potent antitumor activity against glioblastoma by targeting IKKbeta kinase activity and interrupting NF-kappaB/COX-2-mediated signaling cascades. J Exp Clin Cancer Res. 2017;36(1):93. doi:10.1186/s13046-017-0563-8
 © 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.