")
Back to Journals » Neuropsychiatric Disease and Treatment » Volume 11
Brain in flames – animal models of psychosis: utility and limitations
Authors Mattei D, Schweibold R, Wolf S
Received 10 February 2015
Accepted for publication 31 March 2015
Published 27 May 2015 Volume 2015:11 Pages 1313—1329
DOI https://doi.org/10.2147/NDT.S65564
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr Roger Pinder
Daniele Mattei,1 Regina Schweibold,1,2 Susanne A Wolf1
1Department of Cellular Neuroscience, Max-Delbrueck-Center for Molecular Medicine, Berlin, Germany; 2Department of Neurosurgery, Helios Clinics, Berlin, Germany
Abstract: The neurodevelopmental hypothesis of schizophrenia posits that schizophrenia is a psychopathological condition resulting from aberrations in neurodevelopmental processes caused by a combination of environmental and genetic factors which proceed long before the onset of clinical symptoms. Many studies discuss an immunological component in the onset and progression of schizophrenia. We here review studies utilizing animal models of schizophrenia with manipulations of genetic, pharmacologic, and immunological origin. We focus on the immunological component to bridge the studies in terms of evaluation and treatment options of negative, positive, and cognitive symptoms. Throughout the review we link certain aspects of each model to the situation in human schizophrenic patients. In conclusion we suggest a combination of existing models to better represent the human situation. Moreover, we emphasize that animal models represent defined single or multiple symptoms or hallmarks of a given disease.
Keywords: inflammation, schizophrenia, microglia, animal models
Introduction
Over the past 20 years, an increasing amount of evidence supports the involvement of inflammatory processes in the pathophysiology, and more recently in the etiology of schizophrenia. High levels of proinflammatory cytokines are found in the blood and in the cerebrospinal fluid of schizophrenia patients.1,2 Current treatments with classical or atypical antipsychotics have significantly improved the life quality of affected individuals; however, these medications have often severe side effects, especially when taken over a long period of time.3 Moreover, existing antipsychotic drugs, including clozapine, treat only a subset of the symptoms. Existing drugs have little (or no) efficacy against the cognitive and negative symptoms that are responsible for much disability.4–6 The second-generation antipsychotic drugs have far less tendency than the older drugs to cause serious motor system side effects. But the newer drugs carry their own burden of serious side effects, such as significant weight gain and elevated levels of glucose and lipids.7–9 In clinical trials, minocycline, an anti-inflammatory antibiotic has been used with positive outcomes on the negative and cognitive symptoms.10–12
A growing body of literature also supports an increased appreciation for the integrated role of microglia in psychiatric disorders research. Microglia cells are the resident immune competent cells and phagocytes of the brain. Recently, microglia have been identified as potential pathological key player in schizophrenia.13 Evaluating postmortem tissue, an increase in reactive microglia density has been reported in distinct subtypes of schizophrenia and different brain regions.14 Using a ligand for the peripheral benzodiazepine receptor and PET (positron emission tomography) scans, a state of neuro inflammation in vivo in schizophrenic patients have been confirmed.15,16 Interestingly, microglial activation has also been found in animal models such as the maternal immune activation (MIA) that mimics a plethora of schizophrenia-like symptoms and hallmarks. The use of the antibiotic minocycline in animal models of schizophrenia proved the efficacy of treatments targeting microglia, the primary source of reactive oxygen species and proinflammatory cytokines in the brain, on schizophrenia-like manifestations.17–20 Minocycline is also able to decrease glutamate release through the interaction with voltage-dependent Ca2+ and Na+ channels, and this mode of action may also participate in the outcome of such a treatment in this pathology.21
Microglia orchestrates neuroinflammation and thus might become a new target of anti-inflammatory treatments. We here review present animal models in connection to either microglial activation and/or anti-inflammatory treatment outcome to see whether inflammatory processes are bridging the heterogeneous pool of animal models of schizophrenia. In addition, identifying a common inflammatory pattern would rationalize the use of anti-inflammatory drugs as an adjunctive therapeutic option.
When it comes to the development of animal models for a given neuropsychiatric condition, three main validity domains are usually screened to assess the strength of the model in reproducing a given human condition: construct validity, that is how the model was developed in terms of fidelity toward the given natural causes of the disease in humans; face validity, which describes a model’s reliability in terms of symptom array and biological changes pertinent to the disease being studied; predictive validity, how well the model responds to present medication used to treat the given disorder in comparison to how human patient responds and therein how valid the model is for testing of new compounds and biological targets for future research.22 The models described in this review will be addressed to as “models of schizophrenia”. However, schizophrenia is a complex human disorder with a yet unclear etiology and pathophysiology; it is therefore not reproducible in animals. The models used up to date all express several of the known neurochemical and behavioral abnormalities related to the human clinical picture of schizophrenia. Every time we refer to “models of schizophrenia” here, we actually address only a distinct manifestation of symptoms, hallmarks, and traits of the human disease, which will be discussed in detail for each model.
Genetic models
Schizophrenia has been linked to many different gene variants and mutations. Nevertheless, it is more probable that susceptibility genes together with environmental factors influencing the epigenome bring about the neurodevelopmental process leading to a schizophrenic phenotype.23–25 In order to study the effect of genes in the pathophysiology of schizophrenia, many different transgenic mouse models have been developed. Because of the large number of schizophrenia-related transgenic mice, we refer here to the works of Miyamoto and Nitta26 and van den Buuse,27 which thoroughly reviewed the vast array of genetic models in this regard. We decided to discuss briefly only one of the most studied group of genetic mouse models of schizophrenia, the DISC1 (disrupted in schizophrenia-1) transgenic mice. The dominant-negative DISC1 mouse has a forebrain-specific expression of dominant-negative truncated DISC1 under the promoter for calcium/calmodulin-dependent kinase II in C57BL/6 mice, which shows the typical cellular and morphological changes observed in patients, such as decreased parvalbumin (PV)-expressing interneurons and enlarged ventricles. As reviewed by van den Buuse,27 though, not all DISC1 transgenes do show a clear-cut schizophrenic phenotype in terms of sensorimotor deficits, hyperlocomotion, and other symptoms. This reflects the fact that genetic changes in one gene are probably not enough to reproduce the complexity of this disease, and the outcome of any DISC1 transgene may depend on the applied truncation or deletion in the exons of the mouse Disc1 gene.27,28 Changes in microglia cell phenotype and function in this mouse would be of particular interest in light of the fact that DISC1 is expressed in microglia cells (and also the other glia cells, astrocytes, and oligodendrocytes) in both humans and rats.29 In summary, the discovery of susceptibility genes has helped scientists to understand which biological pathways are possibly perturbed in schizophrenic endophenotypes. It is therefore conceivable that the same pathways or genes can be altered also in the absence of genetic mutations, eg, by environmental challenges against which cells respond via epigenetic mechanisms to adapt to changed conditions.23 One example is the changes in reelin expression observed in schizophrenic patients, which prompted scientists to develop reelin KO-mice as a model of schizophrenia, while schizophrenic patients do not present a full deletion of the reelin gene.30 On the other hand, the adult offspring of PolyI:C-challenged pregnant dams (discussed later in the review) presents a naturally diminished reelin expression following MIA, a known environmental risk factor for developing schizophrenia.31,32 In the same line, it appears that prenatal MIA is able to also alter the expression of DISC1 in the hippocampus of adult offspring.33 A final remark on genetic models is the lack of studies testing antipsychotic treatment on the behavioral features displayed by this pool of models. Future research should aim at validating these models for predictive validity so as to assess the effect of antipsychotics on the genetic component of this disease. Table 1 summarizes the major behavioral deficits found in the DISC1 mouse for a better overview.
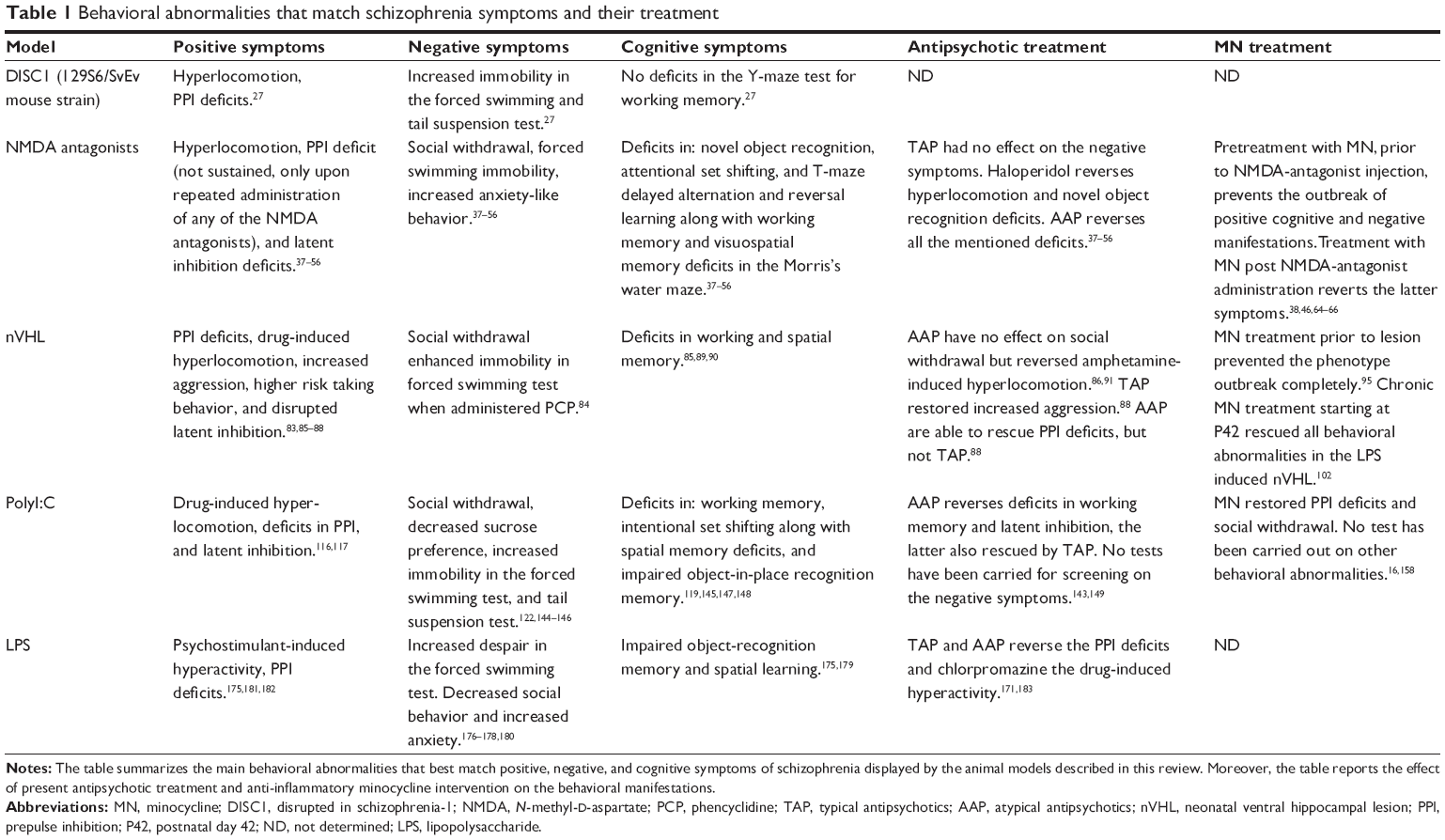 | Table 1 Behavioral abnormalities that match schizophrenia symptoms and their treatment |
Pharmacological models
Pharmacological models are a valuable tool through which specific underlying causes of schizophrenia can be reproduced. One example is the induction of glutamatergic hypoactivity observed in schizophrenic patients by injecting NMDA receptor antagonists into adult mice or rats.34–36 The most widely used and explored NMDA receptor antagonists are phencyclidine (PCP), ketamine, and dizocilpine (MK-801). The use of these compounds to reproduce schizophrenic-like symptoms such as psychosis came from the observation that humans administered with or without abusing these compounds develop features of a wide array of symptoms matching many of the positive, negative, and cognitive symptoms of schizophrenia. Moreover, schizophrenic patients administered with NMDA receptor antagonists experience a worsening of symptoms.37,38 The strength of this model is the simplicity in producing it and the possibility to quickly test new pharmacological interventions. The downside of the model is that it lacks the neurodevelopmental component that is thought to be the basis for the structural and cytoarchitectural abnormalities that result in the symptomatology of schizophrenia.30 On the cognitive symptoms side, ketamine administration has been reported to induce working memory deficits in the Y-maze, but no impairments were observed in the novel object exploration test.39,40 On the negative symptoms side, ketamine increases immobility in the forced swim test, decreases social interaction, and increases anxiety-like behaviors. Signs of positive symptoms include deficits in PPI (prepulse inhibition), PCP/MK-801-induced hyperlocomotion, and disrupted latent inhibition (LI). Comparable to the human clinical situation, atypical antipsychotics are more effective than the typical ones in treating the negative symptoms in this model. Haloperidol (a typical antipsychotic) seems to have no effect on the negative and cognitive symptom manifestations in this model, but it is efficacious against the positive ones, while clozapine (an atypical antipsychotic) has been found to revert both symptomatologies, which means that this model has a good predictive validity to test the efficacy of novel compounds in different symptom domains pertinent to schizophrenia pathology.39–43 PCP induced a huge array of cognitive manifestations, namely, deficits in novel object recognition, attentional set shifting, and T-maze delayed alternation and reversal learning. Moreover, it disrupted working and short-term memory. As for the positive-like symptoms, hyperlocomotion, disrupted PPI and LI have been reported along with negative symptoms such as impaired social behavior and enhanced immobility in the forced swim test. Similar to the ketamine model, haloperidol is able to reverse positive but not cognitive or negative symptoms, whereas clozapine showed positive effects on most behavioral abnormalities.44–52 The same cognitive, negative, and positive symptoms manifestations have also been found upon administration of MK-801, with very similar outcomes of antipsychotic treatment.53–58 In summary, although NMDA antagonism does not represent the perfect construct validity as a model, it shows a very good face validity in terms of schizophrenia-related behavioral abnormalities and an appreciable predictive validity according to the studies testing present antipsychotics.
NMDA antagonism causes acute and reversible miss-wiring of brain areas involved in schizophrenia, leading to schizophrenia endophenotypes and also indirectly to dopaminergic derangements.35,59 Nevertheless, the fact that minocycline pretreatment prevents the induction of positive, negative, and cognitive symptoms associated with NMDA antagonism implies that some form of inflammation may also be involved in this model.40 It has been reported that NMDA antagonism can, depending on the dose and number of administrations, damage neurons and lead to apoptosis and inflammatory responses.39,45,60 This in turn may lead to microglial activation and cytokine production in a brain region specific fashion. In fact, Zhu et al44 reported microglial activation following chronic PCP administration. In this regard, minocycline can be beneficial in two different ways: one is through minocycline’s neuroprotective and antiapoptotic action,61,62 and the other is the prevention of microglial activation.63,64 A study reported necrotic cortical neurons even with a dose of 1 mg/kg of MK-801, with intracellular vacuolization associated with reactive microglia. At higher doses (5 and 10 mg/kg), this response was even more evident.65 Monte et al40 reported that minocycline treatment was efficient in both preventing and treating the ketamine-induced cognitive, negative, and positive schizophrenia endophenotypes. Zhang et al66 showed that minocycline treatment can attenuate MK-801-induced hyperlocomotion and PPI deficits, showing here too that treatment after the administration of NMDA antagonists is effective. Finally, Fujita et al48 found that minocycline treatment successfully improved the cognitive deficits induced by chronic PCP administration. It is believed that oxidative stress is also part of the pathophysiology of schizophrenia.67,68 It is therefore inferable that minocycline acts on other aspects besides the anti-inflammatory action, considering it has scavenger and anti-apoptotic properties and modulating glutamatergic neurotransmission. Table 1 summarizes the major behavioral deficits found in this group of models and the outcome of the various treatments.
Lesion and developmental models
While pharmacological models of schizophrenia were created based on early observations of the neurochemical changes in brains of schizophrenic patients, subsequent models focused more on the theory of a neurodevelopmental hypothesis of schizophrenia. One of the first attempts to reproduce a possible developmental perturbation was the neonatal ventral hippocampal lesion (nVHL) model in rats. In this model, an excitotoxic lesion is provoked in the ventral hippocampus (VH) on postnatal day 7 (P7) by local and bilateral administration of either ibotenic acid (permanent lesion) or tetrodotoxin (temporary inactivation of the VH).69,70 The rats show features of positive, negative, and cognitive correlates of symptoms observed in schizophrenia as they reach late puberty. This is more alike the human clinical situation and hypothesis of a neurodevelopmental course of the disease than the pharmacological induced models.71 In the adult brain, the VH is functionally connected to the striatum, nucleus accumbens (NAc), prefrontal cortex (PFC), and amygdala. The nVHL will result in an aberrant development of connections between the VH and these areas, leading to an aberrant development of the PFC. The latter region will then result to be disinhibited in the adult lesioned rats.72–74 This is believed to be the net result of an absent maturation of dopamine (DA) receptor 2 (D2 receptor)-mediated control of fast-spiking interneurons within this structure, which should occur by the end of puberty. The connections between the aforementioned areas are not fully functional until early adulthood, and it is believed that other areas take over until then, so that any developmental misconnection within these circuits will not appear symptomatically until these areas, in early adulthood, are functionally implemented in the circuitry.75,76 Alterations in the dopaminergic system have been reported in the lesion model, mainly in the form of increased DA levels in hippocampus and an increase in its metabolite dihydroxyphenylacetic acid in the mPFC (medial prefrontal cortex).77 Flores et al78,79 further reported increased binding of DA receptor 3 (D3) in limbic subregions and also increased binding of D1 and D2 receptors in the striatum, suggestive of dopaminergic hyperactivity in this limbic region. Furthermore, decreased binding of glutamate was found along with alterations of the dopaminergic and glutamatergic neurotransmission in adult rats with postnatal ibotenic acid hippocampal lesion.80 Finally, the GABA (γ-aminobutyric acid)ergic interneuron system has also been found to be altered.81,82 Changes in the cortical GABAergic interneuron system have been proposed to play a pivotal role in many aspects of the symptomatology of schizophrenia, since aberrations in this system seem to contribute to the cortical miss-wiring and disinhibition observed in these patients; hence, these findings highlight an important similarity of this model to the clinical piture.30,83 The nVHL model also displays cytoarchitectural abnormalities congruent with the findings in human studies on schizophrenic patients.30 Flores et al84 reported decreased spine density and dendritic length in PFC and NAc medium spiny neurons, in line with the cytoarchitectural abnormalities known in schizophrenia pathology. Also, decreased spinogenesis and neuronal atrophy have been found in the NAc, PFC, and basolateral amygdala of the nVHL rats.85 As the lesioned animals reach early adulthood, they show features of negative symptoms in the form of social withdrawal in the sociability test and enhanced immobility in the forced swimming test when administered with PCP.86 As signs of positive symptoms, they show amphetamine- and PCP-induced hyperactivity, deficits in sensory and sensorimotor gating, increased aggressive behavior and higher risk-taking behavior in the elevated plus maze, and also disrupted LI.85,87–90 The lesioned animals have been found to have deficits in working and spatial memory as measured in the Morris’s water maze and T-maze.91–93 The lesion model is mainly applied in rats, and recently, Naert et al87 carried out a large behavioral screening of this model in C57B1/6J mice. They showed that lesioned mice present with higher spontaneous and amphetamine-induced activity and higher risk-taking behavior in the elevated plus maze. Further, they found deficits in working memory assessed by means of Y-maze (spontaneous alternation) and t-test (online storage of information). Working memory in these tasks is dependent on proper PFC function, and these deficits can be related to the known aberrant PFC function in nVHL rats. Nevertheless, the mouse model of nVHL did not display deficits in spatial memory assessed with the Morris’s water maze, and also the social behavior was intact. Furthermore, these mice had an intact sensorimotor gating and LI. These results shows that the mouse model differs from the rat model which seem to congregate more symptom domains as compared to the mouse model.87 One major advantage of the nVHL mouse model conceived by Naert et al87 is that it can be combined with genetic mouse models of schizophrenia endophenotypes in order to encompass more etiological factors. The lesion model has, in general, good face validity, encompassing many symptom domains of schizophrenia with a neurodevelopmental component and a postpubertal manifestation of symptoms. As for the predictive validity, the outcome of antipsychotic testing has been quite mixed. So far, clozapine and haloperidol have been shown to be able to reverse drug-induced hyperactivity.94 Becker et al90 showed that haloperidol did not restore the deficit in social behavior but decreased the aggressive behavior, which is more related to the positive symptoms of schizophrenia. On the other hand, clozapine did not have any effect on the aggressive behavior but restored a normal social pattern. This is in contrast with the results of another study in which neither clozapine nor risperidone (another atypical antipsychotic) managed to restore a normal social interaction.88 Nevertheless, clozapine, risperidone, and olanzapine improve the deficits in sensorimotor gating, while haloperidol has no effect in this positive symptom-like measure, which is not what is expected in terms of treatment efficacy, considering the human clinical picture.95 Levin and Christopher96 showed that clozapine can worsen the already impaired learning and memory functions in this animal model. The construct validity of this model may be questionable, since schizophrenic patients do not display a direct and profound lesion in the anterior hippocampus, which is the corresponding VH in rodents.97 Somehow, this model shares a component with the viral and bacterial developmental models of schizophrenia (discussed later). The immune response in the mother and consequently in the fetal brain may represent the spark of an initial damage directed against neurodevelopmental processes involving, among other regions, the hippocampus. In this regard, these models have one thing in common: microglia activation and ongoing inflammatory processes that may be the basis of the developmental perturbation leading to the postpubertal outbreak of symptoms. In line with the theory implicating developmental neuroinflammation in the pathophysiology of schizophrenia, Drouin-Ouellet et al98 analyzed in a recent study the role of microglia cells in the nVHL. They reproduced the postpubertal impairments showed by others in this model, including amphetamine-induced hyperlocomotion, deficits in PPI, impaired social interaction, and spatial working memory deficits. They could detect microglial activation following ibotenic acid injection from early after the injection until P35, long after ibotenic acid treatment, but not in adulthood (P56); moreover, interleukin-1 beta (IL-1β) was upregulated in the hippocampus until P14. Interestingly, they observed a selective induction of the metabotropic glutamate receptor 5 only in activated microglia, implicating the glutamatergic system in this immune response. Finally, they showed that treatment with minocycline 12 hours and 30 minutes prior to ibotenic acid administration and for 3 days following surgery prevented microglial activation, the raise in IL-1β, and the outbreak of the aforementioned symptoms. Interestingly, it also preserved to a certain extent the VH neuronal loss following ibotenic acid injections.98 They basically show that microglial activation is largely responsible for the short- and long-term consequences of nVHL with ibotenic acid. Two recent studies confirm these findings, further linking developmental neuroinflammation and microglial activation in the development of a schizophrenic endophenotype in this model. Feleder et al73 combined the lipopolysaccharide (LPS)-induced developmental model (discussed later) with the nVHL model by injecting LPS bilaterally in the VH at P6–P7 in rats. The rationale behind this model is to have a construct that implements a local inflammatory perturbation without any severe and destructive lesion. The authors showed that this kind of LPS challenge does not lead to changes in hippocampal morphology. Then, they demonstrated an increase in IL-1β and IL-2 in the hippocampus, PFC, and NcA, which was persistent and detectable also in adulthood.73 Interestingly, the same persistent increase in brain cytokines has been found in postmortem tissue from schizophrenic patients, suggesting that if any developmental perturbation occurred in pre- or perinatal stages of their lives, it may have given rise to a persistent increase in proinflammatory mediators.99–101 In a recent study, Zhu et al102 reproduced this model, demonstrating that nVHL challenge with LPS also leads to negative and cognitive manifestations in the adult rats which show deficits in social interaction and novel object recognition. Furthermore, they displayed PPI deficits as a sign of positive symptoms, but no locomotor hyperactivity. Here the authors found a marked increase in microglia density in the VH, thalamus, and cortex of the adult rats injected in the VH with LPS at P7. These rats were treated with minocycline and risperidone either alone or in combination, starting from P42 for 14 consecutive days. The authors report that both compounds, either alone or in combination, managed to rescue all the behavioral abnormalities and also the increase in microglia density in the aforementioned brain areas.102 This study shows that minocycline is effective in this model as a treatment and not only as a pretreatment as demonstrated earlier by Feleder et al.73
Both studies demonstrate that a persistent lesion is not required to reproduce a valid schizophrenia endophenotype with neonatal perturbation of the VH, as also demonstrated by the transient VH inactivation model with tetrodotoxin. In conclusion, the latter three studies carried out by Drouin-Ouellet et al,98 Feleder et al73 and Zhu et al102 implicate microglia density and inflammatory processes in the developmental course of this animal model, suggesting that anti-inflammatory intervention can prevent the outbreak of schizophrenia-like symptoms when administered early and also when administered in later pubertal stages. Further studies may address the questions as to why inflammatory processes and microglia cells are activated in regions far from the actual site of lesion/LPS injection, and how this activation can be so persistent. Table 1 summarizes the major behavioral alterations found in this group of models and the outcome of the various treatments.
Maternal immune activation models
Prenatal PolyI:C
Epidemiological studies revealed a correlation between certain viral and bacterial infections during pregnancy and the development of schizophrenia in the offspring.103 This finding was further strengthened by more recent serological studies conducted on the blood samples collected during pregnancy from mothers of children who developed schizophrenia.104,105 Fatemi et al106 during the early nineties showed that inoculation of human influenza virus in pregnant rodent dams caused the offspring to develop naturally a schizophrenia endophenotype as they reached early adulthood, with changes in various gene expression profiles in the brain, affecting also schizophrenia-related symptoms. The model was further developed using more practical means of evoking MIA by avoiding human pathogens. Instead of actual viruses, the double-stranded synthetic RNA PolyI:C is used nowadays.107 Double-stranded RNA is a product of viral replication for most kinds of viruses, and it is recognized by toll-like receptor 3 (TLR3) as a viral component, thereby evoking an innate antiviral immune response that includes the production of proinflammatory cytokines such as IL-1β, IL-6, TNF-α, and type I interferons: INF-α and INF-β.108 It is believed that the maternal immune response interferes with the correct development of the fetal brain in sensitive time-windows of pregnancy.109 In rodents, MIA at gestational day (G)15 gives rise to a reliable schizophrenia-like phenotype in the adult offspring, which will present hallmark structural changes found in schizophrenic patients, including enlarged ventricles and thinning of the hippocampus, which can be blocked by administering atypical antipsychotics during the asymptomatic period in adolescence.110–112 Although G15 is a commonly used gestational day, MIA at G9 and G17 also gives rise to schizophrenia endophenotype pathological components.113,114 To analyze the exact outcome of prenatal PolyI:C on the many different gestational days used is beyond the scope of this review; nevertheless in the discussion that follows, we will specify the day of MIA when relevant in the context of major phenotypical differences. Additionally, the last trimester of pregnancy in humans corresponds to P2–P6 in rodents, and in this case the animals are injected postnatal and the PolyI:C is given once a day for these 5 consecutive days.115 We decided not to elaborate on the neonatal PolyI:C model since it lacks the maternal immune response–fetal interface that reproduces the reported risk factor in a biologically more congruent way.
The adult offspring of dams challenged with PolyI:C (also referred to here as PolyI:C animals) have been found to present many histopathological and neurochemical hallmarks of schizophrenia in regions implicated in this pathology: limbic DA hyperactivity in striatal slices from adult brain, along with structural histopathological abnormalities in hippocampal CA (cornu ammonis) 1 and CA3 regions (PolyI:C at G15),116 increased DA turnover and decreased D2-like receptors binding in the striatum (PolyI:C at G12–G17),117 and reduction in reelin- and PV-expressing prefrontal neurons.113 This is of particular relevance to the PFC disinhibition features also shown by the nVHL model and schizophrenic patients thought to be caused, at least in part, by a malfunctioning GABAergic system.118 Following this line, an increased immunoreactivity for GABA-A receptor in the limbic system119 and reduced expression of glutamic acid decarboxylase 65 and 67 (GAD65/67) isoforms in the PFC and dorsal hippocampus (PolyI:C at G17) were reported in this model, which may contribute to GABAergic dysfunction. GAD is the rate-limiting enzyme in GABA biosynthesis; therefore, we could infer that a challenge in late gestation may decrease GABA levels at least in PFC and dorsal hippocampus.120 Soumiya et al121 also reported decreased GAD67 expression in the upper cortical layers together with decreased synaptophysin immunoreactivity. Winter et al122 screened the content of several neurotransmitters in brain regions relevant to schizophrenia (PolyI:C at G9). They found that this offspring showed in adulthood increased DA levels and its major metabolites in the lateral globus pallidus and PFC and decreased serotonin and its metabolite in the hippocampus, NAc, and lateral globus pallidus. Nevertheless, central glutamate and GABA contents were unchanged. On the other hand, another study reported decreased DA, glutamate, and GABA levels in the PFC and hippocampus (PolyI:Cat G17).123 These two studies reveal that different neurotransmitters are differentially affected in adult PolyI:C offspring, depending on the timing of MIA. Nevertheless, it has to be stressed that dysfunctions in specific neurotransmitter systems may manifest in the form of altered receptor or receptor subunit expression pattern independently of the gestational day of MIA. For instance, a study conducted by Roenker et al124 revealed marked glutamatergic hypofunction in the offspring of dams challenged at G14, which was manifested through decreased NMDA receptor function and elevated extracellular glutamate levels in the PFC. Besides the changes in serotonin and glutamate levels, changes in receptor expression for these two neurotransmitters have also been shown. Holloway et al125 demonstrated that MIA with PolyI:C leads to increased expression of the serotonin receptor 5-HT2A and a decrease in metabotropic glutamate receptor 2 in the frontal cortex of adult offspring. Similarly, Dalton et al126 showed an increase in 5-HT1A receptor in the brain of adult PolyI:C animals, and the increase was greater if these animals were exposed to a cannabinoid during adolescence. These findings are of particular relevance since serotonergic dysfunctions have been described in this disease and medications acting on the serotonergic system are being screened as a possible new treatment strategy.127 Another neurotransmitter system found to be impaired in both human schizophrenic patients and the PolyI:C model is the cholinergic system, with particular focus on the nicotinergic receptor system.128–131 Particular focus has been put on the α7 nicotinic receptor system, with agonists being tested for their efficacy in bettering cognitive symptoms of schizophrenia.130–132 Interestingly, Wu et al found that following MIA with PolyI:C, there was an increase in Chrna7 gene expression, which is the gene encoding for the nicotinic acetylcholine receptor α7 subunit in the fetal brain.133 This was paralleled by an increase in IL-6 expression. Furthermore, Chrna7+/− mice were more susceptible to MIA and displayed a higher increase in fetal brain IL-6.
In addition to the neurochemical changes, cytoarchitectural abnormalities have also been reported in mPFC and dentate gyrus of the hippocampus of PolyI:C animals. These abnormalities consisted of decreased complexity and density of the dendritic spines (PolyI:C at G9.5).134 Interestingly, Soumiya et al121 reported increased presynaptic structure density in the upper cortical layers and a decreased number of synaptophysin- and GAD67-positive puncta surrounding the neuronal bodies in the upper cortical layers, suggestive of an imbalance between excitatory and inhibitory synapses. Therefore, prenatal PolyI:C may determine aberrant spine-density development in a brain region specific way, and dendritic spine pathology is indeed a feature of the pathophysiology of schizophrenia.135 Studies conducted with human material from schizophrenic patients revealed that structural abnormalities are not restricted to dendritic spines but are also reported to include white matter abnormalities in the PFCs and in the tracts linking the PFC to limbic regions, as reviewed by Najjar and Pearlman.136 Importantly, changes in white matter structure correlated with microglial activation, changes in GAD65/67 immunoreactivity, and increased mRNA for IL-1β and IL-6 in different regions of the PFCs. Higher density of neuronal nuclear antigen immunoreactive cells was found at the sites of inflammation as compared to controls, suggesting that correct development and cellular density organization were not properly shepherded.136 White matter pathology in schizophrenic patients and in PolyI:C mice has also been shown in vivo using diffusion tensor imaging, which is a particular kind of magnetic resonance imaging often used to analyze white matter structural integrity.137 Using this method, Alvarado-Alanis et al138 and Li et al139 found white matter abnormalities in frontostriatal–limbic tracts in the adult PolyI:C mice, reflecting the human abnormalities especially in treatment-naive first-episode patients.
Finally, it has been shown that maternal PolyI:C challenge at G15 impairs neuronal synchrony between the mPFC and the hippocampus, and this is a major basis for thought disorder and for deficits in PPI.140,141
The adult offspring of PolyI:C-challenged dams presents with correlates of phenomena pertaining to the positive, negative, and cognitive symptoms of schizophrenia. As signs of positive symptoms, PolyI:C mice display psychostimulant-induced hyperlocomotion, deficits in PPI, and LI.116,117 Concerning the negative symptoms, they have been shown to have deficits in social behavior, decreased sucrose preference (anhedonia), and also increased behavioral despair in the forced swimming test.123,142,143 Zhang and van Praag144 found, moreover, increased immobility in the tail suspension test. The offspring of Poly:IC mice show compromised working memory and also intentional set shifting deficits along with spatial memory deficits in the Morris’s water maze and object-in-place recognition memory deficits.120,145–147 Furthermore, impairments in hippocampal dependent behavioral tests along with decreased hippocampal neurogenesis have also been reported.144 Chronic clozapine treatment has been shown to revert impairments in working memory.148 Zuckerman et al116 found that both clozapine and haloperidol could revert the deficits in LI. Moreover, as reviewed by Dickerson and Bilkey,140 clozapine treatment could efficiently restore the deficits in PPI and the aberrant neural synchrony between the mPFC and the hippocampus. So far, no study has been carried out to test the present antipsychotic treatment on the negative symptoms part in this MIA model. Behavioral outcomes with PolyI:C prenatal exposure seem to be highly dependent on the dam’s individual immune response. Missault et al149 discovered that pregnant dams that lost weight after PolyI:C administration had the highest serum TNF-α levels, and the offspring displayed the most severe behavioral deficits. On the other hand, the offspring of dams that gained weight and showed a minor rise in TNF-α after PolyI:C showed a more blunted phenotype. This may account for some variability in reporting certain behavioral deficits in this model, and perhaps, it may even be a decisive factor for the penetrance of this risk factor in humans, given that the immune response of pregnant mothers can vary. After PolyI:C injection in the mother, there is a rise in several proinflammatory cytokines and chemokines in the fetal brain.150–152 Microglia expresses the TLR3 to which PolyI:C binds, evoking an innate immune response in both rodents and humans, and besides being able to respond to PolyI:C, they are able to sense changes in cytokine levels.153 It is known that microglia during fetal development are controlling and guiding neural precursor cells and axonal and dendritic outgrowth by active phagocytosis.154,155 They may, therefore, not be ready for an immune challenge yet. The interesting part is that this early activation seems to permanently change microglial phenotype throughout development, as highlighted by a study in which they showed that following PolyI:C prenatal challenge in regions relevant to schizophrenia, cytokines follow a precise expression pattern from P0–P60 and that MIA with PolyI:C disrupts this program.156 In this regard, IL6 seems to play a major role in the PolyI:C-induced neurodevelopmental derangement.142,151 In the absence of IL-6 (IL-6 knockout mice), the phenotype will not develop despite PolyI:C injection.142 There are evidences that microglia cells persist in an activated phenotype in the adult PolyI:C animal.18,19,157,158 This is in line with in vivo PET scan studies conducted on schizophrenic patients using the binding of a ligand to the peripheral benzodiazepine receptor as a measure for neuroinflammation. Activated microglia increase the expression of the peripheral benzodiazepine receptor. Thus, the measure of the ligand binding in the brain can be used as an in vivo correlate for microglia activation.15,16 As we reported earlier, anti-inflammatory treatment with minocycline has been proven efficacious in human studies, especially in improving cognitive and negative symptoms, which are the most resistant to present treatment.159,160 Minocycline has been applied in some studies to PolyI:C animals with promising preclinical results. Our group tested it successfully in a chronic treatment regimen, whereupon we showed a restoration of the deficits in PPI accompanied by decreased mRNA levels of IL-1β and TNF-α in microglia cells freshly isolated from the hippocampi of PolyI:C rats. This was accompanied by a restored adult hippocampal neurogenesis.19 Meyer et al148 also showed that PolyI:C animals have decreased hippocampal neurogenesis, but clozapine treatment did not ameliorate the decrease, while we proved that minocycline did while decreasing microglial proinflammatory cytokine production. Van den Eynde et al157 observed that PolyI:C offspring showed increased microglial density in the chronic stage of the model (P180), accompanied by hypolocomotion and social defeat. Minocycline intervention restored social defeat.
A final remark on this model regards its phenotypical similarities to autism spectrum disorders. As reviewed by Brown103 prenatal immune activation can also confer risk for developing autism spectrum disorders. In fact, some components of the negative and cognitive symptom scales are overlapping with aspects of the autism spectrum.103,161 Likewise, behavioral abnormalities in the MIA models, for instance, increased stereotyped behaviors (eg, grooming), increased anxiety, and social defeat, along with several cognitive dysfunctions and PPI deficits, are considered also as correlates of phenomena observed in autism.161,162 As we discussed earlier, microglial activation following MIA may participate in the aberrant formation of brain systems, and they are also a component of the adult aspect of the disease in schizophrenic patients. Given that autism and schizophrenia share an environmental risk factor and some symptoms, they may even have in common persistent microglial activation.163–165 Table 1 summarizes the major behavioral deficits found in this model of schizophrenia and the outcome of the various treatments.
Prenatal LPS
Since bacterial infections during pregnancy have also been linked to the development of schizophrenia in the offspring, some groups use LPS as a “primary hit” during the same time windows.104 LPS binds to TLR4 in immune cells, thereby inducing a systemic innate immune response similar to PolyI:C in terms of proinflammatory cytokine release such as IL-1β, IL-6, and TNF-α, although lacking the interferon-like response typical of viral immune challenge.108 It is difficult to directly compare the PolyI:C- and the LPS-exposed offspring since the studies conducted vary significantly and are inconsistent in the time point, the dose, and the amount of PolyI:C and LPS injections. Meyer107 recently compared in a direct way the general behavioral outcome of, among others, LPS and PolyI:C MIA models, and we refer to this review for a direct LPS vs PolyI:C comparison. As we will discuss, the phenotype of the adult offspring of dams challenged with LPS (also referred to as LPS animals) will present many similarities with the PolyI:C offspring, with several features matching a schizophrenic endophenotype.
Baharnoori et al166 and Santana et al167 demonstrated that LPS offspring display decreased D2 receptor binding potential in PV-positive GABAergic interneurons in the PFC. This is comparable to the lack of maturation in D2 receptor-mediated control of the GABAergic interneuron system in the PFC found in the nVHL model.73,75 Moreover, these findings are conformant with what Meyer107 found in the PolyI:C model. Baharnoori et al166 also showed altered DA transporter binding in limbic regions, a sign of dopaminergic malfunction. Further evidence of anomalies in the DAergic system in this model comes from Borrell et al168 who found increased tyrosine hydroxylase immunoreactivity in the NcA of these animals, along with the findings of increased DA levels in the NcA and increased dihydroxyphenylacetic acid in the striatum by Romero et al.169 In this study, the authors also report increased immunoreactivity for synaptophysin in the frontal cortex and hippocampus, a clear sign of synaptic reorganization in these two areas in adult offspring from LPS challenged dams.169 Further cytoarchitectural abnormalities are found in the pyramidal neurons in the mPFC and CA1 region of the hippocampus.170 Dendritic arbor was found significantly decreased in both the regions, at different postnatal time points, including adulthood, and as we mentioned earlier, dendritic spine pathology is a recognized feature in schizophrenic patients.170 Golan et al171 reported an increase in hippocampal pyramidal and granular cells with a shrinkage in cellular size, restricted to the pyramidal cells, further highlighting the sensitivity of the hippocampus to prenatal inflammatory insult. This model also presents with aberrations in the GABAergic system as it is the case in schizophrenic patients, the nVHL and the PolyI:C models. Basta-Kaim et al172 recently showed diminished total number of PV- and GAD67-positive neurons in the mPFC of adult LPS female animals, while the same changes occurred in the hippocampus of the males, although a decrease in PV-positive axon terminals of GABAergic cells was reported in the mPFC of both sexes. On the other hand, Wischhof et al173 showed a decrease in PV-positive neurons in the mPFC, hippocampus, and entorhinal cortex in both adult male and female LPS animals. The difference between the two studies may be due to the fact that Basta-Kaim et al172 administered LPS every second day, starting from G9 until delivery, whereas Wischhof et al173 administered two injections at G15 and G16. Furthermore, Li et al,139 Wischhof et al,173 and Alvarado-Alanis et al138 also show impaired myelination in cortical and limbic regions in these mice, reflecting the abnormalities previously reported in the Poly:IC model and in schizophrenic patients.
LPS mice also display several schizophrenia endophenotype-related behaviors which show up in early adulthood. Adult LPS mice show negative-like symptoms in the form of increased sensitivity to stress and increased despair in the forced swimming test, and also decreased social behavior and increased anxiety in the elevated plus maze test (Table 1).174–177 Wischhof et al173 demonstrated the presence of cognitive deficits in the form of impaired object recognition memory and also PPI deficits. Impaired spatial learning in the Morris’s water maze was also shown by Chlodzinska et al.178 Moreover, besides the PPI deficits, another sign of positive symptoms found in the LPS rodent model is the psychostimulant-induced hyperlocomotion, as shown by Basta-Kaim et al179 in a study in which they also demonstrate a hyperactive hypothalamic–pituitary–adrenal axis, which might be the reason for the altered stress responsiveness observed in this model by Lin et al.180 When it comes to the predictive validity of this model, only one study has shown that haloperidol treatment reverts the deficit in PPI, as did clozapine but not chlorpromazine, which decreased the amphetamine-induced hyperlocomotion, however.169,181 The LPS model of schizophrenia has till date not been thoroughly investigated in terms of inflammatory processes and microglial activation. We could find two studies addressing this topic, both carried out by Graciarena et al.182,183 These studies showed that prenatal LPS leads to activated microglia cells in the hippocampus of adult offspring, and this is accompanied by a decrease in transforming growth factor beta 1, lower adult hippocampal neurogenesis (in line with what we reported in the PolyI:C model), and deficits in the novel object recognition test.182–184 Furthermore, Cunningham et al155 demonstrated that microglia cells from the embryonic day 15 onward play a fundamental role in regulating the number of neuronal precursor cell (NPC) pool that are meant to build the cerebral cortex by active phagocytosis of the NPCs. LPS challenge in pregnant dams between G15 and G16 can significantly decrease the number of neural precursor cells possibly due to a significantly increased proportion of inducible nitric oxide synthase-positive microglia cells in the subventricular zone. These effects were ablated by anti-inflammatory treatment with doxycycline, a tetracycline, and a close derivative of minocycline.155 This particular function of microglia has been shown to be important also in the adult regulation of neurogenesis in the hippocampus. Therefore, it is not surprising to find decreased neurogenesis in the adult offspring of PolyI:C- and LPS-treated dams, and also that treatment with minocycline improves neurogenesis, as we demonstrated.184–186 For an overview of the major behavioral deficits found in this model of schizophrenia and the outcome of the various treatments we refer to Table 1.
Conclusion
Overall, the construct validity of the MIA models with LPS and PolyI:C show great reliability, being based on human epidemiological observations. Moreover, there is no manipulation in terms of lesions, administration of psychotropic drugs, or knockout/in of genes. On the contrary, they show a natural postpubertal outbreak of symptoms as observed in humans, suggestive of a neurodevelopmental course. As reviewed by Meyer,107 the PolyI:C model seems to have the widest array of schizophrenia-like behavioral aberrations. Moreover, as far as we could find in the literature, it has been more thoroughly characterized as compared to the LPS model. As for the face validity, both MIA models show structural, neurochemical, and behavioral abnormalities, which share a lot of common hallmarks with the human clinical picture. A growing body of evidence is associating schizophrenia to microglial activation and inflammation, with promising outcome of anti-inflammatory intervention. The studies carried out so far in the PolyI:C model show that microglia activation is also a feature of this model, with the neurodevelopmental component and a proven efficacy of minocycline at least in the aspects screened up to date. One major limitation of this model is the fact that full organogenesis, including the brain, is completed after birth, while in humans it occurs in utero, and it is difficult to match the right trimester of rodents and humans.32 To our knowledge, the best way of getting around this issue has been proposed by Ratnayake et al32 by utilizing the spiny mouse strain, which has a complete organogenesis at birth with a gestational term of 39 days. The outcome of maternal PolyI:C exposure seems to be quite the same as for the other mice strain utilized for the purpose, including microglia cell reactivity. Nevertheless, as we mentioned earlier, some groups prefer to inject PolyI:C in early postnatal days (P2–P6) to specifically target the corresponding early second trimester of pregnancy in humans.186 This is normally done in mouse strains that do not reach complete organogenesis at birth. As adults, the treated mice will display behavioral and neurochemical changes similar to those of the prenatally challenged animals, and it is therefore a model worth deeper characterization.186 However, these mice are directly exposed to PolyI:C and therefore lack the interface between MIA, placental and amniotic fluid alterations in cytokine expression, and, consequently, fetal brain changes in cytokine production that characterize the risk factor per se.187,188 A schizophrenic phenotype is most probably the result of susceptibility genes and environmental factors that disturb normal brain development and wiring.189 Therefore, studying this interaction would probably be a very good way of completing our understanding of this pathology and also for the development of novel pharmaceutical targets, eg, targeting more specifically microglia activation. Accordingly, in recent times, several groups started to combine the known etiological factors in animal models. One such example is the MIA with PolyI:C in DISC1 transgenic mice. Lipina et al190 used a heterozygous mutant for a point mutation in the Disc1 gene, which shows mild behavioral abnormalities reminiscent of schizophrenia and induced MIA at G9 with PolyI:C. The resultant adult offspring presented with intense deficits in social interaction, PPI, and LI, accompanied by reduced spatial object recognition, all stronger than those observed in the mutant mouse alone. Interestingly, the development of these phenotype could be blocked by coadministration of anti-IL-6 antibodies together with PolyI:C, highlighting the importance of inflammatory processes also in the gene–environment interaction, as also shown by Lipina et al190 and Smith et al142 in the PolyI:C model alone. DISC1 is also found in adult glia cells, although its function there is not yet known; this model provides a good tool to screen how these cells are affected by an aberrant DISC1 production. Other combinatorial strategies include the “two hit models”, where, eg, a first immunological hit (MIA) is combined with a peri- or a postpubertal second hit, usually involving physical or mental stress. This is based on the observation that the outbreak of schizophrenic symptoms often follows exposure to strong emotional stress that functions as a second hit in predisposed individuals primed earlier in life by either genes or an environmental factor. These studies reveal that the second hit with stress enhances the outbreak of schizophrenia-like symptoms in terms of stress responses, neurochemical and behavioral changes.191–193 These are initial promising studies that pave the way for future research into following models that reproduces more than one etiological factor of schizophrenia, and they may represent the future tool to explore this pathology more in depth. Moreover, animal models will provide us with defined representations of single or multiple symptoms or hallmarks of a given disease. Thus, in the future, we might rather use a combination of different models to mimic a human pathology like schizophrenia in its complexity.
Acknowledgments
SAW is supported by the SFB TR43 granted by the German Research Foundation (DFG).
Disclosure
The authors report no conflicts of interest in this work.
References
Watanabe Y, Someya T, Nawa H. Cytokine hypothesis of schizophrenia pathogenesis: evidence from human studies and animal models. Psychiatry Clin Neurosci. 2010;64(3):217–230. doi:10.1111/j.1440-1819.2010.02094.x. | ||
Horváth S, Mirnics K. Immune system disturbances in schizophrenia. Biol Psychiatry. 2014;75(4):316–323. doi:10.1016/j.biopsych. 2013.06.010. | ||
Leucht S, Barnes TR, Kissling W, Engel RR, Correll C, Kane JM. Relapse prevention in schizophrenia with new-generation antipsychotics: a systematic review and exploratory meta-analysis of randomized, controlled trials. Am J Psychiatry. 2003;160(7):1209–1222. | ||
Keefe RS, Bilder RM, Davis SM, et al. Neurocognitive effects of antipsychotic medications in patients with chronic schizophrenia in the CATIE Trial. Arch Gen Psychiatry. 2007;64(6):633–647. doi:10.1001/archpsyc.64.6.633. | ||
Lesh TA, Niendam TA, Minzenberg MJ, Carter CS. Cognitive control deficits in schizophrenia: mechanisms and meaning. Neuropsychopharmacology. 2011;36(1):316–338. doi:10.1038/npp.2010.156. | ||
Buckley PF, Stahl SM. Pharmacological treatment of negative symptoms of schizophrenia: therapeutic opportunity or cul-de-sac? Acta Psychiatr Scand. 2007;115(2):93–100. doi:10.1111/j.1600-0447.2007.00992.x. | ||
Uçok A, Gaebel W. Side effects of atypical antipsychotics: a brief overview. World Psychiatry. 2008;7(1):58–62. | ||
Muench J, Hamer AM. Adverse effects of antipsychotic medications. Am Fam Physician. 2010;81(5):617–622. | ||
Pakpoor J, Agius M. A review of the adverse side effects associated with antipsychotics as related to their efficacy. Psychiatr Danub. 2014;26(Suppl 1):273–284. | ||
Chaudhry IB, Hallak J, Husain N, et al. Minocycline benefits negative symptoms in early schizophrenia: a randomised double-blind placebo-controlled clinical trial in patients on standard treatment. J Psychopharmacol. 2012;26(9):1185–1193. doi:10.1177/ 0269881112444941. | ||
Jhamnani K, Shivakumar V, Kalmady S, Rao NP, Venkatasubramanian G. Successful use of add-on minocycline for treatment of persistent negative symptoms in schizophrenia. J Neuropsychiatry Clin Neurosci. 2013;25(1):E06–E07. doi:10.1176/appi.neuropsych.11120376. | ||
Levkovitz Y, Mendlovich S, Riwkes S, et al. A double-blind, randomized study of minocycline for the treatment of negative and cognitive symptoms in early-phase schizophrenia. J Clin Psychiatry. 2010;71(2):138–149. doi:10.4088/JCP.08m04666yel. | ||
Frick LR, Williams K, Pittenger C. Microglial dysregulation in psychiatric disease. Clin Dev Immunol. 2013;2013:608654. doi:10.1155/ 2013/608654. | ||
Bernstein H-G, Steiner J, Guest PC, Dobrowolny H, Bogerts B. Glial cells as key players in schizophrenia pathology: recent insights and concepts of therapy. Schizophr Res. 2015;161(1):4–18. doi:10.1016/j.schres.2014.03.035. | ||
Doorduin J, de Vries EF, Willemsen AT, de Groot JC, Dierckx RA, Klein HC. Neuroinflammation in schizophrenia-related psychosis: a PET study. J Nucl Med. 2009;50(11):1801–1807. doi:10.2967/jnumed.109.066647. | ||
Van Berckel BN, Bossong MG, Boellaard R, et al. Microglia activation in recent-onset schizophrenia: a quantitative (R)-[11C]PK11195 positron emission tomography study. Biol Psychiatry. 2008;64(9):820–822. doi:10.1016/j.biopsych.2008.04.025. | ||
Zhu F, Liu Y, Zhao J, Zheng Y. Minocycline alleviates behavioral deficits and inhibits microglial activation induced by intrahippocampal administration of Granulocyte-Macrophage Colony-Stimulating Factor in adult rats. Neuroscience. 2014;266:275–281. doi:10.1016/j.neuroscience.2014.01.021. | ||
Juckel G, Manitz MP, Brüne M, Friebe A, Heneka MT, Wolf RJ. Microglial activation in a neuroinflammational animal model of schizophrenia – a pilot study. Schizophr Res. 2011;131(1–3):96–100. doi:10.1016/j.schres.2011.06.018. | ||
Mattei D, Djodari-Irani A, Hadar R, et al. Minocycline rescues decrease in neurogenesis, increase in microglia cytokines and deficits in sensorimotor gating in an animal model of schizophrenia. Brain Behav Immun. 2014;38:175–184. doi:10.1016/j.bbi.2014.01.019. | ||
Zhang L, Zhao J. Profile of minocycline and its potential in the treatment of schizophrenia. Neuropsychiatr Dis Treat. 2014;10:1103–1111. doi:10.2147/NDT.S64236. | ||
González JC, Egea J, Del Carmen Godino M, et al. Neuroprotectant minocycline depresses glutamatergic neurotransmission and Ca2+ signalling in hippocampal neurons. Eur J Neurosci. 2007;26(9):2481–2495. doi:10.1111/j.1460-9568.2007.05873.x. | ||
Willner P. Validation criteria for animal models of human mental disorders: learned helplessness as a paradigm case. Prog Neuropsychopharmacol Biol Psychiatry. 1986;10(6):677–690. | ||
Smith CL, Bolton A, Nguyen G. Genomic and epigenomic instability, fragile sites, schizophrenia and autism. Curr Genomics. 2010;11(6): 447–469. doi:10.2174/138920210793176001. | ||
Network P. Psychiatric genome-wide association study analyses implicate neuronal, immune and histone pathways. Nat Neurosci. 2015;18(2):199–209. doi:10.1038/nn.3922. | ||
Van Os J, Rutten BP, Poulton R. Gene-environment interactions in schizophrenia: review of epidemiological findings and future directions. Schizophr Bull. 2008;34(6):1066–1082. doi:10.1093/schbul/sbn117. | ||
Miyamoto Y, Nitta A. Behavioral phenotypes for negative symptoms in animal models of schizophrenia. J Pharmacol Sci. 2014;126(4):310–320. doi:10.1254/jphs.14R02CR. | ||
van den Buuse M. Modeling the positive symptoms of schizophrenia in genetically modified mice: pharmacology and methodology aspects. Schizophr Bull. 2010;36(2):246–270. doi:10.1093/schbul/sbp132. | ||
Krueger D, Howell J, Hebert B, Olausson P, Taylor J, Nairn A. Assessment of cognitive function in the heterozygous reeler mouse. Psychopharmacology (Berl). 2006;189(1):95–104. doi:10.1007/s00213-006-0530-0. | ||
Seshadri S, Kamiya A, Yokota Y, et al. Disrupted-in-schizophrenia-1 expression is regulated by beta-site amyloid precursor protein cleaving enzyme-1-neuregulin cascade. Proc Natl Acad Sci U S A. 2010; 107(12): 5622–5627. doi:10.1073/pnas.0909284107. | ||
Jaaro-Peled H, Ayhan Y, Pletnikov MV, Sawa A. Review of pathological hallmarks of schizophrenia: comparison of genetic models with patients and nongenetic models. Schizophr Bull. 2010;36(2):301–313. doi:10.1093/schbul/sbp133. | ||
Harvey L, Boksa P. A stereological comparison of GAD67 and reelin expression in the hippocampal stratum oriens of offspring from two mouse models of maternal inflammation during pregnancy. Neuropharmacology. 2012;62(4):1767–1776. doi:10.1016/j.neuropharm. 2011.11.022. | ||
Ratnayake U, Quinn TA, Castillo-Melendez M, Dickinson H, Walker DW. Behaviour and hippocampus-specific changes in spiny mouse neonates after treatment of the mother with the viral-mimetic Poly I:C at mid-pregnancy. Brain Behav Immun. 2012;26(8):1288–1299. doi:10.1016/j.bbi.2012.08.011. | ||
Tang B, Jia H, Kast RJ, Thomas EA. Epigenetic changes at gene promoters in response to immune activation in utero. Brain Behav Immun. 2013;30(2):168–175. doi:10.1016/j.bbi.2013.01.086. | ||
Marsman A, Van Den Heuvel MP, Klomp DW, Kahn RS, Luijten PR, Hulshoff Pol HE. Glutamate in schizophrenia: a focused review and meta-analysis of 1H-MRS studies. Schizophr Bull. 2013;39(1):120–129. doi:10.1093/schbul/sbr069. | ||
Homayoun H, Moghaddam B. NMDA receptor hypofunction produces opposite effects on prefrontal cortex interneurons and pyramidal neurons. J Neurosci. 2007;27(43):11496–11500. doi:10.1523/JNEUROSCI.2213-07.2007. | ||
Anticevic A, Corlett PR, Cole MW, et al. N-methyl-D-aspartate receptor antagonist effects on prefrontal cortical connectivity better model early than chronic schizophrenia. Biol Psychiatry. 2015;77(6):569–580. doi:10.1016/j.biopsych.2014.07.022. | ||
Bondi C, Matthews M, Moghaddam B. Glutamatergic animal models of schizophrenia. Curr Pharm Des. 2012;18(12):1593–1604. | ||
Moghaddam B, Jackson ME. Glutamatergic animal models of schizophrenia. Ann N Y Acad Sci. 2003;1003(1):131–137. doi:10.1196/annals. 1300.065. | ||
Hou Y, Zhang H, Xie G, et al. Neuronal injury, but not microglia activation, is associated with ketamine-induced experimental schizophrenic model in mice. Prog Neuropsychopharmacol Biol Psychiatry. 2013;45:107–116. doi:10.1016/j.pnpbp.2013.04.006. | ||
Monte AS, de Souza GC, McIntyre RS, et al. Prevention and reversal of ketamine-induced schizophrenia related behavior by minocycline in mice: possible involvement of antioxidant and nitrergic pathways. J Psychopharmacol. 2013;27(11):1032–1043. doi:10.1177/0269881113503506. | ||
Becker A, Peters B, Schroeder H, Mann T, Huether G, Grecksch G. Ketamine-induced changes in rat behaviour: a possible animal model of schizophrenia. Prog Neuropsychopharmacol Biol Psychiatry. 2003;27(4):687–700. doi:10.1016/S0278-5846(03)00080-0. | ||
Silvestre JS, Nadal R, Pallarés M, Ferré N. Acute effects of ketamine in the holeboard, the elevated-plus maze, and the social interaction test in Wistar rats. Depress Anxiety. 1997;5(1):29–33. doi:10.1002/(SICI)1520-6394(1997)5:1<29::AID-DA5>3.0.CO;2-0. | ||
Chindo BA, Adzu B, Yahaya TA, Gamaniel KS. Ketamine-enhanced immobility in forced swim test: a possible animal model for the negative symptoms of schizophrenia. Prog Neuropsychopharmacol Biol Psychiatry. 2012;38(2):310–316. doi:10.1016/j.pnpbp.2012.04.018. | ||
Zhu S, Wang H, Shi R, et al. Chronic phencyclidine induces inflammatory responses and activates GSK3β in mice. Neurochem Res. 2014;39(12):2385–2393. doi:10.1007/s11064-014-1441-9. | ||
Nakki R, Nickolenko J, Chang J, Sagar SM, Sharp FR. Haloperidol prevents ketamine- and phencyclidine-induced hsp70 protein expression but not microglial activation. Exp Neurol. 1996;137(2):234–241. | ||
Noda Y, Yamada K, Furukawa H, Nabeshima T. Enhancement of immobility in a forced swimming test by subacute or repeated treatment with phencyclidine: a new model of schizophrenia. Br J Pharmacol. 1995;116:2531–2537. | ||
Schroeder U, Schroeder H, Schwegler H, Sabel BA. Neuroleptics ameliorate phencyclidine-induced impairments of short-term memory. Br J Pharmacol. 2000;130:33–40. doi:10.1038/sj.bjp.0703171. | ||
Fujita Y, Ishima T, Kunitachi S, et al. Phencyclidine-induced cognitive deficits in mice are improved by subsequent subchronic administration of the antibiotic drug minocycline. Prog Neuropsychopharmacol Biol Psychiatry. 2008;32(2):336–339. doi:10.1016/j.pnpbp.2007.08.031. | ||
Sams-Dodd F. Phencyclidine-induced stereotyped behaviour and social isolation in rats: a possible animal model of schizophrenia. Behav Pharmacol. 1996;7(1):3–23. | ||
Sams-Dodd F. Effect of novel antipsychotic drugs on phencyclidine-induced stereotyped behaviour and social isolation in the rat social interaction test. Behav Pharmacol. 1997;8(2–3):196–215. | ||
Noda A, Noda Y, Kamei H, et al. Phencyclidine impairs latent learning in mice: interaction between glutamatergic systems and sigma1 receptors. Neuropsychopharmacology. 2001;24(4):451–460. doi:10.1016/S0893-133X(00)00192-5. | ||
Fellini L, Kumar G, Gibbs S, Steckler T, Talpos J. Re-evaluating the PCP challenge as a pre-clinical model of impaired cognitive flexibility in schizophrenia. Eur Neuropsychopharmacol. 2014;24(11):1836–1849. doi:10.1016/j.euroneuro.2014.08.012. | ||
Brown JW, Rueter LE, Zhang M. Predictive validity of a MK-801-induced cognitive impairment model in mice: implications on the potential limitations and challenges of modeling cognitive impairment associated with schizophrenia preclinically. Prog Neuropsychopharmacol Biol Psychiatry. 2014;49:53–62. doi:10.1016/j.pnpbp.2013.11.008. | ||
Gaisler-Salomon I, Weiner I. Systemic administration of MK-801 produces an abnormally persistent latent inhibition which is reversed by clozapine but not haloperidol. Psychopharmacology (Berl). 2003;166(4):333–342. doi:10.1007/s00213-002-1311-z. | ||
Rung JP, Carlsson A, Ryden Markinhuhta K, Carlsson ML. (+)-MK-801 induced social withdrawal in rats; a model for negative symptoms of schizophrenia. Prog Neuropsychopharmacol Biol Psychiatry. 2005; 29(5):827–832. doi:10.1016/j.pnpbp.2005.03.004. | ||
Rung JP, Carlsson A, Markinhuhta KR, Carlsson ML. The dopaminergic stabilizers (−)-OSU6162 and ACR16 reverse (+)-MK-801-induced social withdrawal in rats. Prog Neuropsychopharmacol Biol Psychiatry. 2005;29(5):833–839. doi:10.1016/j.pnpbp.2005.03.003. | ||
Yoshimi N, Futamura T, Hashimoto K. Improvement of dizocilpine-induced social recognition de fi cits in mice by brexpiprazole, a novel serotonin – dopamine activity modulator. Eur Neuropsychopharmacol. 2015;25(3):356–364. doi:10.1016/j.euroneuro.2014.12.014. | ||
Levin ED, Bettegowda C, Weaver T, Christopher NC. Nicotine-dizocilpine interactions and working and reference memory performance of rats in the radial-arm maze. Pharmacol Biochem Behav. 1998;61(3):335–340. doi:10.1016/S0091-3057(98)00109-9. | ||
Bubeníková-Valesová V, Horácek J, Vrajová M, Höschl C. Models of schizophrenia in humans and animals based on inhibition of NMDA receptors. Neurosci Biobehav Rev. 2008;32(5):1014–1023. doi:10.1016/ j.neubiorev.2008.03.012. | ||
Horvath ZC, Czopf J, Buzsaki G. MK-801-induced neuronal damage in rats. Brain Res. 1997;753(2):181–195. | ||
Levkovitz Y, Levi U, Braw Y, Cohen H. Minocycline, a second-generation tetracycline, as a neuroprotective agent in an animal model of schizophrenia. Brain Res. 2007;1154:154–162. doi:10.1016/j.brainres. 2007.03.080. | ||
Kraus RL, Pasieczny R, Lariosa-Willingham K, Turner MS, Jiang A, Trauger JW. Antioxidant properties of minocycline: neuroprotection in an oxidative stress assay and direct radical-scavenging activity. J Neurochem. 2005;94:819–827. doi:10.1111/j.1471-4159.2005. 03219.x. | ||
Nikodemova M, Watters JJ, Jackson SJ, Yang SK, Duncan ID. Minocycline down-regulates MHC II expression in microglia and macrophages through inhibition of IRF-1 and protein kinase C (PKC)alpha/betaII. J Biol Chem. 2007;282(20):15208–15216. doi:10.1074/jbc.M611907200. | ||
Kobayashi K, Imagama S, Ohgomori T, et al. Minocycline selectively inhibits M1 polarization of microglia. Cell Death Dis. 2013;4(3):e525. doi:10.1038/cddis.2013.54. | ||
Fix AS, Horn JW, Wightman KA, et al. Neuronal vacuolization and necrosis induced by the noncompetitive N-methyl-D-aspartate (NMDA) antagonist MK(+)801 (dizocilpine maleate): a light and electron microscopic evaluation of the rat retrosplenial cortex. Exp Neurol. 1993;123:204–215. doi:10.1006/exnr.1993.1153. | ||
Zhang L, Shirayama Y, Iyo M, Hashimoto K. Minocycline attenuates hyperlocomotion and prepulse inhibition deficits in mice after administration of the NMDA receptor antagonist dizocilpine. Neuropsychopharmacology. 2007;32(9):2004–2010. doi:10.1038/sj.npp.1301313. | ||
Zhang XY, Chen D-C, Tan Y-L, et al. The interplay between BDNF and oxidative stress in chronic schizophrenia. Psychoneuroendocrinology. 2015;51:201–208. doi:10.1016/j.psyneuen.2014.09.029. | ||
Boskovic M, Vovk T, Kores Plesnicar B, Grabnar I. Oxidative stress in schizophrenia. Curr Neuropharmacol. 2011;9(2):301–312. doi:10.2174/157015911795596595. | ||
Lipska BK, Halim ND, Segal PN, Weinberger DR. Effects of reversible inactivation of the neonatal ventral hippocampus on behavior in the adult rat. J Neurosci. 2002;22(7):2835–2842. doi: 20026195. | ||
Lipska BK, Jaskiw GE, Chrapusta S, Karoum F, Weinberger DR. Ibotenic acid lesion of the ventral hippocampus differentially affects dopamine and its metabolites in the nucleus accumbens and prefrontal cortex in the rat. Brain Res. 1992;585(1–2):1–6. doi:10.1016/0006-8993(92)91184-G. | ||
Lipska BK, Weinberger DR. A neurodevelopmental model of schizophrenia: neonatal disconnection of the hippocampus. Neurotox Res. 2002;4(5–6):469–475. doi:10.1080/1029842021000022089. | ||
Tseng K-Y, Lewis BL, Lipska BK, O’Donnell P. Post-pubertal disruption of medial prefrontal cortical dopamine-glutamate interactions in a developmental animal model of schizophrenia. Biol Psychiatry. 2007;62(7):730–738. doi:10.1016/j.biopsych.2006.10.012. | ||
Feleder C, Tseng KY, Calhoon GG, O’Donnell P. Neonatal intrahippocampal immune challenge alters dopamine modulation of prefrontal cortical interneurons in adult rats. Biol Psychiatry. 2010;67(4):386–392. doi:10.1016/j.biopsych.2009.09.028. | ||
Lipska B, Weinberger D. A neurodevelopmental model of schizophrenia: neonatal disconnection of the hippocampus. Neurotox Res. 2002;4(5–6):469–475. doi:10.1080/1029842021000022089. | ||
Tseng KY, O’Donnell P. Dopamine modulation of prefrontal cortical interneurons changes during adolescence. Cereb Cortex. 2007; 17(5):1235–1240. doi:10.1093/cercor/bhl034. | ||
Weinberger DR, Lipska BK. Cortical maldevelopment, anti-psychotic drugs, and schizophrenia: a search for common ground. Schizophr Res. 1995;16:87–110. doi:10.1016/0920-9964(95)00013-C. | ||
Alquicer G, Silva-Gómez AB, Peralta F, Flores G. Neonatal ventral hippocampus lesion alters the dopamine content in the limbic regions in postpubertal rats. Int J Dev Neurosci. 2004;22(2):103–111. doi:10.1016/j.ijdevneu.2003.12.003. | ||
Flores G, Barbeau D, Quirion R, Srivastava LK. Decreased binding of dopamine D3 receptors in limbic subregions after neonatal bilateral lesion of rat hippocampus. J Neurosci. 1996;16(6):2020–2026. | ||
Flores G, Wood GK, Liang JJ, Quirion R, Srivastava LK. Enhanced amphetamine sensitivity and increased expression of dopamine D2 receptors in postpubertal rats after neonatal excitotoxic lesions of the medial prefrontal cortex. J Neurosci. 1996;16(22):7366–7375. | ||
Schroeder H, Grecksch G, Becker A, Hoellt V, Bogerts B. Alterations of the dopaminergic and glutamatergic neurotransmission in adult rats with postnatal ibotenic acid hippocampal lesion. Psychopharmacology (Berl). 1999;145:61–66. doi:10.1007/s002130051032. | ||
François J, Ferrandon A, Koning E, Angst M-J, Sandner G, Nehlig A. Selective reorganization of GABAergic transmission in neonatal ventral hippocampal-lesioned rats. Int J Neuropsychopharmacol. 2009;12:1097–1110. doi:10.1017/S1461145709009985. | ||
Tseng KY, Lewis BL, Hashimoto T, et al. A neonatal ventral hippocampal lesion causes functional deficits in adult prefrontal cortical interneurons. J Neurosci. 2008;28(48):12691–12699. doi:10.1523/JNEUROSCI.4166-08.2008. | ||
Benes FM, Berretta S. GABAergic interneurons: implications for understanding schizophrenia and bipolar disorder. Neuropsychopharmacology. 2001;25(01):1–27. doi:10.1016/S0893-133X(01)00 225-1. | ||
Flores G, Alquicer G, Silva-Gómez AB, et al. Alterations in dendritic morphology of prefrontal cortical and nucleus accumbens neurons in post-pubertal rats after neonatal excitotoxic lesions of the ventral hippocampus. Neuroscience. 2005;133:463–470. doi:10.1016/j.neuroscience.2005.02.021. | ||
Bringas ME, Morales-Medina JC, Flores-Vivaldo Y, et al. Clozapine administration reverses behavioral, neuronal, and nitric oxide disturbances in the neonatal ventral hippocampus rat. Neuropharmacology. 2012;62(4):1848–1857. doi:10.1016/j.neuropharm.2011.12.008. | ||
Hori T, Subramaniam S, Srivastava LK, Quirion R. Behavioral and neurochemical alterations following repeated phencyclidine administration in rats with neonatal ventral hippocampal lesions. Neuropharmacology. 2000;39:2478–2491. doi:10.1016/S0028-3908(00)00059-9. | ||
Naert A, Gantois I, Laeremans A, et al. Behavioural alterations relevant to developmental brain disorders in mice with neonatally induced ventral hippocampal lesions. Brain Res Bull. 2013;94:71–81. doi:10.1016/j.brainresbull.2013.01.008. | ||
Rueter LE, Ballard ME, Gallagher KB, Basso AM, Curzon P, Kohlhaas KL. Chronic low dose risperidone and clozapine alleviate positive but not negative symptoms in the rat neonatal ventral hippocampal lesion model of schizophrenia. Psychopharmacology (Berl). 2004;176(3–4): 312–319. doi:10.1007/s00213-004-1897-4. | ||
Le Pen G, Kew J, Alberati D, Borroni E, Heitz MP, Moreau JL. Prepulse inhibition deficits of the startle reflex in neonatal ventral hippocampal-lesioned rats: reversal by glycine and a glycine transporter inhibitor. Biol Psychiatry. 2003;54(03):1162–1170. doi:10.1016/S0006-3223(03)00374-3. | ||
Becker A, Grecksch G. Haloperidol and clozapine affect social behaviour in rats postnatally lesioned in the ventral hippocampus. Pharmacol Biochem Behav. 2003;76(1):1–8. doi:10.1016/S0091-3057(03)00139-4. | ||
Le Pen G, Grottick AJ, Higgins GA, Martin JR, Jenck F, Moreau JL. Spatial and associative learning deficits induced by neonatal excitotoxic hippocampal damage in rats: further evaluation of an animal model of schizophrenia. Behav Pharmacol. 2000;11(3–4):257–268. | ||
Beninger RJ, Tuerke KJ, Forsyth JK, et al. Neonatal ventral hippocampal lesions in male and female rats: effects on water maze, locomotor activity, plus-maze and prefrontal cortical GABA and glutamate release in adulthood. Behav Brain Res. 2009;202(2):198–209. doi:10.1016/j.bbr.2009.03.044. | ||
Lecourtier L, Antal M-C, Cosquer B, et al. Intact neurobehavioral development and dramatic impairments of procedural-like memory following neonatal ventral hippocampal lesion in rats. Neuroscience. 2012;207:110–123. doi:10.1016/j.neuroscience.2012.01.040. | ||
Lipska B, Weinberger D. Subchronic treatment with haloperidol and clozapine in rats with neonatal excitotoxic hippocampal damage. Neuropsychopharmacology. 1994;10(3):199–205. | ||
Le Pen G, Moreau JL. Disruption of prepulse inhibition of startle reflex in a neurodevelopmental model of schizophrenia: reversal by clozapine, olanzapine and risperidone but not by haloperidol. Neuropsychopharmacology. 2002;27(1):1–11. doi:10.1016/S0893-133 X(01)00383-9. | ||
Levin ED, Christopher NC. Effects of clozapine on memory function in the rat neonatal hippocampal lesion model of schizophrenia. Prog Neuropsychopharmacol Biol Psychiatry. 2006;30(2):223–229. doi:10.1016/j.pnpbp.2005.10.018. | ||
Lipska BK. Using animal models to test a neurodevelopmental hypothesis of schizophrenia. J Psychiatry Neurosci. 2004;29(4): 282–286. | ||
Drouin-Ouellet J, Brownell AL, Saint-Pierre M, et al. Neuroinflammation is associated with changes in glial mGluR5 expression and the development of neonatal excitotoxic lesions. Glia. 2011;59(2):188–199.doi:10.1002/glia.21086. | ||
Rao JS, Kim H-W, Harry GJ, Rapoport SI, Reese EA. Increased neuroinflammatory and arachidonic acid cascade markers, and reduced synaptic proteins, in the postmortem frontal cortex from schizophrenia patients. Schizophr Res. 2013;147(1):24–31. doi:10.1016/j.schres.2013.02.017. | ||
Steiner J, Bielau H, Brisch R, et al. Immunological aspects in the neurobiology of suicide: elevated microglial density in schizophrenia and depression is associated with suicide. J Psychiatr Res. 2008;42(2):151–157. doi:10.1016/j.jpsychires.2006.10.013. | ||
Monji A, Kato TA, Mizoguchi Y, et al. Neuroinflammation in schizophrenia especially focused on the role of microglia. Prog Neuropsychopharmacol Biol Psychiatry. 2013;42:115–121. doi:10.1016/j.pnpbp.2011.12.002. | ||
Zhu F, Zheng Y, Ding Y-Q, et al. Minocycline and risperidone prevent microglia activation and rescue behavioral deficits induced by neonatal intrahippocampal injection of lipopolysaccharide in rats. PLoS One. 2014;9(4):e93966. doi:10.1371/journal.pone.0093966. | ||
Brown AS. Epidemiologic studies of exposure to prenatal infection and risk of schizophrenia and autism. Dev Neurobiol. 2012;72:1272–1276. doi:10.1002/dneu.22024. | ||
Meyer U, Feldon J. Epidemiology-driven neurodevelopmental animal models of schizophrenia. Prog Neurobiol. 2010;90(3):285–326. doi:10.1016/j.pneurobio.2009.10.018. | ||
Kneeland RE, Fatemi SH. Viral infection, inflammation and schizophrenia. Prog Neuropsychopharmacol Biol Psychiatry. 2013;42:35–48. doi: 10.1016/j.pnpbp.2012.02.001. | ||
Fatemi SH, Folsom TD, Reutiman TJ, et al. Abnormal expression of myelination genes and alterations in white matter fractional anisotropy following prenatal viral influenza infection at E16 in mice. Schizophr Res. 2009;112(1–3):46–53. doi:10.1016/j.schres.2009.04.014. | ||
Meyer U. Prenatal poly(i:C) exposure and other developmental immune activation models in rodent systems. Biol Psychiatry. 2014;75(4):307–315. doi:10.1016/j.biopsych.2013.07.011. | ||
Takeda K, Akira S. Toll-like receptors in innate immunity. Int Immunol. 2005;17(1):1–14. doi:10.1093/intimm/dxh186. | ||
Li Q, Cheung C, Wei R, et al. Prenatal immune challenge is an environmental risk factor for brain and behavior change relevant to schizophrenia: evidence from MRI in a mouse model. PLoS One. 2009;4(7):e6354. doi:10.1371/journal.pone.0006354. | ||
Piontkewitz Y, Arad M, Weiner I. Abnormal trajectories of neurodevelopment and behavior following in utero insult in the rat. Biol Psychiatry. 2011;70(9):842–851. doi:10.1016/j.biopsych.2011.06.007. | ||
Piontkewitz Y, Arad M, Weiner I. Risperidone administered during asymptomatic period of adolescence prevents the emergence of brain structural pathology and behavioral abnormalities in an animal model of schizophrenia. Schizophr Bull. 2011;37(6):1257–1269. doi:10.1093/schbul/sbq040. | ||
Piontkewitz Y, Assaf Y, Weiner I. Clozapine administration in adolescence prevents postpubertal emergence of brain structural pathology in an animal model of schizophrenia. Biol Psychiatry. 2009;66(11):1038–1046. doi:10.1016/j.biopsych.2009.07.005. | ||
Meyer U, Nyffeler M, Yee BK, Knuesel I, Feldon J. Adult brain and behavioral pathological markers of prenatal immune challenge during early/middle and late fetal development in mice. Brain Behav Immun. 2008;22(4):469–486. doi:10.1016/j.bbi.2007.09.012. | ||
Meyer U, Nyffeler M, Engler A, et al. The time of prenatal immune challenge determines the specificity of inflammation-mediated brain and behavioral pathology. J Neurosci. 2006;26(18):4752–4762. doi:10.1523/JNEUROSCI.0099-06.2006. | ||
Ibi D, Nagai T, Kitahara Y, et al. Neonatal polyI:C treatment in mice results in schizophrenia-like behavioral and neurochemical abnormalities in adulthood. Neurosci Res. 2009;64(3):297–305. doi:10.1016/j.neures.2009.03.015. | ||
Zuckerman L, Rehavi M, Nachman R, Weiner I. Immune activation during pregnancy in rats leads to a postpubertal emergence of disrupted latent inhibition, dopaminergic hyperfunction, and altered limbic morphology in the offspring: a novel neurodevelopmental model of schizophrenia. Neuropsychopharmacology. 2003;28(10):1778–1789. doi:10.1038/sj.npp.1300248. | ||
Ozawa K, Hashimoto K, Kishimoto T, Shimizu E, Ishikura H, Iyo M. Immune activation during pregnancy in mice leads to dopaminergic hyperfunction and cognitive impairment in the offspring: a neurodevelopmental animal model of schizophrenia. Biol Psychiatry. 2006;59(6):546–554. doi:10.1016/j.biopsych.2005. 07.031. | ||
Tse MT, Piantadosi PT, Floresco SB. Prefrontal cortical gamma-aminobutyric acid transmission and cognitive function: drawing links to schizophrenia from preclinical research. Biol Psychiatry. 2014:1–11. doi:10.1016/j.biopsych.2014.09.007. | ||
Nyffeler M, Meyer U, Yee BK, Feldon J, Knuesel I. Maternal immune activation during pregnancy increases limbic GABAA receptor immunoreactivity in the adult offspring: implications for schizophrenia. Neuroscience. 2006;143(1):51–62. doi:10.1016/j.neuroscience. 2006.07.029. | ||
Richetto J, Calabrese F, Meyer U, Riva MA. Prenatal versus postnatal maternal factors in the development of infection-induced working memory impairments in mice. Brain Behav Immun. 2013;33:190–200. doi:10.1016/j.bbi.2013.07.006. | ||
Soumiya H, Fukumitsu H, Furukawa S. Prenatal immune challenge compromises development of upper-layer but not deeper-layer neurons of the mouse cerebral cortex. J Neurosci Res. 2011;89(9):1342–1350. doi:10.1002/jnr.22636. | ||
Winter C, Djodari-Irani A, Sohr R, et al. Prenatal immune activation leads to multiple changes in basal neurotransmitter levels in the adult brain: implications for brain disorders of neurodevelopmental origin such as schizophrenia. Int J Neuropsychopharmacol. 2009; 12(4):513–524. doi:10.1017/S1461145708009206. | ||
Bitanihirwe BK, Peleg-Raibstein D, Mouttet F, Feldon J, Meyer U. Late prenatal immune activation in mice leads to behavioral and neurochemical abnormalities relevant to the negative symptoms of schizophrenia. Neuropsychopharmacology. 2010;35(12):2462–2478. doi:10.1038/npp.2010.129. | ||
Roenker NL, Gudelsky G, Ahlbrand R, et al. Effect of paliperidone and risperidone on extracellular glutamate in the prefrontal cortex of rats exposed to prenatal immune activation or MK-801. Neurosci Lett. 2011;500(3):167–171. doi:10.1016/j.neulet.2011.06.011. | ||
Holloway T, Moreno JL, Umali A, et al. Prenatal stress induces schizophrenia-like alterations of serotonin 2A and metabotropic glutamate 2 receptors in the adult offspring: role of maternal immune system. J Neurosci. 2013;33(3):1088–1098. doi:10.1523/JNEUROSCI. 2331-12.2013. | ||
Dalton VS, Verdurand M, Walker A, Hodgson DM, Zavitsanou K. Synergistic effect between maternal infection and adolescent cannabinoid exposure on serotonin 5HT1A receptor binding in the hippocampus: testing the “two hit” hypothesis for the development of schizophrenia. ISRN Psychiatry. 2012;2012:1–9. doi:10.5402/2012/451865. | ||
Selvaraj S, Arnone D, Cappai A, Howes O. Alterations in the serotonin system in schizophrenia: a systematic review and meta-analysis of postmortem and molecular imaging studies. Neurosci Biobehav Rev. 2014;45:233–245. doi:10.1016/j.neubiorev.2014.06.005. | ||
Dalack GW, Healy DJ, Meador-Woodruff JH. Nicotine dependence in schizophrenia: clinical phenomena and laboratory findings. Am J Psychiatry. 1998;155(11):1490–1501. | ||
Holt DJ, Herman MM, Hyde TM, et al. Evidence for a deficit in cholinergic interneurons in the striatum in schizophrenia. Neuroscience. 1999;94(1):21–31. doi:10.1016/S0306-4522(99)00279-1. | ||
Olincy A, Stevens KE. Treating schizophrenia symptoms with an alpha7 nicotinic agonist, from mice to men. Biochem Pharmacol. 2007;74(8):1192–1201. doi:10.1016/j.bcp.2007.07.015. | ||
Berman JA, Talmage DA, Role LW. Cholinergic circuits and signaling in the pathophysiology of schizophrenia. Int Rev Neurobiol. 2007;78:193–223. doi:10.1016/S0074-7742(06)78007-2. | ||
Money TT, Scarr E, Udawela M, et al. Treating schizophrenia: novel targets for the cholinergic system. CNS Neurol Disord Drug Targets. 2010;9(2):241–256. | ||
Wu W-L, Adams CE, Stevens KE, Chow K-H, Freedman R, Patterson PH. The interaction between maternal immune activation and alpha 7 nicotinic acetylcholine receptor in regulating behaviors in the offspring. Brain Behav Immun. 2015;46:192–202. | ||
Li W-Y, Chang Y-C, Lee LJ, Lee L-J. Prenatal infection affects the neuronal architecture and cognitive function in adult mice. Dev Neurosci. 2014;36(5):359–370. doi:10.1159/000362383. | ||
Glausier JR, Lewis DA. Dendritic spine pathology in schizophrenia. Neuroscience. 2013;251:90–107. doi:10.1016/j.neuroscience. 2012.04.044. | ||
Najjar S, Pearlman DM. Neuroinflammation and white matter pathology in schizophrenia: systematic review. Schizophr Res. 2015; 161(1):102–112. doi:10.1016/j.schres.2014.04.041. | ||
Hagmann P, Jonasson L, Maeder P, Thiran J-P, Wedeen VJ, Meuli R. Understanding diffusion MR imaging techniques: from scalar diffusion-weighted imaging to diffusion tensor imaging and beyond. Radiographics. 2006;26(Suppl 1):S205–S223. doi:10.1148/rg.26si065510. | ||
Alvarado-Alanis P, Leon-Ortiz P, Reyes-Madrigal F, et al. Abnormal white matter integrity in antipsychotic-naive first-episode psychosis patients assessed by a DTI principal component analysis. Schizophr Res. 2015;162(1–3):14–21. doi:10.1016/j.schres.2015.01.019. | ||
Li Q, Cheung C, Wei R, et al. Voxel-based analysis of postnatal white matter microstructure in mice exposed to immune challenge in early or late pregnancy. Neuroimage. 2010;52(1):1–8. doi:10.1016/j.neuroimage.2010.04.015. | ||
Dickerson DD, Bilkey DK. Aberrant neural synchrony in the maternal immune activation model: using translatable measures to explore targeted interventions. Front Behav Neurosci. 2013;7(12):217. doi:10.3389/fnbeh.2013.00217. | ||
Dickerson DD, Overeem KA, Wolff AR, Williams JM, Abraham WC, Bilkey DK. Association of aberrant neural synchrony and altered GAD67 expression following exposure to maternal immune activation, a risk factor for schizophrenia. Transl Psychiatry. 2014;4(7):e418. doi:10.1038/tp.2014.64. | ||
Smith SE, Li J, Garbett K, Mirnics K, Patterson PH. Maternal immune activation alters fetal brain development through interleukin-6. J Neurosci. 2007;27(40):10695–10702. doi:10.1523/JNEUROSCI.2178-07.2007. | ||
Khan D, Fernando P, Cicvaric A, et al. Long-term effects of maternal immune activation on depression-like behavior in the mouse. Transl Psychiatry. 2014;4:e363. doi:10.1038/tp.2013.132. | ||
Zhang Z, van Praag H. Maternal immune activation differentially impacts mature and adult-born hippocampal neurons in male mice. Brain Behav Immun. 2015;45:60–70. doi:10.1016/j.bbi.2014.10.010. | ||
Zhang Y, Cazakoff BN, Thai CA, Howland JG. Prenatal exposure to a viral mimetic alters behavioural flexibility in male, but not female, rats. Neuropharmacology. 2012;62(3):1299–1307. doi:10.1016/j.neuropharm.2011.02.022. | ||
Howland JG, Cazakoff BN, Zhang Y. Altered object-in-place recognition memory, prepulse inhibition, and locomotor activity in the offspring of rats exposed to a viral mimetic during pregnancy. Neuroscience. 2012;201:184–198. doi:10.1016/j.neuroscience.2011. 11.011. | ||
Khan D, Fernando P, Cicvaric A, et al. Long-term effects of maternal immune activation on depression-like behavior in the mouse. Transl Psychiatry. 2014;4:e363. doi:10.1038/tp.2013.132. | ||
Meyer U, Knuesel I, Nyffeler M, Feldon J. Chronic clozapine treatment improves prenatal infection-induced working memory deficits without influencing adult hippocampal neurogenesis. Psychopharmacology (Berl). 2010;208(4):531–543. doi:10.1007/s00213-009-1754-6. | ||
Missault S, Van den Eynde K, Vanden Berghe W, et al. The risk for behavioural deficits is determined by the maternal immune response to prenatal immune challenge in a neurodevelopmental model. Brain Behav Immun. 2014;42:138–146. doi:10.1016/j.bbi.2014.06.013. | ||
Arrode-Brusés G, Brusés JL. Maternal immune activation by poly I:C induces expression of cytokines IL-1β and IL-13, chemokine MCP-1 and colony stimulating factor VEGF in fetal mouse brain. J Neuroinflammation. 2012;9(1):83. doi:10.1186/1742-2094-9-83. | ||
Pratt L, Ni L, Ponzio NM, Jonakait GM. Maternal inflammation promotes fetal microglial activation and increased cholinergic expression in the fetal basal forebrain: role of interleukin-6. Pediatr Res. 2013;74(4):393–401. doi:10.1038/pr.2013.126. | ||
Ratnayake U, Quinn T, LaRosa DA, Dickinson H, Walker DW. Prenatal exposure to the viral mimetic poly I:C alters fetal brain cytokine expression and postnatal behaviour. Dev Neurosci. 2014;36(2):83–94. doi:10.1159/000362205. | ||
Jack CS, Arbour N, Manusow J, et al. TLR signaling tailors innate immune responses in human microglia and astrocytes. J Immunol. 2005;175(7):4320–4330. | ||
Kettenmann H, Kirchhoff F, Verkhratsky A. Microglia: new roles for the synaptic stripper. Neuron. 2013;77(1):10–18. doi:10.1016/j.neuron.2012.12.023. | ||
Cunningham CL, Martínez-Cerdeño V, Noctor SC. Microglia regulate the number of neural precursor cells in the developing cerebral cortex. J Neurosci. 2013;33(10):4216–4233. doi:10.1523/JNEUROSCI.3441-12.2013. | ||
Garay PA, Hsiao EY, Patterson PH, Mcallister AK. Maternal immune activation causes age- and region-specific changes in brain cytokines in offspring throughout development. Brain Behav Immun. 2013;31:54–68. doi:10.1016/j.bbi.2012.07.008. | ||
Van den Eynde K, Missault S, Fransen E, et al. Hypolocomotive behaviour associated with increased microglia in a prenatal immune activation model with relevance to schizophrenia. Behav Brain Res. 2014;258:179–186. doi:10.1016/j.bbr.2013.10.005. | ||
Zhu F, Zheng Y, Liu Y, Zhang X, Zhao J. Minocycline alleviates behavioral deficits and inhibits microglial activation in the offspring of pregnant mice after administration of polyriboinosinic-polyribocytidilic acid. Psychiatry Res. 2014;219(3):680–686. doi:10.1016/j.psychres.2014.06.046. | ||
Keller WR, Kum LM, Wehring HJ, Koola MM, Buchanan RW, Kelly DL. A review of anti-inflammatory agents for symptoms of schizophrenia. J Psychopharmacol. 2013;27(4):337–342. doi:10.1177/0269881112467089. | ||
Sommer IE, van Westrhenen R, Begemann MJ, de Witte LD, Leucht S, Kahn RS. Efficacy of anti-inflammatory agents to improve symptoms in patients with schizophrenia: an update. Schizophr Bull. 2014;40(1):181–191. doi:10.1093/schbul/sbt139. | ||
Patterson PH. Modeling autistic features in animals. Pediatr Res. 2011;69(5):34–40. doi:10.1203/PDR.0b013e318212b80f. | ||
Patterson PH. Maternal infection and immune involvement in autism. Trends Mol Med. 2011;17(7):389–394. doi:10.1016/j.molmed.2011. 03.001. | ||
Suzuki K, Sugihara G, Ouchi Y, et al. Microglial activation in young adults with autism spectrum disorder. JAMA Psychiatry. 2013;70:49–58. doi:10.1001/jamapsychiatry.2013.272. | ||
Rodriguez JI, Kern JK. Evidence of microglial activation in autism and its possible role in brain underconnectivity. Neuron Glia Biol. 2012;7:1–9. doi:10.1017/S1740925X12000142. | ||
Theoharides TC, Athanassiou M, Panagiotidou S, Doyle R. Dysregulated brain immunity and neurotrophin signaling in Rett syndrome and autism spectrum disorders. J Neuroimmunol. 2015;279:33–38. doi:10.1016/j.jneuroim.2014.12.003. | ||
Baharnoori M, Bhardwaj SK, Srivastava LK. Effect of maternal lipopolysaccharide administration on the development of dopaminergic receptors and transporter in the rat offspring. PLoS One. 2013;8(1):18–23. doi:10.1371/journal.pone.0054439. | ||
Santana N, Mengod G, Artigas F. Quantitative analysis of the expression of dopamine D1 and D2 receptors in pyramidal and GABAergic neurons of the rat prefrontal cortex. Cereb Cortex. 2009;19(4): 849–860. doi:10.1093/cercor/bhn134. | ||
Borrell J, Vela JM, Arévalo-Martin A, Molina-Holgado E, Guaza C. Prenatal immune challenge disrupts sensorimotor gating in adult rats: implications for the etiopathogenesis of schizophrenia. Neuropsychopharmacology. 2002;26(2):204–215. doi:10.1016/S0893-133X(01)00360-8. | ||
Romero E, Ali C, Molina-Holgado E, Castellano B, Guaza C, Borrell J. Neurobehavioral and immunological consequences of prenatal immune activation in rats. Influence of antipsychotics. Neuropsychopharmacology. 2007;32(8):1791–1804. doi:10.1038/sj.npp.1301292. | ||
Baharnoori M, Brake WG, Srivastava LK. Prenatal immune challenge induces developmental changes in the morphology of pyramidal neurons of the prefrontal cortex and hippocampus in rats. Schizophr Res. 2009;107(1):99–109. doi:10.1016/j.schres.2008.10.003. | ||
Golan HM, Lev V, Hallak M, Sorokin Y, Huleihel M. Specific neurodevelopmental damage in mice offspring following maternal inflammation during pregnancy. Neuropharmacology. 2005;48(6):903–917. doi:10.1016/j.neuropharm.2004.12.023. | ||
Basta-Kaim A, Fijal K, Slusarczyk J, et al. Prenatal administration of lipopolysaccharide induces sex-dependent changes in glutamic acid decarboxylase and parvalbumin in the adult rat brain. Neuroscience. 2015;287:78–92. doi:10.1016/j.neuroscience.2014.12.013. | ||
Wischhof L, Irrsack E, Osorio C, Koch M. Prenatal LPS-exposure – a neurodevelopmental rat model of schizophrenia – differentially affects cognitive functions, myelination and parvalbumin expression in male and female offspring. Prog Neuropsychopharmacol Biol Psychiatry. 2015;57:17–30. doi:10.1016/j.pnpbp.2014.10.004. | ||
Lin Y-L, Wang S. Prenatal lipopolysaccharide exposure increases depression-like behaviors and reduces hippocampal neurogenesis in adult rats. Behav Brain Res. 2014;259:24–34. doi:10.1016/j.bbr.2013.10.034. | ||
Kirsten TB, Taricano M, Maiorka PC, Palermo-Neto J, Bernardi MM. Prenatal lipopolysaccharide reduces social behavior in male offspring. Neuroimmunomodulation. 2010;17(4):240–251. doi:10.1159/000290040. | ||
Foley KA, MacFabe DF, Vaz A, Ossenkopp K-P, Kavaliers M. Sexually dimorphic effects of prenatal exposure to propionic acid and lipopolysaccharide on social behavior in neonatal, adolescent, and adult rats: implications for autism spectrum disorders. Int J Dev Neurosci. 2014;39:68–78. doi:10.1016/j.ijdevneu.2014.04.001. | ||
Hava G, Vered L, Yael M, Mordechai H, Mahoud H. Alterations in behavior in adult offspring mice following maternal inflammation during pregnancy. Dev Psychobiol. 2006;48(2):162–168. doi:10.1002/dev.20116. | ||
Chlodzinska N, Gajerska M, Bartkowska K, Turlejski K, Djavadian RL. Lipopolysaccharide injected to pregnant mice affects behavior of their offspring in adulthood. Acta Neurobiol Exp (Wars). 2011;71(4):519–527. | ||
Basta-Kaim A, Budziszewska B, Leskiewicz M, et al. Hyperactivity of the hypothalamus-pituitary-adrenal axis in lipopolysaccharide-induced neurodevelopmental model of schizophrenia in rats: effects of antipsychotic drugs. Eur J Pharmacol. 2011;650(2–3):586–595. doi:10.1016/j.ejphar.2010.09.083. | ||
Lin Y-L, Lin S-Y, Wang S. Prenatal lipopolysaccharide exposure increases anxiety-like behaviors and enhances stress-induced corticosterone responses in adult rats. Brain Behav Immun. 2012;26(3): 459–468. doi:10.1016/j.bbi.2011.12.003. | ||
Basta-kaim A, Szczêsny E, Leœkiewicz M, et al. Maternal immune activation leads to age-related behavioral and immunological changes in male rat offspring – the effect of antipsychotic drugs. Pharmacol Rep. 2012;64(6):1400–1410. | ||
Graciarena M, Depino AM, Pitossi FJ. Prenatal inflammation impairs adult neurogenesis and memory related behavior through persistent hippocampal TGFbeta1 downregulation. Brain Behav Immun. 2010; 24(8):1301–1309. doi:10.1016/j.bbi.2010.06.005. | ||
Graciarena M, Roca V, Mathieu P, Depino AM, Pitossi FJ. Differential vulnerability of adult neurogenesis by adult and prenatal inflammation: role of TGF-beta1. Brain Behav Immun. 2013;34:17–28. doi:10.1016/j.bbi.2013.05.007. | ||
Battista D, Ferrari CC, Gage FH, Pitossi FJ. Neurogenic niche modulation by activated microglia: transforming growth factor beta increases neurogenesis in the adult dentate gyrus. Eur J Neurosci. 2006;23(1):83–93. doi:10.1111/j.1460-9568.2005.04539.x. | ||
Sierra A, Encinas JM, Deudero JJ, et al. Microglia shape adult hippocampal neurogenesis through apoptosis-coupled phagocytosis. Cell Stem Cell. 2010;7(4):483–495. doi:10.1016/j.stem.2010.08.014. | ||
Ibi D, Nagai T, Kitahara Y, et al. Neonatal polyI:C treatment in mice results in schizophrenia-like behavioral and neurochemical abnormalities in adulthood. Neurosci Res. 2009;64(3):297–305. doi:10.1016/j.neures.2009.03.015. | ||
Urakubo A, Jarskog LF, Lieberman JA, Gilmore JH. Prenatal exposure to maternal infection alters cytokine expression in the placenta, amniotic fluid, and fetal brain. Schizophr Res. 2001;47:27–36. doi:10.1016/S0920-9964(00)00032-3. | ||
Ashdown H, Dumont Y, Ng M, Poole S, Boksa P, Luheshi GN. The role of cytokines in mediating effects of prenatal infection on the fetus: implications for schizophrenia. Mol Psychiatry. 2006;11:47–55. doi:10.1038/sj.mp.4001748. | ||
Maynard TM, Sikich L, Lieberman JA, LaMantia AS. Neural development, cell-cell signaling, and the “two-hit” hypothesis of schizophrenia. Schizophr Bull. 2001;27:457–476. | ||
Lipina TV, Zai C, Hlousek D, Roder JC, Wong AH. Maternal immune activation during gestation interacts with Disc1 point mutation to exacerbate schizophrenia-related behaviors in mice. J Neurosci. 2013;33(18):7654–7666. doi:10.1523/JNEUROSCI.0091-13.2013. | ||
Giovanoli S, Weber L, Meyer U. Single and combined effects of prenatal immune activation and peripubertal stress on parvalbumin and reelin expression in the hippocampal formation. Brain Behav Immun. 2014;40:48–54. | ||
Giovanoli S, Engler H, Engler A, et al. Stress in puberty unmasks latent neuropathological consequences of prenatal immune activation in mice. Science. 2013;339(6123):1095–1099. | ||
Deslauriers J, Larouche A, Sarret P, Grignon S. Combination of prenatal immune challenge and restraint stress affects prepulse inhibition and dopaminergic/GABAergic markers. Prog Neuropsychopharmacol Biol Psychiatry. 2013;45:156–164. |
 © 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms.php
and incorporate the Creative Commons Attribution
- Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms.php
and incorporate the Creative Commons Attribution
- Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.