Back to Journals » OncoTargets and Therapy » Volume 8
BRAF-mutant melanoma: treatment approaches, resistance mechanisms, and diagnostic strategies
Authors Spagnolo F, Ghiorzo P, Orgiano L, Pastorino L, Picasso V, Tornari E, Ottaviano V, Queirolo P
Received 4 September 2014
Accepted for publication 22 October 2014
Published 16 January 2015 Volume 2015:8 Pages 157—168
DOI https://doi.org/10.2147/OTT.S39096
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 4
Editor who approved publication: Dr Faris Farassati
Francesco Spagnolo,1 Paola Ghiorzo,2,3 Laura Orgiano,4 Lorenza Pastorino,2,3 Virginia Picasso,4 Elena Tornari,4 Vincenzo Ottaviano,4 Paola Queirolo4
1Department of Plastic and Reconstructive Surgery, IRCCS Azienda Ospedaliera Universitaria San Martino, IST Istituto Nazionale per la Ricerca sul Cancro, Genova, Italy; 2Department of Internal Medicine and Medical Specialties (DiMI), University of Genoa, Genova, Italy; 3Genetics of Rare Cancers, IRCCS Azienda Ospedaliera Universitaria San Martino, IST Istituto Nazionale per la Ricerca sul Cancro, Genova, Italy; 4Department of Medical Oncology, IRCCS Azienda Ospedaliera Universitaria San Martino, IST Istituto Nazionale per la Ricerca sul Cancro, Genova, Italy
Abstract: BRAF inhibitors vemurafenib and dabrafenib achieved improved overall survival over chemotherapy and have been approved for the treatment of BRAF-mutated metastatic melanoma. More recently, the combination of BRAF inhibitor dabrafenib with MEK inhibitor trametinib has shown improved progression-free survival, compared to dabrafenib monotherapy, in a Phase II study and has received approval by the US Food and Drug Administration. However, even when treated with the combination, most patients develop mechanisms of acquired resistance, and some of them do not achieve tumor regression at all, because of intrinsic resistance to therapy. Along with the development of BRAF inhibitors, immunotherapy made an important step forward: ipilimumab, an anti-CTLA-4 monoclonal antibody, was approved for the treatment of metastatic melanoma; anti-PD-1 agents achieved promising results in Phase I/II trials, and data from Phase III studies will be ready soon. The availability of such drugs, which are effective regardless of BRAF status, has made the therapeutic approach more complex, as first-line treatment with BRAF inhibitors may not be the best choice for all BRAF-mutated patients. The aim of this paper is to review the systemic therapeutic options available today for patients affected by BRAF V600-mutated metastatic melanoma, as well as to summarize the mechanisms of resistance to BRAF inhibitors and discuss the possible strategies to overcome them. Moreover, since the molecular analysis of tumor specimens is now a pivotal and decisional factor in the treatment strategy of metastatic melanoma patients, the advances in the molecular detection techniques for the BRAF V600 mutation will be reported.
Keywords: melanoma, BRAF, vemurafenib, dabrafenib, resistance, BRAF inhibitor
Background
Cutaneous melanoma is the most aggressive form of skin cancer, with a global incidence of about 200,000 new cases per year, likely to increase over the next years. Although melanoma represents only 4% of all types of skin cancers, it is correlated with about 80% of skin cancer-related deaths (about 65,000 per year). Survival rates depend on the clinical stage at the diagnosis, with 5-year survival ranging from 15% to 60% in patients with distant and local metastases, respectively.1
The prognosis of metastatic melanoma has recently changed substantially thanks to the approval of kinase inhibitors vemurafenib,2 dabrafenib,3 and trametinib,4 and the immune checkpoint inhibitor ipilimumab.5,6 More recently, immune checkpoint inhibitors nivolumab7 and pembrolizumab8 have achieved promising results in clinical trials, which will probably lead to the approval of these drugs by the regulatory agencies.
Vemurafenib and dabrafenib are selective inhibitors of BRAF V600, a mutation carried by almost half of melanomas,9 and are approved by the US Food and Drug Administration (FDA) and European Medicines Agency for the treatment of unresectable or metastatic melanoma with mutant BRAF V600. In the Phase III trial of vemurafenib,2 median overall survival (OS) was 13.6 months in the vemurafenib group, compared with 9.7 months in the dacarbazine group; median progression-free survival (PFS) was 6.9 months in the vemurafenib group and only 1.6 months in the dacarbazine group; response rate for vemurafenib was 57%, compared with 9% for dacarbazine. Dabrafenib achieved similar results, with a PFS of 5.1 months in the Phase III study.3
Trametinib is an MEK inhibitor that achieved improved PFS (4.8 versus 1.5 months) compared to chemotherapy in a randomized Phase III study4 and, more importantly, was investigated as combination therapy with dabrafenib. In the Phase II trial comparing trametinib plus dabrafenib with dabrafenib alone,10 the median PFS were 9.4 months and 5.8 months for patients treated with dabrafenib 150 mg twice daily plus trametinib 2 mg daily and dabrafenib monotherapy, respectively. On the basis of these results, at the beginning of 2014 the FDA approved the combination of dabrafenib plus trametinib for the treatment of unresectable or metastatic melanoma with a BRAF V600E or V600K mutation.
Other kinase inhibitors are in late stages of clinical development. LGX818 is a potent and selective BRAF inhibitor with a dissociation half-time about 10 times longer than other BRAF inhibitors; in the Phase I trial a response rate as high as 67% was achieved among BRAF inhibitors-naïve patients.11 A three-arm Phase III trial is currently recruiting participants in order to compare the efficacy and safety of LGX818 monotherapy and LGX818 in combination with MEK inhibitor MEK162 as compared to vemurafenib in patients with locally advanced unresectable or metastatic melanoma with BRAF V600 mutation (NCT01909453). Moreover, a Phase III clinical trial comparing vemurafenib in combination with MEK inhibitor cobimetinib versus vemurafenib monotherapy (NCT01689519) met its primary endpoint (PFS), and these data are planned to be submitted to health authorities.12
Along with the development of BRAF and MEK inhibitors, immunotherapy made some steps forward as well: ipilimumab, a fully human IgG1 monoclonal antibody that blocks cytotoxic T-lymphocyte antigen (CTLA)-4 to elicit antitumor T-cell-mediated responses, was approved for the treatment of metastatic melanoma as it achieved a statistically significant improvement in OS in two different randomized Phase III trials in pretreated5 and in treatment-naïve6 patients with metastatic melanoma; nivolumab and pembrolizumab, monoclonal antibodies targeting the programmed cell death-1 (PD-1) receptor on infiltrating T-cells, which otherwise produces an inhibition of T-cells directed against melanoma antigens, showed promising clinical activity and efficacy, and pembrolizumab was recently approved by the FDA for the treatment of patients with unresectable or metastatic melanoma and disease progression following ipilimumab and, if BRAF V600 mutation positive, a BRAF inhibitor. A compassionate use of both drugs is available in several countries all over the world.
Signaling pathways in BRAF-mutated melanoma
The mitogen-activated protein kinase (MAPK) pathway plays an important role in the pathogenesis of melanoma. This pathway is physiologically activated when extracellular signals bind to their cognate membrane receptor, typically a receptor tyrosine kinase (RTK). RAF kinases are components of the pathway: their activity requires the formation of dimers, which is promoted by RAS activation. Activated RAF kinases phosphorylate and activate MEK1/2, which in turn phosphorylate and activate ERK1/2, leading to cellular proliferation, survival, and differentiation, and to an inhibitory feedback toward upstream components of the pathway.13
About 50% of melanomas harbor an activating mutation in BRAF, the most common being BRAFV600E,9 which renders the kinase constitutively active. In BRAF-mutated melanomas, RAS is negatively regulated by ERK-dependent feedback and BRAFV600E exists mainly as a monomer.13
BRAF inhibitors inhibit ERK signaling only in BRAF-mutated tumors. In wild-type (WT) cells, BRAF and CRAF form homo- and heterodimers on RAS activation; BRAF inhibitors binding to one member of the dimer causes an allosteric transactivation of the drug-free protomer, leading to ERK activation. This is enhanced when RAS, which promotes RAF dimerization, is overexpressed:14 in fact, most cutaneous tumors developing in patients treated with BRAF inhibitors harbor RAS mutations.15
The PI3K/AKT/mTOR pathway is a key regulator of cellular growth and protein synthesis.16,17 The MAPK pathway interacts with the PI3K/AKT/mTOR pathway at multiple points: for example, RAS directly activates PI3K, and the inhibition of a pathway may upregulate the other one.16,18
Diagnostic strategies
Detection of BRAF V600 mutations has recently become mandatory to treat patients with advanced or metastatic melanoma. Several methods have been used to detect BRAF mutations, including Sanger sequencing, mismatch ligation assay, ligase detection reaction, denaturating high-performance liquid chromatography, SNAPshot®, high-resolution melting, mutation-specific polymerase chain reaction (PCR) and mutation-specific real-time (RT) PCR, including EntroGen molecular probes (CE-IVD), pyrosequencing, and mass spectrometry.19–24 Each method has its own sensitivity, specificity, cost, and response delay. Several studies have been performed to compare the methods for detection of these mutations.24–29
Recently, detection of BRAF p.V600E mutation was also performed by immunohistochemistry (IHC) with VE1 antibody, and next-generation sequencing (NGS) technology is currently being used to analyze melanoma specimens in many research institutions. Below, we compare the most commonly used methods for the detection of BRAF mutations in melanomas to determine the method or combination of methods that should be used in diagnostic daily practice (Table 1). Our suggestion is that sequential analysis, with initial detection of p.V600E-positive cases by IHC and/or Sanger sequencing, followed by pyrosequencing or RT-PCR-based tests in negative or uninterpretable cases, is the most efficient method to use in daily practice, in certified laboratories, with validated techniques.
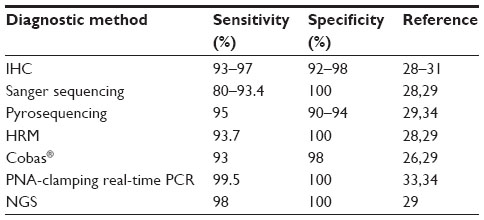 | Table 1 Sensitivity and specificity of the described methods |
Immunohistochemistry
IHC with VE1 monoclonal antibody was found to be efficient to detect p.V600E mutations.30 The advantages of this technique are that only two slides are needed, no specialized equipment is needed, it is cheaper than other techniques, and results are easily obtained within 48 hours. Interestingly, IHC with VE1 monoclonal antibody was reported to have a 100% sensitivity and specificity for detection of p.V600E, even when compared with methods more sensitive than Sanger sequencing.31 However, important disadvantages are that staining interpretation is easy in most, but not all, cases, and that the VE1 antibody is highly specific for the V600E mutation, but other clinically relevant BRAF mutations are missed. For these reasons, it could be used as a cost-effective first-line method for BRAF V600E detection in a daily practice sequential combination of methods.32
Sanger sequencing
For many years, Sanger sequencing has been considered the reference method for identification of acquired mutations in tumors. However, because of its low sensitivity (direct sequencing cannot detect the presence of mutant alleles when the mutant/WT ratio is less than 1:5),28 detection of mutations from tumor DNA requires a high percentage of tumor cells within the samples, a requirement that cannot always be met in routine diagnostic testing of human samples. A recent study compared four methods for the detection of BRAF mutations in metastatic melanomas.31 Microdissection of metastatic melanomas was performed to increase the number of tumor cells to at least 60%. However, 3 of the 40 melanomas with p.V600E mutation detected by other methods were negative with Sanger sequencing. The sensitivity of Sanger sequencing was 92.5% (95% confidence interval [CI], 78.5%–98.0%) for p.V600E detection. Thus, 7.5% of patients eligible for treatment with BRAF inhibitors would have been excluded. In line with this, another recently published series found a false-negative rate of Sanger detection of BRAF mutations of 9.2%.24 Interestingly, false-negative cases with Sanger sequencing corresponded to tumors with a high proportion of tumor cells within the samples. This finding may be related to tumor heterogeneity, with only some subclones containing p.V600E mutation and others being WT. These data indicate that Sanger sequencing failed to detect BRAF mutations in melanomas and therefore should no longer be considered as the reference test, but as a first screening or a confirmation test. Alternatively, in samples negative with Sanger sequencing, one of the tests described below should be performed.
Pyrosequencing
Pyrosequencing is an RT sequencing by synthesis approach which allows the quantification of mutated alleles. It is a rapid and more sensitive method compared with direct sequencing for quantifying the BRAF V600 mutation. In a study comparing different techniques, pyrosequencing has been shown to have 100% sensitivity (95% CI, 92.4%–100%) and specificity (95% CI, 91.6%–100%) for p.V600 detection.31 Another recent study comparing different detection methods found that there was no difference in sensitivity between the high-resolution melting analysis and Sanger sequencing (98%). All mutations down to 6.6% allele frequency could be detected with 100% specificity. In contrast, pyrosequencing detected 100% of the mutations down to 5% allele frequency but exhibited only 90% specificity, being prone to errors without using a customer-designed setup to identify all BRAF mutations at codon V600.29 Overall, pyrosequencing could be performed only on cases not interpretable or negative, corresponding to approximately 50%–60% of samples analyzed by IHC and Sanger sequencing.
RT-PCR-based tests
Several RT-PCR-based tests, certified or not, have been developed, with an overall sensitivity of 97.5% (higher than that of Sanger sequencing) and variable specificity (depending on their design specific for V600E mutation or not, and thus ranging from 87.8% to 100%). The two FDA/CE-IVD-approved tests for BRAF mutations (cobas® 4800 BRAF V600 mutation test and THxID®-BRAF) are both RT-PCR-based assays. Major disadvantages are that they are optimized for the most common BRAF mutation, and less common BRAF mutations that may still be responsive to BRAF inhibitor therapy may be missed. Further, although these assays require only a small amount of DNA, if the specimen contains <10% of tumor cells, these assays may fail to detect the mutation (the cobas® test guidelines recommend a tumor content of 50% in samples).
Recently, the cobas® test was found to detect only 70% of p.V600K, whereas 100% of p.V600E was detected.24 Interestingly, in a study comparing different techniques, RT-PCR detected 39 of the 40 p.V600E mutations and all WT cases. The sensitivity of RT PCR for p.V600E detection was 97.5% (95% CI, 87.1% to 99.6%), and specificity was 87.8% (95% CI, 75.8%–94.3%). Surprisingly, the six cases with p.V600K mutation were also positive, although with a lower signal. The four other BRAF mutations, including p.V600R, were not detected by RT-PCR. The RT-PCR detection of p.V600K was possible only after inhibition of WT allele amplification. This may be the cause of a lower sensitivity.31 Inhibition of the WT allele amplification, as performed with peptide nucleic acids (PNAs) to analyze the mutated allele, is in fact an added value to these techniques.
Recently, CE-IVD RT-PCR tests based on PNA inhibition have been developed to detect all the mutations at codon V600, with a sensitivity comparable to that of pyrosequencing.33 PNA-mediated clamping PCR (PNA-clamping PCR) is based on the principle that PNA inhibits WT by hybridizing normal sequences, and therefore mutant DNA is preferentially amplified.34 Indeed, PNA clamp RT-PCR detected a 0.5% BRAF V600E mutant in the background of the WT with high sensitivity. PNA-clamping PCR may offer a sensitive and reliable alternative method to pyrosequencing, particularly for the detection of a small amount of mutant.
Overall, RT-PCR-based assays, however, provide qualitative information only on BRAF at codon 600; no other genes are characterized, and the results are not quantitative (how much mutated BRAF is present). Further, when they are optimized for the most common BRAF mutation (V600E), less common BRAF mutations that may still be responsive to inhibitor therapy may be missed with the exception of PNA-clamp PCR, which detects all mutations at p.V600 with a sensitivity comparable to that of pyrosequencing.
A recent study comparing different methods for detecting BRAF mutations concluded that in their present setup, the cobas® 4800 BRAF V600 test as well as the therascreen® BRAF Pyro Kit (Qiagen NV, Venlo, the Netherlands) are not sufficient for the European approval of vemurafenib because there is a therapeutic option for melanoma patients with any mutation in codon 600 of the BRAF gene.35 The authors suggest a combination of VE1 antibody staining and high-resolution melting or sequencing for p.V600E mutation analysis, combining the lowest detection limit with a fast, reliable method with 100% sensitivity for routine diagnostics at the moment.
Next-generation sequencing
NGS is currently being used to analyze melanoma specimens in many research institutions. Many platforms are available, but what they have in common is that they are massively parallel sequencing techniques in which relatively small stretches (which may cover an exon, a gene, or the whole genome) of DNA are sequenced many times (typically 20 to several hundred times). NGS has several advantages over RT-PCR techniques, the most important being that it provides far more genetic information: besides mutations, NGS can detect rearrangements, amplifications, and deletions, and can analyze many genes. This will become increasingly important as we discover other actionable mutations/mechanisms of resistance to BRAF inhibition. NGS is also more sensitive than many RT-PCR assays, and can detect mutations even when tumor DNA represents less than 10% of the total DNA. NGS could analyze 100% of the cases with 100% specificity and exhibited 98.6% sensitivity in a recent study comparing different methods.29 Among disadvantages, NGS generally requires more tumor material, has a longer turn-around time, and requires a higher expertise in computational biology than any other established methods. Most of the information obtained is not yet clinically relevant, and the assays are not FDA approved. International validation is ongoing. NGS requires specialized equipment, computers, and bioinformatics, making it more expensive. This makes NGS largely a research tool at this time. In the near future and with growing experiences, it is an inevitable fact that NGS will replace all established methods for molecular diagnostics, in view of the high sensitivity and multiplexing options of this method allowing generation of a molecular profile of each tumor sample analyzed.29 A recent study performing whole cancer genome sequencing by NGS methods states that almost 75% of cancer gene variations may be missed by an approach analyzing only hotspot mutations.36
Treatment approaches
The presence of a BRAF V600 mutation is an important factor to decide the treatment approach that is the best for each patient, but it is not the only one. In fact, first-line treatment with BRAF inhibitors or BRAF inhibitors in combination with MEK inhibitors may not be the best therapeutic strategy for all patients, and the possibility to start with immunotherapy must be considered. FDA approval of ipilimumab includes the first-line treatment of metastatic melanoma, and even if in Europe, initially, the indication was in pretreated patients only, since October 2013 it has been broadened to first-line as well. Moreover, anti-PD-1 agents achieved promising results in clinical trials7,8,37 and may be recommended soon as a first-line treatment.
The role of chemotherapy as a frontline approach for BRAF-mutated metastatic melanoma has been limited by the introduction of targeted therapies. Currently, dacarbazine is the only FDA-approved chemotherapeutic drug for the treatment of metastatic melanoma, and it has not been shown to improve PFS or OS. BRAF inhibitors and BRAF inhibitors in combination with MEK inhibitors achieve tumor regression in a high rate of patients (50%–76%),2,3,10 PFS2,3 and OS2 are improved compared to those seen with chemotherapy, and the onset of tumor regression is early,41 allowing to treat successfully even symptomatic patients with low performance status and a rapidly evolving disease. Even if mechanisms of resistance, which will be discussed in the “Resistance mechanisms” section, arise in most patients, leading to tumor regrowth, there is the chance for long-term survival at least for a subset of patients, with 26% of patients from the Phase I study being alive at 3 years.38 Emerging clinical evidence suggests that extended BRAF inhibition after progression on BRAF inhibitors may prolong survival.42,43 In a series of 114 patients treated with vemurafenib and dabrafenib within clinical trials, continued therapy with BRAF inhibitors after progressive disease was associated with prolonged survival;42 similar results were observed in the Phase I study of vemurafenib.38 However, these data may be biased by patient selection, and prospective randomized trials are needed to investigate the role that prolonged BRAF inhibition may have in the treatment strategies for BRAF-mutated patients. A prospective, single-arm Phase II study was designed to evaluate the activity of treatment after progression during the therapy with vemurafenib with the combination of vemurafenib and fotemustine (NCT01983124).
Preliminary results of the Phase III trial of the combination of dabrafenib and trametinib versus dabrafenib monotherapy were presented at ASCO 2014 annual meeting:39 even if PFS was only slightly better in the combination arm (9.3 versus 8.8 months), dabrafenib and trametinib achieved 29% improvement in response rate and 37% reduction in risk of death over the monotherapy. In addition, the analysis ad interim of the other Phase III trial comparing the combination of dabrafenib and trametinib with vemurafenib monotherapy showed an OS benefit in the combination arm, allowing the crossover to the combination arm for the patients in treatment with vemurafenib.40
In contrast, the response rate and PFS with ipilimumab are lower than those with BRAF/MEK inhibitors and the onset of tumor regression is slow, as it may take time to build an immune antitumor response. However, even if the number of objective responses is relatively low, ipilimumab can induce long-lasting disease control and long-term survival: 18.2% of patients treated within Phase II studies with the approved dose of ipilimumab were alive after 4 years.44
Retrospective clinical data seem to indicate that the activity of ipilimumab is not influenced by BRAF mutational status,45 but the administration of ipilimumab after the failure of BRAF inhibitors may have suboptimal results;46–48 however, no prospective data are available to date. In the ECOG E1612 trial, patients with BRAF-mutated metastatic melanoma were randomized to receive either ipilimumab followed by vemurafenib at progression or vice versa; patients were stratified based on ECOG performance status (0 or 1), stage (III and M1a/b or M1c), and prior treatment (yes or no). ECOG trial E4613 will similarly investigate sequential treatment with the combinations ipilimumab and nivolumab versus dabrafenib and trametinib. Until the availability of prospective data from these studies, the choice of first-line treatment for BRAF-mutated metastatic melanoma patients relies on retrospective data and expert opinion. Patients with poor performance status and rapidly evolving disease, whose estimated life expectancy is less than 3 months, may not benefit from front-line treatment with ipilimumab, as may patients with high LDH levels.49
The presence of brain metastasis is historically associated with lack of efficacy of systemic therapies and poor prognosis.50 Even if ipilimumab showed clinical activity in patients with pretreated, asymptomatic, and not steroid-dependent brain metastasis,51,52 only BRAF inhibitors have evidence of activity in case of active brain metastasis.53–55
The identification of new biomarkers may also help selecting patients who are likely to respond to ipilimumab: for example, CTLA-4 gene polymorphisms seem to influence the response to anti-CTLA-4 antibodies,56 and some immunological signature may predict response to immunotherapy in general.57
Anti-PD-1 agents nivolumab and pembrolizumab achieved higher response rates in Phase I–II studies than did ipilimumab (25%–38%),7,8 and the onset of response was shorter. The duration of response is also impressive, with most responses lasting more than 12 months. Their availability in daily clinical practice may influence the therapeutic strategies, as also patients unfit to be treated with ipilimumab may benefit from treatment with anti-PD-1 drugs. Until then, patients with short life expectancy or who are unlikely to respond to ipilimumab should be treated with BRAF inhibitors or BRAF and MEK inhibitors in combination as a first-line treatment. Two BRAF inhibitors are currently available: vemurafenib and dabrafenib. Data from clinical trials showed that they have substantially the same clinical activity in patients with the BRAF V600E mutation, but they slightly differ in toxicity. Both treatments are well-tolerated, and dose reductions were needed in the range of 28% for dabrafenib3 and 38% for vemurafenib2 in the Phase III studies. Cutaneous side effects, fatigue, arthralgia, and nausea are the most common adverse events shared by the two drugs;2,3 however, vemurafenib causes a higher rate of hepatic transaminitis, photosensitivity, and cutaneous hyperproliferative lesions (including squamous cell carcinomas and keratoacanthomas), whereas pyrexia is more commonly seen with dabrafenib.2,3 As for the BRAF V600K mutation, which is the second most common BRAF V600 mutation in melanoma (19% as compared with 73% for BRAF V600E),58 in the Phase III study, vemurafenib achieved similar PFS and OS irrespective of the mutation;2 in the Phase III trial of dabrafenib,3 patients whose melanoma harbored a V600K mutation were excluded from the study, but some data are available from the Phase II trial:59 only 2 patients with the V600K mutation had a response (13%), compared with 45 patients (59%) harboring V600E. However, median PFS (4.5 versus 6.3 months) and OS (12.9 versus 13.1 months) were similar in the two groups. About 8% of melanomas harbor other genotypes than V600E and V600K,58 such as V600R, and some clinical evidence suggest that vemurafenib and dabrafenib may have clinical activity in this setting.60 The clinical activity of BRAF inhibitors in patients whose melanomas harbor a non-V600E mutation underlines the importance of using a diagnostic tool that detects all BRAF V600 mutations.
The role of trametinib monotherapy may be limited as a first-line treatment of BRAF-mutated melanoma, as well as after the failure of therapy with BRAF inhibitors.61 In the Phase II study of MEK inhibitor trametinib in BRAF-mutated patients,61 there were no confirmed objective responses in the cohort of patients previously treated with a BRAF inhibitor. On the contrary, combination therapy with trametinib and dabrafenib achieved improved clinical activity over dabrafenib monotherapy, but adverse events leading to treatment discontinuation (9% versus 5%), dose reduction (24% versus 13%), and dose interruption (45% versus 30%) were more frequent in patients treated with the combination than in those receiving only dabrafenib.43 Pyrexia was the most notable risk for the combination compared with dabrafenib, while fewer cutaneous hyperproliferative events were observed in the combination arm, consistent with the identification of activating RAS mutations in most skin tumors developing during therapy with BRAF inhibitors.13
Resistance mechanisms
About 15% of patients treated with BRAF inhibitors do not achieve tumor regression, because of intrinsic/primary mechanisms of resistance, and most patients who respond to therapy ultimately develop a mechanism of acquired/secondary resistance, leading to progressive disease.
Mechanisms of primary resistance include RAC1P29S mutations,62 COT overexpression,63 alterations in RTK signaling,64,65 loss of function of NF1,66–69 alterations in the RB1 pathway,70,71 and alterations in the PI3K-AKT-mTOR pathway (loss of function of PTEN) (Table 2).71–74
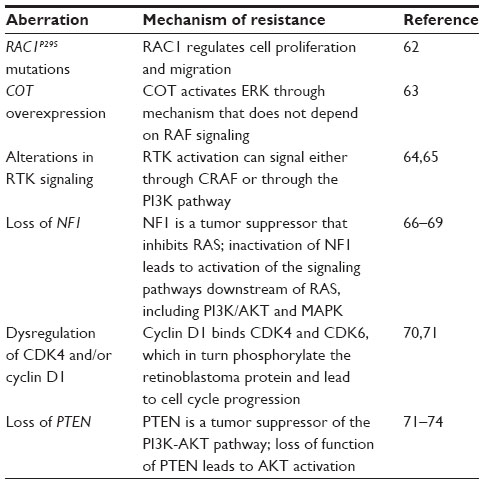 | Table 2 Mechanisms of intrinsic/primary resistance |
The reactivation of the MAPK pathway is the most frequent cause of acquired/secondary resistance; it may be driven by events that occur upstream (upregulation and activation of the RTKs,75–79 NRAS activating mutations75), downstream (activating MEK1/2 mutations62,74,80–83), or at the level of BRAF (alternative splicing of V600E BRAF,84 BRAFV600E copy number amplification,85 elevated CRAF levels86) (Table 3). The PI3K-PTEN-AKT pathway is a second core resistance pathway: AKT1/3 mutations and mutations in PI3K-AKT positive-regulatory and negative-regulatory genes may upregulate this pathway,62,80,87 driving resistance to BRAF inhibitor.
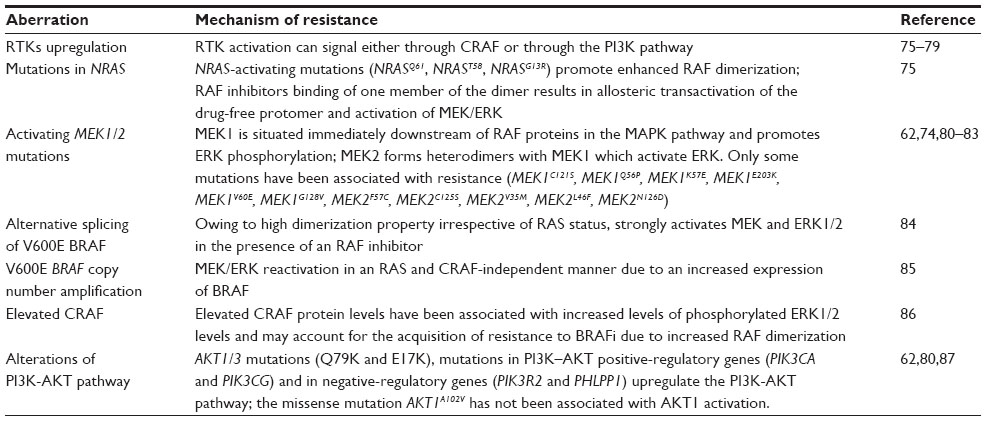 | Table 3 Mechanisms of acquired/secondary resistance |
No association was observed between clinical outcome (best response and PFS) and specific mechanisms of resistance.80 Some tumors develop multiple mechanisms of resistance simultaneously in the same patient (intrapatient heterogeneity) or even in the same lesion (intratumor heterogeneity).60,87
In addition to intrinsic and acquired resistance, mechanisms of adaptive response to BRAF inhibition limit the efficacy of treatment with BRAF inhibitors, leading mostly to partial responses, with complete response rate being in the range of only 3%–6% in the Phase III studies of vemurafenib and dabrafenib.2,3 In BRAF-mutated cells, ERK transcriptional products are upregulated, including negative-feedback components, which suppress RAS activation. As a result, RAS does not promote RAF dimerization and BRAF exists predominantly as an active monomer. Treatment with BRAF inhibitors, in addition to arresting tumor growth, relieves ERK negative feedback, partially restoring the sensitivity to extracellular signaling and the activity of RAS, promoting the formation of RAF dimers. BRAF inhibitors bind to one component of the dimer and cause an allosteric activation of the other one.14 ERK is reactivated and negative-feedback pathways are partially restored over time, leading to the formation of a new steady state of reactivated ERK signaling, which is different among different cell lines.13 The PI3K-AKT-mTOR pathway is also involved in the mechanisms of adaptive resistance: in fact, the inhibition of the MAPK pathway leads to early, adaptive AKT signaling, unleashing a rebound activation of PI3K-AKT pathway.88
Discussion and conclusion
Intrapatient and intratumor heterogeneity of resistance,62,87,89,90 cross-resistance,17,91 and alternative pathways activation88 are a challenge for personalized targeted therapies. Preclinical and clinical evidence suggest that one strategy to overcome resistance to BRAF inhibition may be the combination of multiple inhibitors. The combination of BRAF and MEK inhibitors dabrafenib and trametinib achieved a slightly better PFS over dabrafenib monotherapy in the Phase III study. Nevertheless, most mechanisms of resistance may confer cross-resistance to MEK inhibition and may lead to the activation of additional pathways, such as PI3K-AKT-mTOR, suggesting that multiple pathways may be needed to be targeted to achieve durable responses.17,91 Phase I/II trials are evaluating the safety and clinical activity of such combination regimens in patients (Table 4).
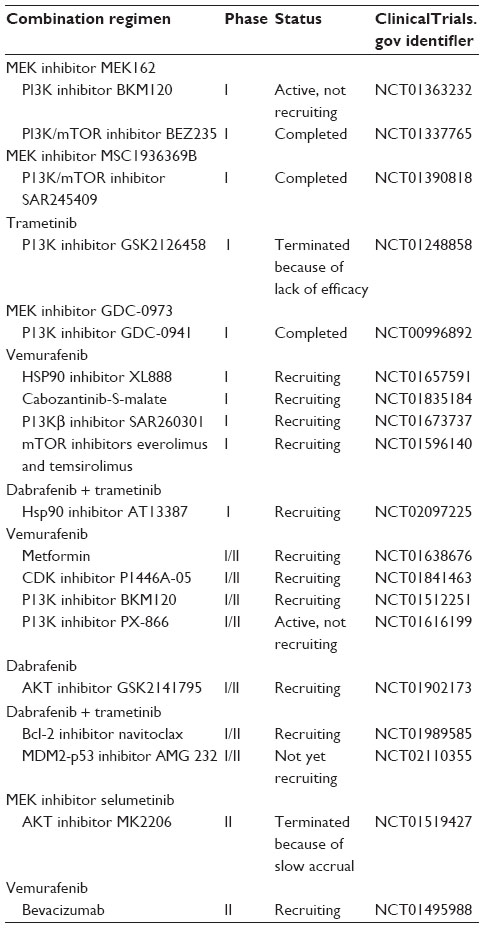 | Table 4 Ongoing Phase l/ll studies investigating multitargeted combinations |
An alternative to combination strategies may be an adaptive sequential approach based on the biopsy of progressing tumors during therapy with BRAF inhibitors. A Phase II study (NCT01820364) is currently recruiting metastatic melanoma patients who progress on treatment with BRAF inhibitor LGX818: resistant tumors will be biopsied and compared with a pretreatment biopsy to identify the mechanism of resistance. On the basis of the alterations identified in the tumor samples, a second agent from a list of MEK, CDK4/6, FGFR, PI3K, and c-MET inhibitors will be added to the regimen. A limitation of this study is that single biopsy may underestimate the tumor genomics landscape due to tumor heterogeneity. In the future, the analysis of circulating tumor cells or circulating tumor-derived DNA may provide a complete genetic profile compared to single tumor biopsies,92 but no standard method for their detection and molecular analysis is currently available.93,94
In addition to inhibiting multiple molecular targets, either in combination or in sequence, the combination of BRAF inhibitors and immunotherapy may be a strategy to provide durable responses in a high rate of metastatic melanoma patients. BRAF inhibitors do not seem to impair the immune system95,96 and, on the contrary, may enhance immune activation.97–102 Combined BRAF inhibitor vemurafenib and anti-CTLA-4 antibody ipilimumab are not tolerated.103 Nevertheless, anti-PD-1 antibodies pembrolizumab and nivolumab seem to be more tolerated than ipilimumab: their safety in combination with BRAFi and BRAFi + MEKi is under investigation in Phase I–II clinical studies. In addition, clinical studies are underway to determine the safety and clinical activity of the combination of BRAFi with other immunotherapeutic agents such as anti-PDL-1 antibodies, interleukin, adoptive cell therapy, and interferon (Table 5).
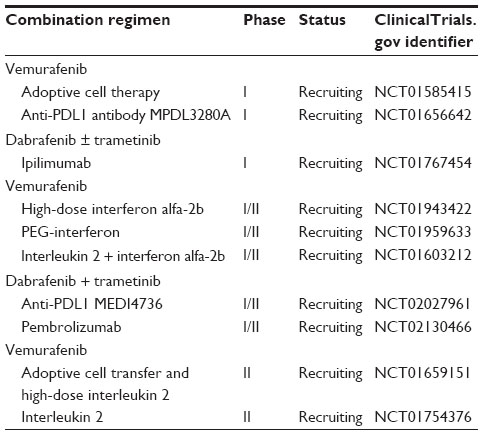 | Table 5 Ongoing Phase I/II studies investigating the combination of MAPK inhibitors with immunotherapy |
As mentioned in the “Treatment approaches” section, clinical evidence suggests that prolonged BRAF inhibition after progression on BRAF inhibitors may prolong survival in a subset of patients.42,43 In contrast with these clinical observations, Das Thakur et al demonstrated that cessation of BRAF inhibition may lead to regression of tumors expressing BRAF alternative splicing variants or amplified BRAF and that, in vivo, a discontinuous dosing strategy delayed the onset of resistance over continuous administration.104–106 The safety and efficacy of an intermittent regimen with BRAF inhibitors will be prospectively evaluated in a Phase II trial (NCT01894672):LGX818 will be administered on a daily schedule dosing for the first 6 weeks; this will be followed by a 2-week break and, thereafter, patients will resume LGX818 on a 2-weeks-on/2-weeks-off schedule.
Until further investigations and availability of new drugs, in clinical practice patients with rapid and extensive progressive disease during treatment with BRAF inhibitors, alone or in combination with MEK inhibitors, are unlikely to benefit from extended MAPK inhibition and should switch to another treatment; patients with isolated progression, on the other hand, could continue treatment with BRAF inhibitors after local treatment of resistant lesions, as resistance mechanisms are not always shared by all metastases.89
Disclosure
The authors have no financial interests in any of the products, devices, or drugs mentioned in this paper. Paola Queirolo has received lecture fees from Bristol-Myers Squibb and Roche and served on advisory boards for Bristol-Myers Squibb, Roche, and GSK; Francesco Spagnolo has received lecture fees from Bristol-Myers Squibb, GSK, and Roche. The authors declare no other conflicts of interest.
References
Siegel R, Ma J, Zou Z, Jemal A. Cancer statistics, 2014. CA Cancer J Clin. 2014;64(1):9–29. | |
McArthur GA, Chapman PB, Robert C, et al. Safety and efficacy of vemurafenib in BRAFV600E and BRAFV600K mutation-positive melanoma (BRIM-3):extended follow-up of a phase 3, randomised, open-label study. Lancet Oncol. 2014;15(3):323–332. | |
Hauschild A, Grob J-J, Demidov LV, et al. Dabrafenib in BRAF-mutated metastatic melanoma: a multicentre, open-label, phase 3 randomised controlled trial. Lancet. 2012;380(9839):358–365. | |
Flaherty KT, Robert C, Hersey P, et al. Improved survival with MEK inhibition in BRAF-mutated melanoma. N Engl J Med. 2012;367(2):107–114. | |
Hodi FS, O’Day SJ, McDermott DF, et al. Improved survival with Ipilimumab in patients with metastatic melanoma. N Engl J Med. 2010;363(8):711–723. | |
Robert C, Thomas L, Bondarenko I, et al. Ipilimumab plus Dacarbazine for previously untreated metastatic melanoma. N Engl J Med. 2011;364(26):2517–2526. | |
Topalian SL, Sznol M, McDermott DF, et al. Survival, durable tumor remission, and long-term safety in patients with advanced melanoma receiving Nivolumab. J Clin Oncol. 2014;32(10):1020–1030. | |
Hamid O, Robert C, Daud A, et al. Safety and tumor responses with Lambrolizumab (Anti–PD-1) in melanoma. N Engl J Med. 2013;369(2):134–144. | |
Davies H, Bignell GR, Cox C, et al. Mutations of the BRAF gene in human cancer. Nature. 2002;417(6892):949–954. | |
Flaherty KT, Infante JR, Daud A, et al. Combined BRAF and MEK inhibition in melanoma with BRAF V600 mutations. N Engl J Med. 2012;367(18):1694–1703. | |
Dummer R, Robert C, Nyakas M et al. Initial results from a phase I, open-label, dose escalation study of the oral BRAF inhibitor LGX818 in patients with BRAF V600 mutant advanced or metastatic melanoma. J Clin Oncol. 2013;31:(suppl; abstr 9028). | |
Larkin J, Ascierto PA, Dréno B et al. Combined vemurafenib and cobimetinib in BRAF-mutated melanoma. N Engl J Med. 2014 Nov 13;371(20):1867–1876. | |
Lito P, Rosen N, Solit DB. Tumor adaptation and resistance to RAF inhibitors. Nat Med. 2013;19(11):1401–1409. | |
Poulikakos PI, Zhang C, Bollag G, Shokat KM, Rosen N. RAF inhibitors transactivate RAF dimers and ERK signalling in cells with wild-type BRAF. Nature. 2010;464(7287):427–430. | |
Su F, Viros A, Milagre C, et al. RAS mutations in cutaneous squamous-cell carcinomas in patients treated with BRAF inhibitors. N Engl J Med. 2012;366(3):207–215. | |
Mendoza MC, Er EE, Blenis J. The Ras-ERK and PI3K-mTOR pathways: cross-talk and compensation. Trends Biochem Sci. 2011;36(6):320–328. | |
Britten CD. PI3K and MEK inhibitor combinations: examining the evidence in selected tumor types. Cancer Chemother Pharmacol. 2013;71(6):1395–1409. | |
Sánchez-Hernández I, Baquero P, Calleros L, Chiloeches A. Dual inhibition of V600EBRAF and the PI3K/AKT/mTOR pathway cooperates to induce apoptosis in melanoma cells through a MEK-independent mechanism. Cancer Lett. 2012;314(2):244–255. | |
Xing M, Tufano RP, Tufaro AP, et al. Detection of BRAF mutation on fine needle aspiration biopsy specimens: a new diagnostic tool for papillary thyroid cancer. J Clin Endocrinol Metab. 2004;89(6):2867–2872. | |
Spittle C, Ward MR, Nathanson KL, et al. Application of a BRAF pyrosequencing assay for mutation detection and copy number analysis in malignant melanoma. J Mol Diagn. 2007;9(4):464–471. | |
Thomas RK, Baker AC, DeBiasi RM, et al. High-throughput oncogene mutation profiling in human cancer. Nat Genet. 2007;39(3):347–351. | |
Pichler M, Balic M, Stadelmeyer E, et al. Evaluation of high-resolution melting analysis as a diagnostic tool to detect the BRAF V600E mutation in colorectal tumors. J Mol Diagn. 2009;11(2):140–147. | |
Jarry A, Masson D, Cassagnau E, Parois S, Laboisse C, Denis MG. Real-time allele-specific amplification for sensitive detection of the BRAF mutation V600E. Mol Cell Probes. 2004;18(5):349–352. | |
Anderson S, Bloom KJ, Vallera DU, et al. Multisite analytic performance studies of a real-time polymerase chain reaction assay for the detection of BRAF V600E mutations in formalin-fixed, paraffin-embedded tissue specimens of malignant melanoma. Arch Pathol Lab Med. 2012;136(11):1385–1391. Accessed September 1, 2014. | |
Ibrahem S, Seth R, O’Sullivan B, Fadhil W, Taniere P, Ilyas M. Comparative analysis of pyrosequencing and QMC-PCR in conjunction with high resolution melting for KRAS/BRAF mutation detection. Int J Exp Pathol. 2010;91(6):500–505. | |
Halait H, DeMartin K, Shah S, et al. Analytical performance of a real-time PCR-based assay for V600 mutations in the BRAF gene, used as the companion diagnostic test for the novel BRAF Inhibitor Vemurafenib in metastatic melanoma. Diagn Mol Pathol. 2012;21(1):1–8. Available at: http://journals.lww.com/molecularpathology/Fulltext/2012/03000/Analytical_Performance_of_a_Real_time_PCR_based.1.aspx. Accessed September 1, 2014. | |
Heideman DAM, Lurkin I, Doeleman M, et al. KRAS and BRAF mutation analysis in routine molecular diagnostics. J Mol Diagn. 14(3):247–255. | |
Carbonell P, Turpin MC, Torres-Moreno D, et al. Comparison of allelic discrimination by dHPLC, HRM, and TaqMan in the detection of BRAF mutation V600E. J Mol Diagn. 2011;13(5):467–473. Accessed September 1, 2014. | |
Ihle M, Fassunke J, Konig K, et al. Comparison of high resolution melting analysis, pyrosequencing, next generation sequencing and immunohistochemistry to conventional Sanger sequencing for the detection of p.V600E and non-p.V600E BRAF mutations. BMC Cancer. 2014;14(1):13. | |
Long GV, Wilmott JS, Capper D, et al. Immunohistochemistry is highly sensitive and specific for the detection of V600E BRAF mutation in melanoma. Am J Surg Pathol. 2013;37(1):61–65. Available at: http://journals.lww.com/ajsp/Fulltext/2013/01000/Immunohistochemistry_Is_Highly_Sensitive_and.7.aspx. Accessed September 1, 2014. | |
Colomba E, Hélias-Rodzewicz Z, Von Deimling A, et al. Detection of BRAF p.V600E mutations in melanomas: comparison of four methods argues for sequential use of immunohistochemistry and pyrosequencing. J Mol Diagn. 2013;15(1):94–100. | |
Boursault L, Haddad V, Vergier B, et al. Tumor homogeneity between primary and metastatic sites for BRAF status in metastatic melanoma determined by immunohistochemical and molecular testing. PLoS ONE. 2013;8(8):e70826. | |
Jeong D, Jeong Y, Park J, et al. BRAF V600E mutation analysis in papillary thyroid carcinomas by peptide nucleic acid clamp real-time PCR. Ann Surg Oncol. 2013;20(3):759–766. | |
Kang S-H, Pyo JY, Yang S-W, Hong SW. Detection of BRAF V600E mutation with thyroid tissue using pyrosequencing: comparison with PNA-clamping and real-time PCR. Am J Clin Pathol. 2013;139(6):759–764. | |
Da Rocha Dias S, Salmonson T, van Zwieten-Boot B, et al. The European Medicines Agency review of vemurafenib (Zelboraf®) for the treatment of adult patients with BRAF V600 mutation-positive unresectable or metastatic melanoma: summary of the scientific assessment of the Committee for Medicinal Products for Human Use. Eur J Cancer. 2013;49(7):1654–1661. | |
Ross JS, Cronin M. Whole cancer genome sequencing by next-generation methods. Am J Clin Pathol. 2011;136(4):527–539. | |
Robert C, Long GV, Brady B et al. Nivolumab in Previously Untreated Melanoma without BRAF Mutation. N Engl J Med. November 16, 2014. [Epub ahead of print]. | |
Kim K, Amaravadi RK, Flaherty K. et al. Significant long-term survival benefit demonstrated with vemurafenib in ongoing Phase I study. Pigment Cell Melanoma Res. 2012;25:866 [abstract]. | |
Long GV, Stroyakovsky DL, Gogas H et al. COMBI-d: A randomized, double-blinded, Phase III study comparing the combination of dabrafenib and trametinib to dabrafenib and trametinib placebo as first-line therapy in patients (pts) with unresectable or metastatic BRAFV600E/K mutation-positive cutaneous melanoma. J Clin Oncol. 32:5s, 2014 (suppl; abstr 9011). | |
Robert C, Karaszewska B, Schachter J et al. Improved Overall Survival in Melanoma with Combined Dabrafenib and Trametinib. N Engl J Med. 2014 Nov 16. [Epub ahead of print]. | |
McArthur GA, Puzanov I, Amaravadi R, et al. Marked, homogeneous, and early [18F]fluorodeoxyglucose–positron emission tomography responses to vemurafenib in BRAF-mutant advanced melanoma. J Clin Oncol. 2012;30(14):1628–1634. | |
Chan MMK, Haydu LE, Menzies AM, et al. The nature and management of metastatic melanoma after progression on BRAF inhibitors: effects of extended BRAF inhibition: BRAF inhibitor progression in melanoma. Cancer. 2014;120(20):3142–3153. | |
Carlino MS, Gowrishankar K, Saunders CAB, et al. Antiproliferative effects of continued mitogen-activated protein kinase pathway inhibition following acquired resistance to BRAF and/or MEK inhibition in melanoma. Mol Cancer Ther. 2013;12(7):1332–1342. | |
Wolchok JD, Weber JS, Maio M, et al. Four-year survival rates for patients with metastatic melanoma who received ipilimumab in phase II clinical trials. Ann Oncol. 2013;24(8):2174–2180. Accessed September 1, 2014. | |
Ascierto P, Simeone E, Sileni V, et al. Clinical experience with ipilimumab 3mg/kg: real-world efficacy and safety data from an expanded access programme cohort. J Transl Med. 2014;12(1):116. | |
Ackerman A, Klein O, McDermott DF, et al. Outcomes of patients with metastatic melanoma treated with immunotherapy prior to or after BRAF inhibitors. Cancer. 2014;120(11):1695–1701. | |
Ascierto PA, Margolin K. Ipilimumab before BRAF inhibitor treatment may be more beneficial than vice versa for the majority of patients with advanced melanoma. Cancer. 2014;120(11):1617–1619. | |
Ascierto PA, Simeone E, Sileni VC, et al. Sequential treatment with ipilimumab and BRAF inhibitors in patients with metastatic melanoma: data from the Italian cohort of the ipilimumab expanded access program. Cancer Invest. 2014;32(4):144–149. | |
Kelderman S, Heemskerk B, van Tinteren H, et al. Lactate dehydrogenase as a selection criterion for ipilimumab treatment in metastatic melanoma. Cancer Immunol Immunother. 2014;63(5):449–458. | |
Vecchio S, Spagnolo F, Merlo DF, et al. The treatment of melanoma brain metastases before the advent of targeted therapies: associations between therapeutic choice, clinical symptoms and outcome with survival. Melanoma Res. 2014;24(1):61–67. Available at: http://journals.lww.com/melanomaresearch/Fulltext/2014/02000/The_treatment_of_melanoma_brain_metastases_before.8.aspx. Accessed September 1, 2014. | |
Queirolo P, Spagnolo F, Ascierto P, et al. Efficacy and safety of ipilimumab in patients with advanced melanoma and brain metastases. J Neurooncol. 2014;118(1):109–116. | |
Margolin K, Ernstoff MS, Hamid O, et al. Ipilimumab in patients with melanoma and brain metastases: an open-label, phase 2 trial. Lancet Oncol. 2012;13(5):459–465. | |
Long GV, Trefzer U, Davies MA, et al. Dabrafenib in patients with Val600Glu or Val600Lys BRAF-mutant melanoma metastatic to the brain (BREAK-MB):a multicentre, open-label, phase 2 trial. Lancet Oncol. 2012;13(11):1087–1095. | |
Dummer R, Goldinger SM, Turtschi CP, et al. Vemurafenib in patients with BRAFV600 mutation-positive melanoma with symptomatic brain metastases: final results of an open-label pilot study. Eur J Cancer. 50(3):611–621. | |
Rochet NM, Kottschade LA, Markovic SN. Vemurafenib for melanoma metastases to the brain. N Engl J Med. 2011;365(25):2439–2441. | |
Queirolo P, Morabito A, Laurent S, et al. Association of CTLA-4 polymorphisms with improved overall survival in melanoma patients treated with CTLA-4 Blockade: a pilot study. Cancer Invest. 2013;31(5):336–345. | |
Gajewski TF. Molecular profiling of melanoma and the evolution of patient-specific therapy. Semin Oncol. 2011;38(2):236–242. | |
Menzies AM, Haydu LE, Visintin L, et al. Distinguishing clinicopathologic features of patients with V600E and V600K BRAF-mutant metastatic melanoma. Clin Cancer Res. 2012;18(12):3242–3249. | |
Ascierto PA, Minor D, Ribas A, et al. Phase II trial (BREAK-2) of the BRAF inhibitor dabrafenib (GSK2118436) in patients with metastatic melanoma. J Clin Oncol. 2013;31(26):3205–3211. | |
Klein O, Clements A, Menzies AM, O’Toole S, Kefford RF, Long GV. BRAF inhibitor activity in V600R metastatic melanoma. Eur J Cancer. 2013;49(5):1073–1079. | |
Kim KB, Kefford R, Pavlick AC, et al. Phase II study of the MEK1/MEK2 inhibitor trametinib in patients with metastatic BRAF-mutant cutaneous melanoma previously treated with or without a BRAF inhibitor. J Clin Oncol. 2013;31(4):482–489. | |
Van Allen EM, Wagle N, Sucker A, et al. The genetic landscape of clinical resistance to RAF inhibition in metastatic melanoma. Cancer Discov. 2014;4(1):94–109. | |
Johannessen CM, Boehm JS, Kim SY, et al. COT drives resistance to RAF inhibition through MAP kinase pathway reactivation. Nature. 2010;468(7326):968–972. | |
Wilson TR, Fridlyand J, Yan Y, et al. Widespread potential for growth-factor-driven resistance to anticancer kinase inhibitors. Nature. 2012;487(7408):505–509. | |
Straussman R, Morikawa T, Shee K, et al. Tumour micro-environment elicits innate resistance to RAF inhibitors through HGF secretion. Nature. 2012;487(7408):500–504. | |
Gibney GT, Smalley KSM. An unholy alliance: cooperation between BRAF and NF1 in melanoma development and BRAF inhibitor resistance. Cancer Discov. 2013;3(3):260–263. | |
Whittaker SR, Theurillat J-P, Van Allen E, et al. A genome-scale RNA interference screen implicates NF1 loss in resistance to RAF inhibition. Cancer Discov. 2013;3(3):350–362. | |
Maertens O, Johnson B, Hollstein P, et al. Elucidating distinct roles for NF1 in melanomagenesis. Cancer Discov. 2013;3(3):338–349. | |
Nissan MH, Pratilas CA, Jones AM, et al. Loss of NF1 in cutaneous melanoma is associated with RAS activation and MEK dependence. Cancer Res. 2014;74(8):2340–2350. | |
Smalley KS, Lioni M, Dalla Palma M, et al. Increased cyclin D1 expression can mediate BRAF inhibitor resistance in BRAF V600E–mutated melanomas. Mol Cancer Ther. 2008;7(9):2876–2883. | |
Nathanson KL, Martin A-M, Wubbenhorst B, et al. Tumor genetic analyses of patients with metastatic melanoma treated with the BRAF inhibitor dabrafenib (GSK2118436). Clin Cancer Res. 2013;19(17):4868–4878. | |
Paraiso KHT, Xiang Y, Rebecca VW, et al. PTEN loss confers BRAF inhibitor resistance to melanoma cells through the suppression of BIM expression. Cancer Res. 2011;71(7):2750–2760. | |
Xing F, Persaud Y, Pratilas CA, et al. Concurrent loss of the PTEN and RB1 tumor suppressors attenuates RAF dependence in melanomas harboring V600EBRAF. Oncogene. 2012;31(4):446–457. | |
Trunzer K, Pavlick AC, Schuchter L, et al. Pharmacodynamic effects and mechanisms of resistance to vemurafenib in patients with metastatic melanoma. J Clin Oncol. 2013;31(14):1767–1774. | |
Nazarian R, Shi H, Wang Q, et al. Melanomas acquire resistance to B-RAF(V600E) inhibition by RTK or N-RAS upregulation. Nature. 2010;468(7326):973–977. | |
Poulikakos PI, Rosen N. Mutant BRAF melanomas – dependence and resistance. Cancer Cell. 2011;19(1):11–15. | |
Shi H, Kong X, Ribas A, Lo RS. Combinatorial treatments that overcome PDGFR -driven resistance of melanoma cells to V600EB-RAF inhibition. Cancer Res. 2011;71(15):5067–5074. | |
Sun C, Wang L, Huang S, et al. Reversible and adaptive resistance to BRAF(V600E) inhibition in melanoma. Nature. 2014;508(7494):118–122. | |
Girotti MR, Pedersen M, Sanchez-Laorden B, et al. Inhibiting EGF receptor or SRC family kinase signaling overcomes BRAF inhibitor resistance in melanoma. Cancer Discov. 2013;3(2):158–167. | |
Rizos H, Menzies AM, Pupo GM, et al. BRAF inhibitor resistance mechanisms in metastatic melanoma: spectrum and clinical impact. Clin Cancer Res. 2014;20(7):1965–1977. | |
Wagle N, Emery C, Berger MF, et al. Dissecting therapeutic resistance to RAF inhibition in melanoma by tumor genomic profiling. J Clin Oncol. 2011;29(22):3085–3096. | |
Shi H, Moriceau G, Kong X, et al. Preexisting MEK1 exon 3 mutations in V600E/KBRAF melanomas do not confer resistance to BRAF inhibitors. Cancer Discov. 2012;2(5):414–424. | |
Emery CM, Vijayendran KG, Zipser MC, et al. MEK1 mutations confer resistance to MEK and B-RAF inhibition. Proc Natl Acad Sci. 2009;106(48):20411–20416. | |
Poulikakos PI, Persaud Y, Janakiraman M, et al. RAF inhibitor resistance is mediated by dimerization of aberrantly spliced BRAF(V600E). Nature. 2011;480(7377):387–390. | |
Shi H, Moriceau G, Kong X, et al. Melanoma whole-exome sequencing identifies V600EB-RAF amplification-mediated acquired B-RAF inhibitor resistance. Nat Commun. 2012;3:724. | |
Montagut C, Sharma SV, Shioda T, et al. Elevated CRAF as a potential mechanism of acquired resistance to BRAF inhibition in melanoma. Cancer Res. 2008;68(12):4853–4861. | |
Shi H, Hugo W, Kong X, et al. Acquired resistance and clonal evolution in melanoma during BRAF inhibitor therapy. Cancer Discov. 2014;4(1):80–93. | |
Shi H, Hong A, Kong X, et al. A novel AKT1 mutant amplifies an adaptive melanoma response to BRAF inhibition. Cancer Discov. 2014;4(1):69–79. | |
Romano E, Pradervand S, Paillusson A, et al. Identification of multiple mechanisms of resistance to vemurafenib in a patient with BRAFV600E-mutated cutaneous melanoma successfully rechallenged after progression. Clin Cancer Res. 2013;19(20):5749–5757. | |
Wilmott JS, Menzies AM, Haydu LE, et al. BRAFV600E protein expression and outcome from BRAF inhibitor treatment in BRAFV600E metastatic melanoma. Br J Cancer. 2013;108(4):924–931. | |
Greger JG, Eastman SD, Zhang V, et al. Combinations of BRAF, MEK, and PI3K/mTOR inhibitors overcome acquired resistance to the BRAF inhibitor GSK2118436 dabrafenib, mediated by NRAS or MEK mutations. Mol Cancer Ther. 2012;11(4):909–920. | |
Smalley KSM, Weber JS. Up close and personal: the challenges of precision medicine in melanoma. JNCI J Natl Cancer Inst. 2014;106(2):djt443. | |
Rodic S, Mihalcioiu C, Saleh RR. Detection methods of circulating tumor cells in cutaneous melanoma: a systematic review. Crit Rev Oncol Hematol. 2014;91(1):74–92. | |
Luo X, Mitra D, Sullivan RJ, et al. Isolation and molecular characterization of circulating melanoma cells. Cell Rep. 2014;7(3):645–653. | |
Hong DS, Vence L, Falchook G, et al. BRAF(V600) inhibitor GSK2118436 targeted inhibition of mutant BRAF in cancer patients does not impair overall immune competency. Clin Cancer Res. 2012;18(8):2326–2335. | |
Comin-Anduix B, Chodon T, Sazegar H, et al. The oncogenic BRAF kinase inhibitor PLX4032/RG7204 does not affect the viability or function of human lymphocytes across a wide range of concentrations. Clin Cancer Res. 2010;16(24):6040–6048. | |
Frederick DT, Piris A, Cogdill AP, et al. BRAF inhibition is associated with enhanced melanoma antigen expression and a more favorable tumor microenvironment in patients with metastatic melanoma. Clin Cancer Res. 2013;19(5):1225–1231. | |
Boni A, Cogdill AP, Dang P, et al. Selective BRAFV600E inhibition enhances T-cell recognition of melanoma without affecting lymphocyte function. Cancer Res. 2010;70(13):5213–5219. | |
Liu C, Peng W, Xu C, et al. BRAF inhibition increases tumor infiltration by T cells and enhances the antitumor activity of adoptive immunotherapy in mice. Clin Cancer Res. 2013;19(2):393–403. | |
Wilmott JS, Long GV, Howle JR, et al. Selective BRAF inhibitors induce marked T-cell infiltration into human metastatic melanoma. Clin Cancer Res. 2012;18(5):1386–1394. | |
Cooper ZA, Juneja VR, Sage PT, et al. Response to BRAF inhibition in melanoma is enhanced when combined with immune checkpoint blockade. Cancer Immunol Res. 2014;2(7):643–654. | |
Callahan MK, Masters G, Pratilas CA, et al. Paradoxical activation of T cells via augmented ERK signaling mediated by a RAF inhibitor. Cancer Immunol Res. 2014;2(1):70–79. | |
Ribas A, Hodi FS, Callahan M, Konto C, Wolchok J. Hepatotoxicity with combination of vemurafenib and ipilimumab. N Engl J Med. 2013;368(14):1365–1366. | |
Das Thakur M, Stuart DD. The evolution of melanoma resistance reveals therapeutic opportunities. Cancer Res. 2013;73(20):6106–6110. | |
Das Thakur M, Salangsang F, Landman AS, et al. Modelling vemurafenib resistance in melanoma reveals a strategy to forestall drug resistance. Nature. 2013;494(7436):251–255. | |
Das Thakur M, Stuart DD. Molecular pathways: response and resistance to BRAF and MEK inhibitors in BRAFV600E tumors. Clin Cancer Res. 2014;20(5):1074–1080. |
 © 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.