Back to Journals » Drug Design, Development and Therapy » Volume 14
Botulinum Toxin Type A Possibly Affects Cav3.2 Calcium Channel Subunit in Rats with Spinal Cord Injury-Induced Muscle Spasticity
Authors Ma K, Zhu D, Zhang C, Lv L
Received 3 April 2020
Accepted for publication 26 June 2020
Published 28 July 2020 Volume 2020:14 Pages 3029—3041
DOI https://doi.org/10.2147/DDDT.S256814
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Qiongyu Guo
Kening Ma,1 Dan Zhu,2 Chunguo Zhang,1 Lijie Lv3
1Department of Pain Medicine, The First Hospital of Jilin University, Changchun 130021, People’s Republic of China; 2Department of Neurologic Medicine, The First Hospital of Jilin University, Changchun 130021, People’s Republic of China; 3Department of Medicine and Pension, The First Hospital of Jilin University, Changchun 130021, People’s Republic of China
Correspondence: Lijie Lv
Department of Medicine and Pension, The First Hospital of Jilin University, No. 71 Xinmin Street, Changchun 130021, People’s Republic of China
Tel +86-431-88782032
Email [email protected]
Introduction: Spinal cord injury (SCI) often causes muscle spasticity, which can be inhibited by using calcium channel blocker. Botulinum toxin type A (BoT-A) shows therapeutic efficacy on spasticity and may exert inhibitory effects on the calcium channel.
Methods: A rat model with muscle spasticity was established after SCI via contusion and compression. Different concentrations (0, 1, 3 and 6 U/kg) of BoT-A Botox were injected in the extensor digitorum longus (EDL) muscles of the right hindlimb in the muscle spasticity model. The changes of muscle spasticity and calcium level in EDL muscles were measured after the establishment of SCI-induced spasticity. Cav 3.2 calcium channel subunit and its mutant (M1560V) were analyzed using Western blot before (input) or after immunoprecipitation with anti-FLAG antibody, and their currents were measured in motoneurons by using whole-cell voltage clamp recordings.
Results: SCI induced muscle spasticity, whereas calcium level in EDL muscles and expression of Cav 3.2 was increased in the SCI model when compared with the sham group (p < 0.05). BoT-A Botox treatment significantly reduced muscle spasticity and calcium level in EDL muscles and Cav 3.2 expression in a dose-dependent way (p < 0.05). The ratio of biotinylated to total Cav 3.2 was reduced in the mutant (M1560V) of Cav 3.2 and lower than that in the wild Cav 3.2. BoT-A Botox intervention also reduced the current values of calcium channel and the ratio in a dose-dependent way (p < 0.05).
Discussion: BoT-A Botox possibly attenuates SCI-induced muscle spasticity by affecting the expression of Cav 3.2 calcium channel subunit in the rat models. There may be multiple mechanisms for the function of BoT-A Botox. Further work is needed to be done to address these issues.
Keywords: botulinum toxin type A, muscle spasticity, Cav 3.2 calcium channel, rat model, spinal cord injuries
Introduction
Spinal cord injury (SCI) is a global dilemma and its incidence is estimated as 40–80 new cases per million population per year.1 The immediate consequences of SCI are motor and sensory deficits with spasticity, such as initiate and coordinate muscle spasticity.2 Muscle spasticity can lead to uncontrolled body movements,3 sustained abnormal postures of the affected body part4 and even muscle damage and rhabdomyolysis sometimes.5
Muscle spasticity is usually caused by neuromuscular excitability6 and represents a complicated array of neurological motor disorders.7 Enhancing neurite growth and circuit plasticity by using some inhibitors has become as a potential approach for improving motor function after SCI injury.8,9 The calcium channel plays an important role in the risk of spasticity,10 and Ca2+ release modulates muscle contraction11 and is involved with the plasticity activity.12 Cav3.2 is a calcium channel subunit of the T-type that is involved in the activity of skeletal muscle13,14 and may be associated with spasticity. Furthermore, Cav3.2 is expressed in the neurons of the spinal cord.15 Calcium channel blockers are thought effective to prevent muscle spasticity.16
Botulinum toxins type A (BoT-A) is produced from Clostridium botulinum type A and its main function is the cleavage of neuronal synaptosomal-associated protein of 25 kDa (SNAP-25), a protein involved in vesicular membrane fusion with neuronal plasma membrane.17,18 BoT-A is intended for intramuscular, intradetrusor and intradermal use, and has been increasingly used in clinical care for many neural disorders that include spasticity,19 and other abnormal movement diseases.20 Therefore, BoT-A is supposed to treat SCI-induced muscle spasticity. The calcium level plays an important role in the spasticity risk21 and modulates the activity of calcium channel.22 The prevention of the activity of calcium channel can reduce spasticity.16 BoT-A may prevent SCI-induced muscle spasticity as a kind of calcium channel blocker or by affecting calcium release. In the present work, muscle spasticity was characterized by measuring swimming activities in spinal cord-injured rats injected with different doses of BoT-A and the related molecules Cav3.2 subunit (which underlies the functional T-type calcium channel23) and its mutant were measured.
Materials and Methods
Experimental Animals
All surgical procedures and postoperative care were in accordance with relevant guidelines and regulations of NIH, and performed following the guidelines of the Animal Care and Use Committee of The First Hospital of Jilin University (approval No. AJLU28-18, Changchun, China). Every effort was made to minimize the number and suffering of animals used in the present experiment. Forty 8-week-old Wistar rats (200 ± 20 g) were purchased from the Animal Center of Jilin University (License No: SCXK-Ji 2008–0005). All rats were housed under a 12 h: 12 h light-dark cycle and had ad libitum access to food and water.
Establishment of SCI-Induced Muscle Spasticity
All surgical procedures were performed under protocol anesthesia (intraperitoneally, 2 mg/kg, Solarbio Science & Technology Co., Ltd., Beijing, China). The rat was fixed with a stereotaxic instrument. The dorsal, dorsolateral, and ventral funiculus bilaterally was interrupted via T-shaped lesions of the thoracic spinal cord (T9) by delivering 50 kdyn or 75 kdyn force contusions with 20 sec of sustained compression.24 Muscle spasm will be developed after SCI induction.16 Contusion was induced on laminectomized animals and muscle spasticity was determined by evaluating muscle tone and muscle spasm according to the Ashworth and spasm–frequency scales.25 Post-operative care was performed including manually assisted extrusion and analgesia. BoT-A Botox was diluted with 2 mL of saline and a solution was prepared with 6 U of toxin per 0.1 mL. The highest dose of BoT-A was 6 U/kg (https://allergan-web-cdn-prod.azureedge.net›products›pms) and some model rats were injected with Botox in the extensor digitorum longus (EDL) muscles of the right hindlimb with different concentrations (0, 1, 3 and 6 U/kg). All rats were assigned into five groups: CG (sham surgery group), WG (model group without BoT-A Botox treatment), LG (1 U/kg BoT-A Botox), MG (3 U/kg BoT-A Botox) and HG (6 U/kg BoT-A Botox).
Electromyographic (EMG) Recordings of Muscle Spasticity
EMG can be used to evaluate muscle spasticity.26 The rats were anesthetized by using ketamine cocktail and a surgical level of anesthesia was determined with the loss of paw withdrawal activities to strong foot pinch. Bipolar intramuscular EMG electrodes were implanted bilaterally into the muscles of tibialis anterior (TA) and unilaterally into the muscles of left rectus abdominis (RA). EMG signal was measured by using an EMG telemeter system (BioLog DL-5000, S&ME Co., Japan). The pre-amplified signal was subsequently demodulated at 1-kHz-sampling rate, amplified by 1000 times, and high-pass filtered (30 Hz). EMG activity was evaluated by using biological analysis software (BIMUTAS-Video, Kissei Comtec Co., Ltd., Japan).
Analysis of Muscle Spasticity via a Swimming Test
The rats swam in a rectangular water pool (200×50×20 cm, 22°C, unless otherwise indicated). The rats were placed at the end of the pool and swam to the plate on the other end. They were scored while swimming at 0.6 m in the middle of the pool (after one run).27 The degree of body flexion was evaluated during spasticity, and then examined whether the rats were still in flexed position after 0.6-m swimming. The rats were scored 1 or 2 if their swimming was in the slightly flexed position (minor spasticity, each hip was unilaterally flexed to ≤90°) for < or > half a run. They were scored 3 or 4 if their swimming was in the strong flexed position (major spasticity, each hip was unilaterally flexed to ≥90°) for < or > half a run. All data were the average when 5 runs were performed. According to the previous report, temperature affected muscle spasticity.28 After 1-month SCI establishment, the effects of water temperature on muscle spasticity were evaluated at 15°C, 22°C, and 37°C for successive 3 d. The activities of the animals were recorded by a camera (positioned in a certain way relative to the testing arena) and the parameters were measured subsequently by an automated method.
Muscle Calcium Assay
EDL muscles of the right hindlimb were excised, taken as much as 10 mg, and ground using a mortar and pestle (Nitrogen Technologies Inc., Pittsburgh, USA). The ground muscles were resuspended in 1 mL ddH2O and centrifuged at 12,000×g for 10 min. The supernatants were obtained to measure calcium level by using atomic absorption spectrophotometry in a Perkin–Elmer Model 2380 (Norwalk, CT, USA).
Site-Directed Mutagenesis of Cav3.2 Calcium Channel α1 Subunit
The Cav3.2 calcium channel α1 subunit was mutant in pCDNA-3 using the quick-change mutagenesis kit (Stratagene, CA, USA).29 For the Cav3.2 mutant (M1560V), the Cav3.2-pCDNA-3 plasmid was used as a template, and the mutant gene was amplified according to a previous report.30
Motor Neuron Isolation and Transfection
After 1-month SCI, all rats were anesthetized with enflurane: oxygen: nitrous oxide (1:33:66) and spinal cord sections were isolated by using sterile surgery and placed in a sterile Eppendorf tube. Motor neurons were isolated from the spinal cord and cultured in the medium with DMEM/F12, 1% N2, 0.5% L-glutamine, 0.5% glucose, and 0.0016% 2-mercaptoethanol.31 To prevent non-neuronal cell growth, Fluorodeoxyuridine (FDU) (10 mM, Sigma Aldrich, St. Louis Missouri, MO, USA) was added to the medium. The cells were cultured at 37°C in an incubator with 60% humidity and 5% CO2. The cells were dissociated with trypsin (0.5%) before and plated on glass coverslips.
M1560V substitution of Cav3.2 channel α1 subunits (XP_017452441.1) resulted in impaired channel inactivation and increased intracellular Ca2+ concentrations.32 Mutant (M1560V) and wild-type Cav3.2 channel α1 subunits (10 μg) DNA were transfected into cells using Lipofectamine 2000 (Invitrogen) according to the manufacturer’s protocol. The cells were cultured at 28°C, and recordings were conducted after 2-day transfection.
Electrophysiology
Whole-cell currents of Cav3.2 channel in motoneurons were measured by using the voltage clamp recordings at 22°C. Current signals were amplified using an A-M Systems model 2400 patch-clamp amplifier (Sequim, WA, USA), sampled at 100 kHz and a higher frequency range was filtered out by the digital low-pass filter at 10 kHz. Data were obtained using a data acquisition system (Digidata1440A) and analyzed using pClamp 10 software (Molecular Devices, CA, USA). The resistance of patch pipettes was 2.0–4 MΩ and series resistance was compensated by >80%.
External and internal solution was prepared according to a previous report.33 Holding potential was set at −120 mV and current-density was calculated using the ratio of the peak current and cell capacitance. The voltage-dependent activation was measured using fitting currents, and generated by the steps from −80 to 40 mV in 5 mV increments with a Boltzmann equation.34 Steady-state inactivation was evaluated by using a 20 ms test pulse to −20 mV after a 500 ms of pre-pulse to varied voltages from −140 to −45 mV in 5 mV steps. Steady-state inactivation (SSI) curves were fitted with Boltzmann equation.35 Inactivation recovery was calculated using 1 s of conditioning pulse to −20 mV followed by a test pulse to −20 mV after 24 ms with 1 ms of step at −120 mV. Time constants for inactivation recovery were calculated by using one exponential equation.36 Waveform’s current decay was analyzed by using Levenberg-Marquardt algorithm.37
Western Blot Analysis
The antibodies for Cav3.2 (PA5-77,313) and Goat anti-Rabbit IgG (H+L) secondary antibody, HRP conjugate (Product # 31,460) were from Thermo Fisher Scientific, Inc. (Waltham, MA, USA). The total protein from spinal cord was isolated by using the protein isolate kit (Invent, Plymouth, MN, USA). Western blot was performed according to a previous report.38
CaM Pull-Down Assay
The antibody for γ-tubulin was from Sigma (MA1-850 T5192). The antibody for Na+K+ATPase (ST0533) was from Thermo Fisher Scientific, Inc. (Waltham, MA, USA) and CaM was from Merck Millipore (05–173, MA, USA). The motor neuron cells, infected or non-infected by mutant (M1560V), were incubated with 1 mg/mL EZ-Link Sulfo-NHS-SS-Biotin (Thermo Scientific, Rockford, IL, USA) in PBS (20 mM, pH 8) with 1 mM Ca2+ and 1 mM Mg2+ (PBS-CM) on ice for half an hour. The reaction was stopped in 50 mM glycine in PBS-CM on ice for 5 min. The cells were scraped and washed with PBS-CM containing 1 mM PMSF on ice and transferred to lysis buffer (20 mM Tris–HCl pH 8, 100 mM NaCl, 1 mM EGTA, and 1% NP40, 20 mM β-glycerol phosphate and 2 mM Na3VO4) at 4°C for half an hour. Thirty μg of the sample was used for input. The protein from each group was incubated with streptavidin-agarose beads (Invitrogen, Waltham, MA, USA) for 1 h on ice. Samples were washed with PBS buffer with 1 mM PMSF, and proteins were eluted by using PBS buffer with 10 mM DTT and then treated at 65°C for 10 min. Cav3.2 channel protein intensities were compared among different groups. CaM agarose beads were washed with 4-bed volumes of PBS buffer, then were incubated with an equal volume of total protein. The samples were washed with 20 mM Tris–HCl pH 7.5, 100 mM NaCl and 1% Triton with 1 mM PMSF.
Statistical Analysis
All data were presented as mean values ± S.D. (standard deviation) and analyzed by using SPSS software 20.0 (SPSS Inc., Chicago, Ill, USA). A two-way repeated measure analysis of variance (ANOVA) was performed to assess the effects of BoT-A Botox on the rat models (0 and 1 month after SCI-induced spasticity). ANOVA followed by post hoc analyses, a defined post hoc test for multiple comparisons within the treatment groups (e.g. Tukey, Bonferroni), was performed to evaluate the effects of water temperature on muscle spasticity. Pearson’s correlation coefficient test was used to explore the relationship between relative protein levels of Cav3.2 and calcium levels.
Results
Characterization of SCI
After surgery operation, the spinal cord tissue was destructed by the range between 50% and 65% of the spinal cord cross section. The surgery resulted in tissue damage, including the symptoms of bleeding, swelling, ischemia, and inflammation. The dorsal, dorsolateral, and ventromedial funiculi were successfully disrupted, which triggered secondary damage and extensive non-resolving inflammation, aggravated neuronal loss and hindered recovery. The statistical difference for the percentage of spinal cord destruction was insignificant among all the model groups (p > 0.05). After 2-day SCI, most rats dragged their hind limbs with extensive movements in other organisms, such as hip, knee, and ankle. After 1-week SCI, most model rats exhibited sweeping activity with hind limbs and difficult for weight-supporting. After 2-week SCI, the symptoms were improved and the rats exhibited weight-supporting with a little difficult. Intact rats swam using the hind limbs and tail. The rats held the forelimbs under the chin and steered by using forelimbs. After 1–3 days of SCI, most rats used their forelimbs for propulsion and only made hind limb strokes. After 1–2 weeks of SCI, most rats recovered alternating hind limb usage but still used their forelimbs. Muscle spasticity was shown with a bent, ventroflexed posture and an erected tail.
BoT-A Botox Reduced Muscle Spasticity
For intact rats, the EMG signals showed that they exhibited fast left and right alternating TA activity and no activity in the RA (Figure 1A). After 1-week SCI, the rats showed uncommon left and right TA activities and no RA activity (Figure 1B). After 1-month SCI, the rats in the WG group still showed left–right alternating TA muscle with lower frequency than that in the CG group. Comparatively, the RA muscle showed sustained EMG signals during spasm activity (Figure 1C). Meanwhile, left and right TA activities were with normal signals, suggesting the hind limb movements recovered. Comparing with the rats in the WG group, low-dose BoT-A Botox treatment reduced the range of spasm activity, and the frequency of left and right TA activities increased (Figure 1D). Middle-dose BoT-A Botox intervention further reduced the range of spasticity activity and the frequency of left and right TA activities further increased (Figure 1E). The range of spasticity activity reduced to the lowest level and frequencies increased to the highest level in the rat models (Figure 1F). All the results suggest that BoT-A Botox treatment improves muscle spasticity.
 |
Figure 1 Electromyographic (EMG) recordings of the effects of BoT-A Botox on SCI-induced muscle spasticity. (A) Intact rat without muscle spasticity. (B) The rats without muscle spasticity after immediate SCI establishment. (C) The rats developed muscle spasticity after 1-month SCI model establishment. (D) the muscle spasticity was reduced with 1 U/L of BoT-A Botox treatment. (E) The muscle spasticity was further reduced with 3 U/L of BoT-A Botox treatments. (F) The muscle spasticity was reached the lowest level with 6 U/L of BoT-A Botox treatments. n = 8 for each group. |
The swimming test showed similar EMG signals among all the groups. The spasm scores were zero among all groups before SCI model establishment and the spasm scores in the WG group were higher than those in other model groups treated by BoT-A Botox and the spasm scores were reduced with the increase in the dose of BoT-A Botox usage after 1-month SCI establishment (Figure 2A, p < 0.05). In similar cases, the prevalence of major spasticity in the WG was higher than that in other model groups treated by BoT-A Botox and the prevalence of major spasticity was reduced with the increase in the dose of BoT-A Botox usage after 1-month SCI establishment (Figure 2B, p < 0.05).
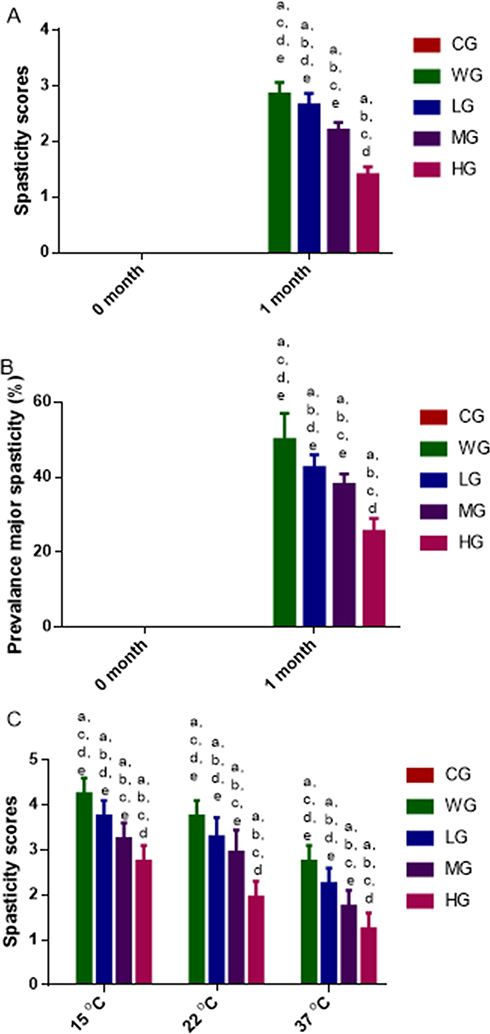 |
Figure 2 Swimming analysis of the effects of BoT-A Botox on muscle spasticity. (A) Muscle spasticity scores after different time of SCI-induced muscle spasticity. (B) The prevalence of major muscle spasticity after different time of SCI-induced muscle spasticity. (C) The effects of water temperature on muscle spasticity after 1-month SCI-induced muscle spasticity. n = 8 for each group. ap < 0.05 vs the CG group, bp < 0.05 vs the WG group, cp < 0.05 vs the LG group, dp < 0.05 vs the MG group and ep < 0.05 vs the HG group. |
Water Temperature Affected the Severity of Muscle Spasticity
Water temperature showed significant effects on spasm scores whereas low temperature increased spasm scores, and high temperature reduced spasm scores (Figure 2C, p < 0.05). The results suggest that water temperature affects the severity of muscle spasticity.
BoT-A Botox Reduced Calcium Release in EDL Muscles
The calcium level in EDL muscles was lowest in the CG group and highest in the WG group. BoT-A Botox treatment reduced calcium level in EDL muscles when compared with the WG group and reached the low level in the HG group (Figure 3, p<0.05). The results suggest that BoT-A Botox reduces calcium release in EDL muscles in a dose-dependent way. There may be other molecular mechanisms for the function of BoT-A Botox in the prevention of SCI-induced muscle spasticity, just like a kind of calcium channel blocker. Therefore, the level of calcium channel was measured in the next step.
 |
Figure 3 The effects of BoT-A Botox on the calcium level in EDL muscles after 1-month SCI-induced muscle spasticity. n = 8 for each group. ap < 0.05 vs the CG group, bp < 0.05 vs the WG group, cp < 0.05 vs the LG group, dp < 0.05 vs the MG group and ep < 0.05 vs the HG group. |
BoT-A Reduced the Expression of Cav3.2
Relative protein level of Cav3.2 in the CG group was lower than that in the WG group. BoT-A Botox treatment further reduced relative protein levels of Cav3.2 when compared with the WG group and reached the lowest level in the HG group (Figure 4 and Figure S1, p < 0.05). The results suggest that BoT-A Botox reduces relative protein levels of Cav3.2 in a dose-dependent way, which may be the main mechanism for the function of BoT-A Botox in the prevention of SCI-induced muscle spasticity.
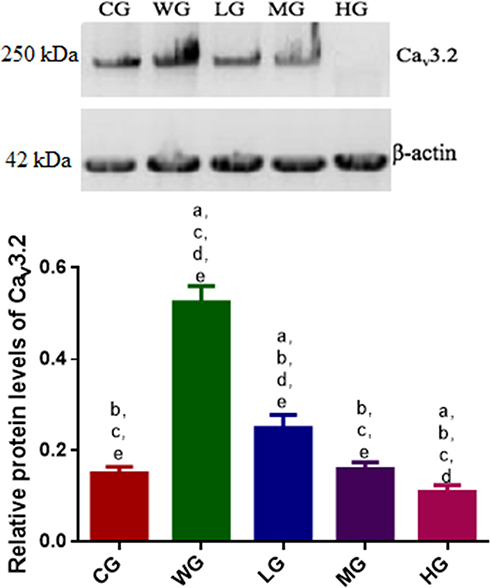 |
Figure 4 The effects of BoT-A Botox on the protein levels of Cav3.2 after 1-month SCI-induced muscle spasticity. n = 8 for each group. ap < 0.05 vs the CG group, bp < 0.05 vs the WG group, cp < 0.05 vs the LG group, dp < 0.05 vs the MG group and ep < 0.05 vs the HG group. |
Relative protein levels of Cav3.2 had a strong relationship with the calcium level. Pearson’s correlation coefficient test showed that the increase in relative protein levels of Cav3.2 resulted in an increase in the calcium levels. Relative protein levels of Cav3.2 had a strong relationship with the calcium levels (Figure 5, rho = 0.712 and p < 0.001). The result suggests that the calcium level may be affected by the expression level of Cav3.2 calcium channel subunits.
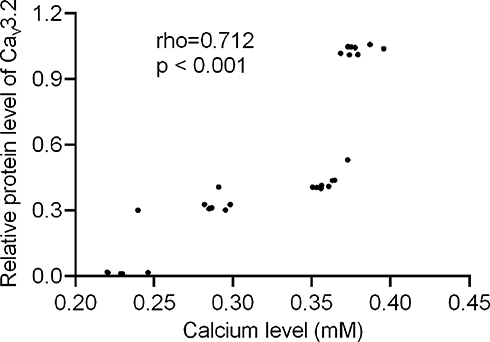 |
Figure 5 Pearson’s correlation coefficient test of the relationship between relative protein levels of Cav3.2 and calcium level. There was a strong relationship if rho > 0.5. |
Electrophysiology
Whole-cell patch clamp results showed that the current signals of mutant M1560V Cav3.2 channels were significantly reduced compared to wild-type channels, whereas the signals in the group with the overexpression of Cav3.2 were higher than those in the wild-type channels (Figure 6A, p < 0.05). The signals were reduced with the increase in the dose of BoT-A Botox (Figure 6A, p < 0.05). In similar cases, the peak current values of mutant M1560V Cav3.2 channels were significantly reduced compared to wild-type channels whereas the peak values in the group with the overexpression of Cav3.2 were higher than the wild-type channels (Figure 6B and C, p < 0.05). The peak current values were reduced with the increase in the dose of BoT-A Botox (Figure 6B and C, p < 0.05). The results suggest that BoT-A Botox may reduce the current density via Cav3.2.
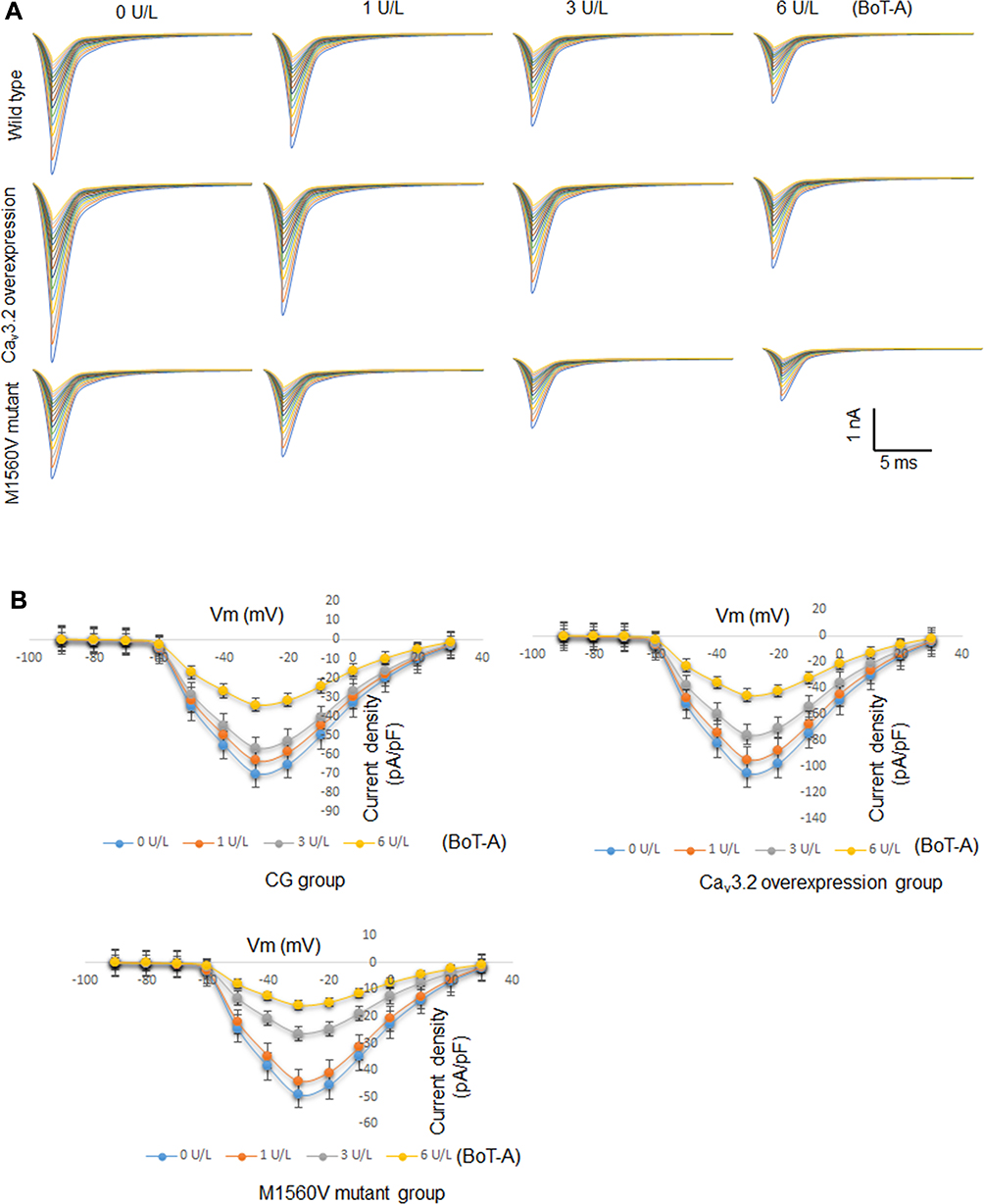 |
Figure 6 Continued. |
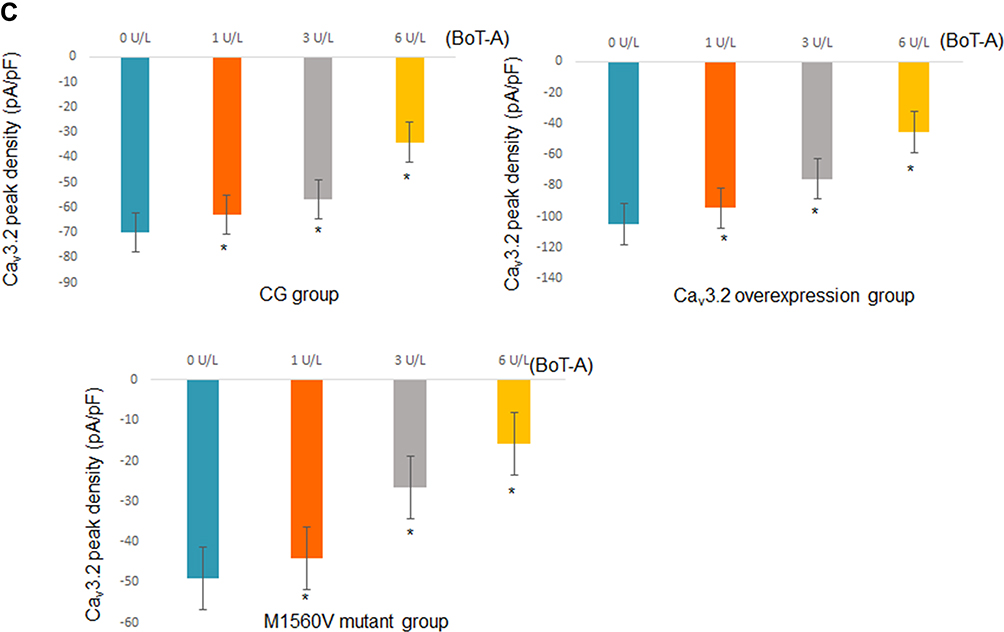 |
Figure 6 The effects of BoT-A Botox on the whole-cell currents of Cav3.2 channel measured in motoneurons by using the voltage clamp technique. (A) Current signals among different groups. (B) Current curves among different groups. (C) The peak current densities among different groups. n = 5 for each group. *p < 0.05 vs the group without BoT-A Botox treatment. |
BoT-A Botox Reduced the Relative Ratio of Biotinylated to Total (Input) Cav3.2
The relative ratio of biotinylated to total (input) Cav3.2 of wild-type channels (Figure 7A and Figure S2) was lower than that in the group with overexpression of Cav3.2 channels (Figure 7B and Figure S2), whereas the ratio in the group with the M1560V mutant of Cav3.2 (Figure 7C and Figure S2) was lower than that in the wild-type channels (Figure 7A, p < 0.05). BoT-A Botox treatment reduced the relative ratio of biotinylated to the total (input) Cav3.2 in the groups with the wild-type Cav3.2 channel and overexpression of Cav3.2 (Figure 7A and B, p < 0.05). In the mutant group, BoT-A Botox treatment increased the ratio (Figure 7C, p < 0.05). The un-normal changes may be associated with mutant M1560V. The results further suggest that BoT-A Botox may reduce the current density via Cav3.2.
 |
Figure 7 The effects of BoT-A Botox on %trafficking of biotinylated/input of Cav3.2. (A) wild-type calcium channel Cav3.2. (B) The overexpression of calcium channel Cav3.2. C, the mutant M1560V of calcium channel Cav3.2. n = 5 for each group. ap < 0.05 vs without BoT-A Botox treatment, bp < 0.05 vs with 1 U/L BoT-A Botox treatment, cp < 0.05 vs with 3 U/L BoT-A Botox treatment, and dp < 0.05 vs with 6 U/L BoT-A Botox treatment. |
Discussion
After the establishment of SCI-induced muscle spasm model, all the rats exhibited the symptoms of muscle spasticity, which were observed by using swimming tests. BoT-A Botox treatment improved the symptoms in a dose-dependent manner, and spasm scores or the prevalence of major spasticity were gradually reduced with the increase in the dose of BoT-A Botox. Furthermore, the spasticity was affected by water temperature and high temperature inhibited the incidence of muscle spasticity.
Food and Drug Administration (FDA) has approved BoT-A for the treatment of strabismus, eyelid spasm, neck dystonia, and cosmetic disorders. A large number of clinical randomized controlled trials and retrospective analyses have demonstrated that BoT-A injection reduces muscle tone,39 expands joint mobility of affected joints,40,41 and indirectly improves motor function of patients,42 which makes BoT-A suitable in the prevention of muscle spasticity.43 Toxin is a neurotoxin produced by Clostridium botulinum in anaerobic environment. There are different bacterial strains producing different subtypes of neurotoxin. Among them, BoT-A is the most toxic and the most stable. The present findings suggest that BoT-A Botox exerts its function by affecting the expression of Cav3.2 calcium channel subunit and may prevent calcium release.44
The normal mechanism for the effects of BoT-A Botox on calcium release has not been reported yet. The calcium level in EDL muscles was increased in the SCI-induced model with muscle spasticity and could be inhibited by BoT-A Botox (Figure 3). The results suggest that calcium level in EDL muscles may be the main reason for causing spasticity since its level in the CG group was lower than that in the WG group, whereas BoT-A Botox may prevent spasticity by inhibiting calcium release. The inhibition of calcium release from the sarcoplasmic reticulum may be a potential approach to reduce spasticity by uncoupling electrical excitation from contraction.45 However, the reverse results have also been reported that the increase in the muscle calcium will reduce spasticity severity.10 Much work is needed to address the important issues.
Furthermore, we explored other possible mechanisms according to calcium channel blockers in the therapy of muscle spasticity. Just as we supposed, the expression of Cav3.2 was lower in the sham surgery group than that in the SCI-induced muscle spasticity group (Figure 4). BoT-A Botox treatment reduced the protein level of Cav3.2 in a dose-dependent way. The results suggest BoT-A Botox may prevent the risk of SCI-induced muscle spasticity via the calcium channel.
The present findings also demonstrated that the current changes were caused by Cav3.2 calcium channel subunit via the mutant M1560V (Figures 5 and 6). Meanwhile, the BoT-A Botox treatment also reduced the peak current density of supposed calcium channel and relative ratio of biotinylated to total Cav3.2 in a dose-dependent way. The results further suggest that BoT-A Botox may prevent the risk of SCI-induced muscle spasticity via the calcium channel subunit Cav3.2. Figures 4–6 showed that the BoT-A Botox treatment dose-dependently decreased the expression of the channel subunit protein in the spinal cord while the in vitro tests on transfected motoneurons suggested a direct inhibitory effect of BoT-A Botox on Ca2+ currents. These are two fundamentally different effects, whereas the calcium-binding dynamics of the channel protein will affect intracellular Ca.2+46
It is generally accepted that BoT-A exerts its function by inhibiting the release of acetylcholine at neuromuscular junction leading to a reduction in muscle tone.47 Furthermore, BoT-A suppresses the muscle spindle discharge which is a major excitatory input to the spinal cord and this may be another mechanism affecting spasticity.48 The present results showed that BoT-A Botox treatment reduced the expression level of Cav3.2 (Figure 4). Cav3.2 mediates calcium influx49 while calcium level will also affect acetylcholine release from motor nerve terminals,50 resulting in the change of muscle activity. The calcium level is maintained by the calcium channels51 and the decrease in the expression of calcium channel subunit will result in low-level calcium. The reduced calcium release reduces acetylcholine release from motor neurons, and thus relaxed the muscle. It may be a novel mechanism for BoT-A Botox in the treatment of spasticity via affecting the expression of T-type calcium channel subunit Cav3.2. Therefore, there may be multiple mechanisms for the effects of BoT-A on muscle spasticity. Much work is still needed to be done to confirm the present conclusion.
There were some limitations to the present study. BoT-A Botox injection can last 6-month effect and has to be repeated used once every half a year. In the present study, only 1-month or less duration was explored in the animal model. The common side effects of BoT-A include urinary tract infections,52 respiratory tract infection,53 fatigue and weakness54 and constipation.55 The related symptoms were not investigated in the present work.
In summary, the rat model with SCI-induced muscle spasticity is a useful model to explore the effects of BoT-A Botox on the progression of muscle spasticity and BoT-A Botox is a potential drug to treat muscle spasticity by affecting calcium channel Cav3.2.
Data Sharing Statement
The data generated during and/or analyzed during the current study are available from the corresponding author upon reasonable request.
Ethical Approval
All surgical procedures and postoperative care were performed following the guidelines of the Animal Care and Use Committee of The First Hospital of Jilin University (approval No. AJLU28-18, Changchun, China). All experiments were performed in accordance with relevant guidelines and regulations of the Animal Experiment Ethics Committee of The First Hospital of Jilin University. Every effort was made to minimize the number and suffering of animals used in the present experiment.
Disclosure
The authors declare no competing interests.
References
1. Castany S, Codony X, Zamanillo D, et al. Repeated sigma-1 receptor antagonist MR309 administration modulates central neuropathic pain development after spinal cord injury in mice. Front Pharmacol. 2019;10:222. doi:10.3389/fphar.2019.00222
2. Lin S, Li Y, Lucas-Osma AM, et al. Locomotor-related V3 interneurons initiate and coordinate muscles spasms after spinal cord injury. J Neurophysiol. 2019;121(4):1352–1367. doi:10.1152/jn.00776.2018
3. Levy J, Hartley S, Mauruc-Soubirac E, et al. Spasticity or periodic limb movements? Eur J Phys Rehabil Med. 2018;54(5):698–704. doi:10.23736/S1973-9087.17.04886-9
4. Buddhdev P, Fry NR, LePage R, et al. Abnormality of standing posture improves in patients with bilateral spastic cerebral palsy following lower limb surgery. Gait Posture. 2017;54:255–258. doi:10.1016/j.gaitpost.2017.03.014
5. Ko H-Y. Management and Rehabilitation of Spinal Cord Injuries. Springer; 2019:399–412.
6. Kumar KR, Ng K. Reduced facial nerve hyperexcitability from contralateral cerebral stroke in hemifacial spasm. Mov Disord. 2010;25(9):1310–1312. doi:10.1002/mds.23094
7. McDowell MM, Zhu X, Hughes MA, Sekula RF. Facial spasms, but not hemifacial spasm: a case report and review of literature. Child Nerv Syst. 2016;32(9):1735–1739. doi:10.1007/s00381-016-3057-7
8. Sugaya T, Kanno H, Matsuda M, et al. B-RAF(V600E) inhibitor dabrafenib attenuates RIPK3-Mediated necroptosis and promotes functional recovery after spinal cord injury. Cells. 2019;8(12):1582. doi:10.3390/cells8121582
9. Lei F, He W, Tian X, et al. GSK-3 inhibitor promotes neuronal cell regeneration and functional recovery in a rat model of spinal cord injury. Biomed Res Int. 2019;2019:9628065. doi:10.1155/2019/9628065
10. Indriasari M, Kustiyah L, Priosoeryanto BP, Idris FH. Effect of calcium consumption on the spasticity in the spastic rats. Food Nutr Sci. 2018;10:37–50. doi:10.4236/fns.2019.101004
11. Xiyuan Z, Fink RHA, Mosqueira M. NO-sGC pathway modulates Ca(2+) release and muscle contraction in zebrafish skeletal muscle. Front Physiol. 2017;8:607. doi:10.3389/fphys.2017.00607
12. Li Y, Gorassini MA, Bennett DJ. Role of persistent sodium and calcium currents in motoneuron firing and spasticity in chronic spinal rats. J Neurophysiol. 2004;91(2):767–783. doi:10.1152/jn.00788.2003
13. Perez-Reyes E. Molecular physiology of low-voltage-activated t-type calcium channels. Physiol Rev. 2003;83(1):117–161. doi:10.1152/physrev.00018.2002
14. Strube C. Absence of regulation of the T-type calcium current by Cav1.1, beta1a and gamma1 dihydropyridine receptor subunits in skeletal muscle cells. Pflugers Arch. 2008;455(5):921–927. doi:10.1007/s00424-007-0345-9
15. Candelas M, Reynders A, Arango-Lievano M, et al. Cav3.2 T-type calcium channels shape electrical firing in mouse lamina II neurons. Sci Rep. 2019;9(1):3112. doi:10.1038/s41598-019-39703-3
16. Marcantoni M, Fuchs A, Low P, Kiehn O, Bellardita C. Nimodipine prevents the development of spasticity after spinal cord injury. BioRxiv. 2019:639211.
17. Chien CT, Lee HM, Wu CC, Li PC. Inhibitory effect of botulinum toxin type A on the NANC system in rat respiratory models of neurogenic inflammation. Arch Biochem Biophys. 2012;524:106–113. doi:10.1016/j.abb.2012.05.016
18. Charoensook SN, Williams DJ, Chakraborty S, Leong KW, Vunjak-Novakovic G. Bioreactor model of neuromuscular junction with electrical stimulation for pharmacological potency testing. Integr Biol. 2017;9(12):956–967. doi:10.1039/c7ib00144d
19. Yadav S, Chand S, Majumdar R, Sud A. Effect of botulinum toxin type-A in spasticity and functional outcome of upper limbs in cerebral palsy. J Clin Orthopaed Trauma. 2020;11(2):208–212. doi:10.1016/j.jcot.2020.01.002
20. Briand -M-M, Boudier-Réverêt M, Rodrigue X, Sirois G, Chang MC. A moving residual limb: botulinum toxin to the rescue. Transl Neurosci. 2020;11(1):34–37. doi:10.1515/tnsci-2020-0006
21. Li Y, Gorassini MA, Bennett DJ. Role of persistent sodium and calcium currents in motoneuron firing and spasticity in chronic spinal rats. J Neurophysiol. 2004;91(2):767–783. doi:10.1152/jn.00788.2003
22. Tülümen E, Borggrefe M. Antiarrhythmic Drugs. Springer; 2020:233–264.
23. Niwa N, Yasui K, Opthof T, et al. Cav3. 2 subunit underlies the functional T-type Ca2+ channel in murine hearts during the embryonic period. Am J Physiol Heart Circul Physiol. 2004;286:H2257–H2263. doi:10.1152/ajpheart.01043.2003
24. Orr MB, Simkin J, Bailey WM, et al. Compression decreases anatomical and functional recovery and alters inflammation after contusive spinal cord injury. J Neurotrauma. 2017;34(15):2342–2352. doi:10.1089/neu.2016.4915
25. Livshits A, Rappaport Z, Livshits V, Gepstein R. Surgical treatment of painful spasticity after spinal cord injury. Spinal Cord. 2002;40(4):161–166. doi:10.1038/sj.sc.3101236
26. Beattie C, Gormley M, Wervey R, Wendorf H. An electromyographic protocol that distinguishes spasticity from dystonia. J Pediatr Rehabil Med. 2016;9(2):125–132. doi:10.3233/PRM-160373
27. Gonzenbach RR, Gasser P, Zörner B, et al. Nogo-A antibodies and training reduce muscle spasms in spinal cord-injured rats. Ann Neurol. 2010;68(1):48–57. doi:10.1002/ana.22009
28. Ostrowski J, Herb CC, Scifers J, et al. Comparison of muscle temperature increases produced by moist hot pack and thermostim probe. J Sport Rehabil. 2019;28(5):459–463. doi:10.1123/jsr.2017-0294
29. McRory JE, Santi CM, Hamming KSC, et al. Molecular and functional characterization of a family of rat brain T-type calcium channels. J Biol Chem. 2001;276(6):3999–4011. doi:10.1074/jbc.M008215200
30. Scholl UI, Stölting G, Nelson-Williams C, et al. Recurrent gain of function mutation in calcium channel CACNA1H causes early-onset hypertension with primary aldosteronism. Elife. 2015;4:e06315.
31. Glajch KE, Ferraiuolo L, Mueller KA, et al. MicroNeurotrophins improve survival in motor neuron-astrocyte co-cultures but do not improve disease phenotypes in a mutant SOD1 mouse model of amyotrophic lateral sclerosis. PLoS One. 2016;11(10):e0164103. doi:10.1371/journal.pone.0164103
32. Scholl UI, Stölting G, Nelson-Williams C, et al. Recurrent gain of function mutation in calcium channel CACNA1H causes early-onset hypertension with primary aldosteronism. Elife. 2015;4. doi:10.7554/eLife.06315
33. Kisilevsky AE, Zamponi GW. D2 dopamine receptors interact directly with N-type calcium channels and regulate channel surface expression levels. Channels. 2008;2(4):269–277. doi:10.4161/chan.2.4.6402
34. Liu JL, Eisenberg B. Correlated ions in a calcium channel model: a Poisson-Fermi theory. J Phys Chem B. 2013;117(40):12051–12058. doi:10.1021/jp408330f
35. Camara AK, Begic Z, Kwok WM, Bosnjak ZJ. Differential modulation of the cardiac L- and T-type calcium channel currents by isoflurane. Anesthesiology. 2001;95(2):515–524. doi:10.1097/00000542-200108000-00038
36. Serrano JR, Perez-Reyes E, Jones SW. State-dependent inactivation of the alpha1G T-type calcium channel. J Gen Physiol. 1999;114(2):185–201. doi:10.1085/jgp.114.2.185
37. Ozeki T, Kobayashi T, Nakayama K, Iwasaki K, Sakamoto T. Polarization-mode dispersion measurement by an optical time-domain reflectometer with polarimetry assuming backscattering by randomly oriented nonspherical particles. Opt Lett. 2003;28(15):1293–1295. doi:10.1364/ol.28.001293
38. De Sevilla Muller LP, Liu J, Solomon A, Rodriguez A, Brecha NC. Expression of voltage-gated calcium channel alpha(2)delta(4) subunits in the mouse and rat retina. J Comp Neurol. 2013;521(11):2486–2501. doi:10.1002/cne.23294
39. Olver J, Esquenazi A, Fung VSC, et al. Botulinum toxin assessment, intervention and aftercare for lower limb disorders of movement and muscle tone in adults: international consensus statement. Eur J Neurol. 2010;17 Suppl 2:57–73. doi:10.1111/j.1468-1331.2010.03128.x
40. Martino Cinnera A, Pucello A, Lupo A, et al. Upper limb motor improvement in chronic stroke after combining botulinum toxin A injection and multi-joints robot-assisted therapy: a case report. Oxf Med Case Rep. 2019;2019(10):omz097. doi:10.1093/omcr/omz097
41. Cimolin V, Galli M, Crivellini M, Albertini G. Quantitative effects on proximal joints of botulinum toxin treatment for gastrocnemius spasticity: a 4-year-old case study. Case Rep Med. 2009;2009:985717. doi:10.1155/2009/985717
42. Fujimura K, Kagaya H, Onaka H, et al. Upper limb motor function affects the outcome after treatment with botulinum toxin A. Eur Neurol. 2019;81(1–2):30–36. doi:10.1159/000499907
43. Caravia LG, Nica A, Mologhianu G, et al. The action mechanism of botulinum toxin in reducing muscle spasticity. Acta Med Marisiensis. 2019;65.
44. Suko J, Wyskovsky W, Hohenegger M, et al. Calcium channel mediated calcium release from the rabbit skeletal muscle sarcoplasmic reticulum vesicle. Wien Klin Wochenschr. 1990;102(20):616–621.
45. Patel DR, Soyode O. Pharmacologic interventions for reducing spasticity in cerebral palsy. Indian J Pediatr. 2005;72(10):869–872. doi:10.1007/BF02731118
46. Ying YJ, Huang ZQ. Effects of time delay on intracellular Ca2+ concentration oscillations. Chinese Phys Lett. 2001;18(5):695–697. doi:10.1088/0256-307X/18/5/324
47. Ridgeway B, Wallace M, Gerayli A. Ziconotide for the treatment of severe spasticity after spinal cord injury. Pain. 2000;85(1):287–289. doi:10.1016/S0304-3959(99)00255-9
48. Filippi GM, Errico P, Santarelli R, Bagolini B, Manni E. Botulinum A toxin effects on rat jaw muscle spindles. Acta Otolaryngol. 1993;113(3):400–404. doi:10.3109/00016489309135834
49. Anderson D, Mehaffey WH, Iftinca M, et al. Regulation of neuronal activity by Cav3-Kv4 channel signaling complexes. Nat Neurosci. 2010;13(3):333–337. doi:10.1038/nn.2493
50. Katz B, Miledi R. The effect of calcium on acetylcholine release from motor nerve terminals. Proce R Soc London Series B Biol Sci. 1965;161:496–503.
51. Saravanaraman P, Chinnadurai RK, Boopathy R. Why calcium channel blockers could be an elite choice in the treatment of alzheimer’s disease: a comprehensive review of evidences. Rev Neurosci. 2014;25(2):231–246. doi:10.1515/revneuro-2013-0056
52. Jamnagerwalla J, Houman J, Patel D, Wood L, Anger JT, Eilber KS. Defining the rate and predictors of urinary tract infection after botox injection in patients receiving prophylactic antibiotics. Neurourol Urodynam. 2016;35:S105–S105.
53. Kirke DN, Kaye R, Blitzer A. Impact of an upper respiratory tract infection on botulinum toxin efficacy in spasmodic dysphonia patients. Laryngoscope. 2019. doi:10.1002/lary.28283
54. Varghese-Kroll E, Elovic EP. Contralateral weakness and fatigue after high-dose botulinum toxin injection for management of poststroke spasticity. Am J Phys Med Rehab. 2009;88(6):495–499. doi:10.1097/PHM.0b013e3181a5b056
55. Vles GF, Vles JSH. Constipation as an adverse event after botulinum toxin A treatment in children with cerebral palsy. Dev Med Child Neurol. 2010;52(10):. doi:10.1111/j.1469-8749.2010.03696.x
 © 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.