Back to Journals » Journal of Multidisciplinary Healthcare » Volume 17
Bone Disease among Children with Sickle Cell Disease: A Scoping Review of Incidence and Interventions
Authors Mulyana AM ![]() , Rakhmawati W
, Rakhmawati W ![]() , Pramukti I
, Pramukti I ![]() , Lukman M, Wartakusumah R, Mediani HS
, Lukman M, Wartakusumah R, Mediani HS ![]()
Received 24 April 2024
Accepted for publication 26 June 2024
Published 9 July 2024 Volume 2024:17 Pages 3235—3246
DOI https://doi.org/10.2147/JMDH.S475371
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Scott Fraser
Aep Maulid Mulyana,1,* Windy Rakhmawati,2,* Iqbal Pramukti,3,* Mamat Lukman,3,* Riki Wartakusumah,4,* Henny Suzana Mediani2,*
1Faculty of Nursing, Universitas Padjadjaran, Bandung, West Java, 40132, Indonesia; 2Department of Pediatric Nursing, Faculty of Nursing, Universitas Padjadjaran, Bandung, West Java, 40132, Indonesia; 3Department of Community Health Nursing, Faculty of Nursing, Universitas Padjadjaran, Bandung, West Java, 40132, Indonesia; 4Department of Nursing Science, Faculty of Medicine Public Health, and Nursing, Universitas Gadjah Mada, Sleman, Yogyakarta, 55284, Indonesia
*These authors contributed equally to this work
Correspondence: Henny Suzana Mediani, Department of Pediatric Nursing, Faculty of Nursing, Universitas Padjadjaran, Bandung, West Java, 40132, Indonesia, Tel +62 822-1739-1965, Email [email protected]
Background: Children with sickle cell disease (SCD) are more likely to have deficient serum levels of vitamin D for bone metabolism. However, appropriate interventions are essential to prevent its progression and alleviate symptoms.
Purpose: The aim of this study is to determine interventions for managing bone disease in children with SCD.
Methods: This study uses a scoping review. A literature review was conducted using PubMed, CINAHL, ScienceDirect, Scopus, and Google Scholar search engines. The study was eligible for inclusion if it included articles published from 2013 to 2023, full-text, and original study design. Study quality was assessed using the Joanna Briggs Institute (JBI) appraisal tool.
Results: This review identified six studies and 114 children with SCD, including 57 boys (50%) and 57 girls (50%). The majority of SCD types experienced were HbSS (86.84%), HbS-B0 Thal (7.01%), HbSC (5.27%), and the HbSS Arab-Indian haplotype (0.88%). Bone disease problems that often arise in children with SCD include Avascular Necrosis (AVN) (78.08%), Osteonecrosis of the Femoral Head (ONFH) (18.42%), and other bone problems (3.50%). Meanwhile, four types of intervention findings were used in managing bone disease among children with SCD: 1). Surgical procedures 53 (41.09%) included total hip arthroplasty (THA), Osteotomy, and Multiple epiphyseal drilling with Autologous Bone Marrow Implantation (AMBI); 2). Invasive procedures 67 (51.93%) included intravenous bisphosphonates, hydroxyurea (HU), and core decompression (CD) with bone marrow aspirate concentrate injection: 3). Oral pharmacological or Vitamin D3 (cholecalciferol) 4 (3.10%); 4). Non-pharmacology or physical therapy 5 (3.88%).
Conclusion: Our findings highlight that surgical, invasive, pharmacological, and physical therapy interventions positively impact increasing bone mineral density (BMD) and functional improvement of bone disease among children with SCD. The interventions provided excellent functional outcomes with minimal complications and no life-threatening side effects.
Keywords: bone disease, children, intervention, incidence, sickle cell disease
Introduction
Sickle cell disease (SCD) arises due to a genetic mutation in the gene that encodes the β-globin protein. This mutation leads to an anomalous haemoglobin structure, haemoglobin S (HbS), and dysfunctional red blood cells.1 SCD is an inherited haemoglobinopathy leading to chronic anaemia and microvascular occlusion.2 Sickle cell disease is the most prevalent inherited blood disorder in the human population, impacting millions of people worldwide.3 Although statistical data may differ globally, approximately 4.4 million individuals are affected by sickle cell disease.4 Every year, an estimated 300,000 babies are born with this condition worldwide.5
People who have sickle cell disease and carry two copies of the βS gene mutation are at the highest risk of experiencing the typical common symptoms and complications of the condition.6 Children with SCD are more likely to have deficient serum levels of vitamin D for bone metabolism.7 Previous studies showed that of 306 children and adolescents, 31% had low areal BMD.8
In recent years, the problem of bone disease has continued to be of concern. Recent studies shows that children with SCD often experience severe vitamin D deficiency, as a result of which children can experience bone complications that manifest as bone deformities, Avascular Necrosis (AVN), vaso-occlusive bone pain crisis, infarction, osteoporosis, osteopenia, osteonecrosis, dactylitis, or osteomyelitis.7,9 Bone disease is a common orthopedic complication associated with sickle cell disease.10 The incidence has been documented in children as young as 5 years old and increases with age, and the prevalence rate varies from 3.2% to 39.4%, which mostly includes children and adults with SCD.11
Bone disease in pediatric with SCD, requires careful and prompt treatment to prevent its progression and alleviate symptoms. Some interventions that can be used in the management of bone-related issues. Nonoperative treatment is the first line of treatment: pain control, physiotherapy, and modification of activities to restrict overhead activities and manual labour, including administering vitamin D,12 and operative management such as intravenous bisphosphonates, stem cell implantation, and bone marrow transplantation.13–15 In implementing these interventions, several barriers have been encountered, such as the lack of recent or specific references, resulting in different guidelines for each location. As a result, comprehensive and up-to-date guidance can be challenging to obtain, potentially leading to variations in practice across various regions.12,14
Studies regarding bone disease interventions in children with SCD need to be carried out comprehensively and systematically including available interventions, outcomes, and long-term implications. However, this research is essential for improving and optimizing healthcare strategies for children with SCD. Therefore, this study’s aim is to determine interventions for managing bone disease in children with SCD.
Materials and Methods
Study Design
This study uses a systematic scoping review and follows the framework by Arksey and O’Malley.16,17 This method is suitable for this study to explore comprehensive and relevant studies on one topic. The framework consists of five stages: 1) Identifying research questions; 2) Identifying relevant research results; 3) Screening articles according to criteria; 4) Analyzing, organizing and summarising the included articles; and 5) Reporting results.18
Search Strategy
Search study using PRISMA-ScR guidelines for a systematic scoping review.19 A literature review was conducted using four databases, including PubMed, CINAHL, ScienceDirect, Scopus, and Google Scholar search engine. The keyword adjusts the medical subject heading (MeSH) using boolean OR dan AND, including
SCD, sickle cell disease, sickle cell, sickle cell anemias, sickle cell disorders, intervention, management, treatment, prevention, avascular necrosis, AVN, death of bone tissue, bone necrosis, osteonecrosis, bone aseptic necrosis, osteoporosis, bone diseases, bone mineral disease, children, child, pediatric, kids.
Eligibility Criteria
The criteria in this study are based on the PICO question framework: Participants/population: Children with SCD; Intervention(s): Intervention or treatment; Comparator(s): Not applicable; Outcome: Incidence and intervention in bone disease among children with sickle cell disease. A study was eligible for inclusion if it included articles published from 2013 to 2023, full-text, and original study design. Studies were excluded if they were not in English and the adult population. All researchers independently screen articles, including duplicate topics, titles, abstracts, full text, and appraised study quality.
Quality Appraisal
Study quality was assessed using critical appraisal checklist tools for cohort study from the Joanna Briggs Institute (JBI).20 The evaluation for the cohort study was consistent with eleven questions and four categories: yes, no, unclear, and not applicable. Score 0 for “No” and 1 for “Yes”, with a total quality score ranging from 0 to 11.
Data Extraction and Analysis
Data extraction was displayed using the tabulation method in Microsoft Excel (Microsoft Corp., New York, USA). The extracted items included study characteristics (author, year, study design, country, population, and sample); participant characteristics and SCD (age, gender, SCD type, incidence, type of bone disease); intervention and outcomes (intervention, type of intervention, instruments, intervention procedures, follow-up, intervention effects, and results).
Results
Description of Study Selection
The article search results obtained were 2084 articles from four databases and grey literature. After checking for duplicating the collected articles, filtering the collected titles, and limiting the years, 44 articles remained. Then, after selection based on inclusion criteria, 14 articles remained. Therefore, after screening based on inclusion, six articles were included in this study (Figure 1). Then, the articles were analyzed using the JBI Critical Appraisal Tool assessment for the Retrospective and Prospective cohorts (Table 1).
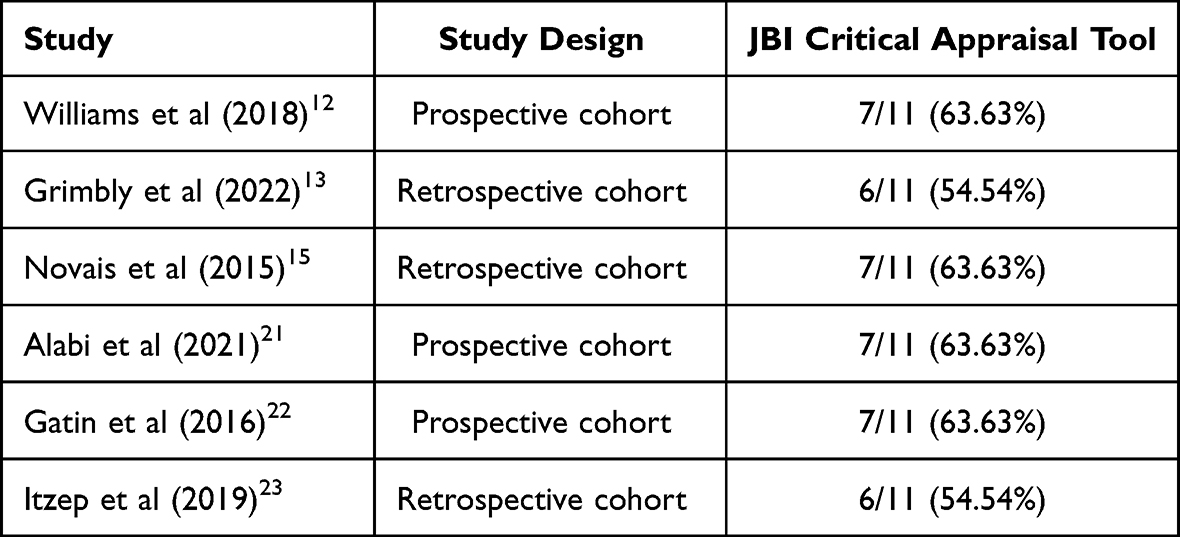 |
Table 1 Critical Appraisal Tool |
 |
Figure 1 PRISMA Flow diagram. Adapted from Page MJ, McKenzie JE, Bossuyt PM, et al. The PRISMA 2020 statement: an updated :guideline for reporting systematic reviews. BMJ. 2021;372:n71. Creative Commons. |
Characteristics of Study
This study included six reviewed articles. This study’s total sample of participants was 114 children with SCD, boys and girls aged 4–19 years. The study designs included a retrospective cohort (n = 3) and a prospective cohort (n = 3). The research conducted included the USA (n = 3), Nigeria (n = 1), Canada (n = 1), and France (n = 1). The SCD types in this review are HbSS, HbS-B0, HbSC, and HbSS Arab-Indian haplotype. Bone disease problems in children with SCD analyzed included AVN (n = 3), ONFH (n = 2), and Chronic arthritis, non-displaced fracture, Central end plate depression of thoracic, and Joint bony infarct (n = 1) (Table 2).
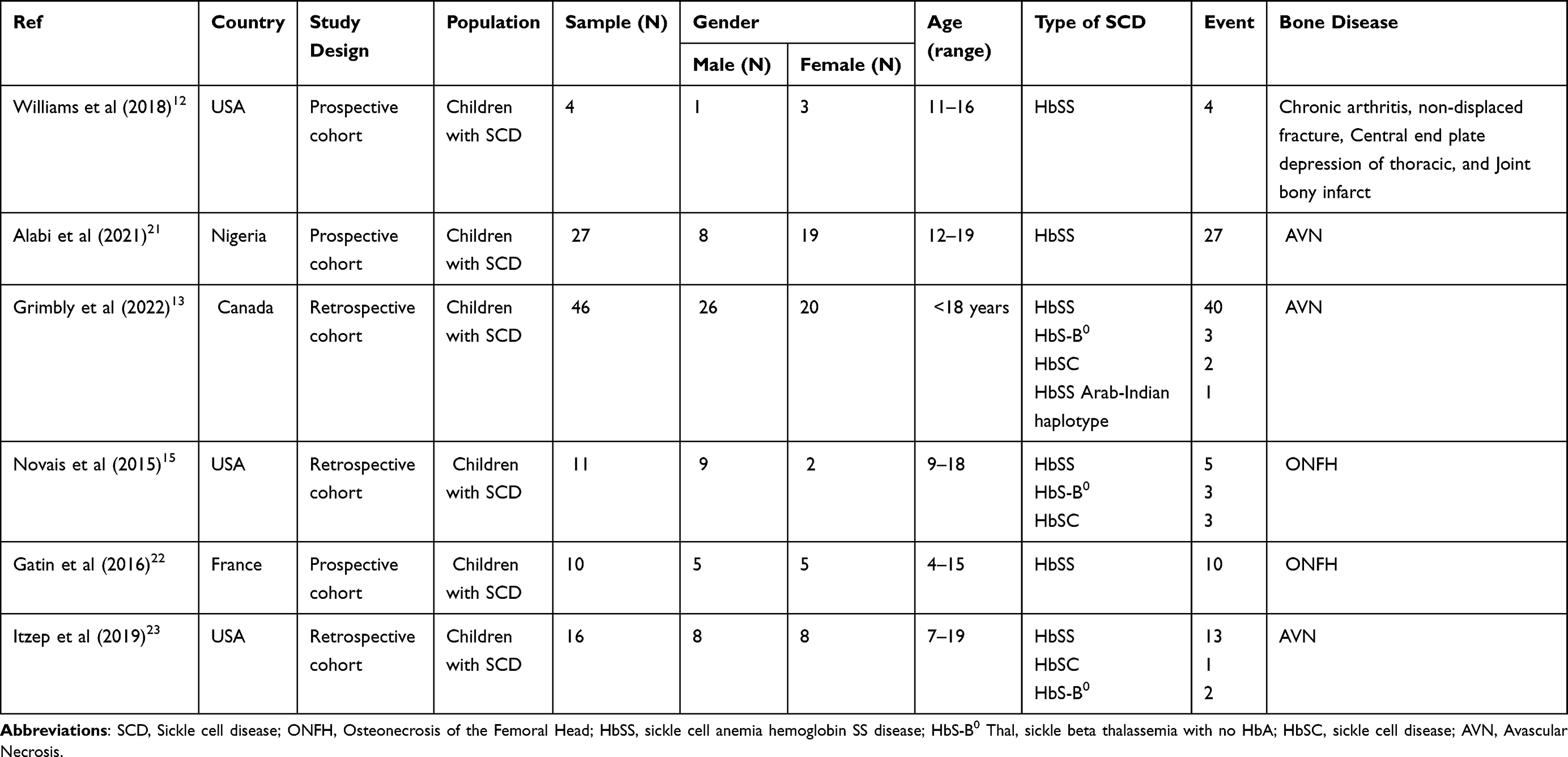 |
Table 2 Characteristics of Studies Reporting Bone Disease Management of Children with SCD (n = 6) |
Incidence of Bone Disease in Children with SCD
This study found 114 children with SCD, including 57 boys (50%) and 57 girls (50%). The majority of SCD types experienced were HbSS (86.84%), HbS-B0 (7.01%), and HbSC (5.27%), and the HbSS Arab-Indian haplotype only occurred in one person from the total sample included. Bone disease problems that often occur in children with SCD are AVN (78.08%), ONFH (18.42%), and other bone problems (3.50%). The incidence of children with SCD was found on three continents, including America (67.54%), Africa (23.69%), and Europe (8.77%). Meanwhile, four types of intervention findings used in the management of children with SCD who experience bone disease include surgical procedures 53 (41.09%), invasive 67 (51.93%), oral pharmacology 4 (3.10%), and non-pharmacology 5 (3.88%) (Table 3).
 |
Table 3 Percentage Analysis by Demographic Variable in Selected Studies |
Interventions and Outcomes in the Management of Bone Disease in Children with SCD
Surgical Interventions
We found four studies that included surgical intervention in the management of bone disease in SCD children, namely total hip arthroplasty (THA), Osteotomy, Multiple epiphyseal drilling with Autologous Bone Marrow Implantation (AMBI), and Surgical.15,21–23 THA can be given to children with SCD aged 12–19 with HbSS type who experience AVN; osteotomy and multiple epiphyseal drilling with AMBI can be performed on children with SCD type HbSS, HbS-B0, HbSC aged 4–18 years who experience ONFH.15,21,22
THA, osteotomy, and multiple epiphyseal drilling with AMBI intervention in SCD children with AVN and ONFH, there were no life-threatening side effects, considered safe and beneficial, although challenging. The intervention provided excellent functional results with minimal complications.21 The study by Itzep et al revealed that the results of surgical management alone did not show radiographic improvement in SCD children with AVN.23
Invasive Interventions
We found two studies that included three invasive interventions in managing bone disease in SCD children, namely IV bisphosphonates, HU, and CD with bone marrow aspirate concentrate injection.13,23 Positive results from these interventions were increased 25(OH)D levels, areal bone mineral density (aBMD), and radiographic improvement in AVN. Overall, there were no reports of complications with the invasive treatments used. However, after the IV bisphosphonates procedure, there was one episode of symptomatic hypocalcemia that was treatable with oral calcium and calcitriol for 5 days,13 and none showed results in radiographic improvement after the CD procedure with bone marrow aspirate concentrate injection.23
Invasive IV bisphosphonates intervention can be given to SCD children with AVN problems aged <18 years, and HU and CD interventions with bone marrow aspirate concentrate injection can be used for SCD children with AVN problems aged 7–19 years. The types of SCD include sickle cell anemia hemoglobin SS disease (HbSS), sickle beta thalassemia with no HbA (HbS-B0), sickle cell disease (HbSC), and HbSS Arab-Indian haplotype.
Oral Pharmacological Interventions
We found that one study using Vitamin D3 (cholecalciferol) gave positive results on bone strength in children with SCD, including increased serum 25(OH)D levels, BMD, and normalization of PTH. The use of Vitamin D3 (cholecalciferol) can be given to children with sickle cell anemia hemoglobin SS disease (HbSS) aged 11–16 years who have bone disease problems such as chronic arthritis, non-displaced fracture, central end plate depression of the thoracic, and joint bony infarct.12
Non-Pharmacological Interventions
Physical therapy can be done as an effort to treat children with SCD with types HbSS, HbSC and HbS-B0, aged 7–19, and experiencing AVN. However, physical therapy did not provide significant results in improving radiographic AVN.23 (Table 4).
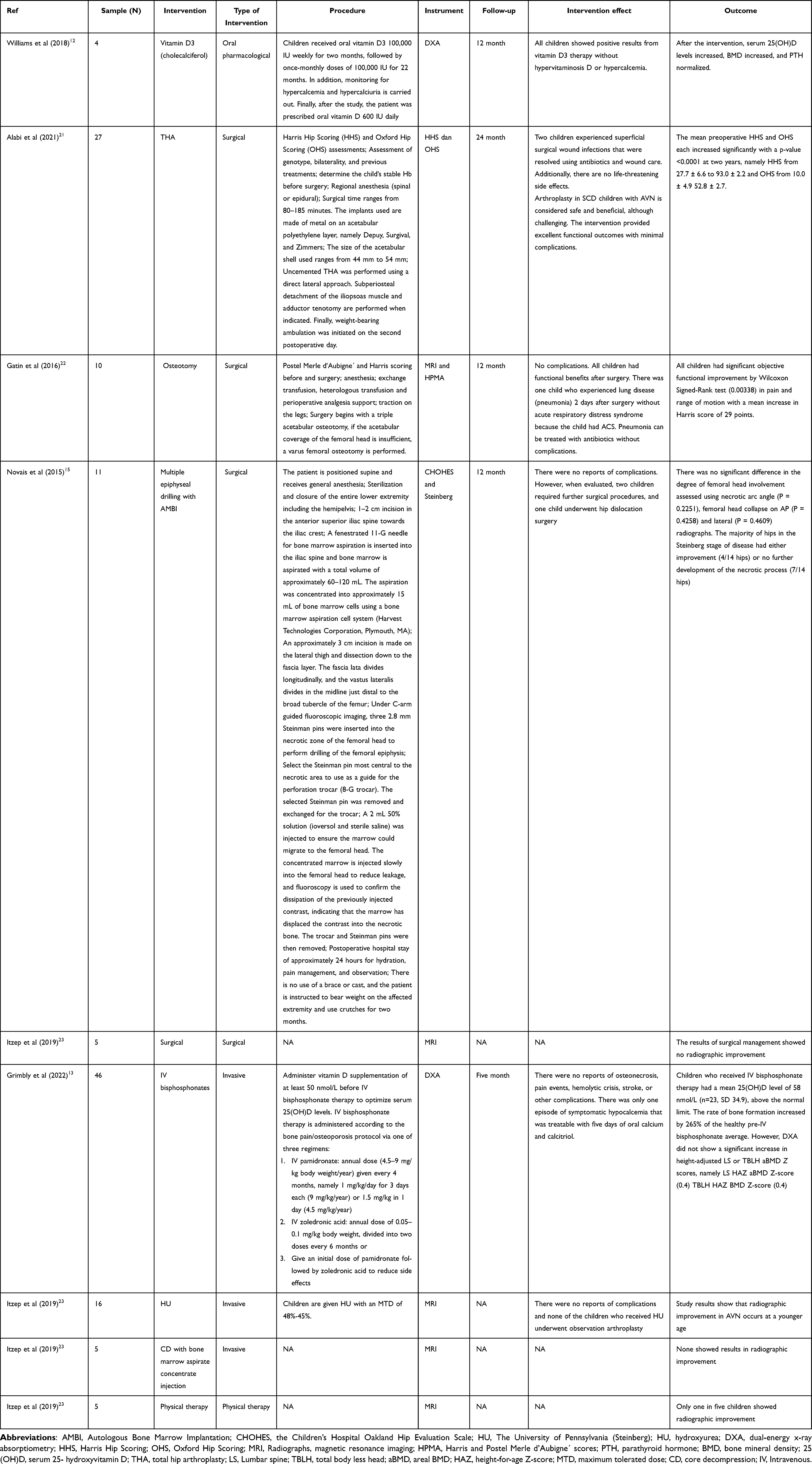 |
Table 4 Interventions in the Management of Bone Disease in Children with SCD (n = 6) |
Discussion
Principal Finding
This study aims to conduct a scoping review to determine interventions for managing bone disease in children with Sickle cell disease (SCD). This scoping review obtained several findings, including incidence, interventions, and outcomes in children with SCD who have bone disease.
Sickle cell disease is an inherited blood disorder characterized by phenotypic diversity.24 Previous studies have shown that SCD causes bone loss and changes in bone anatomy.25 Low serum vitamin D3 levels are a risk for abnormal BMD in SCD.26,27 SCD patients have very low bone mass density (BMD).27,28
This study found 114 children with SCD boys (50%) and girls (50%) aged 4–19 years who had bone disease. Another study revealed that 39 SCD children (23 boys and 16 girls) with an average age of 10.3 years had bone disease.29 This study highlights that age and type of malignancy contribute to the incidence of bone disease in SCD children.
Bone disease in patients with SCD varies from manifestations to more chronic and debilitating complications, including osteonecrosis, osteoporosis, osteopenia, growth disorders, chronic infections,27 and other bone-related complications such as vaso-occlusive pain, ischemic damage, osteomyelitis, and bone marrow hyperplasia or sickle bone disease (SBD).25 Findings from this study showed several bone disease problems in children with SCD analyzed, including AVN (n = 3), ONFH (n = 2), and chronic arthritis, non-displaced fracture, central end plate depression of thoracic, and joint bony infarct. The most frequent occurrence of bone disease in SCD children was AVN 89 (78.08%). In line with the previous study, out of 33 patients with SCD and low BMD, including osteoporosis (36%) and (24%).27
Previous studies have revealed that the majority of children with SCD and bone disease have the HbSS genotype.13 Meanwhile, the previous study shows that SCD types SS and SC have more bone disease.25,28 Findings from the six studies we analyzed revealed that several types of SCD in children with bone disease include HbSS, HbS-B0, HbSC, and HbSS Arab-Indian haplotype. However, children with HbSS-type SCD had the highest rate of bone disease 99 (86.84%).
Sickle cell disease has a very high morbidity and mortality rate.30 In developed countries, cutting-edge technology has driven the development of treatments for SCD. However, in developing countries, medical management of SCD is still limited, relying on the use of hydroxyurea, blood transfusions and analgesics.24 Although bone complications may not directly contribute to increased mortality, SCD can be a significant source of comorbidities and adverse impact on quality of life. Findings in the six studies analyzed showed four types of intervention findings used in the management of children with SCD who experience bone disease, including surgical procedures 53 (41.09%), invasive 67 (51.93%), oral pharmacology 4 (3.10%), and non-pharmacology 5 (3.88%). This study highlighted that the interventions analyzed positively improved and increased BMD and functional improvement in the bones of children with SCD.
Our findings in the current study are supported by previous studies. Several interventions effectively support SCD patients with bone disease: nutritional interventions,24 ω-3 fatty acid supplementation,31 high-dose oral cholecalciferol,1,12 and adding hip core decompression to physical therapy.32 Another study revealed a reduction in morbidity and mortality in children with SCD due in part to the availability of a comprehensive care program that includes immunizations and vaccinations, prophylactic therapy, vitamin supplements, and empowerment of patients and families through education.30
Strength and Limitations
This study has several limitations that could potentially bias the findings. The article search was limited to four databases and one search engine, making it possible that there was still literature in other databases and causing the literature to be incomplete. Some studies did not describe intervention procedures, and follow-up and the sample sizes involved in the studies were small. In addition, the quality assessment of articles using JBI only involved two people, which potentially biased the quality assessment results, not being able to access some full-text articles, and limiting references to only English may have limited the research results.
However, despite its limitations, several benefits of this research must also be acknowledged. First, this study implemented a comprehensive search strategy, strict inclusion and exclusion criteria, systematic data extraction, and quality assessment of articles. Second, focus on interventions in managing bone disease in children with SCD. This study is the first scoping review to identify bone disease in children with SCD, including incidence and interventions. Third, this study involves countries worldwide, including America, Europe and Africa. Although the available evidence is still limited, the findings of this study provide strong indications regarding the applicability of interventions used in the management of children with CSD who experience bone dissection. However, the findings of this study represent three continents and can be valuable information for healthcare providers as a basis for policy-making and strategic interventions.
Implications for Clinical Practice
The implications of our study findings suggest that several types of interventions used in managing bone disease problems may have benefits in reducing bone disease problems in children with SCD. This review offers a broader range of intervention methods, allowing nurses, child health specialists, medical personnel, or other healthcare providers to determine appropriate intervention methods in managing bone disease in children with SCD. The findings of this study support that intervention methods that have multiple benefits with minimal adverse effects should be adopted to meet the healthcare needs of children with SCD. In addition, this study provides an overview of how government and health service facilities can collaborate to adopt safe control and management system policies in managing bone disease for children with SCD.
Conclusion
Based on the results of this scoping review, six articles describe the interventions used in managing bone disease problems in children with SCD. Our study found 114 children with SCD from America, Africa and Europe, and most of the types of SCD experienced were HbSS, bone disease problems that often occur in children with SCD were AVN and ONFH. Our study has identified four types of intervention: pharmacological, namely Vitamin D3 (cholecalciferol); Surgical, including THA, Osteotomy, Multiple epiphyseal drilling with AMBI, and Surgical only; Invasive, including IV bisphosphonates, HU, CD with bone marrow aspirate concentrate injection; non-pharmacological, namely physical therapy. Overall, the interventions provided excellent functional outcomes with minimal complications and no life-threatening side effects, safe and beneficial, although challenging.
This study highlights that surgical, invasive, pharmacological, and physical therapy interventions positively impact bone mineral density and functional improvement in children with SCD who experience bone disease. This research can provide additional information for further research on a similar topic. As a direction for future research, a meta-analysis of the efficacy of each intervention in helping improve the quality of life of pediatric patients with SCD who experience bone disease is needed. A meta-analysis study with a larger sample could strengthen the current findings.
Acknowledgments
The authors appreciated the Universitas Padjadjaran for supporting and facilitating the database search.
Disclosure
The authors declare no conflicts of interest in this work.
References
1. Grégoire-Pelchat P, Alos N, Ribault V, Pastore Y, Robitaille N, Mailhot G. Vitamin D intake and status of children with sickle cell disease in Montreal, Canada. J Pediatr Hematol Oncol. 2018;40(8):e531–e536. doi:10.1097/MPH.0000000000001306
2. Piccin A, Fleming P, Eakins E, McGovern E, Smith OP, McMahon C. Sickle cell disease and dental treatment. J Ir Dent Assoc. 2008;54(2):24–28.
3. Rees DC, Williams TN, Gladwin MT. Sickle-cell disease. Lancet. 2010;376(9757):2018–2031. doi:10.1016/S0140-6736(10)61029-X
4. Vos T, Allen C, Arora M, et al. Global, regional, and national incidence, prevalence, and years lived with disability for 310 diseases and injuries, 1990–2015: a systematic analysis for the Global Burden of Disease Study 2015. Lancet. 2016;388(10053):1545–1602. doi:10.1016/S0140-6736(16)31678-6
5. Kato GJ, Piel FB, Reid CD, et al. Sickle cell disease. Nat Rev Dis Primers. 2018;4:1–22. doi:10.1038/nrdp.2018.10
6. Kavanagh PL, Fasipe TA, Wun T. Sickle Cell Disease: a Review. JAMA. 2022;328(1):57–68. doi:10.1001/jama.2022.10233
7. de Oliveira JF, Vicente NG, Santos JPP, Weffort VRS. Vitamin D in children and adolescents with sickle cell disease: an integrative review. Rev Paul Pediatr. 2015;33(3):349–354. doi:10.1016/j.rppede.2015.06.015
8. Oyebimpe O, Lynne D, Hematologi D, et al. Osteonekrosis pada penyakit sel sabit: pembaruan pada faktor risiko, diagnosis, dan manajemen; 2019:351–358.
9. Garrido C, Bardón-Cancho EJ, Fajardo-Sánchez V, et al. Evaluation of the effectiveness of prophylactic oral vitamin D (cholecalciferol) in children with sickle cell disease. Bone. 2020;133:115228. doi:10.1016/j.bone.2020.115228
10. Silva Junior GB, Daher ED, Rocha FA. Osteoarticular involvement in sickle cell disease. Rev Bras Hematol Hemoter. 2012;34(2):156–164. doi:10.5581/1516-8484.20120036
11. Matos MA, Carrasco J, Lisle L, Castelar M. Avascular necrosis of the femoral head in sickle cell disease in pediatric patients suffering from Hip dysfunction. Rev Salud Pública. 2016;18(6):986. doi:10.15446/rsap.v18n6.50069
12. Williams KM, Lee MT, Licursi M, Brittenham GM, Fennoy I. Response to long-term vitamin D therapy for bone disease in children with sickle cell disease. J Pediatr Hematol Oncol. 2018;40(6):458–461. doi:10.1097/MPH.0000000000001155
13. Grimbly C, Escagedo PD, Jaremko JL, et al. Sickle cell bone disease and response to intravenous bisphosphonates in children. Osteoporos Int. 2022;33(11):2397–2408. doi:10.1007/s00198-022-06455-2
14. Daltro GC, Fortuna V, De Souza ES, et al. Efficacy of autologous stem cell-based therapy for osteonecrosis of the femoral head in sickle cell disease: a five-year follow-up study. Stem Cell Res Ther. 2015;6(1):1–18. doi:10.1186/s13287-015-0105-2
15. Novais EN, Sankar WN, Wells L, Carry PM, Kim YJ. Preliminary results of multiple epiphyseal drilling and autologous bone marrow implantation for osteonecrosis of the femoral head secondary to sickle cell disease in children. J Pediatr Orthop. 2015;35(8):810–815. doi:10.1097/bpo.0000000000000381
16. Levac D, Colquhoun H, O’Brien KK. Scoping studies: advancing the methodology. Implement Sci. 2010;5(69):2–9. doi:10.1017/cbo9780511814563.003
17. Arksey H, O’Malley L. Scoping studies: towards a methodological framework. Int J Soc Res Methodol Theory Pract. 2005;8(1):19–32. doi:10.1080/1364557032000119616
18. Bradbury-Jones C, Aveyard H, Herber OR, Isham L, Taylor J, O’Malley L. Scoping reviews: the PAGER framework for improving the quality of reporting. Int J Soc Res Methodol. 2022;25(4):457–470. doi:10.1080/13645579.2021.1899596
19. Tricco AC, Lillie E, Zarin W, et al. PRISMA extension for scoping reviews (PRISMA-ScR): checklist and explanation. Ann Intern Med. 2018;169(7):467–473. doi:10.7326/M18-0850
20. JBI. The Joanna Briggs institute critical appraisal tools for use in JBI systematic reviews 2017. The Joanna Briggs Institute Critical; 2017. Available from: https://jbi.global/critical-appraisal-tools.
21. Alabi I, Salihu M, Arojuraye S, et al. Functional outcome of total hip arthroplasty in teenagers with end stage avascular necrosis of the head of femur resulting from sickle cell disease. J Orthop Rheum. 2021;5(1):15–20. doi:10.36959/479/439
22. Gatin L, De MAR, Mary P, Vialle R, Gatin L. Osteonecrosis of the femoral head: a proposed new treatment in homozygous sickle cell disease osteonecrosis of the femoral head: a proposed new treatment in homozygous sickle cell disease. Hemoglobin. 2016;40(1):1–9. doi:10.3109/03630269.2015.1092159
23. Itzep NP, Jadhav SP, Kanne CK, Sheehan VA. Spontaneous healing of avascular necrosis of the femoral head in sickle cell disease. Am J Hematol. 2019;94(6):E160–E162. doi:10.1002/ajh.25453
24. Bell V, Varzakas T, Psaltopoulou T, Fernandes T. Sickle cell disease update: new treatments and challenging nutritional interventions. Nutrients. 2024;16(2):1–23. doi:10.3390/nu16020258
25. Selma J, Song H, Rivera C, et al. Sickle cell disease promotes sex-dependent pathological bone loss through enhanced cathepsin proteolytic activity in mice. Blood Adv. 2022;6(5):1381–1393. doi:10.1182/bloodadvances.2021004615
26. Sukmawati S, Hermayanti Y, Fadlyana E, Mulyana AM, Nurhakim F, Mediani HS. Supplementation of prenatal Vitamin D to prevent children’s stunting: a literature review. Int J Womens Health. 2023;15:1637–1650. doi:10.2147/IJWH.S431616
27. Garadah TS, Hassan AB, Jaradat AA, et al. Predictors of abnormal bone mass density in adult patients with homozygous sickle-cell disease. Clin Med Insights Endocrinol Diabetes. 2015;8:35–40. doi:10.4137/CMED.S24501
28. Lee MT, Licursi M, McMahon DJ. Vitamin D deficiency and acute vaso-occlusive complications in children with sickle cell disease. Pediatr Blood Cancer. 2015;62(4):643–647. doi:10.1002/pbc.25399
29. Ceglie G, Di Mauro M, Tarissi De Jacobis I, et al. Gender-related differences in sickle cell disease in a pediatric cohort: a single-center retrospective study. Front Mol Biosci. 2019;6:1–5. doi:10.3389/fmolb.2019.00140
30. Mulumba LL, Wilson L. Sickle cell disease among children in Africa: an integrative literature review and global recommendations. Int J Africa Nurs Sci. 2015;3(2015):56–64. doi:10.1016/j.ijans.2015.08.002
31. Valenti MT, Mattè A, Federti E, et al. Dietary ω-3 fatty acid supplementation improves murine sickle cell bone disease and reprograms adipogenesis. Antioxidants. 2021;10(5):1–15. doi:10.3390/antiox10050799
32. Martí-Carvajal AJ, Solà I, Agreda-Pérez LH. Treatment for avascular necrosis of bone in people with sickle cell disease. Cochrane Database Syst Rev. 2019;2019(12). doi:10.1002/14651858.CD004344.pub7
 © 2024 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2024 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
Recommended articles
Practical Guidance for the Use of Voxelotor in the Management of Sickle Cell Disease
Barriteau CM, Badawy SM
Journal of Blood Medicine 2022, 13:739-745
Published Date: 29 November 2022
Sickle Cell Disease With Ulcerative Colitis in An Ethiopian Child
Tamire AH, Million T
International Medical Case Reports Journal 2024, 17:521-525
Published Date: 21 May 2024
Prevalence of Gallstones and Associated Factors Among Children and Adolescents with Sickle Cell Disease in Eastern Uganda
Okejomoe AO, Chebet M, Tegu C, Abeso J, Achiro K, Hakizimana T, Katuramu R, Pitua I, Adoch CO, Mupere E, Olupot-Olupot P
Pediatric Health, Medicine and Therapeutics 2026, 17:571634
Published Date: 27 March 2026