")
Back to Journals » International Journal of Nanomedicine » Volume 16
Bombesin Peptide Conjugated Water-Soluble Chitosan Gallate—A New Nanopharmaceutical Architecture for the Rapid One-Pot Synthesis of Prostate Tumor Targeted Gold Nanoparticles
Authors Tangthong T, Piroonpan T, Thipe VC , Khoobchandani M, Katti K, Katti KV, Pasanphan W
Received 29 June 2021
Accepted for publication 17 September 2021
Published 13 October 2021 Volume 2021:16 Pages 6957—6981
DOI https://doi.org/10.2147/IJN.S327045
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Phong A Tran
Theeranan Tangthong,1,2 Thananchai Piroonpan,2 Velaphi C Thipe,3,4 Menka Khoobchandani,4,5 Kavita Katti,4,5 Kattesh V Katti,4– 6 Wanvimol Pasanphan1,2
1Department of Materials Science, Faculty of Science, Kasetsart University, Chatuchak, Bangkok, 10900, Thailand; 2Center of Radiation Processing for Polymer Modification and Nanotechnology (CRPN), Department of Materials Science, Faculty of Science, Kasetsart University Chatuchak, Bangkok, 10900, Thailand; 3Department of Chemistry, University of Missouri, Columbia, MO, 65211, USA; 4Institute of Green Nanotechnology, University of Missouri, Columbia, MO, 65211, USA; 5Department of Radiology, University of Missouri, Columbia, MO, 65211, USA; 6Department of Physics, University of Missouri, Columbia, MO, 65211, USA
Correspondence: Wanvimol Pasanphan
Department of Materials Science, Faculty of Science, Kasetsart University, 50 Ngam Wong Wan Road, Lat Yao, Chatuchak, Bangkok, 10900, Thailand
Tel +66 2562 5555 (Ext. 646518)
Email [email protected]
Kattesh V Katti
Institute of Green Nanotechnology, University of Missouri, Columbia, MO, 65211, USA
Tel +1 573 882-5656
Fax +1 573 884-5679
Email [email protected]
Purpose: We report herein bombesin peptide conjugated water-soluble chitosan gallate as a template for rapid one-pot synthesis of gold nanoparticles (AuNPs) with capabilities to target receptors on prostate cancer cells.
Methods: Water-soluble chitosan (WCS), anchored with gallic acid (GA) and LyslLys3 (1,4,7,10-tetraazacyclo dodecane-1,4,7,10-tetraacetic acid) bombesin 1– 14 (DBBN) peptide, provides a tumor targeting nanomedicine agent. WCS nanoplatforms provide attractive strategies with built-in capabilities to reduce gold (III) to gold nanoparticles with stabilizing and tumor-targeting capabilities. WCS-GA-DBBN encapsulation around gold nanoparticles affords optimum in vitro stability.
Results: The DBBN content in the WCS-GA-DBBN sample was ∼ 27%w/w. The antioxidant activities of WCS-GA and WCS-GA-DBBN nanocolloids were enhanced by 12 times as compared to the nascent WCS. AuNPs with a desirable hydrodynamic diameter range of 40– 60 nm have been efficiently synthesized using WCS-GA and WCS-GA-DBBN platforms. The AuNPs were stable over 4 days after preparation and ∼ 3 days after subjecting to all relevant biological fluids. The AuNPs capped with WCS-GA-DBBN peptide exhibited superior cellular internalization into prostate tumor (PC-3) cells with evidence of receptor mediated endocytosis.
Conclusion: The AuNPs capped with WCS-GA-DBBN exhibited selective affinity toward prostate cancer cells. AuNPs conjugated with WCS-GA-DBBN serve as a new generation of theranostic agents for treating various neoplastic diseases, thus opening-up new applications in oncology.
Keywords: water-soluble chitosan, gallic acid, bombesin, one-pot synthesis, gold nanoparticle, prostate cancer
Introduction
The application of nanotechnology in tumor diagnostics and therapy continues to attract considerable attention.1–33 In this context, we have been exploring the utility of green nanotechnology in the design and architecture of tumor-avid gold nanoparticles.6,9,11,12,14,20,23,26,34 Tumor avidity and selective accumulation of optimum doses of therapeutic gold nanoparticles is of paramount importance in drug delivery.35,36 Specifically, tumor selectivity and cytotoxicity of NPs depend on many factors, such as particle size, surface chemistry and biological target.37 Firstly, the particle size should be small enough to escape capture by fixed macrophages that are lodged in the reticuloendothelial system (liver and spleen) but should be large enough to prevent their rapid leakage into blood capillaries.38 The optimal NP size is considered to be in the range of 10 to 100 nm.39 Secondly, the effect of surface charge on toxicity and cellular internalization are important considerations in the design and development of nanomedicine agents. As reported herein, functionalization of gold nanoparticles with positive effective surface charges are ideal for disrupting the negatively charged cellular membrane through electrostatic interactions.40,41 Thirdly, the targeting agents should have the ability to be selective toward tumor cells to reduce off target systemic toxicity.35 Gold nanoparticles (AuNPs) possess outstanding properties because of high surface area-to-volume ratio and their unique ability to anchor or conjugate specific ligands with myriad functions on their surface—all aimed at improving the effective targeting capabilities that are required in diagnosis and therapy.42 The surface properties of AuNPs can be changed by conjugating with active targeting molecules, such as tumor-avid small molecules, proteins, peptides, and monoclonal antibodies.43 For cancer treatment, AuNPs can easily permeate, accumulate, and remain in tumors due to their permeability and retention (EPR) effects, thus resulting in enhanced uptake of diagnostic/therapeutic cargo.44 Although AuNPs enjoy several advantages for their use in nanomedicine, the synthesis of AuNPs generally involves chemical reduction reactions in the presence of highly toxic reducing agents, such as hexadecyltrimethyl ammonium bromide (CTAB),45,46 sodium borohydride (NaBH4) and formaldehyde, and sodium hydrazine (N2H4).47 Toxicities of representative examples of reducing agents, capping agents and solvents have been reviewed in the literature.47 Takahashi et al 41 have shown that AuNPs extracted from CTAB exhibited higher biocompatibility when compared to CTAP-capped AuNPs. AuNPs stabilized with CTAB showed strong cytotoxicity at 0.05 mM (Au atom) due to unreacted free CTAB.48 It has been reported that polyethylene glycol (PEG) capped-AuNPs would help in enhancing the biodistribution and also to reduce systemic toxicity of CTAB.48 Therefore, the development of efficient, eco-friendly, and straightforward one-pot synthetic protocols to prepare AuNPs in a water-based system is of paramount importance. Many natural compounds,13,15 such as those derived from phytochemicals,49,50 ascorbic acid,51 sugar,52–54 starch,55,56 alginate,57 gum arabic,15,23 fibroin16 and chitosan17,58–60 have been reported for green synthesis of non-toxic AuNPs. The pioneering efforts of Katti et al have demonstrated the extensive utility of electron-rich phytochemicals from tea, cinnamon, mango and various other plant species for the development of tumor-specific diagnostic and therapeutic gold nanoparticles.26,31–33
Chitosan (CS) is a biopolymer composing of D-glucosamine and N-acetyl-D-glucosamine units linked by β (1, 4)-glycosidic linkages. It has excellent biocompatible and biodegradable characteristics with low toxicity and immune stimulating properties, including excellent antioxidant activity.60 The development of chitosan and its derivatives is of interest in many areas, especially biomedicine. When considering biological molecules such as lipids, proteins, enzymes, DNA, and RNA, it is important to recognize how free radical reactions of these substances are involved in human health issues, including aging, atherosclerosis, and carcinogenesis.61,62 Chitosan has been exploited as an alternative natural antioxidant.63,64 Due to the abundance of amino (– NH2) and hydroxyl (–OH) groups, acting as electron transfer agents, CS has attracted considerable attention for its utility as a natural antioxidant65–67 and as a reducing agent for phytochemical-mediated synthesis of AuNPs.17 Although the –OH and –NH2 groups of CS present several advantages, CS still has some limitations of non-reactivity and insolubility in water. Aqueous solutions of CS exhibit pH >6.5 and while in organic solvents the insolubility is dictated by strong inter- and intra-molecular hydrogen bonds.68 This has limited the use of CS in aqueous solutions at neutral pH profiles relevant in biological media. CS gallate derivative was developed and proposed as a novel and potential antioxidant due to synergistic effects of CS and gallic acid function.67,69 This novel antioxidant was developed in our laboratory and in this paper we are exploring its utility as an effective reducing agent in the form of a new nano-template for the rapid and efficient one-pot synthesis of functionalized AuNPs.
The CS gallate derivatives promote free radical scavenging by trapping hydroxyl radicals, carbon center radicals and also the 2,2-diphenyl-1-picrylhydrazyl (DPPH) radicals. In particular, CS gallate can suppress the unwanted pro-oxidant metals by chelating agents. Since the first establishment of CS-GA derivative,67,69 it has been used as an antioxidant in various applications, for example in food packaging films,70,71 wound healing,72 tissue engineering,73,74 nanocomposite of contact lenses,75 inhibition of hydrogen peroxide-induced oxidative damage,76 prevention of oxidative stress in Chang liver cells,77 drug delivery system18,22,78 and synthesis of silver NPs.5 In continuation of efforts to capitalize on the antioxidant and reducing capacity of CS, water-soluble chitosan (WCS) has been developed through the green radiolysis processes.17 Based on the chemical structure of WCS, it is susceptible to depolymerization by radiolysis to form smaller units of D-glucosamine, a derivative of glucose.28 Therefore, the WCS nanocolloids, without functionalization, serve as efficient CS-containing antioxidants with built-in capabilities, including reducing power and stabilizing efficiency—all serving vital roles in the green synthesis of AuNPs.8
We hypothesized that WCS gallate would be a good candidate for not only its role as a reducing and stabilizing agent in the green synthesis of AuNPs, but also for providing antioxidant functions for use in cancer treatment. It is known that gallic acid (GA) is an antioxidant extractable from plants19 which is widely used in foods, drugs, and cosmetics. This antioxidant effect could prove beneficial to address numerous disease states, including cardiovascular disease. On the medical applications front, GA exhibits no cytotoxicity against normal fibroblast and endothelial cells. Therefore, the development of synthetic gallic acid derivatives has attracted considerable attention as a potent chemotherapeutic agent. Polyphenol compounds are well known as an important class of chemopreventive agents because of their acidity and the presence of delocalized π-electrons of benzene rings, leading to effective quenching of free radicals in cancer cells.21
Our continued interest in exploring the targeting potential of bombesin (BBN) peptide stems from the fact that it is structurally similar to human gastrin-releasing peptide (GRP).25,79,80 GRP receptors are over-expressed in many neoplastic diseases, including breast, lung and prostate cancers.27 Several analogues of BBN have been synthesized to target BBN receptors in humans for imaging, diagnosis and treatment of cancer.1,23,30 Peptide conjugated gold nanoparticles are able to localize on tumor cells with high selectivity with subsequent penetration through interstitial barriers to reach the desired tumor tissues and cells with minimal loss of their activity in the blood circulation.35 We are, therefore, focusing our efforts on the applications of BBN conjugated gold nanoceuticals for targeting the primary and peripheral tumor targets.29,81
In this paper, we report the preparation of water-soluble chitosan gallate (WCS-GA) derivative and its conjugation with LyslLys3 (1,4,7,10-tetraazacyclododecane-1,4,7,10-tetraacetic acid)-bombesin 1–14 (DOTA-BBN or DBBN) peptide. The successful conjugation of WCS-GA with DBBN (CS-GA-DBBN) has been fully elucidated using FT-IR, 1H-NMR, XRD and SEM-EDX. Nanoparticulate formation and identity has been inferred through a combination of techniques including transmission electron microscopy (TEM) and dynamic light scattering (DLS). The antioxidant activity of WCS-GA-DBBN has been measured using DPPH radical assay. This report also provides full details on the ability of WCS-GA-DBBN to be used as a reducing and stabilizing agent for the one-pot template synthesis of gold nanoparticles (AuNPs). The nanostructure architecture of AuNPs, in the presence of WCS-GA-DBBN, has been further elucidated through UV-Vis, Zetasizer, TEM, HR-TEM, XRD and SEM-EDX. Our investigations have further included in vitro stability of AuNPs-WCS-GA-DBBN toward myriad of biological media through detailed monitoring of hydrodynamic sizes. Prostate tumor receptor avidity of AuNP-WCS-GA-DBBN has been established using PC-3 and LNCaP cell lines. Confocal microscopy and TEM studies have inferred the selective cellular uptake and cytotoxicity of the targeted AuNPs-WCS-GA-DBBN in comparison with the non-targeted AuNPs standards.
Materials and Methods
Chemicals and Materials
Chitosan (CS) with a weight-average molecular weight (Mw) of 258.8 kDa and degree of deacetylation (%DD) of 80 was supplied by Seafresh Chitosan (Lab) Co., Ltd. (Thailand). LyslLys3 (1,4,7,10-tetraazacyclodode cane-1,4,7,10-tetraacetic acid)-Bombesin1-14 (DOTA-BBN or DBBN) was obtained from CSBio Co., Ltd. (USA) and supported by the International Atomic Energy Agency (IAEA), Vienna (Austria). 1-Ethyl-3-(3ʹ-dimethylamino propyl) carbodiimide (EDC) was purchased from TCI Co., Ltd. (Japan). N-Hydroxysuccinimide (NHS) was obtained from Tokyo Kasei Kogyo Co., Ltd. (Japan). Sodium hydroxide (NaOH) was purchased from Carlo Erba Reagent (Italy) and acetic acid (AR grade) was purchased from Lab Scan, Co., Ltd. (Thailand). Dialysis membrane (MWCO = 1000Da) was purchased from Membrane Filtration Products, Inc. (USA). Poly (ethylene oxide) GPC standards were obtained from PSS Polymer Standards Service GmbH, Mainz (Germany). Gallic acid (GA), hydrochloroauric acid (HAuCl4), 2.2-diphenyl-1-picrylhydrazyl (DPPH) standard free radicals, sulphur (S) standard solution (1000 mg/L) in water with a high purity, bovine serum albumin (BSA) and human serum albumin (HSA), Roswell Park Memorial Institute Medium (RPMI), MTT (3-(4,5-dimethylthiazol-2-yl)-2,5 diphenyl tetrazolium bromide)), dimethyl sulfoxide anhydrous (DMSO), wheat germ agglutinin (WGA) and Dulbecco’s Phosphate Buffered Saline x1 (DPBS) were obtained from Sigma-Aldrich (USA). Cysteine and histidine were purchased from Acros Organics (Belgium). Sodium chloride (NaCl) and phosphate buffer solution (pH 5, 7 and 12) were purchased from Fisher Scientific (USA). PC-3 and LNCaP cells were obtained from the American Type Culture Collection (ATCC; Manassas, VA). PC-3 cells were maintained in RPMI medium 1640 (1X) supplemented with 4.5 g/L D-glucose, 2.383 g/L HEPES, 0.11 g/L sodium pyruvate, 1.5 g/L sodium bicarbonate, 2 mM L-glutamine, and 10% fetal bovine serum (FBS) and antibiotics. LNCaP cells were maintained in RPMI-1640 medium supplemented with 4.5 g/L D-glucose, 10 mM HEPES, 1 mM sodium pyruvate, 1.5 g/L sodium bicarbonate, 2 mM L-glutamine, and 10% fetal bovine serum (FBS). Fetal bovine serum and (FBS) and TryplE were purchased from Gibco BRL (USA). Paraformaldehyde (PFA) was bought from Electron Microscopy Sciences. Gentamicin antibiotic was from APP Pharmaceuticals LLC. All pure water with a resistivity of 18.2 MΩ cm was obtained from Milli-Q Academic purification set from Millipore (USA).
Instruments and Characterization
The 60Co from a Gamma cell 220 irradiator was used as a γ-ray source with an absorbed dose rate of 3.3 kGy/h at room temperature. Chemical structure was characterized using Fourier transform infrared spectrophotometer (FT-IR) in a Bruker Tensor 27 (USA) with 32 scans at 2 cm−1 resolution in a frequency range of 4000–400 cm−1. Proton nuclear magnetic resonance 1H NMR were determined on a Bruker Ultrashield 500 Plus (500 MHz) using D2O as solvent. Elemental composition of the conjugated sample was measured using a scanning electron microscope with an energy dispersive X-ray spectroscopy (SEM/EDX) and the elemental mappings were collected on a FET QUANTA 450 (Netherlands). The signals are quantified and plotted against the concentrations of the standard sulfur (S) added into the sample for quantifying the amount of DBBN conjugate. The standard sulfur solution with different concentrations of 0, 0.02, 0.04, 0.06, 0.08 and 0.10 μg/mL were added to the sample solution (7.5 μg/mL, 25 μL) for preparing addition standardization. The set of sample solutions with different sulfur standard concentrations was dropped onto a copper grid and determined using SEM/EDX. Powder X-ray diffraction (XRD) patterns were taken on a Bruker AXS (Germany) D8 Advance X-ray diffractometer with CuKα radiation, operating at 50 kV and 100 mA. The powder of the samples was placed on a polymethyl methacrylate (PMMA) specimen-holder ring. The pattern of the samples was recorded from 10° to 80°, 0.0389° = 2θ. The optical extinction spectra of AuNPs formation was operated using a 1-cm path length quartz cuvette with a Varian Cary 50 UV-Vis spectrophotometer, Shimadzu (USA). The intensity-average hydrodynamic diameter and zeta potentials were measured using Zetasizer Nano S90, Malvern Instruments Ltd. (USA) with a 659 nm diode laser source. The morphologies of the dried samples were taken by transmission electron microscope (TEM) on a JEOL JEM 1400 TEM and operated at 120 kV. The AuNPs sample was prepared by centrifuging at 15,000 rpm, 15 °C for 15 min. The supernatant was discarded and the solution was brought to initial volume with deionized water. The sample was sonicated for 15 min and dropped onto a carbon coated copper grid. The sample was allowed to dry at ambient temperature before TEM measurement. Cellular internalization was investigated using a Confocal Laser Scanning Microscope from Leica Microsystems (USA). In vitro cytotoxicity evaluation was investigated using a bright field microscope and SpectraMax M2 microplate reader from Molecular Devices LLC (USA).
Preparation of Water-Soluble Chitosan Nanoparticles (WCS)
WCS was originally prepared following a procedure reported by Pasanphan et al.17 Briefly, CS solution (1% v/v, 1000 mL) was prepared by dissolving CS powder into aqueous acetic acid solution (1% v/v, 1000 mL) and stirring overnight at room temperature. The CS solution was γ-rays irradiated with an absorbed dose of 80 kGy under ambient temperature in air. The samples were neutralized in NaOH solution (1% w/v, 600 mL) by precipitation method. The sample was dialyzed in a dialysis bag (MWCO = 1000 Da) in deionized water for 24 h. Finally, the CS solution was frozen and lyophilized to obtain water-soluble chitosan nanoparticle (WCS) powder.
FT-IR (KBr, cm−1) for WCS: 670 (N-H stretching), 898 (pyranose ring), 1020 and 1029 (C-O-C bridge and C-O stretching of C3, C6), 1423 (C-N stretching), 1562 (N-H stretching, amide II), 1630 (C=O stretching of acetyl group, amide l), 2900 (C-H stretching) and 3350 (O-H stretching).
1H NMR (ppm) for WCS: 2.0 (H-Ac of −NHCOCH3), 3.0–3.2 (H-2 of −CHNH2), 3.6−3.9 (H-3 to H-6 of −CH−OH), 5.4 (H-1 of −CHCHO2).
Functionalization of WCS with GA
WCS (0.169 g, 1 mol) was dissolved in deionized water (20 mL) and stirred for 15 min. GA (0.510 g, 3 mole, 3 mole equiv. to WCS) was dissolved in ethanol (30 mL) and EDC (1.725 g, 3 mole equivalent to GA) was reacted with GA. NSH (1.036 g, 3 mole equivalent to GA) was further added. The reaction was stirred in an ice bath for 1 h in ethanol to obtain reactive GA/EDC/NSH system. The WCS solution was added into GA/EDC/NHS solution and the reaction was carried out in an ice bath for 15 min and further performed at room temperature for 24 h with magnetic stirring. The sample was dialyzed using a dialysis membrane (MWCO 3500 Da) at room temperature for 24 h and lyophilized to obtain WCS-GA powder.
FT-IR (KBr, cm−1) for WCS-GA: 572 (ring bending), 663 (benzene ring), 1026, 1074 (C-O-C bridge and C-O stretch of C3, C6), 1238, 1375 (aliphatic C-H bending vibrations), 1564 (N-H stretching, amide II), 1652 (C=O stretching of acetyl group, amide l), 1705 (C=O of ester bond), 2936 (C-H stretching) and 3494 (O-H stretching). Found for GA: 468–767 (C-H out-of-plane bending of benzene), 1027, 1257 (C-H in-plane bending), 1311 (C-C stretching), 1431, 1610, 1670 (C-C stretching vibration in the aromatic ring), 1702 (C=O stretching), 3296 and 3502 (O-H stretching).
1H NMR (ppm) for WCS-GA (DSGA/WCS = 19.84%): 2.1 (H-Ac of −NHCOCH3), 3.1–3.2 (H-2 of −CHNH2), 3.6−3.9 (H-3 to H-6 of −CH−OH), 5.4 (H-1 of −CHCHO2), 7.1 (H-a of GA).
Conjugation of DBBN Peptide Onto WCS-GA
DBBN (11.78 mg, 1 mol equiv. to WCS-GA) was stirred in deionized water (1 mL) with EDC (1.14 mg, 1 mol equiv. to WCS-GA), NHS (0.68 mg, 1 mol equiv. to WCS-GA). The reaction was stirred in an ice bath for 15 min. The WCS-GA solution (1 mg/mL, 1 mL) was added into DBBN/EDC/NSH solution and carried out under ambient temperature for 24 h. The solution was dialyzed (MWCO 3500 Da) in the deionized water for 24 h. The solution was dried using a freeze dryer to obtain WCS-GA-DBBN powder.
FT-IR (KBr, cm−1) for WCS-GA-DBBN: 657 (aromatic ring of BBN), 900 (pyranose ring), 1078 (C-O stretching of C-3, C-6), 1382 (C-N stretching), 1566 (N-H stretching, amide II), 1658 (C=O stretching of acetyl group, amide I), 1766 (C=O of ester bond), 2920 (C-H stretching) and 3492 (O-H stretching).
1H NMR (ppm) for WCS-GA-DBBN (DSGA/WCS = 19.80%, DSDBBN/WCS = 0.1%): 1.7–1.9 (H-g of −CHCH2CH−), (H-h of −CHCH3), (H-j of −CHNH2), (H-I of −CH2CH(CH3)2), 2.3–2.9 (H-b of R2N−CH2), (H-d of −NHCH2CO−), (H-e of −NH−CHCH2CO−), (H-I of −NCH2CO−), (H-m of NH2CH2CH2−), (H-m’ of −CH2CH2NH−), (H-n of −CH2CH2CONH2), (H-n’ of −CHCH2CO−), (H-o of −CCHNH−), (H-o’ of −CHCH2NH−), (H-p of −CH3SCH2−), (H-p’ of −CH2SCH3), (H-q of −NHCHN−), 7.0 (H-a of −PhH2(OH)−), 7.1 (H-f of −PhH−), 8.3 (H-k of −C(O)NH2), 8.4 (H-c of −C(O)NH−CH−). Found for WCS: 2.0 (H-Ac of -NHCOCH3), 3.0–3.2 (H-2 of −CHNH2), 3.6−3.9 (H-3 to H-6 of −CH−OH).
The Antioxidant Activity of WCS-GA-DBBN
The 2,2-diphenyl-1-picrylhydrazyl (DPPH) free radical scavenging capacity was measured according to a previous protocol.25 Briefly, the aqueous solutions of WCS-GA-DBBN (0.04, 0.12, 0.2, 0.4, 0.6, 0.8, 10, 16, 2.0, 3.0 and 4.0 mg/mL, 500 µL) were added to methanolic DPPH solution (300 µM, 500 µL). The mixture was stirred for 30 min in a dark at room temperature, followed by absorbance measurement at 515 nm by spectrophotometer. The percentage of DPPH free radical scavenging ability was calculated using the following equation: [A0-Ac/A0] × 100; where A0 and Ac are absorbances of the bank solution and the sample solution at different concentrations, respectively. Similarly, study of the antioxidant activity of WCS, GA, WCS-GA samples was also carried out using the same procedure.
One-Pot Synthesis of AuNPs in WCS-GA-DBBN
One-pot synthesis of AuNPs was carried out via reduction reaction of HAuCl4 1.0 mM using different concentrations of polymer and reaction times. HAuCl4 (1.0 mM, 1 mL) was prepared in aqueous solution and the solution was added into WCS-GA-DBBN aqueous solution (0, 1, 2, 3, 4 mg/mL, 1 mL). The mixture was then stirred for 0, 1, 2, 3, 4, 5, 6, 24, 48, 72 and 96 h at ambient temperature and atmosphere. Similarly, the WCS-GA aqueous solution was mixed with HAuCl4 aqueous solution using the above-described protocol. The AuNPs solution was quantified using UV-vis spectrophotometer in a range of the visible light (400–800 nm). The selected sample of AuNPs-WCS-GA-DBBN was purified by ultracentrifugation at 15,000 rpm at 15 °C for 15 min and re-dispersed in distilled water before further characterization.
In vitro Stability Assessment of AuNPs
In vitro stabilities of the AuNPs-WCS-GA and AuNPs-WCS-GA-DBBN were carried out in the biologically relevant solutions, ie, 1% NaCl, 0.5% cysteine (Cys), 0.2 M histidine (His), 0.5% bovine serum albumin (BSA), 0.5% human serum albumin (HSA), and phosphate buffer saline (PBS) with different pH values (pH 5, pH 7 and pH 12) including deionized (DI) water. The AuNPs aqueous solution was prepared using HAuCl4 (1.0 mM, 10 mL) mixed with WCS-GA and WCS-GA-DBBN (1.0 mg/mL, 10 mL). The mixture was left for 72 h to obtain stationary state of AuNPs-WCS-GA and AuNPs-WCS-GA-DBBN stock solution, respectively. The aqueous solution of 1 mL AuNPs (0.5 mM HAuCl4 in 0.5 mg/mL WCS-GA and WCS-GA-DBBN) was added into each biological media (1 mL). The mixtures were incubated for 24 to 72 h before measuring surface plasmon resonance using a UV-vis spectrophotometer. The alterations in mean hydrodynamic diameter (DH) were measured to evaluate the in vitro stability of AuNPs using the DLS technique in a zetasizer.
Cellular Internalization
Cell internalization of AuNPs into prostate cancer cells was determined by a confocal laser scanning microscope and transmission electron microscope. Human prostate cancer cell line (PC-3) and lymph node carcinoma of the prostate cell line (LNCaP) were sub-cultured in media RPMI and RPMI customized, respectively. To evaluate cellular internalization of AuNPs, PC-3 cells were placed at 4 × 104 cells/mL in a 6-well plate and let to adhere overnight. The cells were then treated with AuNPs (50 µg/mL) and incubated for 6 h. The treated cells were washed twice with PBS and fixed in 2% glutaraldehyde (2% v/v in 0.1 M PBS). The PC-3 cells were stained with wheat germ agglutinin (WGA) stain solution (1 µg/mL) and washed with Hank’s balanced salt solution (HBSS) and Triton solution. The cells were placed onto a glass slide and covered with a cover slip. The images of PC-3 cells were detected with a confocal laser scanning microscope. For TEM measurement, the PC-3 cells were placed at 4 × 104 cells/mL in a 6-well plate, and let to adhere overnight. The cells were then treated with AuNPs (50 µg/mL) and incubated for 6 h. The PC-3 cells were washed twice with PBS and fixed in glutaraldehyde (2% v/v in 0.1 M PBS). The samples were then rinsed twice in buffer solution, post-fixed in 1% osmium tetroxide in 0.1 M PBS for 1 h and dehydrated through a graded ethanol series. The cells were cut on a Leica UCT ultramicrotometer and transferred to copper grids. The TEM images of the cells were acquired with TEM. Similarly, LNCaP cells were prepared and measured using the same procedure.
Cytotoxicity of AuNPs to Prostate Cancer Cells
Human prostate cancer cell lines, ie, PC-3 and LNCaP, were sub-cultured in media RPMI and RPMI customized, respectively supplemented with 10% FBS and gentamicin and stored in a CO2 incubator at 37oC in tissue culture flasks. A concentration of 4 × 104 cells/mL was placed into each well of a 96-well plate. The cells were incubated overnight to allow cell adherence onto the surface of the plate and the media was then removed. Stock solution of the AuNPs-WCS-GA-DBBN sample was prepared using HAuCl4 (0.5 mM) in WCS-GA-DBBN (0.5 mg/mL) reacted for 1 h. The cells (1 mL) were treated with AuNPs-WCS-GA-DBBN (0, 25.36, 50.75, 101.50, 203 and 406 µL) to achieve different concentrations of 0, 6.25, 12.5, 25, 50 and 100 µg/mL. The samples were incubated for 24, 48 and 72 h. Cellular morphologies were investigated using bright-field microscopy. After cellular investigation, MTT dye in PBS solution (5 mg/mL, 10 µL) was added into the cell samples and incubated at 37oC for 3 h. The formazan crystals in the MTT solution were then removed by adding 100 µL of DMSO into the plates and the sample was further incubated for 20 min. The absorbance of the plates was recorded at a wavelength of 570 nm using a SpectraMax M2 microplate reader.
Results and Discussion
Functionalization and Characterization of GA Onto WCS
WCS-GA was prepared by 2 steps. Firstly, CS was depolymerized into small molecular weight and small polymeric colloids in aqueous solution. WCS can be dissolved in nearly neutral solution due to the high abundance of -NH2 functionalities (pH ∼6.5). Because of the chain scission, through high energetic radiation, the original WCS contains -NHCOCH3 of the chitin unit and -NH2 and -OH of CS units. Additional functionalities include C-O-C bonds of the glycosidic linkage and the carbonyl (C=O) group of its degraded chain end. After preparation, a detailed characterization of the WCS nanocolloids was performed to gain full insights into its structure prior to functionalization. As a first observation, the chemical structures were characterized by FT-IR and 1H NMR. The FTIR spectrum of WCS (Figure 1A(a)) shows the characteristic peaks at 898, 1020, 1029, 1423, 1562, 1630, 2900 and 3350 cm−1, which are attributed to pyranose ring, C-O-C bridge and C-O stretching of C3 and C6, C-N stretching, N-H stretching (amide ll), C=O stretching of acetyl group (amide l), C-H stretching and O-H stretching, respectively. The chemical structure of WCS was also established using 1H-NMR. As shown in Figure 1B, the characteristic peaks of WCS exhibited a signal at 2.1 ppm (H-Ac, three protons of the N-acetyl group), 3.3 ppm (H at carbon-2 position), multiple peaks around 3.7–3.9 ppm (H at carbon-3 and carbon-6 position) and a peak at 5.4 ppm (H at carbon-1 position). The FT-IR and 1H-NMR results confirmed the chemical structure of WCS, which has aided our efforts for further chemical functionalization and biomolecular conjugations, as discussed in the following sections.
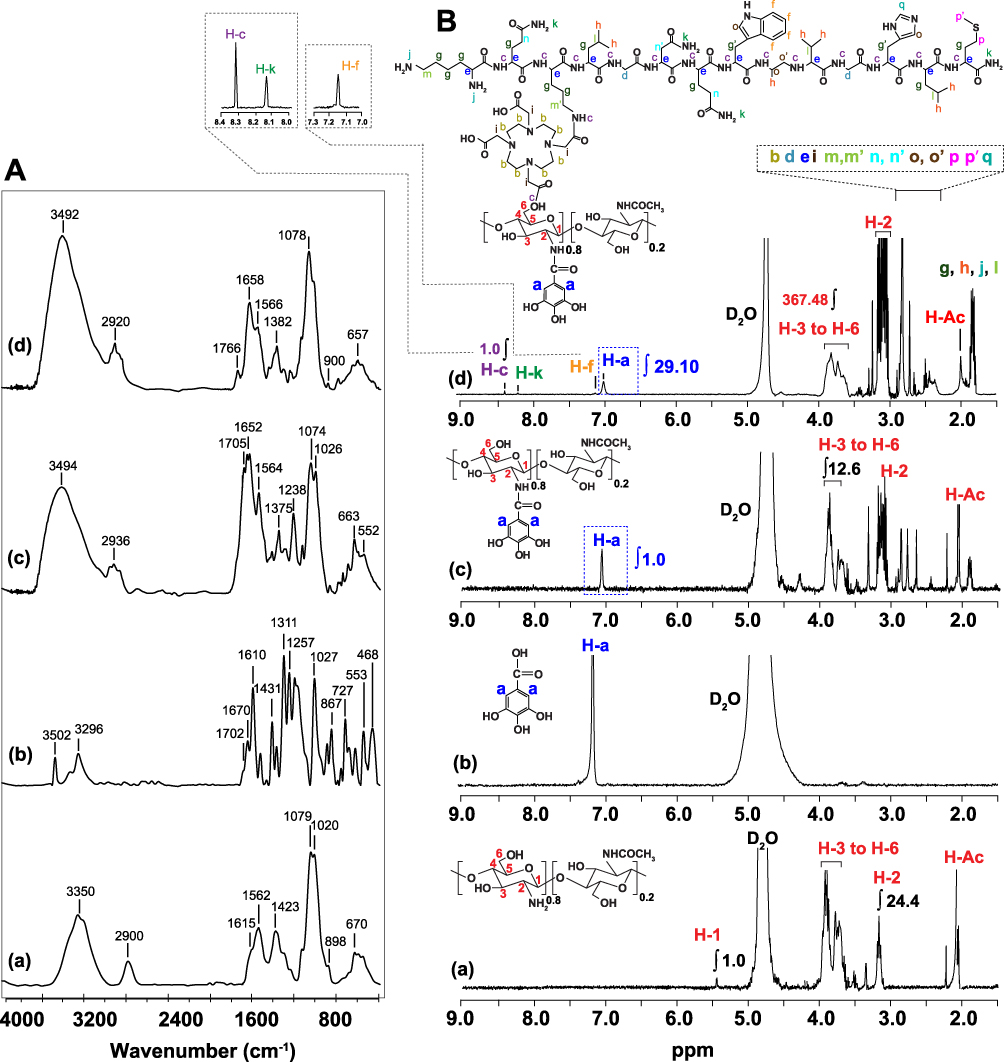 |
Figure 1 (A) FT-IR and (B) 1H NMR spectra of (a) WCS, (b) GA, (c) WCS-GA and (d) WCS-GA-DBBN. |
To improve the antioxidant activity and thereby enhance the reducing capacity of WCS, we anchored the polyphenolic group onto WCS. This approach affords the development of an effective reducing and stabilizing agent for rapid one-pot synthesis of gold nanoparticles (AuNPs). In this way, gallic acid derived polyphenol (GA) polyphenol was functionalized onto WCS. GA was functionalized onto WCS via amide and/or ester linkages using EDC/NHS coupling reaction. The EDC/NHS initiated the reactive ester species on GA and the nucleophilic reaction possibly occurs at the NH2- group at C-2 and the -OH group at C-6 and C-3 on WCS. In the presence of NHS, the long-lived GA-NHS ester is formed to improve the efficiency of EDC coupling reactions with WCS to form WCS-GA.69
Functionalization of GA onto WCS was further confirmed by FT-IR. The FTIR spectra (Figure 1A(c)). FT-IR spectrum of WCS-GA shows peaks of amide bond at 1643 cm−1 implying the formation of amide bonds between -NH2 of WCS and -COOH of GA. In addition, new peaks at 1761 cm−1 were found due to the ester linkage formation between the -OH group of WCS and the -COOH group of GA. In addition, the peaks around 700–900 cm−1 were assigned to the aromatic ring of GA. The FT-IR results agree with the conjugation of GA onto CS as indicated in previous literature.82,83
Further characterization of WCS-GA was performed by 1H NMR.69 The 1H NMR spectrum of WCS-GA (Figure 1B(c)) exhibited all peaks of WCS and an additional new signal at 7.1 ppm (assigned as H-a’) corresponds to non-H-atom substituents on the benzene ring. The 1H NMR spectrum of GA revealed a chemical shift at 7.6 ppm (assigned as H-a) belonging to two protons at the ortho positions on benzene ring of GA at a position (Figure 1B(b)). The shifted peaks from 7.6 (H-a) to 7.1 ppm (H-a’) confirmed the functionalized GA on WCS via amide and ester bonds. The degree of substitution (DS) of GA on WCS-GA was calculated from DS (%) = (IH-a/H-a)/(IH-3 to H-6/H-3 to H-6) × 100 = 19.84%. The DS of GA on WCS was comparable to that reported in the previous work when GA was functionalized on CS (DS ~15%).67,69 The DS of GA on WCS in the present work is slightly higher than that of GA on CS in the previous report owing presumably to higher reactivity of the short WCS chains than the longer CS chains.
Conjugation and Characterization of DBBN Onto WCS-GA
In order to conjugate tumor targeting biomolecules on to WCS-GA, DBBN was conjugated onto WCS-GA via the EDC/NSH coupling system (Scheme 1). The chemical structure of WCS allows the reactions of its -OH as well as -NH2 groups with the COOH on DOTA in DBBN. EDC reacts with the -COOH group of DOTA, forming a reactive ester intermediate. However, this intermediate is prone to hydrolysis and might convert back into the parent carboxylic group. Hence, NHS was added which replaces the previous unstable ester into a much more stable NHS-ester species. With the conjugation on WCS-GA, the NHS-ester species then reacted with the primary -NH2 or -OH groups to form an amide or ester bond, respectively.
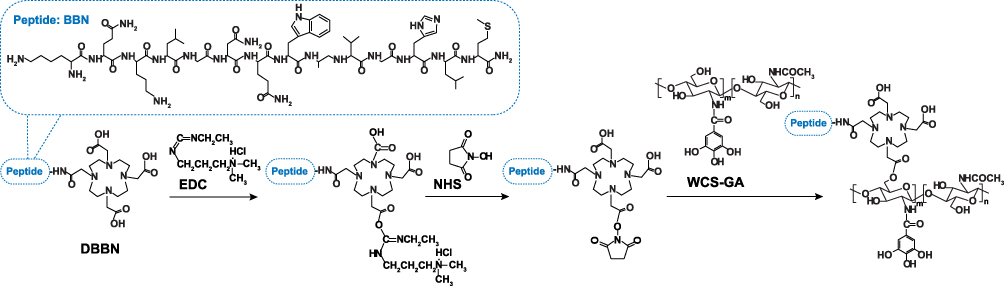 |
Scheme 1 Conjugation of DBBN onto WCS-GA. |
Confirmation of the conjugation of DBBN on WCS-GA has come from detailed analysis of the FTIR (Figure 1A) and 1H NMR (Figure 1B) spectra of WCS-GA-DBBN and comparisons of signals with WCS, GA and WCS-GA. The FT-IR spectrum of WCS-GA-DBBN showed an increase in the peak at 1658 cm−1 and an appearance of a new peak at 1766 cm−1. These two peaks can be attributed to the amide and ester linkages due to the conjugation of DBBN on WCS. Moreover, the peaks of ester linkage of DBBN conjugated product shifted from 1766 cm−1 to 1705 cm−1 due to an ester peak of GA functionalized on WCS. Therefore, FTIR is vital in inferring strong evidence for conjugation of DBBN onto WCS-GA.
The 1H NMR of WCS-GA-DBBN, as shown in Figure 1B(d), depicts characteristic peaks attributable to the DBBN structure. The chemical shift at 7.1 ppm was interpreted as a characteristic proton on the benzene ring (−PhH−) at f position. The chemical shift at 8.3 ppm is due to the proton in the amide (−C(O)NH2) side group at k position and the chemical shift at 8.4 ppm is attributed to the proton in the amide bond (−C(O)NH−CH−) backbone at c position. New signals around 2.3–2.9 ppm (assigned as proton at b, c, d, e, i, l, m, m’, n, n’, o, o’, p, p’ and q) appeared after conjugation. These groups of peaks corresponded to the methine protons of the nearby amine (−NHCH−), carbonyl (−CHC(O)), or sulfide (RS-CH) functions of the DBBN peptide. In addition, we also observed the chemical shifts around 1.7–1.9 ppm assigned to the methine protons (−CHCH2CH−), methyl protons (−CHCH3) and proton on the primary amine (−RNH2) at g, h, and j positions, respectively. The DS of DBBN on WCS was calculated from DS (%) = (IH-c/H-c)/(IH-3 to H-6/H-3 to H-6) × 100. The DS was calculated to be 0.1%. In addition, to confirm the conjugation of WCS-GA, the DS of GA on WCS was again calculated from the 1H NMR spectrum of WCS-GA-DBBN (Figure 1B(d)). It was confirmed that the DS of GA was ~19.80%, agreeing with the above calculations. Our BBN conjugation results from 1H NMR also agree with previous literature. For example, the BBN conjugated N-acetyl histidine-glycol chitosan nanoparticles (BC-NAHis-GC NPs) also showed chemicals shift around 7.5–9.0 ppm attributed to the protons of the aromatic regions of BBN, confirming the conjugation of BBN onto NPs.84
The chemical analysis of WCS-GA-DBBN was further performed by the energy dispersive X-ray (SEM-EDX) (Figure 2A). The WCS-GA-DBBN showed the characteristic X-ray of S at 2.3 keV) as attributed to a Kα X-ray of S in the BBN peptide structure, while WCS-GA did not show an S peak. The S peak was measured from several focal areas under a longer period of measurement with the SEM-EDX. In order to quantify the DBBN content in the WCS-GA-DBBN sample, we used a standard addition technique through SEM-EDX measurements. A series of S standardizations was prepared for determining the S amount in DBBN in the WCS-DBBN sample. The amount of DBBN peptide in the conjugated sample was calculated to be 27.04% (w/w). The S peak and its weight percentage found in the SEM-EDX result demonstrated that DBBN was successfully conjugated with WCS. The S peak and its weight percentage, determined from the SEM-EDX data, also confirmed the conjugation of DBBN peptide on WCS-GA. Our results corroborate with findings from recent reports showing the presence of S, C, N and O in the 177Lu-dendrimer conjugated to BBN with AuNPs.85
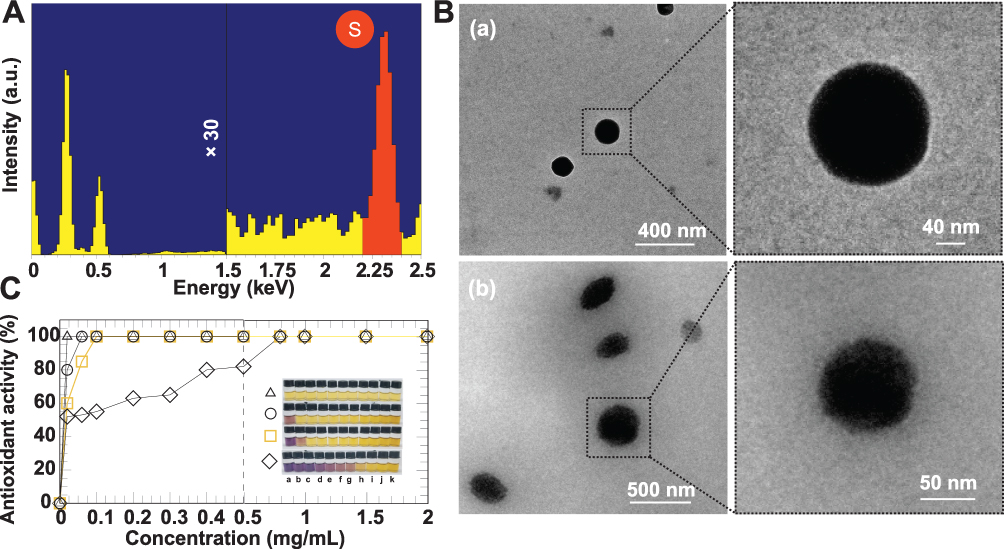 |
Figure 2 (A) SEM-EDX spectrum of WCS-GA-DBBN indicating sulfur elemental composition of WCS-GA-DBBN, (B) TEM images of (a) WCS-GA and (b) WCS-GA-DBBN, (C) DPPH radical scavenging abilities of WCS (◊), WCS-GA-DBBN (□), WCS-GA (○), and GA (Δ) at various concentrations of (a) 0.02, (b) 0.06, (c) 0.1, (d) 0.2, (e) 0.3, (f) 0.4, (g) 0.5, (h) 0.8, (i) 1.0, (j) 1.5 (k) 2.0 mg/mL and their corresponding physical appearance of the DPPH color fading from purple to yellow (insert). |
Morphology and Particles Size of WCS-GA-DBBN
The morphological information of WCS-GA-DBBN was elucidated through TEM. Both WCS-GA (Figure 2B(a)) and WCS-GA-DBBN (Figure 2B(b)) revealed spherical shapes and uniform size distribution (Figure 2B(b)), with average sizes of 113±3 and 154±5 nm, respectively. Conjugations with DBBN peptide resulted in increases in sizes, as seen in WCS-DBBN. The weight and number average molecular weights of WCS were determined by GPC. The Mw and Mn of WCS were 4635 and 1844 Da, respectively (PDI = 2.52). The hydrodynamic diameter (DH) of WCS colloids was 176 nm (PDI = 5.29) and zeta potential was observed to be +1.21 mV. Upon functionalization of GA on WCS, we observed an increase in size for the DH of WCS-GA to 193 nm (PDI = 5.84) while the zeta potential changed to negative charge of -2.11 mV. These changes in the DH and surface charges of WCS-GA implied the incorporation of GA moieties on WCS. With biomolecule conjugation, the WCS-GA-DBBN also became larger, with the DH of 202 nm (PDI = 5.19) and the zeta-potential shifting to more negative (-5.21 mV) values. Considering the swelling in solution, the particle sizes measured by DLS were usually larger than those observed from the TEM images. Peptide conjugation brought about more negatively charged surfaces because of the DBBN moiety containing several nitrogen and oxygen atoms from the amide bonds of the BBN peptide. Increases in the DH and decreases of zeta potentials, as observed, confirmed that the DBBN was conjugated on the WCS-GA structure.
Antioxidant Activity of WCS-GA-DBBN
Based on our previous work, it has been found that functionalization of a radical scavenging group (ie, GA) onto the high molecular weight CS is beneficial to enhance not only solubility but also antioxidant efficiency.67,69 Even with 15% of GA moiety on CS, the galloyl group provided effective transferring of H-atom and subsequently the forming of stable semiquinone radicals. It is important to note that an equivalent GA moiety of CS–GA could quench hydroxyl radical and exhibited synergistic function of CS with GA to retard the pro-oxidation of free molecular GA in the Fenton reaction. Besides CS gallate derivative, short chain WCS nanocolloids also showed superior antioxidant activity than the conventional CS in aqueous acetic acid solution.17 WCS also has powerful reducing capacity to assist the template synthesis of AuNPs.8 The antioxidant activities of WCS-GA and WCS-GA-BBN were determined in order to compare them with free WCS and GA molecules. Using DPPH standard free radical assay, it is possible to measure their antioxidant activities due to both hydrogen and electron transfer mechanisms.86,87 The interaction of samples with DPPH radicals resulted in fast fading of the DPPH solution color from purple, corresponding to the UV−vis spectrum at 515 nm, to yellow (Figure 2C in set).88 Figure 2C shows a plot of the DPPH free radical scavenging activities of WCS, WCS-GA, WCS-GA-DBBN and GA against a wide range of concentrations. The WCS reached 100% radical scavenging activity at its concentration of 0.8 mg/mL. The EC50 values are expressed as an effective antioxidant concentration to reduce the radicals by 50%. The EC50 of WCS was ~0.025 mg/mL. The EC50 of WCS agrees with our previous report17 and this also supports the high reproducibility of radiation-induced depolymerization of CS to create water-soluble nanocolloids of WCS. Compared to the WCS, the WCS-GA showed a significant enhancement in antioxidant activity reaching a maximum at a concentration of 0.02 mg/mL. The EC50 of GA, WCS-GA, WCS-GA-DBBN and WCS were nearly identical and they were around 0.010, 0.012, 0.017 and 0.025 mg/mL, respectively. The antioxidant activity of WCS-GA exhibited 5 times higher than the non-functionalized WCS. It is important to note that the non-functionalized WCS also exhibited ~5 times more powerful antioxidant and reducing activities over the conventional CS in aqueous acetic acid.17 These results corroborate well with our previous work when GA was functionalized on insoluble CS under dissolution in an acetic acid condition.17 It has also been reported that the DPPH-scavenging activities were positively correlated with polyphenol contents in the chitosan-based conjugates. In addition, formation of ester or amide bonds of the synthetic GA derivatives could adapt to increase the antioxidant efficiency of the galloyl group based on a push–pull effect.89 Our present work proves that GA moieties potentiate antioxidant activity of WCS. The EC50 of CS-GA in our previous work was 0.14 mg/mL whereas the EC50 of WCS-GA in the present work is 0.012 mg/mL. The result suggests an enhancement in the antioxidant potential of WCS to almost 12 times greater antioxidant capacity than our original CS-GA reported in 2008 and 2010.67,69
We then focused on the effects of BBN conjugation on antioxidant capacity. We found that DBBN peptide conjugation onto WCS slightly reduced the antioxidant capacity of WCS-GA. This may be attributed to the presence of polyphenol groups in GA, and the amino acid groups in the DBBN peptide. Our findings of antioxidant capacities, as outlined above, are rationalized in terms of the antioxidant capacities of polyphenol-protein complexes, which are always lower than the sum of the antioxidant capacities of the individual components.90
One-Pot Synthesis of AuNPs
Green and one-pot synthesis of AuNPs were carried out using the WCS-GA and WCS-GA-DBBN as a template for reducing and stabilizing nanoparticles. The WCS plays dual roles of reduction and transformation of Au(III) to the corresponding Au nanoparticles, while at the same time the corona of the chitosan polymer stabilizes the surface atoms of gold nanoparticles from a plethora of its -NH2 and -OH groups through electron transfer mechanism. Besides the main functional groups of CS, the WCS prepared from radiation-induced chain scission process also contains C=O ended groups. Considering the GA function on WCS (see in Scheme 2A) on the overall reduction of Au(III) to the corresponding nanoparticles, three main mechanisms of the polyphenolic antioxidant processes are important: the first one involves H atom transfer (route b1); the second one involves single electron transfer (route b2), and the third one involves metals chelation (route b3). We infer that all three mechanisms act in synergy in the overall reduction of Au(III) to the WCS stabilized gold nanoparticles.91,92 Various functional groups of the WCS structure, ie, -NH2 (a1), -OH (a2), -C-O-C- (a3) and -C=O (a4) (see Scheme 2A) support the stabilization of surface atoms on AuNPs through strong electrostatic (see in Scheme 2A(a)) and steric mechanisms.28 We also suggest that WCS chains possibly form complexation with Au(III) ions thus controlling the particle size of AuNPs in the overall polymer template effect.8,93
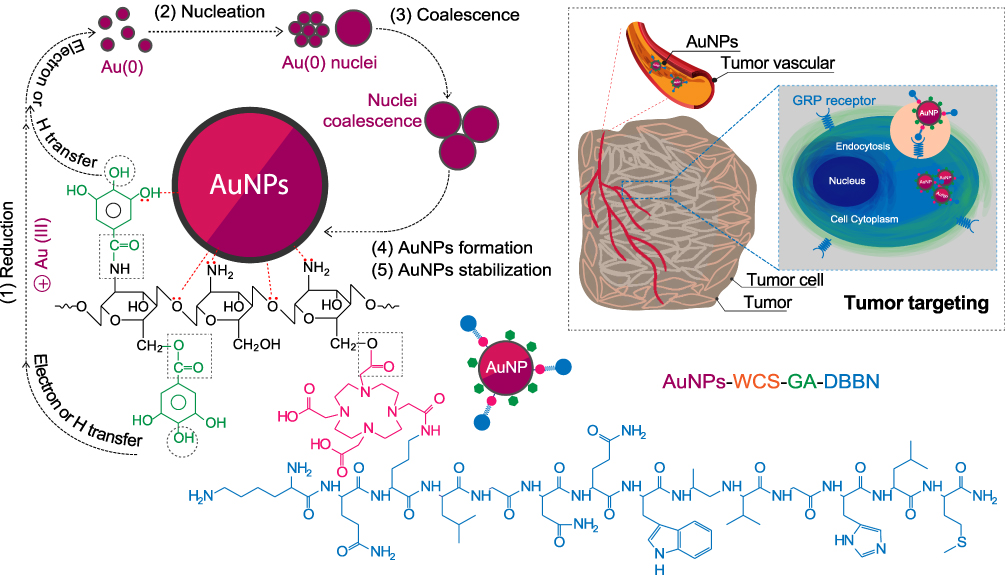 |
Scheme 2 Route of AuNPs formation and stabilization by WCS-GA-DBBN including tumor targeting mechanism. |
To demonstrate the reduction capacity of WCS-GA due to GA moiety, AuNPs were synthesized in WCS-GA and the results compared with WCS as a synthetic template (Figure 3A). The representative UV-vis spectra of AuNPs prepared in WCS-GA and WCS at 6 h are shown in Figure 3A. At 6 h, the AuNPs prepared in WCS-GA exhibited the characteristic surface plasmon resonance (SPR) at 530 nm, indicating successful formation of AuNPs. It is clear that WCS-GA exhibited superior reducing potential over WCS. In order to probe whether the WCS-GA could create AuNPs faster than non-functionalized WCS, the kinetic plot between the absorbance at maximum wavelength (λmax) and reaction times was plotted (Figure 3B). We observed that AuNPs were rapidly formed and almost reached the steady state at a short reaction time of 15 min. The AuNPs formation through the WCS-GA solution was evident within 15 min and reached a steady state at 1 h. The AuNPs formation also reached a steady state when they were prepared in non-functionalized WCS aqueous solution; however, it took a longer duration, of 24 h.94 Considering the first steady state point, the ability of WCS-GA to promote rapid creation of AuNPs was 24-fold faster than WCS. As expected, the result confirms the significant ability of GA moiety on WCS to assist rapid reduction of Au(III) to AuNPs in this green one-pot nanotechnology synthesis route.
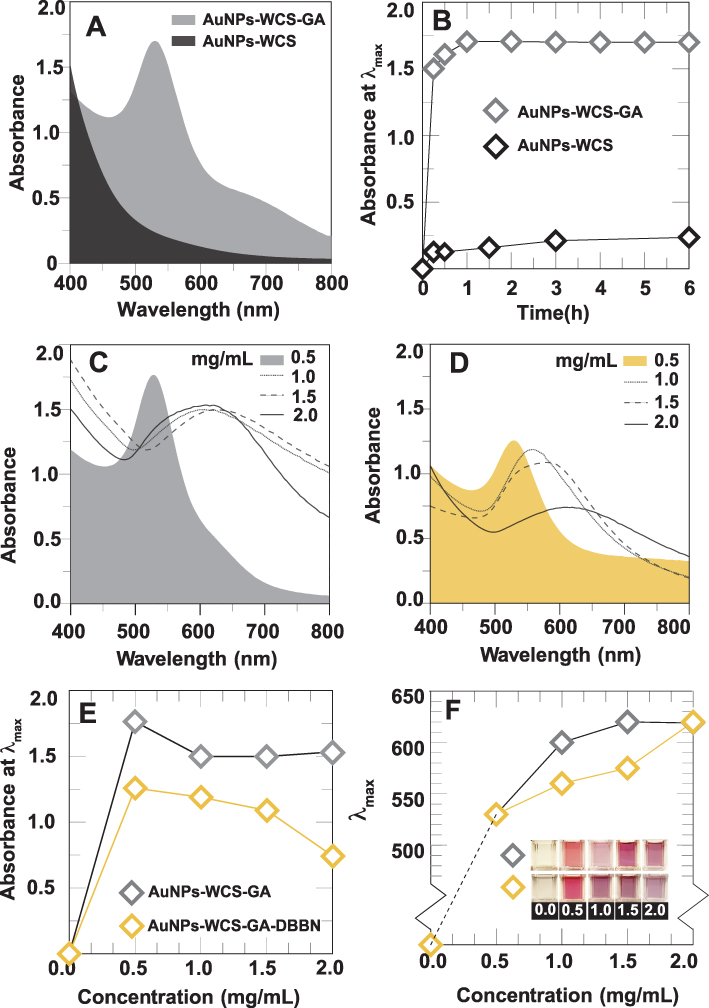 |
Figure 3 (A) Representative UV-vis spectra of AuNPs-WCS and AuNPs-WCS-GA at 6 h. (B) Absorbances at λ max of AuNPs-WCS and AuNPs-WCS-GA formation at different reaction times. UV-vis spectra representing AuNPs formation at 1 h in different concentrations of (C) WCS-GA and (D) WCS-GA-DBBN. (E) Plots of absorbances at λmax against concentrations of samples as in (B). (F) Plot of λmax against concentrations of WCS-GA (◊) and WCS-GA-DBBN (◊) as in (E) and their corresponding physical appearances of the sample solution (in set). |
Formation of AuNPs using WCS-GA and WCS-GA-DBBN as templates was also assessed using UV-vis absorption (Figure 3C and D). At a concentration of 0.5 mg/mL (filled color) of WCS (in gray) and WCS-GA-DBBN (in yellow), the narrow UV-vis peaks corresponding to the surface plasmon resonance (SPR) were found at 530 nm. An SPR absorption band at ~530 nm implied spherical particles of AuNPs.95 When the concentrations increased to 1.0–2.0 mg/mL, the UV signal intensity reduced and the UV-vis spectra of both WCS-GA (Figure 3C) and WCS-GA-DBBN (Figure 3D) became broader and red shifted to a longer wavelength of ~560-620 nm. Figure 3E reveals the plot of the absorbance at λmax against the concentrations of WCS-GA and WCS-GA-DBBN. These observations suggest that higher concentrations of WCS-GA and WCS-GA-DBBN (over 0.5 mg/mL) brought about a decrease in absorption intensities as a result of AuNPs agglomeration. Compared with the absorption intensity of AuNPs-WCS-GA, the absorption intensity of AuNPs-WCS-GA-DBBN was lower. Since the antioxidant activity of WCS-GA-DBBN was a little bit lower than WCS-GA (Figure 2C), the reduction capacity of WCS-GA-DBBN to create AuNPs was also lower than WCS-GA. The lower kinetics of AuNPs production, in WCS-GA-DBBN solution, is consistent with the lower antioxidant reduction capacity of WCS-GA-DBBN as determined by DPPH assay. Effects of changes in the λmax at various concentrations of WCS-GA and WCS-GA-DBBN are shown in Figure 3F. The λmax increased from 530 nm to longer wavelengths of 600–620 nm for AuNPs prepared in WCS-GA at 1.0 and 1.5–2.0 mg/mL, respectively. Similarly, the λmax of AuNPs prepared in WCS-GA-DBBN also gradually increased from 530 nm to longer wavelengths of 560–620 nm when the concentrations increased from 0.5 to 1.0–2.0 mg/mL. The physical appearance of AuNPs solutions are depicted in Figure 3D (in set). The solution color changed from red wine appearance to almost purple, indicating the larger particle size of AuNPs.17 The red color of gold colloid is attributed to SPR arising from the collective oscillation of free conduction electrons induced by an interacting electromagnetic field.96 The UV absorption characteristic of metallic nanoparticles provides accurate insights on changes in particle sizes of AuNPs.97 It is known that the position and intensity of the band depends on the size, surface morphology and aspect ratio of nanostructures.98 The particle size is large due to agglomeration of AuNPs when prepared using high concentrations of reducing and stabilizing functionalized and conjugated polymers. It is well known that certain biomolecules not only act as reducing agents, but also provide capping corona for the stabilization of AuNP surface atoms. When the concentrations of Au(III) ions are high, the strong reducing ability of WCS-GA and WCS-GA-DBBN results in a rapid formation of numerous small metallic particles. The lack of polymeric biomolecules, needed for capping surface atoms, results in a continuous assembly of small nanoparticulates causing their aggregation.99
Particle Morphology and Sizes of AuNPs
Morphologies and particle sizes of AuNPs were visualized using TEM, HR-TEM and SEM-EDX. The TEM images and histograms of AuNPs size distribution are illustrated in Figures 4A and B. Most of the AuNPs prepared in WCS-GA (Figure 4A(a)) and AuNPs-WCS-GA-DBBN (Figure 4A(b)) exhibited spherical and uniform shapes with the equivalent average sizes of 21±5 and 19±4 nm, respectively. AuNPs with the other shapes, ie, round, triangle, pentagon, hexagonal, diamond and rod, were also observed in minor quantities, especially with WCS-GA-DBBN peptide conjugate template. The different particle shape of AuNPs is due to molecular template effects presented by the WCS-GA-DBBN structures. The template-based rapid synthesis, as described above, has produced well-defined spherical-shaped AuNPs. Spherical-shaped AuNPs are taken up by cells more efficiently than other-shaped nanostructures.100
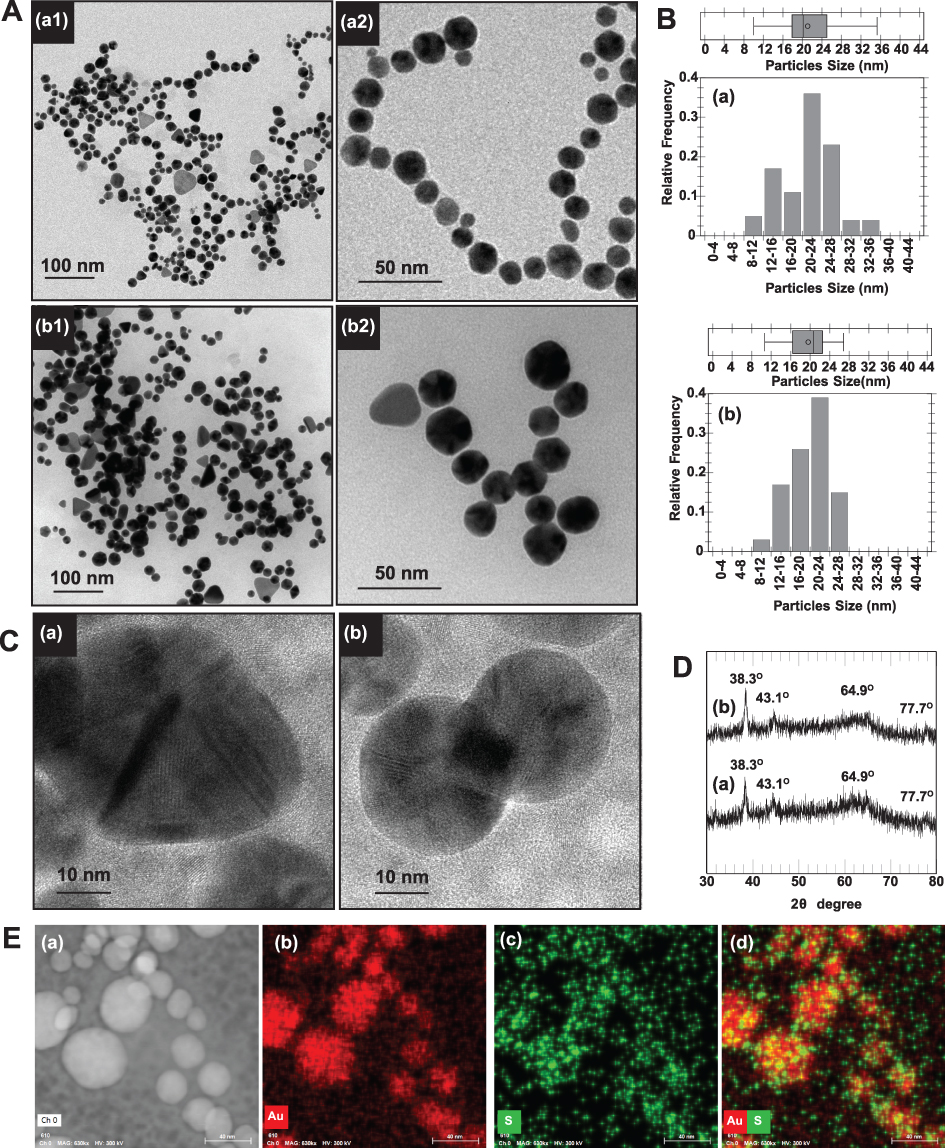 |
Figure 4 (A) TEM images and their close-up images (a2 and b2) and (B) particle size distribution calculated from TEM images (n = 200) of (a) AuNPs-WCS-GA and (b) AuNPs-WCS-GA-DBBN. (C) Representative HR-TEM images of Au nanocrystal of AuNPs-WCS-GA-DBBN as in (b). (D) XRD patterns of (a) AuNPs-WCS-GA and (b) AuNPs-WCS-GA-DBBN. (E) SEM-EDX analysis presenting SEM image of (a) AuNPs-WCS-GA-DBBN and SEM-EDX mapping micrograph of (b) Au, (c) S and (d) mixed Au and S mappings of the AuNPs-WCS-GA-DBBN. |
AuNPs were also analyzed using HRTEM coupled with XRD to confirm Au nanostructures constructed within the WCS-GA and WCS-GA-DBBN templates. The representative HRTEM images clearly showed the Au nanostructure (Figure 4C). The XRD patterns of both AuNPs, prepared through WCS-GA and WCS-GA-DBBN, exhibited four distinct diffraction peaks at scattering angles (°2θ) of 38.3, 43.1, 64.9 and 77.7 (Figure 4D). These are attributed to the planes (111), (200), (220) and (311) of the face-centered cubic Au, respectively, in agreement with previously reported data.24 The high intensity diffraction peak at 38.3° is attributed to the Au crystal lattice plane (111) with an inter-planar distance of ~2.3 °A.
In order to establish the conjugation of sulfur from the BBN peptide on the surface of AuNPs through Au-S bonding, we used the SEM-EDX technique. Figure 4E(a) provides the overall morphology of AuNPs-WCS-GA-DBBN. The mapping profile of the Au and S elements including their overlay are presented in Figure 4E(b-d). A characteristic spectrum of Au Lα X-rays at 9.711 keV and S Kα X-rays at 2.309 keV were observed and used to formulate their mapping profiles. The red color profile reveals the Au element of the AuNPs, whereas the green profile indicates the S element of the BBN structure. As shown in the overlay elemental mapping (Figure 4E(d)), the AuNPs containing the S element on the surface implies that the BBN targeting peptide was anchored with the AuNPs through the WCS-DOTA-DBBN conjugate polymer template. The overall SEM-EDX results demonstrated successful formation of AuNPs conjugated to the tumor-specific BBN peptide for applications as targeted diagnostic, therapeutic or theranostic agents in treating tumors.
Stability of AuNPs
In order to explore further utility of BBN bound AuNPs in imaging and therapy, we studied the kinetic behavior of AuNPs conjugated to WCS-GA and WCS-GA-DBBN. The stability was measured as a function of surface plasmon resonance by monitoring λmax, DH as well as zeta potential in various biologically relevant media over a 0–96 h time period (Figure 5A and B). The AuNPs prepared using 0.5 mM of HAuCl4 and 0.5 mg/mL of WCS-GA (or WCS-GA-DBBN) and the reactions conducted for various time periods and several batches of AuNPs were subjected to stability measurements. Figure 5A shows that the SPR absorbance λmax dramatically increased when the duration of reactions increased from an initial time interval of 15 min to 1 h. At 15 min, the formation of AuNPs using the WCS-GA template was ~3-fold faster than the WCS-GA-DBBN template. At the steady state, the amount of AuNPs-WCS-GA is ~1.5-fold higher than that of AuNPs-WCS-GA-DBBN. Although the amount of AuNPs when using the WCS-GA-DBBN was lower than that produced through the WCS-GA template, the rate of formation of AuNPs through the WCS-GA-DBBN template was significantly higher than that of the WCS-GA template. The results suggested that the lower amount of AuNPs in WCS-GA-DBBN was due to the lower reducing power of WCS-GA-DBBN than the WCS-GA template, as described above.
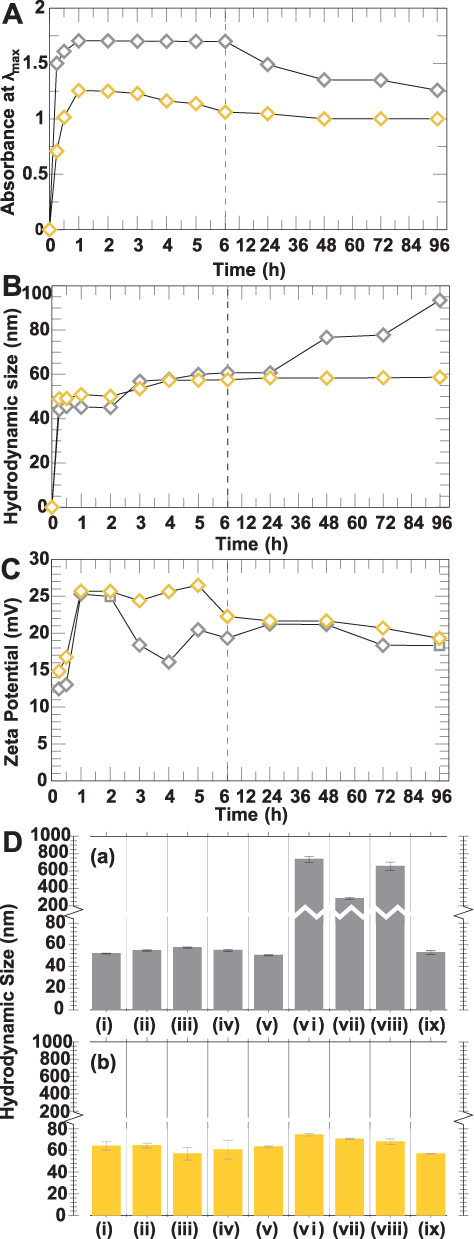 |
Figure 5 (A) UV-vis absorbance at λmax, (B) hydrodynamic size and (C) zeta potential of AuNPs synthesized in WCS-GA (gray mark, ◊) and WCS-GA-DBBN (yellow mark, ◊) at different times. (D) In vitro stability observed via change of the hydrodynamic sizes of (a) AuNPs-WCS-GA and (b) AuNPs-WCS-GA-DBBN in different aqueous solutions of (i) DI, (ii) 0.5% Cys, (iii) 0.2 M His, (iv) 0.5% BSA, (v) 0.5% HSA, (vi) 1% NaCl, (vii) PBS pH 5, (viii) PBS pH 7, and (ix) PBS pH 12 after incubation for 72 h. |
The yields of AuNPs from both WCS-GA and WCS-GA-DBBN templates reached a maximum amount at around 1 h. After 1 h, the intensities of the SPR bands of AuNPs-WCS-GA remained unchanged when aliquots of reaction mixtures at 2, 4 and 24 h were monitored, thus suggesting complete transformation of Au (III) ions in to the corresponding AuNPs within 1 h. Our results corroborate with previous investigations where it has been reported that the SPR wavelength and maximum absorbance of AuNPs synthesized in aqueous extract oils from palm leaves reached a plateau after 1 h, indicating complete reduction of Au(III) precursor to AuNPs.94 We observed that the AuNPs prepared using the WCS-GA template were stable within the time intervals of 1–6 h and the stability gradually reduced by ~30% at 96 h. In the case of AuNPs prepared using the WCS-GA-DBBN template, although the absorbances at λmax gradually declined after 1 h, their stability reduced by only ~8% at 96 h. Overall, our extensive investigations of stability of nanoparticles revealed that the AuNPs-WCS-GA-DBBN nanoparticles exhibited prolonged stability for over 3 days. The results suggested that WCS-GA-DBBN corona on AuNPs exert significantly higher stability than that of the WCS-GA corona.
Figure 5B shows the DH results of AuNPs-WCS-GA and AuNPs-WCS-GA-DBBN within the same time intervals of 15 min to 96 h. The increase in DH (Figure 5B) corresponded with the maximum absorbance λmax data (Figure 5A). Generally, the DH increased because of the particle agglomeration leading to a decrease in the particulate amounts which was reflected in the reduction of absorbance. The DH analyses over a period of time indicated minimal changes in the average DH of AuNPs at various time points. At the reaction time of 15 min, the DH of AuNP-WCS-GA and AuNPs-WCS-GA-DBBN were around 44±1 and 49±1 nm, respectively. At the steady state of the completed AuNP formation (1 h), the DH of AuNP-WCS-GA and AuNPs-WCS-GA-DBBN were around 45±1 and, 51±1 nm, respectively. The AuNPs with the WCS-GA-DBBN corona showed excellent stability, with an average DH ranging from 48 to 60 nm. For AuNPs with the WCS-GA corona, the DH were in the range of 40–60 nm within the time intervals of 15 min to 6 h. Beyond 24 h, the DH of AuNPs-WCS-GA increased up to 80–100 nm. However, the DH of both AuNPs-WCS-GA-DBBN and AuNPs-WCS-GA exhibited sizes in the range of 40–100 nm within the time interval of 3 days. The size of the NPs is of pivotal significance as they dictate blood circulation time and tumor accumulation. The NPs smaller than 20 nm are better candidates for deep tumor penetration, but they are easily removed from blood circulation.101,102 The size of NPs used in any drug delivery system should be small enough to escape capture by fixed macrophages that are lodged in the reticuloendothelial system, such as the liver and spleen, but should be large enough to prevent their rapid leakage into blood capillaries. The size of the sinusoid in the spleen and fenestra of the Kupffer cells in the liver varies from 150 to 200 nm.23 Thus, the size of AuNPs should be appropriate not exceeding 150 nm. In addition, NPs of about 100 nm have longer blood circulation and better tumor accumulation due to the enhanced permeability and retention effects. These data, taken together, suggest that the AuNPs-WCS-GA-DBBN exhibiting DH in the range of 40–60 nm are highly suited to a myriad of applications as diagnostic and therapeutic nanomedicine agents.
Furthermore, zeta potential is an important indicator for assessing the stability of nanoparticles.103 Therefore, zeta potentials of AuNPs were measured to investigate their surface charges and stabilities (Figure 5C). At 15–30 min, the zeta potential of AuNPs-WCS-GA was +12-13 mV while for the AuNPs-WCS-GA-DBBN it was +15-17 mV. For AuNPs-WCS-GA-DBBN, the zeta potential was +25 mV within the time intervals of 1–5 h and approximately +20 mV within the time intervals of 6–96 h. The results suggest that AuNPs which were developed rapidly at 15 min exhibited lower stability than those formed at 1 h. It is therefore important to recognize that the AuNPs-WCS-GA-DBBN exhibited optimum in vitro stability lasting up to 3 days. Generally, those AuNPs that exhibit zeta potentials greater than +20 mV or less than -20 mV possess sufficient electrostatic repulsions to remain stable in solutions for extended periods.104 The positive zeta potential values from the highly charged biomolecular corona on the surface of AuNPs enhance electrostatic repulsive forces between AuNPs and also exert steric effects, thus causing an overall increase in the in vitro stability of AuNPs.105 This is clearly reflected for AuNPs-WCS-GA-DBBN which exhibit enhanced in vitro stability attributable to the WCS-GA-DBBN biomolecular corona that avoids nanoparticles from aggregation.
In vitro Stability of AuNPs
In the present study, we are interested in the development of highly stable AuNPs for use in biomedical applications. Therefore, we have examined the in vitro stability of AuNPs-WCS-GA-DBBN in distilled water in various biologically relevant media, including NaCl, Cysteine (Cys), Histidine (His), BSA, HSA and phosphate buffer at pH 5, 7 and 12. The in vitro stability of AuNPs in these biological fluids was monitored through changes in DH (Figure 5D). The AuNPs solution samples were incubated in different biological media for 72 h before measurement. The DH of AuNPs-WCS-GA was 50 nm in DI water, Cys, His, BSA, HSA and PBS (pH 12) (Figure 5D(a)), whereas the DH of AuNPs-WCS-GA dramatically increased in NaCl, PBS pH 5 and pH 7 solutions. The AuNPs-WCS-GA underwent agglomeration as reflected in their enhanced particle sizes of 735, 284 and 655 nm when incubated in NaCl (Figure 5D(vi)), PBS pH 5 (Figure 5D(vii)), and pH 7 (Figure 5D(viii)), respectively. In contrast, the DH of AuNPs-WCS-GA-DBBN (Figure 5D(b)) was 55–75 nm in all biological media. These results unequivocally suggest that AuNPs-WCS-GA-DBBN are stable in all biological media for 72 h with no observable changes in DH. It is thus conceivable that the WCS-GA-DBBN is not only creating AuNPs efficiently but also exerts stabilization architecture around the AuNPs in various biological fluids. Nanoparticles (NPs) of metal readily aggregate because of their relatively high surface energy and low atomic coordination number. NPs aggregation strongly depends on various parameters, including chemical composition, size, shape, surface coating, and physicochemical characteristics.106 As shown in our investigations, AuNPs-WCS-GA-DBBN exhibited optimum stabilities in all biologically relevant media, whereas the AuNPs-WCS-GA showed instability in NaCl, PBS pH 5 and pH 7. It may be noted that functionalized gold nanoparticles in PBS buffer, with relatively high ionic strength, exhibit particle aggregation causing dispersion behavior of NPs and thus leading to destabilization and adhesion to surrounding substrates.107 The solution pH also strongly affects the surface charge of AuNPs, which can alter the inter-relationship between the van der Waals attraction forces and electrostatic repulsive forces.106 Therefore, nanoparticles solution pH is an important consideration for various pH-responsive cationic nanoparticles.108 In this context, it is well known that NaCl plays an important role in increasing the particle size of CS-based NPs.109 Additionally, CS can be deprotonated at pH 12, thus rendering CS-GA strongly capped and thus stabilizing the corresponding AuNPs. In the presence of DBBN peptide, the protonation of CS is hindered, and the steric effects of AuNPs-WCS-CS-GA appear to be significant, resulting in greater stability for these NPs. The outstanding stability of AuNPs-WCS-GA-DBBN is rationalized in terms of synergistic effects of GA and DBBN functions. The pKa of phenolic hydrogens of GA has been reported to be ~ 5,110,111 thus the phenolic -OH of semiquinone is ionized at pH >5.111 The phenolic -OH and their ionized forms would help in stabilizing additional H+ at pH 5, thus precluding H+ from protonation of CS. At higher pH of 7 and 12, the ionized phenolic -OH groups would cause electrostatic repulsions between individual NPs—thus aiding excellent stability in vitro and in vivo. Overall, the optimum nanoparticulate size and outstanding in vitro stability, as exhibited by the AuNPs-WCS-GA-DBBN, makes them ideal nanoplatforms for use in molecular imaging and therapy.
Cellular Uptake of AuNPs
A major advantage of gold nanoparticles is that they can be engineered to sizes to facilitate tumor cell-specific selectivity and thereby achieve tumor targeting. Toward this objective, we used bombesin peptide (BBN), which has selective and high affinity toward gastrin-releasing peptide (GRP) receptors that are overexpressed in a host of tumors, including breast, small cell lung, prostate and pancreatic cancers. The BBN conjugated AuNP-WCS-GA-DBBN nanoconjugate has been tested for its efficacy in targeting two different prostate cancer cells, the PC-3 and LNCaP. We treated these cancer cells with AuNPs (50 μg/mL) for 6 h and the cellular uptake and localization of AuNPs were visualized using confocal microscopic images and TEM images. The uptake of AuNPs-WCS-GA-DBBN in PC-3 and LNCaP cell lines is illustrated in Figure 6. Figure 6A shows the images of PC-3 cells before and after exposing them to AuNPs-WCS-GA (Figure 6A(b) and AuNPs-WCS-GA-DBBN (Figure 6A(c). The fluorescence micrograph, depicting the cell cytoplasm and nuclei, are indicated in green (515 nm), blue (450 nm) and yellow (561 nm) channels, respectively. At 6 h, a limited number of AuNPs-WCS-GA are internalized into PC-3 cells. In comparison, we observed highly efficient internalization of AuNPs-WCS-GA-DBBN at 6 h, as shown in Figure 6A(c). This image shows that AuNPs have surrounded the nucleus and are bound by the plasma membrane. High magnification of cellular internalization, as shown in Figure 6A(c), indicated the presence of a significant amount of AuNPs in the PC-3 cells, as seen in position numbers 1, 2, 3 and 4 and presented in yellow spots. It is conceivable that AuNPs-WCS-DBBN internalization in the prostate cancer cells is occurring through GRP receptor mediated endocytosis (Scheme 2B) because prostate tumor cells have an abundance of GRP surface receptors, and BBN targets such receptors. It is interesting to note that AuNPs-WCS-GA-DBBN showed a lower propensity to internalize the LNCaP prostate tumor cells as compared to the PC3 cells. It is well known that AuNPs in sizes less than 200 nm are internalized into the cells through micropinocytosis (clathrin-mediated, caveolae/lipid raft-mediated, and clathrin/caveolae-independent).112 In general, AuNPs are internalized from the cell surface through endocytosis and confined to the endosome (Scheme 2). The uptake of AuNPs in PC-3 cells is predominantly due to the surface chemistry of the targeted AuNPs correlating to the number of GRP receptors on the cells.113 This can occur through either receptor-mediated endocytosis, which was expected for the targeted AuNPs, or through pinocytosis, which is known for the nonfunctionalized AuNPs. On the other hand, the particles can avoid the endosomal pathway by being taken up directly into the cytosol using cell-penetrating peptides. However, the BBN peptide used is not a cell-penetrating peptide. Therefore, we infer the mechanism of cell internalization to be receptor-mediated endocytosis for our AuNPs. The highly efficient cellular uptake of AuNPs demonstrates GRP receptor affinity and validates our hypothesis on the creation of prostate tumor-specific AuNPs via a BBN peptide conjugated WCS biopolymer nanocolloid template.
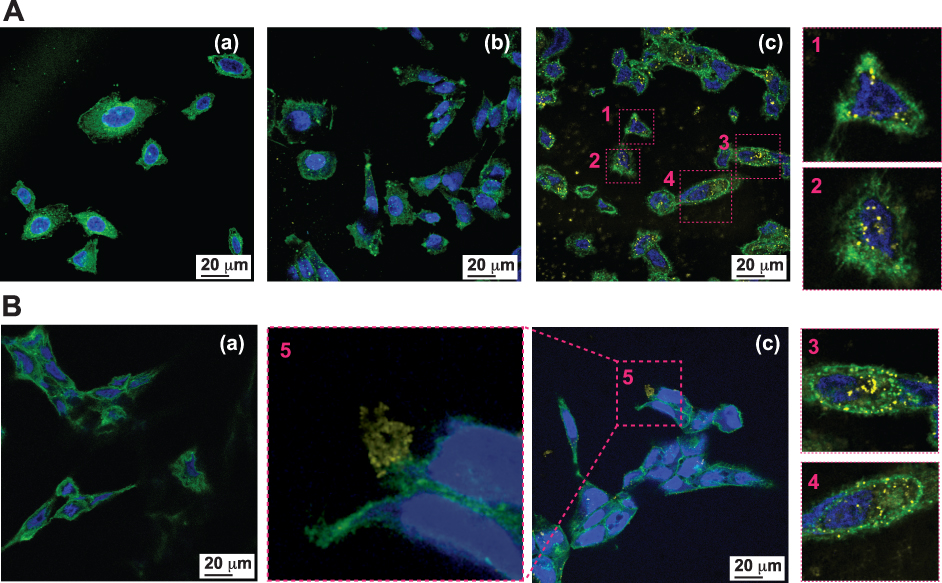 |
Figure 6 Cellular internalization of AuNPs into representative prostate cancer cell lines. Confocal microscopic images of (A) PC-3, (B) LNCaP cells (a) without treatment, (b) treated with AuNPs-WCS-GA (50 μg/mL) for 6 h, and (c) treated with AuNPs-WCS-GA-DBBN (50 μg/mL) for 6 h and the close-up images as in (c) at (1), (2), (3), (4) and (5) positions. |
Detailed TEM images have been analyzed in order to elucidate the uptake of AuNPs into the cells, and gain insights into the mechanism of cell internalization (Figure 7). We observed dark spots corresponding to AuNPs (eg, areas (a) and (b)) in the cytoplasm of both PC-3 (Figure 7A) and LNCaP (Figure 7B) cells. AuNPs were found surrounding the nucleus and bound by the plasma membrane. With a high magnification of a 100 nm scale, we observed a significant amount of AuNPs as dark spots in the areas (a) and (b) in Figure 7 in both PC-3 and LNCaP cells. The particle shapes and sizes of AuNPs corresponded to those observed by TEM, as described in Figure 4, suggesting that the particles retained their size (~20 nm) and shape after internalization in tumor cells.
 |
Figure 7 Cellular internalization of AuNPs into representative prostate cancer cell lines. TEM images of (A) PC-3 cell and (B) LNCaP cell treated with AuNPs-WCS-GA-DBBN (50 μg/mL) at 6 h and close-up morphologies of AuNPs in cell at (a) and (b) positions. |
In vitro Cytotoxicity Evaluation of AuNPs on Prostate Cancer Cells
In our efforts to understand the effects of composition of various AuNPs on tumor cells, we investigated dose-dependent cytotoxicity of various AuNPs on prostate cancer cell viability. Our objective was to evaluate cell viability of AuNPs conjugated WCS with and without GA functions. We performed MTT assays on PC-3 (Figure 8A) and LNCaP (Figure 8B) cells with both AuNPs-WCS and WCS-GA solutions at various concentrations (6.25, 12.5, 25, 50 and 100 µg/mL). The results indicated that AuNPs-WCS-GA exhibited significant reduction of cell viability as compared to AuNPs-WCS, suggesting that the GA functional group exerts significant effects on damaging of PC-3 prostate cancer cells. The inhibition concentration to reduce cell viability by 50% (IC50) of AuNPs-WCS-GA was found to be 6.13 µg/mL. For AuNPs-WCS, no IC50 value and no significant reduction of PC-3 cells viability was observed within the concentration range of 6.25–100 µg/mL.
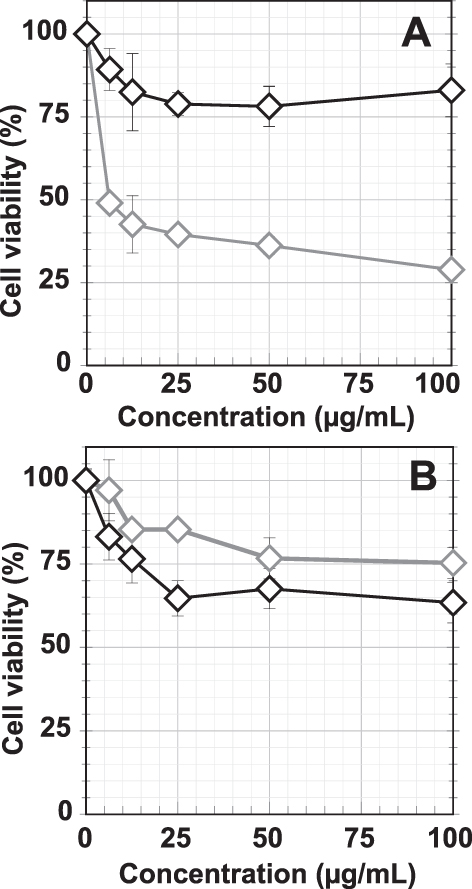 |
Figure 8 Cellular viabilities of (A) PC-3 and (B) LNCaP cells after treating with AuNPs-WCS (◊, gray symbol) and AuNPs-WCS-GA (◊, black symbol) (50 μg/mL) for 24 h. |
The IC50 of GA is approximately 150 μM (10.45 µg/mL) and 50 μM (3.48 µg/mL) for human breast cancer cells for 24 and 48 h.114 In a similar study using the PC-3 cells, it was found that the IC50 calculated for GA was 50 μM (3.48 µg/mL) at 48 h. These results suggest that GA is a potent anticancer agent in the treatment of prostate cancer.115 Although pure GA exhibited a 1.70-fold higher efficiency in damaging PC-3 cells than the AuNPs-WCS-GA-DBBN (IC50 = 5.94 µg/mL) nanoconjugate, it is important to note that WCS-GA contained only ~15% GA conjugated on the WCS.115 In addition, it has been reported that free GA may also promote prooxidation and that the CS gallate derivative provides synergistic effects for antioxidation which may result in reduction of toxicity or side effects in the healthy cells.67
In order to verify how the BBN targeting peptide might influence prostate cancer cell viability, we performed MTT assays using AuNPs with and without the BBN peptide (Figure 9). Various concentrations of AuNPs-WCS-GA and AuNPs-WCS-GA-DBBN were treated with both PC-3 and LNCaP cells, as shown in Figures 9A and B, respectively. For PC-3 cells, we observed that the IC50 values of AuNPs-WCS-GA-DBBN were 5.05, 5.94 and 3.65 µg/mL. These values were lower as compared to the AuNPs-WCS-GA, which showed IC50 values of 6.13, 12.05 and 8.75 µg/mL at 24, 48 and 72 h, respectively. At 24 h, both AuNPs-WCS-GA and AuNPs-WCS-GA-DBBN showed significant damage to both PC-3 and LNCaP cells. It was found that the PC-3 cell viability dramatically reduced even at a low concentration of 6.25 µg/mL, whereas the LNCaP cell viability showed no significant change even at concentrations higher than 6.25 µg/mL. The cell viability significantly reduced at 72 h for both types of AuNPs. Compared to AuNPs-WCS-GA (IC50: 8.74 µg/mL), AuNPs-WCS-GA-DBBN showed a 2.4-fold lower IC50 (3.65 µg/mL), thus supporting measurable effects of the targeted BBN peptide conjugate.
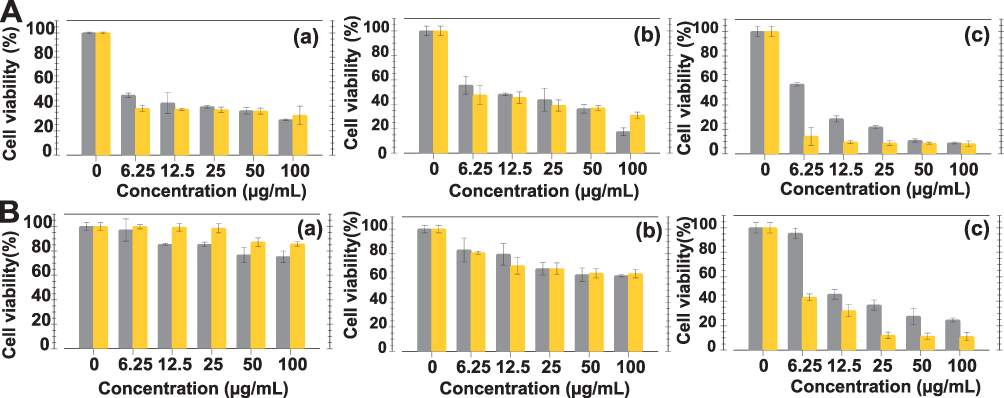 |
Figure 9 Cellular viabilities of (A) PC-3 and (B) LNCaP cells after treating with AuNPs-WCS-GA (gray histogram) and AuNPs-WCS-GA-DBBN (yellow histogram) (50 μg/mL) for (a) 24, (b) 48 and (c) 72 h. |
In our investigations with LNCaP cells (Figure 9B), we observed that the cell viability for AuNPs-WCS-GA was lower as compared to the AuNPs-WCS-GA-DBBN nanoconjugate at the initial time of 24 h and showed no significant difference in cell viability at 48 h. However, no IC50 was observed for AuNPs-WCS-GA at 24 and 48 h. Compared to AuNPs-WCS-GA, considering cell viability at 72 h (Figure 9B(c)), AuNPs-WCS-GA-DBBN showed more impact in damaging LNCaP cells with IC50 5.49 µg/mL. It is important to note that AuNPs-WCS-GA exhibited no IC50 at all incubation times studied.
Overall, our MTT assay results infer that BBN, in the AuNPs-WCS-GA-DBBN nanoconjugate, exerted significant damage to both the PC-3 and LNCaP prostate cancer cells as compared to the AuNPs-WCS-GA which has no BBN peptide. This enhanced tumor cell killing effect may be rationalized in terms of tumor cell receptor specificity of the BBN peptide functionalized AuNPs-WCS-GA-DBBN nanomedicine agent, which enhances internalization of AuNPs into tumor cells more than does the non-BBN conjugate AuNPs-WCS-GA. The potent anticancer activities, against the PC-3 and LNCaP cell lines, for AuNPs-WCS-GA-DBBN is presumably occurring via induction of apoptosis because the cationic NPs are able to bind the negatively charged DNA. Moreover, CS also exerts a strong electrostatic interaction with the negative charge of the tumor cells through the positive charge of amine groups of CS. When DNA binds to highly positive NPs, it wraps around the NPs and bends. This bending generally causes the DNA damage.
Conclusions
Our discovery, as outlined in this paper, encompasses the creation of a multifunctional WCS-GA-DBBN nanomedicine agent containing: (i) antioxidant/reducing functions due to WCS and polyphenolic GA moieties, (ii) gold nanoparticle stabilizable functions due to the steric hindrance of the cationic biopolymer of WCS, and (iii) tumor cell specific targeting function afforded by the BBN peptide vector in AuNPs-WCS-GA-DBBN. The WCS-GA and WCS-GA-DBBN templates exhibited a propensity for the rapid, efficient and reproducible synthesis of functionalized AuNPs within 15 min and achieved a steady state at 1 h. We discovered that incorporation of gallic acid (GA) functionality in WCS-GA and WCS-GA-DBBN enhanced the reduction power to transform Au(III) to the corresponding functionalized biocompatible gold nanoparticles.
Our investigations have unequivocally demonstrated that incorporation of tumor cell specific bombesin (BBN) peptide within the AuNPs-WCS-GA-DBBN nanomedicine agent enhanced intracellular uptake selectively with uniform distribution within the cytoplasm of prostate cancer cells (PC-3 and LNCaP). The higher density of GRP receptors in PC-3 cells resulted in an efficient endocytosis of AuNPs-WCS-GA-DBBN within tumor cells. Overall, our detailed investigations on the discovery of a new nanomedicine architecture consisting of a chitosan platform conjugated to both theranostic gold nanoparticles and a tumor-specific BBN peptide, AuNPs-WCS-GA-DBBN, will provide new opportunities for the selective and efficient delivery of antitumor nanopharmaceuticals for applications in oncology.
Acknowledgments
W.P. and K.V.K gratefully acknowledge the Coordinated Research Project (CRP, No. 18316), International Atomic Energy Agency (IAEA), United Nations, Vienna (Austria) and National Research Council of Thailand (NCRT) for DOTA-BBN peptide and research financial support. T.T. acknowledges the Ministry of Higher Education, Science, Research and Innovation for a graduate scholarship. We also appreciate the Center of Radiation Processing for Polymer Modification and Nanotechnology (CRPN) at Kasetsart University (Thailand) (under the THA1010, THA1011, THA 1014 IAEA Technical Cooperation Programme) for facilities support. K.V.K. thanks the University of Missouri and the Green Nanotechnology Institute within the Medical School for providing all the research logistics and facilities for undertaking joint collaborations with W.P. and T.T.
Disclosure
The authors report no conflicts of interest for this work.
References
1. Tangthong T, Piroonpan T, Thipe VC, et al. Water-soluble chitosan conjugated DOTA-Bombesin peptide capped gold nanoparticles as a targeted therapeutic agent for prostate cancer. Nanotechnol Sci Appl. 2021;14:69. doi:10.2147/NSA.S301942
2. Sibuyi NRS, Thipe VC, Panjtan-Amiri K, Meyer M, Katti KV. Green synthesis of gold nanoparticles using Acai berry and Elderberry extracts and investigation of their effect on prostate and pancreatic cancer cells. Nanobiomedicine. 2021;8:1849543521995310.
3. Seniwal B, Freitas LF, Mendes BM, Lugão AB, Katti KV, Fonseca TC. In silico dosimetry of low-dose rate brachytherapy using radioactive nanoparticles. Phys Med Biol. 2021;66(4):045016.
4. Khoobchandani M, Khan A, Katti K, et al. Green Nanotechnology of MGF-AuNPs for Immunomodulatory Intervention in Prostate Cancer Therapy; 2021. Sci Rep. 2021;11:16797.
5. Lunkov A, Shagdarova B, Konovalova M, et al. Synthesis of silver nanoparticles using gallic acid-conjugated chitosan derivatives. Carbohydr Polym. 2020;234:115916.
6. Khoobchandani M, Katti KK, Karikachery AR, et al. New approaches in breast cancer therapy through green nanotechnology and nano-ayurvedic medicine–pre-clinical and pilot human clinical investigations. Int J Nanomed. 2020;15:181.
7. Thipe VC, Bloebaum P, Khoobchandani M, Karikachery AR, Katti KK, Katti KV. Green nanotechnology: nanoformulations against toxigenic fungi to limit mycotoxin production. In: Rai M, Abd-Elsalam KA, editors. Nanomycotoxicology,Cambridge: Academic Press; 2020:155–188.
8. Piroonpan T, Katemake P, Pasanphan W. Comparative study of different chitosan solutions to assist the green synthesis of gold nanoparticles under irradiation. Radiat Phys Chem. 2020;169:108250.
9. Al-Yasiri AY, White NE, Katti KV, Loyalka SK. Estimation of tumor and local tissue dose in gold nanoparticles radiotherapy for prostate cancer. Rep Pract Oncol Radiother. 2019;24(3):288–293.
10. Thipe VC, Amiri KP, Bloebaum P, et al. Development of resveratrol-conjugated gold nanoparticles: interrelationship of increased resveratrol Corona on anti-tumor efficacy against breast, pancreatic and prostate cancers. Int J Nanomed. 2019;14:4413. doi:10.2147/IJN.S204443
11. Khoobchandani M, Katti KK, Karikachery AR, Thipe VC, Bloebaum PL, Katti KV. Targeted phytochemical-conjugated gold nanoparticles in cancer treatment. In: Khoobchandani M, Saxena A, editors. Biotechnology Products in Everyday Life. Basel: Springer, Cham; 2019:37–52.
12. Santos JJ, Leal J, Dias LA, et al. Bovine serum albumin conjugated gold-198 nanoparticles as model to evaluate damage caused by ionizing radiation to biomolecules. ACS Appl Nano Mat. 2018;1(9):5062–5070.
13. Katti K, Khoobchandani M, Thipe V, et al. Prostate tumor therapy advances in nuclear medicine: green nanotechnology toward the design of tumor specific radioactive gold nanoparticles. J Radioanal Nucl Chem. 2018;318(3):1737–1747.
14. Geraldes AN, da Silva AA, Leal J, et al. Green nanotechnology from plant extracts: synthesis and characterization of gold nanoparticles. ANP. 2016;5(03):176.
15. Gamal-Eldeen AM, Moustafa D, El-Daly SM, et al. Photothermal therapy mediated by gum Arabic-conjugated gold nanoparticles suppresses liver preneoplastic lesions in mice. J Photochem Photobiol B Biol. 2016;163:47–56.
16. Wongkrongsak S, Tangthong T, Pasanphan W. Electron beam induced water-soluble silk fibroin nanoparticles as a natural antioxidant and reducing agent for a green synthesis of gold nanocolloid. Radiat Phys Chem. 2016;118:27–34.
17. Pasanphan W, Rattanawongwiboon T, Choofong S, Güven O, Katti KK. Irradiated chitosan nanoparticle as a water-based antioxidant and reducing agent for a green synthesis of gold nanoplatforms. Radiat Phys Chem. 2015;106:360–370.
18. Hu B, Wang Y, Xie M, Hu G, Ma F, Zeng X. Polymer nanoparticles composed with gallic acid grafted chitosan and bioactive peptides combined antioxidant, anticancer activities and improved delivery property for labile polyphenols. J Funct Foods. 2015;15:593–603.
19. Su Y-L, Cheng S-H. Sensitive and selective determination of gallic acid in green tea samples based on an electrochemical platform of poly (melamine) film. Anal Chim Acta. 2015;901:41–50.
20. Shukla R, Chanda N, Zambre A, et al. Laminin receptor specific therapeutic gold nanoparticles (198AuNP-EGCg) show efficacy in treating prostate cancer. PNAS. 2012;109(31):12426–12431.
21. Khaledi H, Alhadi AA, Yehye WA, Ali HM, Abdulla MA, Hassandarvish P. Antioxidant, cytotoxic activities, and structure–activity relationship of gallic acid‐based indole derivatives. Arch Pharm. 2011;344(11):703–709.
22. Yu S-H, Mi F-L, Pang J-C, et al. Preparation and characterization of radical and pH-responsive chitosan–gallic acid conjugate drug carriers. Carbohydr Polym. 2011;84(2):794–802.
23. Chanda N, Kattumuri V, Shukla R, et al. Bombesin functionalized gold nanoparticles show in vitro and in vivo cancer receptor specificity. PNAS. 2010;107(19):8760–8765.
24. Chanda N, Shukla R, Katti KV, Kannan R. Gastrin releasing protein receptor specific gold nanorods: breast and prostate tumor avid nanovectors for molecular imaging. Nano Lett. 2009;9(5):1798–1805.
25. Okarvi SM, Al Jammaz I. Synthesis and evaluation of a technetium-99m labeled cytotoxic bombesin peptide conjugate for targeting bombesin receptor-expressing tumors. Nucl Med Biol. 2010;37(3):277–288.
26. Nune SK, Chanda N, Shukla R, et al. Green nanotechnology from tea: phytochemicals in tea as building blocks for production of biocompatible gold nanoparticles. J Mater Chem. 2009;19(19):2912–2920.
27. Faintuch BL, Teodoro R, Duatti A, Muramoto E, Faintuch S, Smith CJ. Radiolabeled bombesin analogs for prostate cancer diagnosis: preclinical studies. Nucl Med Biol. 2008;35(4):401–411.
28. Huang H, Yang X. Synthesis of chitosan-stabilized gold nanoparticles in the absence/presence of tripolyphosphate. Biomacromolecules. 2004;5(6):2340–2346.
29. Reubi JC. Peptide receptors as molecular targets for cancer diagnosis and therapy. Endocr Rev. 2003;24(4):389–427.
30. Karra SR, Schibli R, Gali H, et al. 99mTc-labeling and in vivo studies of a bombesin analogue with a novel water-soluble dithiadiphosphine-based bifunctional chelating agent. Bioconjugate Chem. 1999;10(2):254–260.
31. Shukla R, Nune SK, Chanda N, et al. Soybeans as a phytochemical reservoir for the production and stabilization of biocompatible gold nanoparticles. Small. 2008;4(9):1425–1436.
32. Kattumuri V, Katti K, Bhaskaran S, et al. Gum arabic as a phytochemical construct for the stabilization of gold nanoparticles: in vivo pharmacokinetics and X‐ray‐contrast‐imaging studies. Small. 2007;3(2):333–341.
33. Chanda N, Shukla R, Zambre A, et al. An effective strategy for the synthesis of biocompatible gold nanoparticles using cinnamon phytochemicals for phantom CT imaging and photoacoustic detection of cancerous cells. Pharm Res. 2011;28(2):279–291.
34. Pu S, Li J, Sun L, Zhong L, Ma Q. An in vitro comparison of the antioxidant activities of chitosan and green synthesized gold nanoparticles. Carbohydr Polym. 2019;211:161–172.
35. Dadwal A, Baldi A, Kumar Narang R. Nanoparticles as carriers for drug delivery in cancer. Artif Cells Nanomed Biotechnol. 2018;46(sup2):295–305.
36. Al-Hamadani MY, Alzahrani AM, Yousef MI, Kamel MA, El-Sayed WM. Gold nanoparticles perturb drug-metabolizing enzymes and antioxidants in the livers of male rats: potential impact on drug interactions. Int J Nanomed. 2020;15:5005.
37. Wang S, Lawson R, Ray PC, Yu H. Toxic effects of gold nanoparticles on Salmonella typhimurium bacteria. Toxicol Ind Health. 2011;27(6):547–554.
38. Wisse E, Braet F, Luo D, et al. Structure and function of sinusoidal lining cells in the liver. Toxicol. 1996;24(1):100–111.
39. Rivera-Gil P, Jimenez De Aberasturi D, Wulf V, et al. The challenge to relate the physicochemical properties of colloidal nanoparticles to their cytotoxicity. Acc Chem Res. 2013;46(3):743–749.
40. Aldewachi H, Woodroofe N, Gardiner P. Study of the stability of functionalized gold nanoparticles for the colorimetric detection of dipeptidyl peptidase IV. Appl Sci. 2018;8(12):2589.
41. Takahashi H, Niidome Y, Niidome T, Kaneko K, Kawasaki H, Yamada S. Modification of gold nanorods using phospatidylcholine to reduce cytotoxicity. Langmuir. 2006;22:2–5.
42. Love JC, Estroff LA, Kriebel JK, Nuzzo RG, Whitesides GM. Self-assembled monolayers of thiolates on metals as a form of nanotechnology. Chem Rev. 2005;105(4):1103–1170.
43. Liu R, Kay BK, Jiang S, Chen S. Nanoparticle delivery: targeting and nonspecific binding. MRS Bull. 2009;34(6):432–440.
44. Wang AZ, Langer R, Farokhzad OC. Nanoparticle delivery of cancer drugs. Annu Rev Med. 2012;63:185–198.
45. Carnovale C, Bryant G, Shukla R, Bansal V. Identifying trends in gold nanoparticle toxicity and uptake: size, shape, capping ligand, and biological Corona. ACS Omega. 2019;4(1):242–256.
46. Wang S, Lu W, Tovmachenko O, Rai US, Yu H, Ray PC. Challenge in understanding size and shape dependent toxicity of gold nanomaterials in human skin keratinocytes. Chem Phys Lett. 2008;463(1–3):145–149.
47. Duan H, Wang D, Li Y. Green chemistry for nanoparticle synthesis. Chem Soc Rev. 2015;44(16):5778–5792.
48. Niidome T, Yamagata M, Okamoto Y, et al. PEG-modified gold nanorods with a stealth character for in vivo applications. J Controlled Release. 2006;114(3):343–347.
49. Elavazhagan T, Arunachalam KD. Memecylon edule leaf extract mediated green synthesis of silver and gold nanoparticles. Int J Nanomed. 2011;6:1265.
50. Pérez ZEJ, Ramya Mathiyalagan JM, Kim Y-J, et al. Ginseng-berry-mediated gold and silver nanoparticle synthesis and evaluation of their in vitro antioxidant, antimicrobial, and cytotoxicity effects on human dermal fibroblast and murine melanoma skin cell lines. Int J Nanomed. 2017;12:709.
51. Malassis L, Dreyfus R, Murphy RJ, Hough LA, Donnio B, Murray CB. One-step green synthesis of gold and silver nanoparticles with ascorbic acid and their versatile surface post-functionalization. RSC Adv. 2016;6(39):33092–33100.
52. Ribeiro CA, Albuquerque LJ, de Castro CE, et al. One-pot synthesis of sugar-decorated gold nanoparticles with reduced cytotoxicity and enhanced cellular uptake. Colloids Surf a Physicochem Eng Asp. 2019;580:123690.
53. Ayala G, de Oliveira Vercik LC, Menezes TAV, Vercik A. A simple and green method for synthesis of Ag and Au nanoparticles using biopolymers and sugars as reducing agent.
54. Pinheiro T, Ferrão J, Marques AC, et al. Based in-situ gold nanoparticle synthesis for colorimetric, non-enzymatic glucose level determination. Nanomaterials. 2020;10(10):2027.
55. Abdelghany A, Oraby A, Asnag G. Structural, thermal and electrical studies of polyethylene oxide/starch blend containing green synthesized gold nanoparticles. J Mol Struct. 2019;1180:15–25.
56. Vasileva P, Donkova B, Karadjova I, Dushkin C. Synthesis of starch-stabilized silver nanoparticles and their application as a surface plasmon resonance-based sensor of hydrogen peroxide. Colloids Surf a Physicochem Eng Asp. 2011;382(1–3):203–210.
57. Anh NT, Van Phu D, Duy NN, Du BD, Hien NQ. Synthesis of alginate stabilized gold nanoparticles by γ-irradiation with controllable size using different Au3+ concentration and seed particles enlargement. Radiat Phys Chem. 2010;79(4):405–408.
58. Pestov A, Nazirov A, Privar Y, Modin E, Bratskaya S. Role of Au (III) coordination by polymer in□ green□ synthesis of gold nanoparticles using chitosan derivatives. Int J Biol Macromol. 2016;91:457–464.
59. Di Carlo G, Curulli A, Toro RG, et al. Green synthesis of gold–chitosan nanocomposites for caffeic acid sensing. Langmuir. 2012;28(12):5471–5479.
60. Anraku M, Tomida H, Michihara A, et al. Antioxidant and renoprotective activity of chitosan in nephrectomized rats. Carbohydr Polym. 2012;89(1):302–304.
61. Fiorentino A, D’Abrosca B, Pacifico S, Cefarelli G, Uzzo P, Monaco P. Natural dibenzoxazepinones from leaves of Carex distachya: structural elucidation and radical scavenging activity. Bioorg Med Chem Lett. 2007;17(3):636–639.
62. Hussain SP, Hofseth LJ, Harris CC. Radical causes of cancer. Nat Rev Cancer. 2003;3(4):276–285.
63. Bhattarai N, Ramay HR, Chou S-H, Zhang M. Chitosan and lactic acid-grafted chitosan nanoparticles as carriers for prolonged drug delivery. Int J Nanomed. 2006;1(2):181.
64. Fernandes JC, Qiu X, Winnik FM, et al. Low molecular weight chitosan conjugated with folate for siRNA delivery in vitro: optimization studies. Int J Nanomed. 2012;7:5833.
65. Xing R, Liu S, Yu H, Guo Z, Li Z, Li P. Preparation of high-molecular weight and high-sulfate content chitosans and their potential antioxidant activity in vitro. Carbohydr Polym. 2005;61(2):148–154.
66. Feng T, Du Y, Li J, Hu Y, Kennedy JF. Enhancement of antioxidant activity of chitosan by irradiation. Carbohydr Polym. 2008;73(1):126–132.
67. Pasanphan W, Buettner GR, Chirachanchai S. Chitosan gallate as a novel potential polysaccharide antioxidant: an EPR study. Carbohydr Res. 2010;345(1):132–140.
68. Kubota N, Tatsumoto N, Sano T, Toya K. A simple preparation of half N-acetylated chitosan highly soluble in water and aqueous organic solvents. Carbohydr Res. 2000;324(4):268–274.
69. Pasanphan W, Chirachanchai S. Conjugation of gallic acid onto chitosan: an approach for green and water-based antioxidant. Carbohydr Polym. 2008;72(1):169–177.
70. Xie M, Hu B, Yan Y, Zhou L, Ou S, Zeng X. Rheological properties of gallic acid-grafted-chitosans with different substitution degrees. LWT. 2016;74:472–479.
71. Li K, Guan G, Zhu J, Wu H, Sun Q. Antibacterial activity and mechanism of a laccase-catalyzed chitosan–gallic acid derivative against Escherichia coli and Staphylococcus aureus. Food Control. 2019;96:234–243.
72. Kang B, Vales TP, Cho B-K, Kim J-K, Kim H-J. Development of gallic acid-modified hydrogels using interpenetrating chitosan network and evaluation of their antioxidant activity. Molecules. 2017;22(11):1976.
73. Bam P, Bhatta A, Krishnamoorthy G. Design of biostable scaffold based on collagen crosslinked by dialdehyde chitosan with presence of gallic acid. Int J Biol Macromol. 2019;130:836–844.
74. Thangavel P, Ramachandran B, Muthuvijayan V. Fabrication of chitosan/gallic acid 3D microporous scaffold for tissue engineering applications. J Biomed Mater Res Part B Appl Biomater. 2016;104(4):750–760.
75. Hoyo J, Ivanova K, Guaus E, Tzanov T. Multifunctional ZnO NPs-chitosan-gallic acid hybrid nanocoating to overcome contact lenses associated conditions and discomfort. J Colloid Interface Sci. 2019;543:114–121.
76. Bai R, Yong H, Zhang X, Liu J, Liu J. Structural characterization and protective effect of gallic acid grafted O-carboxymethyl chitosan against hydrogen peroxide-induced oxidative damage. Int J Biol Macromol. 2020;143:49–59.
77. Senevirathne M, Jeon Y-J, Kim Y-T, et al. Prevention of oxidative stress in Chang liver cells by gallic acid-grafted-chitosans. Carbohydr Polym. 2012;87(1):876–880.
78. Mitsou E, Pletsa V, Sotiroudis GT, Panine P, Zoumpanioti M, Xenakis A. Development of a microemulsion for encapsulation and delivery of gallic acid. The role of chitosan. Colloids Surf B. 2020;190:110974.
79. Accardo A, Salsano G, Morisco A, et al. Peptide-modified liposomes for selective targeting of bombesin receptors overexpressed by cancer cells: a potential theranostic agent. Int J Nanomed. 2012;7:2007.
80. Hajiramezanali M, Atyabi F, Mosayebnia M, et al. 68Ga-radiolabeled bombesin-conjugated to trimethyl chitosan-coated superparamagnetic nanoparticles for molecular imaging: preparation, characterization and biological evaluation. Int J Nanomed. 2019;14:2591.
81. Wang M, Thanou M. Targeting nanoparticles to cancer. Pharmacol Res. 2010;62(2):90–99.
82. Xie M, Hu B, Wang Y, Zeng X. Grafting of gallic acid onto chitosan enhances antioxidant activities and alters rheological properties of the copolymer. J Agric Food Chem. 2014;62(37):9128–9136.
83. Zheng M, Zhang C, Zhou Y, et al. Preparation of gallic acid-grafted chitosan using recombinant bacterial laccase and its application in chilled meat preservation. Front Microbiol. 2018;9:1729.
84. Lee C-M, Jeong H-J, Cheong S-J, et al. Prostate cancer-targeted imaging using magnetofluorescent polymeric nanoparticles functionalized with bombesin. Pharm Res. 2010;27(4):712–721.
85. Mendoza-Nava H, Ferro-Flores G, Ramírez F, et al. 177Lu-dendrimer conjugated to folate and bombesin with gold nanoparticles in the dendritic cavity: a potential theranostic radiopharmaceutical. J Nanomater. 2016;2016.
86. Huang D, Ou B, Prior RL. The chemistry behind antioxidant capacity assays. J Agric Food Chem. 2005;53(6):1841–1856.
87. Liang N, Kitts DD. Antioxidant property of coffee components: assessment of methods that define mechanisms of action. Molecules. 2014;19(11):19180–19208.
88. Mishra K, Ojha H, Chaudhury NK. Estimation of antiradical properties of antioxidants using DPPH assay: a critical review and results. Food Chem. 2012;130(4):1036–1043.
89. Belin F, Barthélémy P, Ruiz K, Lacombe JM, Pucci B. Synthetic gallic acid derivatives as models for a comprehensive study of antioxidant activity. Helv Chim Acta. 2003;86(2):247–265.
90. Rohn S, Rawel HM, Kroll J. Antioxidant activity of protein-bound quercetin. J Agric Food Chem. 2004;52(15):4725–4729.
91. Leopoldini M, Russo N, Toscano M. The molecular basis of working mechanism of natural polyphenolic antioxidants. Food Chem. 2011;125(2):288–306.
92. Liu Y-S, Chang Y-C, Chen H-H. Silver nanoparticle biosynthesis by using phenolic acids in rice husk extract as reducing agents and dispersants. J Food Drug Anal. 2018;26(2):649–656.
93. Vo KDN, Guillon E, Dupont L, Kowandy C, Coqueret X. Influence of Au (III) interactions with chitosan on gold nanoparticle formation. J Phys Chem C. 2014;118(8):4465–4474.
94. Balasubramani G, Ramkumar R, Raja RK, Aiswarya D, Rajthilak C, Perumal P. Albizia amara Roxb. mediated gold nanoparticles and evaluation of their antioxidant, antibacterial and cytotoxic properties. J Clust Sci. 2017;28(1):259–275.
95. Kowalska E, Mahaney OOP, Abe R, Ohtani B. Visible-light-induced photocatalysis through surface plasmon excitation of gold on titania surfaces. Phys Chem Chem Phys. 2010;12(10):2344–2355.
96. Philip D. Rapid green synthesis of spherical gold nanoparticles using Mangifera indica leaf. Spectrochim Acta A Mol Biomol Spectrosc. 2010;77(4):807–810.
97. Amanulla B, Perumal KN, Ramaraj SK. Chitosan functionalized gold nanoparticles assembled on sulphur doped graphitic carbon nitride as a new platform for colorimetric detection of trace Hg2+. Sensor Actuat B Chem. 2019;281:281–287.
98. Noguez C. Surface plasmons on metal nanoparticles: the influence of shape and physical environment. J Phys Chem C. 2007;111(10):3806–3819.
99. Komenek S, Luesakul U, Ekgasit S, et al. Nanogold-gallate chitosan-targeted pulmonary delivery for treatment of lung cancer. AAPS PharmSciTech. 2017;18(4):1104–1115.
100. Chen H, Dorrigan A, Saad S, Hare DJ, Cortie MB, Valenzuela SM. In vivo study of spherical gold nanoparticles: inflammatory effects and distribution in mice. PLoS One. 2013;8(2):e58208. doi:10.1371/journal.pone.0058208
101. Sun Q, Zhou Z, Qiu N, Shen Y. Rational design of cancer nanomedicine: nanoproperty integration and synchronization. Adv Mater. 2017;29(14):1606628. doi:10.1002/adma.201606628
102. Cui L, Liu W, Liu H, et al. pH-triggered charge-reversal mesoporous silica nanoparticles stabilized by chitosan oligosaccharide/carboxymethyl chitosan hybrids for effective intracellular delivery of doxorubicin. ACS Appl Bio Mater. 2019;2(5):1907–1919. doi:10.1021/acsabm.8b00830
103. Kumar PS, Jeyalatha MV, Malathi J, Ignacimuthu S. Anticancer effects of one-pot synthesized biogenic gold nanoparticles (Mc-AuNps) against laryngeal carcinoma. J Drug Deliv Sci Technol. 2018;44:118–128. doi:10.1016/j.jddst.2017.12.008
104. Keshvari F, Bahram M, Farshid AA. Gold nanoparticles biofunctionalized (grafted) with chiral amino acids: a practical approach to determining the enantiomeric percentage of racemic mixtures. Anal Methods. 2015;7(11):4560–4567. doi:10.1039/C5AY00900F
105. Khoshnamvand M, Huo C, Liu J. Silver nanoparticles synthesized using Allium ampeloprasum L. leaf extract: characterization and performance in catalytic reduction of 4-nitrophenol and antioxidant activity. J Mol Struct. 2019;1175:90–96.
106. Yue K, Jin X, Tang J, Tian X, Tan H, Zhang X. Factors influencing aggregation of gold nanoparticles in whole blood. J Nanosci Nanotechnol. 2019;19(7):3762–3771.
107. Du S, Kendall K, Toloueinia P, Mehrabadi Y, Gupta G, Newton J. Aggregation and adhesion of gold nanoparticles in phosphate buffered saline. J Nanoparticle Res. 2012;14(3):1–14.
108. Popat A, Liu J, Lu GQM, Qiao SZ. A pH-responsive drug delivery system based on chitosan coated mesoporous silica nanoparticles. J Mater Chem. 2012;22(22):11173–11178.
109. Sawtarie N, Cai Y, Lapitsky Y. Preparation of chitosan/tripolyphosphate nanoparticles with highly tunable size and low polydispersity. Colloids Surf B Biointerfaces. 2017;157:110–117.
110. Jovanovic SV, Hara Y, Steenken S, Simic MG. Antioxidant potential of gallocatechins. A pulse radiolysis and laser photolysis study. J Am Chem Soc. 1995;117(39):9881–9888.
111. Eslami AC, Pasanphan W, Wagner BA, Buettner GR. Free radicals produced by the oxidation of gallic acid: an electron paramagnetic resonance study. Chem Cent J. 2010;4(1):1–4.
112. Kumari S, Swetha M, Mayor S. Endocytosis unplugged: multiple ways to enter the cell. Cell Res. 2010;20(3):256–275.
113. Nativo P, Prior IA, Brust M. Uptake and intracellular fate of surface-modified gold nanoparticles. ACS Nano. 2008;2(8):1639–1644.
114. Moghtaderi H, Sepehri H, Delphi L, Attari F. Gallic acid and curcumin induce cytotoxicity and apoptosis in human breast cancer cell MDA-MB-231. BI. 2018;8(3):185.
115. Heidarian E, Keloushadi M, Ghatreh-Samani K, Valipour P. The reduction of IL-6 gene expression, pAKT, pERK1/2, pSTAT3 signaling pathways and invasion activity by gallic acid in prostate cancer PC3 cells. Biomed. 2016;84:264–269.
 © 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.