Back to Journals » Infection and Drug Resistance » Volume 8
Bloodstream infections in intensive care unit patients: distribution and antibiotic resistance of bacteria
Authors Russotto V, Cortegiani A, Graziano G, Saporito L, Raineri SM, Mammina C, Giarratano A
Received 31 May 2015
Accepted for publication 9 July 2015
Published 10 August 2015 Volume 2015:8 Pages 287—296
DOI https://doi.org/10.2147/IDR.S48810
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 4
Editor who approved publication: Professor Suresh Antony
Vincenzo Russotto,1 Andrea Cortegiani,1 Giorgio Graziano,2 Laura Saporito,2 Santi Maurizio Raineri,1 Caterina Mammina,2 Antonino Giarratano1
1Department of Biopathology and Medical Biotechnologies (DIBIMED), Section of Anaesthesia, Analgesia, Intensive Care and Emergency, Paolo Giaccone University Hospital, University of Palermo, Palermo, Italy; 2Department of Sciences for Health Promotion and Mother-Child Care, University of Palermo, Palermo, Italy
Abstract: Bloodstream infections (BSIs) are among the leading infections in critically ill patients. The case-fatality rate associated with BSIs in patients admitted to intensive care units (ICUs) reaches 35%–50%. The emergence and diffusion of bacteria with resistance to antibiotics is a global health problem. Multidrug-resistant bacteria were detected in 50.7% of patients with BSIs in a recently published international observational study, with methicillin resistance detected in 48% of Staphylococcus aureus strains, carbapenem resistance detected in 69% of Acinetobacter spp., in 38% of Klebsiella pneumoniae, and in 37% of Pseudomonas spp. Prior hospitalization and antibiotic exposure have been identified as risk factors for infections caused by resistant bacteria in different studies. Patients with BSIs caused by resistant strains showed an increased risk of mortality, which may be explained by a higher incidence of inappropriate empirical therapy in different studies. The molecular genetic characterization of resistant bacteria allows the understanding of the most common mechanisms underlying their resistance and the adoption of surveillance measures. Knowledge of epidemiology, risk factors, mechanisms of resistance, and outcomes of BSIs caused by resistant bacteria may have a major influence on global management of ICU patients. The aim of this review is to provide the clinician an update on BSIs caused by resistant bacteria in ICU patients.
Keywords: bloodstream infections, multidrug resistant, antibiotic, intensive care unit, MDR, ICU
Introduction
Bloodstream infections (BSIs) are among the leading acquired infections in intensive care unit (ICU) patients.1 BSIs may be the consequence of the bloodstream diffusion of bacteria from a localized infection (secondary BSI) or may be the only identifiable infectious process (primary BSI). Antimicrobial therapy is the mainstay of treatment of BSIs, along with management of severe sepsis and septic shock that may eventually develop.2 During these last few years, clinicians have witnessed a growing incidence of BSIs by bacteria with resistance against commonly used antimicrobials.3 A multidrug-resistant (MDR) microorganism has been defined as an isolate with nonsusceptibility to at least one agent in three or more antimicrobial categories. Extensive drug-resistant (DR) microorganisms exhibit susceptibility to only one or two antimicrobial categories, whereas pan-DR isolates are those microorganisms resistant to all agents from all antimicrobial categories.4 Emergence of resistance among bacteria is considered a public health problem worldwide.5 BSIs caused by MDR microorganisms are associated with excess mortality, which may be attributed, at least in part, to the delay in appropriate therapy institution.6–8 Emergence of resistant bacteria in the ICU implies an even higher challenge for the clinician, dealing with seriously ill patients, who need prompt institution of effective antimicrobial therapy in a setting of higher prevalence of resistant isolates, broad-spectrum antibiotic, use and ease of cross-transmission of resistant microorganisms.2,9,10 The clinician should be aware of the main risk factors for BSIs sustained by resistant bacteria in the ICU, the most commonly met resistant species according to different geographic areas, and their mechanisms of resistance. The aim of this review is to provide the clinician an update on BSI caused by resistant bacteria in ICU patients.
Epidemiology of BSIs caused by resistant bacteria
BSIs are among the leading infections in ICUs. In an international study of the prevalence and outcomes of infections in ICUs, BSIs were observed in 15% of infected patients and represented the third-most commonly met infection.1 The traditional classification of BSIs between community-acquired and hospital-acquired is being reconsidered in light of the evolving health care-system organization. A growing proportion of patients, with older age and several comorbidities, are treated as outpatients, with a shift of health care services from hospitals to the community with different out-of-hospital facilities.11,12 In this sense, health care-associated BSIs have been defined as those occurring in patients living in a nursing home or long-term care facilities, receiving health care at home, under hemodialysis, intravenous chemotherapy, wound care, or enteral nutrition, or who recently accessed an acute care hospital.11 Approximately one in five BSIs diagnosed at ICU admission may be classified as health care-associated BSIs.13 Pathogens responsible for health care-associated BSIs are similar to those isolated from hospital-acquired BSIs, with a higher risk of resistant isolates compared to community-acquired BSIs. A higher incidence of inappropriate empirical therapy and a trend of increased mortality has also been detected in different studies.13,14 A recently published international observational study provided information about hospital-acquired BSIs from 1,156 ICU patients worldwide.10 Among total microorganisms causing BSI, 57.6% were Gram-negative and 33.4% were Gram-positive. MDR bacteria caused BSIs in 50.7% of patients, and extensive DR bacteria were detected in 22% of patients. Among Gram-negative bacteria, carbapenem resistance was detected in 69% of Acinetobacter spp, 38% of Klebsiella pneumoniae, and 37% of Pseudomonas spp. Among Gram-positive bacteria, 48% of Staphylococcus aureus isolates were methicillin-resistant Staphylococcus aureus (MRSA), and 23% of Enterococcus faecium isolates were vancomycin-resistant enterococci (VRE). With regard to the source of BSIs, in 21.1% they was attributed to the respiratory tract, and in 21.4% BSIs were catheter-related. In 23.7% of total patients, no clear source of infection was identified. These data are similar to those of the recently published French national network REA RAISIN of ICU-acquired bacteremia (29.2% catheter-related, 18% lower respiratory tract infections).10,15 According to a large observational study investigating the trend of BSIs caused by resistant bacteria over a period of 10 years, resistant isolates represented an additional burden of disease rather than substituting susceptible bacteria as the cause of BSIs.3 In the modern globalized world, antibiotic-resistant bacteria can be easily distributed across countries by human travelers, animal and insect vectors, agricultural products, and water.16 Analysis of genetic material encoding antibiotic resistance has been used for tracing the diffusion of resistant strains both at a local and international level.17 An example is the worldwide distribution of MDR staphylococci from very few resistant clones.18 Rapid detection of resistant bacteria and their genetic characterization through molecular techniques is therefore a pivotal component of surveillance and control of diffusion of resistant bacteria from a single institution to a global level.19,20
Risk factors for BSI caused by resistant bacteria
Patients in ICUs have different risk factors for BSI development, including greater severity of illness, disruption of anatomical barriers (ie, use of invasive devices, surgery), and impaired immunological response.2 Patients with a central vein catheter (CVC) have additional risk factors for catheter-related BSIs (inadequate adoption of a sterile technique, inexperience of the operator, site of insertion, colonization of the insertion site, contamination of the catheter hub, and duration of catheter placement).21–23 Different observational studies have described risk factors for BSIs caused by resistant bacteria.10–12,24 In general, prior antibiotic exposure and hospitalization, residency in nursing home and in long-term care facilities, and other described risk factors for health care-related BSIs are associated with an increased risk of developing a BSI caused by resistant microorganisms.13,25–27 In a retrospective study of patients with severe sepsis and septic shock attributed to bacteremia caused by Gram-negative microorganisms, prior antibiotic exposure was associated with a reduced susceptibility to cefepime, piperacillin/tazobactam, carbapenems, ciprofloxacin, and gentamicin. Moreover, patients with recent antibiotic exposure more frequently received inappropriate empirical antimicrobial therapy and had higher hospital mortality.26 Patients with BSIs caused by carbapenem-resistant K. pneumoniae more frequently had a CVC or urinary catheter compared to patients with BSIs caused by susceptible microorganisms, received renal replacement therapy or prior antibiotic treatment, and were more likely hospitalized in a ward with other resistant isolates during the 30 days preceding the BSI diagnosis.25 Colonization was identified as a significant risk factor for subsequent development of bacteremia by Acinetobacter baumannii in different studies.9,28,29 Patients were frequently colonized by MDR A. baumannii after approximately 7 days from ICU admission, with respiratory and urinary tracts the most commonly detected colonized sites.9 Reported risk factors for subsequent development of bacteremia by MDR A. baumannii in previously colonized patients were respiratory failure at ICU admission treated with invasive mechanical ventilation, recent CVC insertion, prior antibiotic therapy, and development of bacteremia caused by other microorganisms after MDR A. baumannii colonization.29 Knowledge of risk factors for BSIs caused by resistant bacteria may have a major influence on global management in ICUs, from source-control and surveillance measures to appropriate antibiotic treatment.19
Mechanisms of antibacterial drug resistance
Antibiotic resistance is a complex phenomenon that encompasses recognized in vitro mechanisms and their phenotypic expression, but also – at the clinical level – infection site, pharmacodynamic/pharmacokinetic properties of the specific antibiotic, modification of these pharmacological profiles in the critically ill patient, and immune status.30,31 Resistance patterns of bacteria at a cellular level may be generally classified as follows: altered target site, namely modification of the molecule that is the target of the antimicrobial action; decreased antimicrobial uptake through decreased permeability to the antimicrobial or its elimination by efflux pumps; bypass pathways, which overcome the inhibition caused by the antibiotic molecule; and inactivation of the antimicrobial molecule through the production of enzymes that inactivate the drug.31 Antibiotic resistance may represent an intrinsic property of different bacteria (eg, low permeability to some molecules), or rather may be acquired as a consequence of antibiotic pressure or mobile genetic element transmission.32,33
Table 1 summarizes the most commonly described resistance mechanisms of bacteria against frequently used antibacterial agents in ICU, with the corresponding genes identified to date.
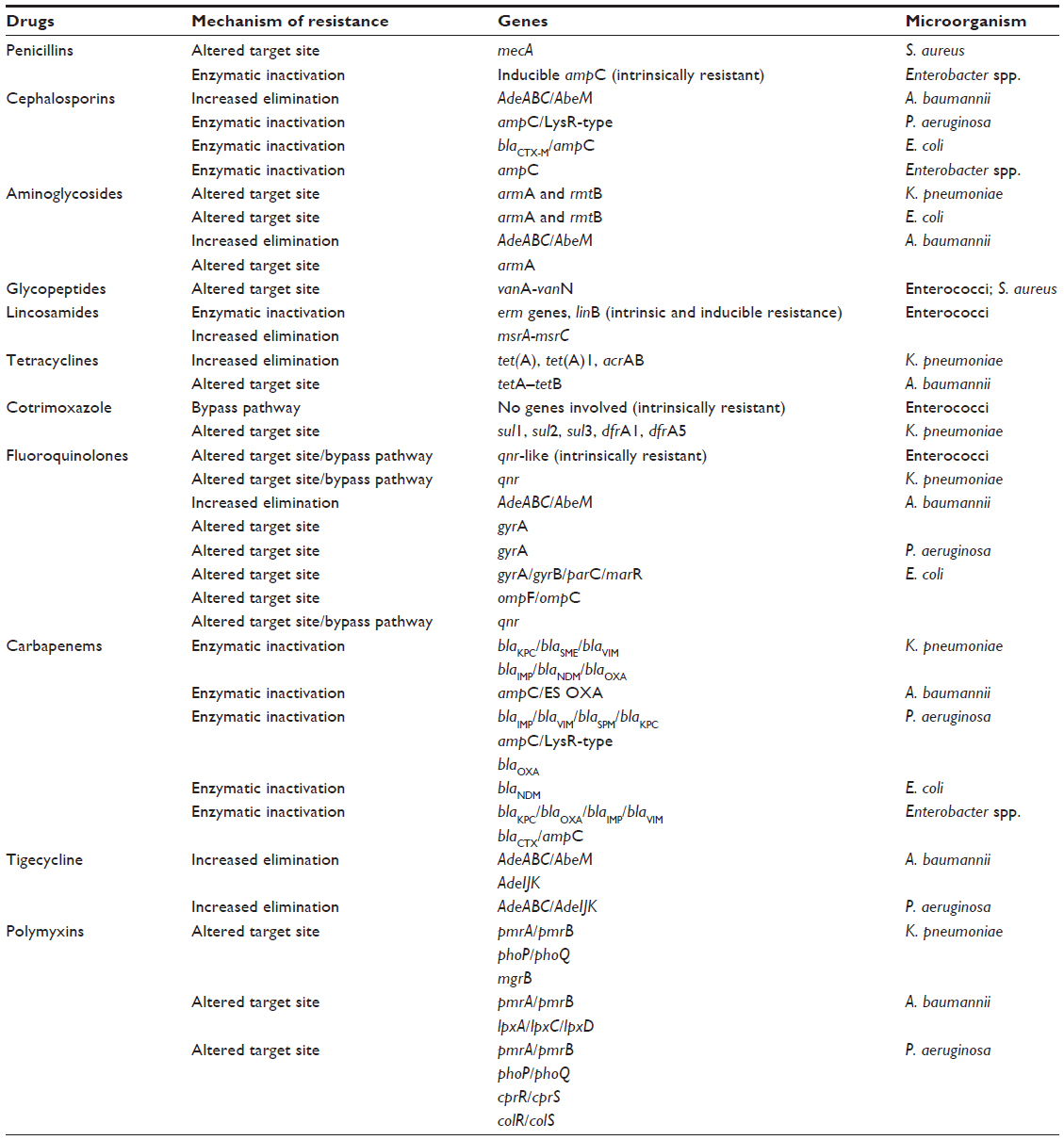 | Table 1 Mechanisms of resistance against most commonly used antimicrobial agents and corresponding genes expressed by bacteria |
Enterococcus spp.
Enterococci are Gram-positive, facultatively anaerobic, opportunistic bacteria. E. faecalis and E. faecium are the leading species responsible of human disease. VRE, which emerged during the late 1980s,34 are among the major health care-associated MDR organisms.10 Enterococci are normal components of the human intestinal flora, but may become responsible for serious infections, such as BSIs and intra-abdominal and surgical site infections, especially among immunocompromised patients.35 These microorganisms represent the third-most prevalent nosocomial pathogens worldwide, and acquired resistance to penicillin/ampicillin, aminoglycosides, and glycopeptides are reported in an increasing number of isolates, causing a serious problem in choosing an appropriate therapy.36 All Enterococcus spp., including E. faecalis and E. faecium, are naturally resistant to several antibiotics, such as cephalosporins, clindamycin, cotrimoxazole, and aminoglycosides.37 Mechanisms of glycopeptide resistance in enterococci have been well investigated, and to date we know of nine mobile gene clusters that mediate the resistance: VanA, VanB, VanC, VanD, VanE, VanG, VanL, VanM, and VanN. VanA resistance is the most prevalent, and confers high-level resistance to all glycopeptides by altering the terminal sequence of cell-wall precursors, thus lowering their binding affinity, while the VanB phenotype is only resistant to vancomycin.37 VRE isolates exhibit a genetic “pathogenicity island”, coding for a putative enterococcal surface protein.38 This protein produces thicker and DR biofilms, and its expression in E. faecium is regulated by temperature, promoting initial colonization and aiding in biofilm formation. Thanks to the ability to colonize gut, skin, and inanimate surfaces, this trait is likely to promote the infection of indwelling medical devices, of which enterococci are frequently associated.39 Of note, vancomycin-resistance genes can be transferred from enterococci to other organisms, causing the emergence of vancomycin-resistant S. aureus (VRSA).40
S. aureus
S. aureus is a Gram-positive, facultative anaerobic pathogen, with both hospital- and community-acquired strains. It is part of the microbiota of the skin, and it is most commonly isolated from moist areas, such as the anterior nares and axillae.41 Traditionally opportunistic, many S. aureus strains are now aggressively pathogenic, causing a wide range of diseases, from skin and wound infections to severe illnesses, such as BSIs and soft-tissue and bone infections.41,42 As enterococci, S. aureus also frequently causes chronic infections by forming biofilms, and this is the leading cause of chronic infections associated with indwelling medical devices.43 MRSA is a major pathogen in both health care and community settings, and its resistance is conferred by the acquisition of one of several staphylococcal cassette chromosome mec elements that carry a gene (mecA) that encodes a penicillin-binding protein (PBP2a) with low affinity for β-lactam antibiotics.44,45 One of the most challenging issues in the evolution of MRSA epidemiology is the emergence and spread of community-acquired MRSA. These strains are generally susceptible to a wider spectrum of non-β-lactam antibiotics compared to hospital-acquired MRSA, but they often carry the genes for Panton–Valentine leukocidin.46,47 First-line treatment for MRSA infections is glycopeptide antibiotics (ie, vancomycin or teicoplanin). However, the intensive selective pressure caused the emergence of vancomycin-intermediate S. aureus and VRSA.42 Vancomycin-intermediate S. aureus strains have evolved as a result of fundamental changes in cell-wall thickness and composition, trapping vancomycin and reducing its permeation to the site of action. VRSA is far less common, and is characterized by an acquired resistance from the VRE vanA gene, which alters the terminal sequence of cell-wall precursors, making them poor substrates for vancomycin and teicoplanin. MDR VRSA isolates exhibit both mecA and vanA resistance genes, with resistance being conferred through the same mechanisms as MRSA and VRE, respectively.48
Enterobacteriaceae
The emergence of resistance in Enterobacteriaceae is considered an alarming health threat.49 During these last few years, a growing number of K. pneumoniae, E. coli, and Enterobacter spp. have developed resistance against third-generation cephalosporin, due to extended-spectrum β-lactamases (ESBLs). ESBLs evolved from BLs, which are frequently expressed in Gram-negative bacteria (TEM-1, TEM-2, SHV-1) and are responsible for resistance against ampicillin, amoxicillin, and early generations of cephalosporins.49,50 Mutations in genes encoding these BLs led to the evolution of new enzymes, able to also hydrolyze third-generation cephalosporins and aztreonam. More than 700 ESBLs variants have been identified to date, some of which have spread rapidly worldwide.50,51 Different classifications of BLs have been proposed, though Bush functional classification52 and Ampler molecular classification are the most commonly used.53 The diffusion of coding genes for ESBLs through mobile genetic elements is responsible for their ease of diffusion, their transmission in bacteria different from Enterobacteriaceae (ie, nonfermenting, Gram-negative rods, such as Pseudomonas aeruginosa), and cotransmission of genes responsible of resistance against other classes of molecules (eg, aminoglycosides).50 Carbapenems are the first line treatment of ESBLs producing bacteria, and the emergence of carbapenem-resistant isolates leaves limited therapeutic options.54,55 Metallo-BLs (MBLs), extended-spectrum oxacillinases, and clavulanic acid-inhibited BLs have all been isolated in Enterobacteriaceae.50 MBLs belonging to class B of the Ambler classification (Verona imipenemase [VIM], imipenemase [IMP]) have been detected in different countries.55 Class D OXA-48 β-lactamases have been detected in K. pneumoniae isolates from Turkey, Lebanon, and Belgium.55–57 Among class A β-lactamases, the most commonly identified enzyme is KPC.55 The first KPC-producing K. pneumoniae isolate was detected in North Carolina, USA, in 1996.58 This first discovery was followed by several other variants (KPC-2 to KPC-7).55 Of note, several KPC-producing K. pneumoniae with susceptibility to carbapenems have been reported, underlying the need of an additional mechanism (eg, poor drug penetration) for full expression of drug resistance.59 Apart from enzymatic inactivation of drug molecules, different mechanisms of virulence and additional antibiotic-resistance mechanisms have been described in Enterobacteriaceae.49 K. pneumoniae is intrinsically virulent and capable of invasive infections, due to fimbrial adhesins and a thick capsule, which acts as a putative antiphagocytic factor.60 Pan-DR strains of KPC-producing K. pneumoniae have been reported in the literature, and isolates were resistant to all antibiotic agents tested, including carbapenems, polymyxin B, and tigecycline.55 Polymyxins are considered the last resort for treatment of infections with carbapenem-resistant Gram-negative bacteria.61 However, resistance to these compounds has begun to emerge, though infrequently. Selective pressure, by the increased use of colistin, and clonal expansion through horizontal transmission, have generated clusters of cases infected with multiresistant K. pneumoniae strains that have been generally attributed to the international epidemic clone ST258. Mono- or multiclonal outbreaks of colistin-resistant, KPC-producing K. pneumoniae have been described in hospitals in many countries, for example Greece, South Korea, the USA, and Italy.62,63 Altered outer-membrane composition with the phosphate groups of lipid A containing five times more l-Ara4N than susceptible strains is known to lower the negative charge of the outer membrane of K. pneumoniae, leading to the reduced interaction of this membrane with polymyxins.64 A molecular characterization of the structural alterations of lipopolysaccharide (LPS) in K. pneumoniae with regard to polymyxin resistance has shown the involvement of the phoP/phoQ and pmrA/pmrB genes.65 One important molecular mechanism that leads to the emergence of colistin resistance in K. pneumoniae that has recently been discovered is the mutation/inactivation of the mgrB gene.66 Disruption of the mgrB gene in K. pneumoniae has been identified to play an important role in polymyxin resistance in this bacterium.67 Polymyxins remain most often active against MDR bacteria, and the emergence of colistin resistance in relation to increased usage is worrisome, since polymyxins are the last remaining therapeutic option in many cases.68
E. coli are Gram-negative, facultative anaerobes, most commonly commensal, but can also become pathogenic, producing potentially deadly toxins, including enterohemorrhagic verotoxins, such as E. coli O157:H7, which causes hemolytic uremic syndrome and renal failure.69 E. coli has been one of the most widely antibiotic-susceptible microorganisms of the Enterobacteriaceae family, but recently horizontal gene transfer has allowed for the rise of highly resistant strains.70 This spread of resistance has major relevance, because these strains are among the most common Gram-negative bacteria causing infections in humans. Strains carrying ESBLs and with high coresistance to fluoroquinolones and gentamicin have been rising in Europe.71 Even if still uncommonly, in multiple continents E. coli have also acquired the NDM-1 enzyme from K. pneumoniae, which confers a broad resistance to all BLs, including carbapenems, with the exception of the monobactam aztreonam.72 In polymyxin-resistant E. coli, lipid A is typically modified with 2-aminoethanol and also with l-Ara4N.73
Enterobacter is a genus of Gram-negative, facultative anaerobic, opportunistic pathogens, possessing endotoxins and antibiotic resistance through expression of an extensive variety of ESBLs and carbapenemases, including KPC, OXA, and MBLs.74 Of note, Enterobacter spp. are intrinsically resistant to aminopenicillins, cefazolin, and cefoxitin, due to the production of constitutive chromosomal AmpC BLs.75
Nonfermenting Gram-negative bacteria
A. baumannii
The most common resistant Acinetobacter species is A. baumannii, a Gram-negative, facultative anaerobic, opportunistic pathogen. It has a thick cell wall that enables it to resist dry conditions, high temperature, and pH and nutrient changes, surviving for up to 5 months on inanimate objects.76 A. baumannii is naturally resistant to many antibiotics, due to both poor membrane penetration and active efflux pumps. Overexpression of the AdeABC and AbeM efflux pumps causes broad resistance to cephalosporins, fluoroquinolones, aminoglycoside, and tigecycline, and also provides resistance to ammonia-based disinfectants.77 Furthermore, A. baumannii isolates produce an exopolysaccharide, leading to biofilm formation, and express a powerful, epithelial cell-targeting cytotoxin that facilitates colonization.78 MDR A. baumannii expresses a variety of BLs, including ESBLs and carbapenemases, such as imipenem MBLs and oxacillinases.79 Among naturally occurring BLs, an AmpC-type cephalosporinase and OXA-51/69 variants have been identified. These enzymes, once overexpressed, may be involved in reduced susceptibility to carbapenems. Most acquired BLs belong to class B or class D of the Ambler classification. OXA-23-like enzymes are the most widespread in A. baumannii worldwide, and these bacteria are the most common sources of nosocomial outbreaks with carbapenem-resistant A. baumannii.80 A. baumannii worldwide shows high-level resistance to all aminoglycosides by decreased outer-membrane permeability, active efflux pumps, and amino acid substitutions in ribosomal proteins.31 In P. aeruginosa and A. baumannii, a single mutation in the gyrA gene encoding DNA gyrase is sufficient to confer clinically high-level resistance to fluoroquinolones. Of note, decreased susceptibility to this class of molecules is also due to low permeability and efflux-pump activity.81 Resistance to tigecycline has been noted on several occasions, and might be due to upregulation of the AdeABC and AdeIJK multidrug efflux-pump systems.82 Tigecycline-resistance levels in A. baumannii isolates may increase during therapy in cases of brief exposure to the drug, compromising its efficacy.82,83 Two mechanisms of resistance to colistin have been described in A. baumannii. The first consists of alterations of the lipid A component of LPS resulting from mutations in the PmrAB two-component system.32,84 The second resistance mechanism is the complete loss of LPS production resulting from mutations in the lpxA, lpxC, and lpxD genes, encoding the enzymes that catalyze the first steps in LPS biosynthesis.85
P. aeruginosa
P. aeruginosa is a Gram-negative, rod-shaped bacterium, a facultative anaerobic, opportunistic pathogen surviving in microaerobic conditions, such as the thick mucus of lungs of cystic fibrosis patients. The ubiquity and survival capacity in extreme environments, like antimicrobial solutions of acetate-buffered benzalkonium chloride, are major determinants of outbreaks of nosocomial infections. It has a high propensity to form biofilms. Its outer-membrane porins make it impermeable and resistant to many antibiotics.86,87 P. aeruginosa is notoriously resistant to fluoroquinolones as a result of target mutations on DNA gyrase and/or topoisomerase IV.33 Many BLs, encoded by plasmids or chromosomally integrated transposons, have been reported (eg, IMP, VIM, KPC, and SPM metallocarbapenemases).88 An acquired ceftazidimase ESBL called PER-1 is widespread in P. aeruginosa isolated in Turkey.89 IMP and VIM enzymes are integron-associated and can be plasmid-mediated and transferable, although chromosomal gene integration is common.88 IMP-1 was first reported in Enterobacteriaceae and P. aeruginosa in Japan and is now globally distributed, suggesting horizontal transfer of blaIMP-1 between unrelated Gram-negative species.79 VIM-1 was the first MBL identified in P. aeruginosa and has been reported in several European countries. However, VIM-2 is now the most widespread MBL as a source of multiple outbreaks.90 The BL SPM is quite different from VIM and IMP, and represents a new subfamily of MBLs, highly resistant to all anti-Gram-negative antibiotics except colistin.91 Intrinsic to P. aeruginosa is a chromosomal gene encoding an AmpC-type cephalosporinase, associated with a LysR-type regulatory gene with which some BL molecules may interact, leading to overexpression of the AmpC gene. Selection of mutants overproducing the AmpC gene is frequently observed in P. aeruginosa, leading to acquired resistance to ticarcillin, piperacillin, and broad-spectrum cephalosporins (ceftazidime).83,92 P. aeruginosa produces a naturally occurring class D BL, OXA-50, that does not contribute to the overall BL-resistance pattern of P. aeruginosa, except for latamoxef.93 Most of the class D BLs able to hydrolyze expanded-spectrum cephalosporins have been identified, and there are two main types of expanded-spectrum class D BLs (ES-OXAs).94 OXA-18, which is inhibited by clavulanic acid, was the first identified ES-OXA in a P. aeruginosa isolate in Paris, from a patient previously hospitalized in Sicily.95 Only two acquired class D BLs, compromising the efficacy of carbapenems, have been reported from P. aeruginosa: OXA-40 and OXA-198.96,97 Resistance to tigecycline has been noted on several occasions, and might be due to upregulation of the AdeABC and AdeIJK multidrug efflux pumps acting synergistically.98 Acquisition of polymyxin resistance is mostly related to modifications of the biosynthesis of the lipid A component of LPS. This resistance is mediated by a complicated regulatory genetic network, such as PmrAB, PhoPQ, ParRS, CprRS, and ColRS, which is currently under investigation worldwide.61
Outcomes
The overall case-fatality rate associated with BSIs is 15%–20%, but reaches 35%–50% when patients with ICU admission are considered.2 In most observational studies investigating the epidemiology and outcomes of BSIs due to resistant bacteria, inappropriate antimicrobial therapy is independently associated with an increased mortality after correction for severity of illness and other potential confounders.8,10,25,99 The risk of inappropriate empirical therapy increases with the prevalence of resistant microorganisms.25 It is generally accepted that the increased risk of mortality attributable to BSIs due to resistant isolates is related to inappropriate therapeutic choices, rather than intrinsic enhanced virulence of resistant microorganisms.25 The overall mortality of patients with BSIs due to carbapenem-resistant K. pneumoniae was 23% at 7 days and 60% at the end of hospitalization in a retrospective observational study. Lack of microbiologic eradication at 7 days was associated with an increased 30-day mortality.100 A higher infection-related mortality was detected in patients with BSIs caused by carbapenem-resistant K. pneumoniae compared to those with ESBL K. pneumoniae or susceptible strains. This increased mortality may be explained by the higher proportion of patients with ineffective antimicrobial therapy in the carbapenem-resistant group.25 Apart from previously reported risk factors (recent history of hospitalization, critical illness at presentation, inappropriate empirical therapy), postantibiogram antimicrobial regimens had an influence on 30-day mortality from carbapenem-resistant K. pneumoniae BSIs, with a higher risk of mortality observed in monotherapy-treated patients compared to patients treated with combination therapy, especially when the combination regimen included a carbapenem.54 The unexpected observation of improved outcomes when carbapenemase-producing isolates of K. pneumoniae are treated with carbapenems confirmed previously reported data from case reports and retrospective studies.101,102
Conclusion
BSIs are among the leading causes of infections in ICU patients. An increasing number of BSIs are caused by resistant bacteria in the ICU setting. The clinician should be aware of the risk factors for BSIs caused by resistant bacteria, common resistance mechanisms, and their diffusion for the global management of critically ill patients, from surveillance measures to source control and appropriate antibiotic treatment.
Disclosure
The authors report no conflicts of interest in this work.
References
Vincent JL, Rello J, Marshall J, et al. International study of the prevalence and outcomes of infection in intensive care units. JAMA. 2009; 302(21):2323–2329. | |
Timsit JF, Laupland KB. Update on bloodstream infections in ICUs. Curr Opin Crit Care. 2012;18(5):479–486. | |
Ammerlaan H, Harbarth S, Buiting A, et al. Secular trends in nosocomial bloodstream infections: antibiotic-resistant bacteria increase the total burden of infection. Clinical Infect Dis. 2013;56(6):798–805. | |
Magiorakos AP, Srinivasan A, Carey R, et al. Multidrug-resistant, extensively drug-resistant and pandrug-resistant bacteria: an international expert proposal for interim standard definitions for acquired resistance. Clin Microbiol Infect. 2012;18(3):268–281. | |
Carlet J, Collignon P, Goldmann D, et al. Society’s failure to protect a precious resource: antibiotics. Lancet. 2011;378(9788):369–371. | |
Retamar P, Portillo MM, López-Prieto MD, et al. Impact of inadequate empirical therapy on the mortality of patients with bloodstream infections: a propensity score-based analysis. Antimicrob Agents Chemother. 2012;56(1):472–478. | |
Zarkotou O, Pournaras S, Tselioti P, et al. Predictors of mortality in patients with bloodstream infections caused by KPC-producing Klebsiella pneumoniae and impact of appropriate antimicrobial treatment. Clin Microbiol Infect. 2011;17(12):1798–1803. | |
Kang CI, Kim SH, Park WB, et al. Bloodstream infections caused by antibiotic-resistant Gram-negative bacilli: risk factors for mortality and impact of inappropriate initial antimicrobial therapy on outcome. Antimicrob Agents Chemother. 2005;49(2):760–766. | |
Jung JY, Park MS, Kim SE, et al. Risk factors for multi-drug resistant Acinetobacter baumannii bacteremia in patients with colonization in the intensive care unit. BMC Infect Dis. 2010;10:228. | |
Tabah A, Koulenti D, Laupland K, et al. Characteristics and determinants of outcome of hospital-acquired bloodstream infections in intensive care units: the EUROBACT International Cohort Study. Intensive Care Med. 2012;38(12):1930–1945. | |
Rodríguez-Baño J, López-Prieto MD, Portillo MM, et al. Epidemiology and clinical features of community-acquired, healthcare-associated and nosocomial bloodstream infections in tertiary-care and community hospitals. Clin Microbiol Infect. 2010;16(9):1408–1413. | |
Kollef MH, Zilberberg MD, Shorr AF, et al. Epidemiology, microbiology and outcomes of healthcare-associated and community-acquired bacteremia: a multicenter cohort study. J Infect. 2011;62(2):130–135. | |
Vallés J, Alvarez-Lerma F, Palomar M, et al. Health-care-associated bloodstream infections at admission to the ICU. Chest. 2011;139(4):810–815. | |
McDonald JR, Friedman ND, Stout JE, Sexton DJ, Kaye KS. Risk factors for ineffective therapy in patients with bloodstream infection. Arch Intern Med. 2005;165(3):308–313. | |
Timsit JF, Soubirou JF, Voiriot G, et al. Treatment of bloodstream infections in ICUs. BMC Infect Dis. 2014;14:489. | |
Okeke IN, Edelman R. Dissemination of antibiotic-resistant bacteria across geographic borders. Clin Infect Dis. 2001;33(3):364–369. | |
Al Naiemi N, Duim B, Savelkoul PH, et al. Widespread transfer of resistance genes between bacterial species in an intensive care unit: implications for hospital epidemiology. J Clin Microbiol. 2005;43(9):4862–4864. | |
Ayliffe G. The progressive intercontinental spread of methicillin-resistant Staphylococcus aureus. Clin Infect Dis. 1997;24 Suppl 1:S74–S79. | |
Montero JG, Lerma FÁ, Galleymore PR, et al. Combatting resistance in intensive care: the multimodal approach of the Spanish ICU “Zero Resistance” program. Crit Care. 2015;19:114. | |
Cortegiani A, Russotto V, Capuano P, et al. Use of Cepheid Xpert Carba-R® for rapid detection of carbapenemase-producing bacteria in critically ill, abdominal surgical patients: first report of an observational study. Crit Care. 2015;19 Suppl 1:108. | |
Pronovost P, Needham D, Berenholtz S, et al. An intervention to decrease catheter-related bloodstream infections in the ICU. N Engl J Med. 2006;355(26):2725–2732. | |
Berenholtz SM, Pronovost PJ, Lipsett PA, et al. Eliminating catheter-related bloodstream infections in the intensive care unit. Crit Care Med. 2004;32(10):2014–2020. | |
Fridkin SK, Pear SM, Williamson TH, Galgiani JN, Jarvis WR. The role of understaffing in central venous catheter-associated bloodstream infection. Infect Control Hosp Epidemiol. 1996;17(3):150–158. | |
Elemam A, Rahimian J, Mandell W. Infection with panresistant Klebsiella pneumoniae: a report of 2 cases and a brief review of the literature. Clin Infect Dis. 2009;49(2):271–274. | |
Ben-David D, Kordevani R, Keller N, et al. Outcome of carbapenem resistant Klebsiella pneumoniae bloodstream infections. Clin Microbiol Infect. 2012;18(1):54–60. | |
Johnson MT, Reichley R, Hoppe-Bauer J, Dunne WM, Micek S, Kollef M. Impact of previous antibiotic therapy on outcome of Gram-negative severe sepsis. Crit Care Med. 2011;39(8):1859–1865. | |
Marschall J, Fraser VJ, Doherty J, Warren DK. Between community and hospital: healthcare-associated Gram-negative bacteremia among hospitalized patients. Infect Control Hosp Epidemiol. 2009;30(11):1050–1056. | |
Shih MJ, Lee NY, Lee HC, et al. Risk factors of multidrug resistance in nosocomial bacteremia due to Acinetobacter baumannii: a case-control study. J Microbiol Immunol Infect. 2008;41(2):118–123. | |
Jang TN, Lee SH, Huang CH, Lee CL, Chen WY. Risk factors and impact of nosocomial Acinetobacter baumannii bloodstream infections in the adult intensive care unit: a case-control study. J Hosp Infect. 2009;73(2):143–150. | |
Thomas JK, Forrest A, Bhavnani SM, et al. Pharmacodynamic evaluation of factors associated with the development of bacterial resistance in acutely ill patients during therapy. Antimicrob Agents Chemother. 1998;42(3):521–527. | |
Hatcher J, Dhillon R, Azadian BS. Antibiotic resistance mechanisms in the intensive care unit. J Intensive Care Soc. 2012;13(4):297–303. | |
Olaitan AO, Morand S, Rolain JM. Mechanisms of polymyxin resistance: acquired and intrinsic resistance in bacteria. Front Microbiol. 2014;5:643. | |
Livermore DM. Multiple mechanisms of antimicrobial resistance in Pseudomonas aeruginosa: our worst nightmare? Clin Infect Dis. 2002; 34(5):634–640. | |
Uttley AC, Collins C, Naidoo J, George R. Vancomycin-resistant enterococci. Lancet. 1988;331(8575):57–58. | |
Elsner HA, Sobottka I, Mack D, Laufs R, Claussen M, Wirth R. Virulence factors of Enterococcus faecalis and Enterococcus faecium blood culture isolates. Eur J Clin Microbiol Infect Dis. 2000;19(1):39–42. | |
Arias C, Murray B. Emergence and management of drug-resistant enterococcal infections. Expert Rev Anti Infect Ther. 2008;6(5):637–655. | |
Hollenbeck BL, Rice LB. Intrinsic and acquired resistance mechanisms in enterococcus. Virulence. 2012;3(5):421–433. | |
Shankar N, Baghdayan AS, Gilmore MS. Modulation of virulence within a pathogenicity island in vancomycin-resistant Enterococcus faecalis. Nature. 2002;417(6890):746–750. | |
Van Wamel WJ, Hendrickx AP, Bonten MJ, Top J, Posthuma G, Willems RJ. Growth condition-dependent Esp expression by Enterococcus faecium affects initial adherence and biofilm formation. Infect Immun. 2007;75(2):924–931. | |
Leclercq R. Epidemiological and resistance issues in multidrug-resistant staphylococci and enterococci. Clin Microbiol Infect. 2009;15(3):224–2231. | |
Lowy F. Staphylococcus aureus infections. N Engl J Med. 1998;339(8):520–532. | |
Chambers HF, DeLeo FR. Waves of resistance: Staphylococcus aureus in the antibiotic era. Nat Re Microbiol. 2009;7(9):629–641. | |
Costerton J, Stewart PS, Greenberg E. Bacterial biofilms: a common cause of persistent infections. Science. 1999;284(5418):1318–1322. | |
Katayama Y, Ito T, Hiramatsu K. A new class of genetic element, Staphylococcus cassette chromosome mec, encodes methicillin resistance in Staphylococcus aureus. Antimicrob Agents Chemother. 2000;44(6):1549–1555. | |
Utsui Y, Yokota T. Role of an altered penicillin-binding protein in methicillin- and cephem-resistant Staphylococcus aureus. Antimicrob Agents Chemother. 1985;28(3):397–403. | |
Geraci D, Bonura C, Giuffrè M, et al. tst1-Positive ST22-MRSA-IVa in healthy Italian preschool children. Infection. 2014;42(3):535–538. | |
Lamaro-Cardoso J, de Lencastre H, Kipnis A, et al. Molecular epidemiology and risk factors for nasal carriage of Staphylococcus aureus and methicillin-resistant S. aureus in infants attending day care centers in Brazil. J Clin Microbiol. 2009;47(12):3991–3997. | |
Appelbaum PC. Reduced glycopeptide susceptibility in methicillin-resistant Staphylococcus aureus (MRSA). Int J Antimicrob Agents. 2007;30(5):398–408. | |
Paterson DL. Resistance in gram-negative bacteria: Enterobacteriaceae. Am J Med. 2006;119(6 Suppl 1):S20–S28. | |
Pitout JD, Laupland KB. Extended-spectrum β-lactamase-producing Enterobacteriaceae: an emerging public-health concern. Lancet Infect Dis. 2008;8(3):159–166. | |
Hawser SP, Bouchillon SK, Hoban DJ, Badal RE, Hsueh PR, Paterson DL. Emergence of high levels of extended-spectrum-β-lactamase-producing Gram-negative bacilli in the Asia-Pacific region: data from the Study for Monitoring Antimicrobial Resistance Trends (SMART) program, 2007. Antimicrob Agents Chemother. 2009;53(8):3280–3284. | |
Bush K, Jacoby GA, Medeiros AA. A functional classification scheme for beta-lactamases and its correlation with molecular structure. Antimicrob Agents Chemother. 1995;39(6):1211–1233. | |
Hall BG, Barlow M. Revised Ambler classification of β-lactamases. J Antimicrob Chemother. 2005;55(6):1050–1051. | |
Tumbarello M, Viale P, Viscoli C, et al. Predictors of mortality in bloodstream infections caused by Klebsiella pneumoniae carbapenemase-producing K. pneumoniae: importance of combination therapy. Clin Infect Dis. 2012;55(7):943–950. | |
Nordmann P, Cuzon G, Naas T. The real threat of Klebsiella pneumoniae carbapenemase-producing bacteria. Lancet Infect Dis. 2009;9(4):228–236. | |
Matar G, Cuzon G, Araj G, et al. Oxacillinase-mediated resistance to carbapenems in Klebsiella pneumoniae from Lebanon. Clin Microbiol Infect. 2008;14(9):887–888. | |
Cuzon G, Naas T, Bogaerts P, Glupczynski Y, Huang TD, Nordmann P. Plasmid-encoded carbapenem-hydrolyzing β-lactamase OXA-48 in an imipenem-susceptible Klebsiella pneumoniae strain from Belgium. Antimicrob Agents Chemother. 2008;52(9):3463–3464. | |
Yigit H, Queenan AM, Anderson GJ, et al. Novel carbapenem-hydrolyzing β-lactamase, KPC-1, from a carbapenem-resistant strain of Klebsiella pneumoniae. Antimicrob Agents Chemother. 2001;45(4):1151–1161. | |
Zhang R, Yang L, Cai JC, Zhou HW, Chen GX. High-level carbapenem resistance in a Citrobacter freundii clinical isolate is due to a combination of KPC-2 production and decreased porin expression. J Med Microbiol. 2008;57(3):332–337. | |
Podschun R, Ullmann U. Klebsiella spp. as nosocomial pathogens: epidemiology, taxonomy, typing methods, and pathogenicity factors. Clin Microbiol Rev. 1998;11(4):589–603. | |
Gutu AD, Sgambati N, Strasbourger P, et al. Polymyxin resistance of Pseudomonas aeruginosa phoQ mutants is dependent on additional two-component regulatory systems. Antimicrob Agents Chemother. 2013;57(5):2204–2215. | |
Mezzatesta M, Gona F, Caio C, et al. Outbreak of KPC-3-producing, and colistin-resistant, Klebsiella pneumoniae infections in two Sicilian hospitals. Clin Microbiol Infect. 2011;17(9):1444–1447. | |
Mammina C, Bonura C, Di Bernardo F, et al. Ongoing spread of colistin-resistant Klebsiella pneumoniae in different wards of an acute general hospital, Italy, June to December 2011. Euro Surveill. 2012;17(33):1–4. | |
Velkov T, Deris ZZ, Huang JX, et al. Surface changes and polymyxin interactions with a resistant strain of Klebsiella pneumoniae. Innate Immun. 2014;20(4):350–363. | |
Cheng HY, Chen YF, Peng HL. Molecular characterization of the PhoPQ-PmrD-PmrAB mediated pathway regulating polymyxin B resistance in Klebsiella pneumoniae CG43. J Biomed Sci. 2010;17:60. | |
Cannatelli A, D’Andrea MM, Giani T, et al. In vivo emergence of colistin resistance in Klebsiella pneumoniae producing KPC-type carbapenemases mediated by insertional inactivation of the PhoQ/PhoP mgrB regulator. Antimicrob Agents Chemother. 2013;57(11):5521–5526. | |
Olaitan AO, Diene SM, Kempf M, et al. Worldwide emergence of colistin resistance in Klebsiella pneumoniae from healthy humans and patients in Lao PDR, Thailand, Israel, Nigeria and France owing to inactivation of the PhoP/PhoQ regulator mgrB: an epidemiological and molecular study. Int J Antimicrob Agents. 2014;44(6):500–507. | |
Carlet J, Mainardi JL. Antibacterial agents: back to the future? Can we live with only colistin, co-trimoxazole and fosfomycin? Clin Microbiol Infect. 2012;18(1):1–3. | |
Kaper JB, Nataro JP, Mobley HL. Pathogenic Escherichia coli. Nat Rev Microbiol. 2004;2(2):123–140. | |
Livermore DM, Hope R, Brick G, Lillie M, Reynolds R. Non-susceptibility trends among Enterobacteriaceae from bacteraemias in the UK and Ireland, 2001– 2006. J Antimicrob Chemother. 2008;62 Suppl 2:ii41–ii54. | |
Höjgård S. Antibiotic resistance – why is the problem so difficult to solve? Infect Ecol Epidemiol. 2012;2:18165. | |
Solé M, Pitart C, Roca I, et al. First description of an Escherichia coli strain producing NDM-1 carbapenemase in Spain. Antimicrob Agents Chemother. 2011;55(9):4402–4404. | |
Nummila K, Kilpeläinen I, Zähringer U, Vaara M, Helander IM. Lipopolysaccharides of polymyxin B-resistant mutants of Escherichia coli are extensively substituted by 2-aminoethyl pyrophosphate and contain aminoarabinose in lipid A. Mol Microbiol. 1995;16(2):271–278. | |
Sanders W, Sanders CC. Enterobacter spp.: pathogens poised to flourish at the turn of the century. Clin Microbiol Rev. 1997;10(2):220–241. | |
Mezzatesta ML, Gona F, Stefani S. Enterobacter cloacae complex: clinical impact and emerging antibiotic resistance. Future Microbiol. 2012;7(7):887–902. | |
Kramer A, Schwebke I, Kampf G. How long do nosocomial pathogens persist on inanimate surfaces? A systematic review. BMC Infect Dis. 2006;6:130. | |
Vila J, Martí S, Sánchez-Céspedes J. Porins, efflux pumps and multidrug resistance in Acinetobacter baumannii. J Antimicrob Chemother. 2007;59(6):1210–1215. | |
Joly-Guillou ML. Clinical impact and pathogenicity of Acinetobacter. Clin Microbiol Infect. 2005;11(11):868–873. | |
Queenan AM, Bush K. Carbapenemases: the versatile β-lactamases. Clin Microbiol Rev. 2007;20(3):440–458. | |
Poirel L, Nordmann P. Carbapenem resistance in Acinetobacter baumannii: mechanisms and epidemiology. Clin Microbiol Infect. 2006;12(9):826–836. | |
Potron A, Poirel L, Nordmann P. Emerging broad-spectrum resistance in Pseudomonas aeruginosa and Acinetobacter baumannii: mechanisms and epidemiology. Int J Antimicrob Agents. 2015;45(6):568–585. | |
Ruzin A, Keeney D, Bradford PA. AdeABC multidrug efflux pump is associated with decreased susceptibility to tigecycline in Acinetobacter calcoaceticus–Acinetobacter baumannii complex. J Antimicrob Chemother. 2007;59(5):1001–1004. | |
Bonomo RA, Szabo D. Mechanisms of multidrug resistance in Acinetobacter species and Pseudomonas aeruginosa. Clin Infect Dis. 2006;43(Suppl 2):S49–S56. | |
Adams MD, Nickel GC, Bajaksouzian S, et al. Resistance to colistin in Acinetobacter baumannii associated with mutations in the PmrAB two-component system. Antimicrob Agents Chemother. 2009;53(9):3628–3634. | |
Moffatt JH, Harper M, Harrison P, et al. Colistin resistance in Acinetobacter baumannii is mediated by complete loss of lipopolysaccharide production. Antimicrob Agents Chemother. 2010;54(12):4971–4977. | |
Fothergill JL, Winstanley C, James CE. Novel therapeutic strategies to counter Pseudomonas aeruginosa infections. Expert Rev Anti Infect Ther. 2012;10(2):219–235. | |
Adair FW, Geftic SG, Gelzer J. Resistance of Pseudomonas to quaternary ammonium compounds. I. Growth in benzalkonium chloride solution. Appl Microbiol. 1969;18(3):299–302. | |
Livermore DM, Woodford N. The β-lactamase threat in Enterobacteriaceae, Pseudomonas and Acinetobacter. Trends Microbiol. 2006;14(9):413–420. | |
Vahaboglu H, Oztürk R, Aygün G, et al. Widespread detection of PER-1-type extended-spectrum β-lactamases among nosocomial Acinetobacter and Pseudomonas aeruginosa isolates in Turkey: a nationwide multicenter study. Antimicrob Agents Chemother. 1997; 41(10):2265–2269. | |
Lauretti L, Riccio ML, Mazzariol A, et al. Cloning and characterization of blaVIM, a new integron-borne metallo-β-lactamase gene from a Pseudomonas aeruginosa clinical isolate. Antimicrob Agents Chemother. 1999;43(7):1584–1590. | |
Poirel L, Magalhaes M, Lopes M, Nordmann P. Molecular analysis of metallo-β-lactamase gene bla(SPM-1)-surrounding sequences from disseminated Pseudomonas aeruginosa isolates in Recife, Brazil. Antimicrob Agents Chemother. 2004;48(4):1406–1409. | |
Rodríguez-Martínez J-M, Poirel L, Nordmann P. Extended-spectrum cephalosporinases in Pseudomonas aeruginosa. Antimicrob Agents Chemother. 2009;53(5):1766–1771. | |
Girlich D, Naas T, Nordmann P. Biochemical characterization of the naturally occurring oxacillinase OXA-50 of Pseudomonas aeruginosa. Antimicrob Agents Chemother. 2004;48(6):2043–2048. | |
Poirel L, Naas T, Nordmann P. Diversity, epidemiology, and genetics of class D β-lactamases. Antimicrob Agents Chemother. 2010;54(1):24–38. | |
Philippon LN, Naas T, Bouthors AT, Barakett V, Nordmann P. OXA-18, a class D clavulanic acid-inhibited extended-spectrum β-lactamase from Pseudomonas aeruginosa. Antimicrob Agents Chemother. 1997;41(10):2188–2195. | |
Sevillano E, Gallego L, García-Lobo J. First detection of the OXA-40 carbapenemase in P. aeruginosa isolates, located on a plasmid also found in A. baumannii. Pathol Biol (Paris). 2009;57(6):493–495. | |
El Garch F, Bogaerts P, Bebrone C, Galleni M, Glupczynski Y. OXA-198, an acquired carbapenem-hydrolyzing class D β-lactamase from Pseudomonas aeruginosa. Antimicrob Agents Chemother. 2011;55(10):4828–4833. | |
Hawkey P, Finch R. Tigecycline: in-vitro performance as a predictor of clinical efficacy. Clin Microbiol Infect. 2007;13(4):354–362. | |
Schwaber MJ, Carmeli Y. Mortality and delay in effective therapy associated with extended-spectrum β-lactamase production in Enterobacteriaceae bacteraemia: a systematic review and meta-analysis. J Antimicrob Chemother. 2007;60(5):913–920. | |
Nguyen M, Eschenauer GA, Bryan M, et al. Carbapenem-resistant Klebsiella pneumoniae bacteremia: factors correlated with clinical and microbiologic outcomes. Diagn Microbiol Infect Dis. 2010;67(2):180–184. | |
Qureshi ZA, Paterson DL, Potoski BA, et al. Treatment outcome of bacteremia due to KPC-producing Klebsiella pneumoniae: superiority of combination antimicrobial regimens. Antimicrob Agents Chemother. 2012;56(4):2108–2113. | |
Daikos G, Markogiannakis A. Carbapenemase-producing Klebsiella pneumoniae: (when) might we still consider treating with carbapenems? Clin Microbiol Infect. 2011;17(8):1135–1141. |
 © 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.