Back to Journals » International Journal of Chronic Obstructive Pulmonary Disease » Volume 18
Blocking CD40 Alleviates Th1 and Th17 Cell Responses in Elastin Peptide-Induced Murine Emphysema
Authors Ma T, Zhang H, Weng Y, Tang S ![]() , Mao J, Feng X, Zhang Y, Zhang J
, Mao J, Feng X, Zhang Y, Zhang J
Received 29 July 2023
Accepted for publication 9 November 2023
Published 23 November 2023 Volume 2023:18 Pages 2687—2698
DOI https://doi.org/10.2147/COPD.S428832
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Prof. Dr. Richard Russell
Tingting Ma,1,2,* Hui Zhang,3,* Yuqing Weng,2,* Shudan Tang,3 Jinshan Mao,2 Xin Feng,3 Yuxin Zhang,4 Jianquan Zhang1
1Department of Respiratory and Critical Medicine, The Eighth Affiliated Hospital, Sun Yat-sen University, Shenzhen, Guangdong, 518000, People’s Republic of China; 2Department of Respiratory and Critical Medicine, Zhuhai People’s Hospital (Zhuhai Hospital Affiliated with Jinan University), Zhuhai, Guangdong, 519000, People’s Republic of China; 3Department of Respiratory Medicine, The First Affiliated Hospital of Guangxi Medical University, Nanning, Guangxi, 530021, People’s Republic of China; 4The First Clinical Medical College, Southern Medical University, Guangzhou, Guangdong, 510515, People’s Republic of China
*These authors contributed equally to this work
Correspondence: Jianquan Zhang, Department of Respiratory and Critical Medicine, The Eighth Affiliated Hospital, Sun Yat-sen University, Shenzhen, Guangdong, 518000, People’s Republic of China, Tel +8613978123845, Fax +86755-23482484, Email [email protected]
Purpose: To investigate the role of the CD40-CD40 ligand (CD40L) pathway in the regulation of Th1, Th17, and regulatory T (Treg)-cell responses in an elastin peptide (EP)-induced autoimmune emphysema mouse model.
Methods: BALB/c mice were transnasally treated with EP on day 0, injected intravenously with anti-CD40 antibody via the tail vein on day 33, and sacrificed on day 40. The severity of emphysema was evaluated by determining the mean linear intercept (MLI) and destructive index (DI) from lung sections. The proportions of myeloid dendritic cells (mDCs) and Th1, Th17, and Treg cells in the blood, spleen, and lungs were determined via flow cytometry. The levels of the cytokines interleukin (IL)-6, IL-17, interferon (IFN)-γ, and transforming growth factor (TGF)-β were detected via enzyme-linked immunosorbent assay. Ifnγ, IL17a, Rorγt and Foxp3 transcription levels were detected via polymerase chain reaction.
Results: CD40+ mDCs accumulated in the lungs of EP-stimulated mice. Blocking the CD40-CD40L pathway with an anti-CD40 antibody alleviated Th1 and Th17 responses; increased the proportion of Treg cells; decreased MLI and DI; reduced the levels of cytokines IL-6, IL-17, and IFN-γ as well as the transcription levels of Ifnγ, IL17a, and Rorγt; and upregulated the expression of TGF-β and Foxp3.
Conclusion: The CD40-CD40L pathway could play a critical role in Th1, Th17 and Treg cell dysregulation in EP-mediated emphysema and could be a potential therapeutic target.
Keywords: elastin peptide, dendritic cell, Cluster of Differentiation 40, CD40, Th cell, emphysema
Introduction
Chronic obstructive pulmonary disease (COPD) is a heterogeneous lung condition characterized by persistent respiratory symptoms and incompletely reversible airflow limitation due to airway abnormalities1. COPD affects more than 300 million people and has become the third major cause of death globally.2 However, current treatment modalities for COPD lack effectiveness and are less ineffective in controlling airway inflammation and halting the decline in lung function. Thus, more effective treatment modalities and targets need to be identified.
The two traditional clinical phenotypes of COPD are emphysema and chronic bronchitis. Emphysema, which is characterized by abnormal and permanent expansion of distal airspaces and destruction of alveolar walls, is a major pathological feature of COPD.3 As the most important risk factors for COPD, cigarette smoke exposure can cause direct structural damage to the alveoli and epithelial cells in the airways.4 In addition, cigarette smoke exposure can also trigger the activation of innate and adaptive immune cells in the lungs, which results in persistent amplifying inflammation and contributes to the destruction of the lung parenchyma.5 The activation and dysregulation of adaptive immune cells results in irreversible airway inflammation and emphysema in patients with COPD and experimental animal models.6,7 Notably, chronic cigarette smoke exposure induces the release of damage-associated molecular patterns (DAMPs), such as lung elastin fragments, which can promote autoimmunity and become involved in COPD pathogenesis.8–11
Elastin peptide (EP), which is produced by neutrophil and macrophage elastase in lung tissue through the decomposition of elastin fibers, accumulates significantly in the lungs of patients with COPD and healthy smokers.12,13 Hexapeptides with a VGVAPG structure have the broadest range of biological activity.14 The amount of EP positively correlates with the severity of emphysema and even predicts poor outcomes in COPD.12,15 EP can not only cause emphysema-related changes in the lungs, but also promote dysregulation of adaptive immune cells in the airway and even airflow limitations, making it an ideal agent for inducing COPD in animals.5,11,14,16,17
The interaction between EP and the elastin receptor S-gal could be one of the mechanisms by which EP promotes T-cell activation,18 but the mechanisms for regulating T-cell differentiation remain unclear. It has been reported that the CD40-CD40 ligand (CD40L) costimulatory signaling pathway plays a critical role in myeloid dendritic cell (mDC)-dependent activation of Th-cell immunity and in the development of smoking-associated COPD/emphysema.19,20 The CD40 receptor on the surface of DCs binds with CD40L on the surface of T cells and then regulates Th cell activation and differentiation.21 CD40 plays a crucial role in promoting the polarization of Th17 cells.22,23 However, whether the CD40-CD40L axis could serve as a therapeutic target in EP-induced emphysema is unclear.
In this study, we used the anti-CD40 antibody HM40-3 to competitively inhibit the interaction between CD40 and CD40L in EP-induced emphysema mice and observed Th1, Th17, and Treg cell responses. This study aimed to further investigate the autoimmunity mechanisms of EP-mediated emphysema.
Materials and Methods
Animals and Ethics
BALB/c mice (6–8 weeks of age) weighing 18–22 g were purchased from the Guangxi Medical University Laboratory Animal Center (Nanning, China). All mice were housed in standard laboratory cages with free access to clean food and water with a 12-h light-dark cycle. They were randomly divided into three groups, namely, the control group, EP group, and EP+anti-CD40 group, with eight mice per group, as described below. All experimental protocols were approved by the Animal Research Care Committee for Animal Studies of Guangxi Medical University, China. Animal ethics review followed the Guiding Opinions on the Treatment of Laboratory Animals issued by the Ministry of Science and Technology of the People’s Republic of China and the Laboratory Animal Guideline for Ethical Review of Animal Welfare issued by the National Standard of the People’s Republic of China (GB/T35892-2018).
EP-Induced Emphysema Models and CD40 Blockade
EP (VGVAPG, Cat#GPS2957, Genepep, Prades-le-Lez, Paris, France) powder was dissolved in deionized water following the manufacturer’s instructions and then diluted in phosphate-buffered saline (PBS, Cat#10010023, Thermo Fisher Scientific, Massachusetts, USA) before use. According to a method described previously with minor adjustments,20 mice in the EP group and the EP+anti-CD40 group received intranasal instillations of EP suspensions (20 μg EP/50 μL PBS) after appropriate sedation on day 0. On day 33, the mice in the EP+anti-CD40 group were injected with anti-CD40 antibody (0.25 mL, 1 mg/mL, HM40-3, Cat#553721, BD Pharmingen, Franklin Lakes, NJ, USA) via the tail vein, while mice in the EP group were injected with an identical volume of PBS. Mice not receiving EP treatment that received an equal volume of PBS without EP via the tail vein were used as the control group. On day 40, all mice were anesthetized with 10% chloral hydrate (0.3 mL/100 g) and sacrificed. Blood was sampled from the posterior orbital vein after anesthesia. Peripheral blood mononuclear cells were isolated using Ficoll-Hypaque (Cat#P8620, Solarbio, Beijing, China) gradient centrifugation, as previously described.24 All animal experiments were replicated three times.
Single-Cell Suspension Processing
The spleens were chopped as finely as possible, dissolved as single-cell suspensions and filtered through a nylon mesh. Then, these cell suspensions were collected and centrifuged (300×g, 10 min). The supernatants were discarded, and the residual erythrocytes in the cell pellets were lysed for 5 min using 3 mL of RBC lysis buffer (Cat#R1010, Solarbio) at room temperature, preferably in the dark. Finally, the cells were washed twice with PBS by centrifugation.
Lung single-cell suspensions were obtained as described previously.17 Briefly, the lungs were cleaned with PBS, and PBS was flushed through the right ventricle. The lungs were then thoroughly chopped and digested at 37°C in RPMI 1640 medium (Cat#11875093, Gibco, Thermo Fisher Scientific, Massachusetts, USA) with 1 mg/mL type IV collagenase (Cat#C4-BIOC, Sigma–Aldrich, St. Louis, MO, USA) for 30–45 min. Next, the lung suspensions were ground with a syringe plunger and filtered through a strainer. The cell suspension was washed with PBS.
Histology
The left lung of each mouse was fixed with 10% formalin, embedded in paraffin, sectioned at a thickness of 5 μm, stained with hematoxylin-eosin (H&E), and analyzed histologically. We quantified the severity of emphysema by calculating the mean linear intercept (MLI) and destructive index (DI) as previously described.25,26 For each mouse, ten different regions were randomly collected from lung sections. Two investigators blinded to the specific groups measured and calculated the MLI and DI independently.
Bronchoalveolar Lavage Fluid Collection
For bronchoalveolar lavage fluid (BALF) collection, mice were first anesthetized, and then a tracheal cannula for PBS installation and recovery was inserted four times (0.25 mL each time). The collected liquid was centrifuged (300×g, 10 min). The supernatants were aspirated and stored at −80°C before use in enzyme-linked immunosorbent assays (ELISAs).
Flow Cytometry
The proportions of mDCs and Th1, Th17, and Treg cells from the blood, spleen, and lungs were determined via flow cytometry using the flow antibodies against the following molecules: CD11c (FITC; Cat#11-0114-82, eBioscience, San Diego, CA, USA), MHC-II (2 mg/mL, PerCP-eFluor® 710; Cat#46-5321-82, eBioscience), CD40 (PE; Cat#553791, BD Pharmingen, San Diego, CA, USA), CD4 (PerCP; Cat#553052, BD Pharmingen), CD25 (PE; Cat#MA5-17495, eBioscience), FOXP3 (APC; Cat#17-5773-82, eBioscience), interferon (IFN)-γ (APC; Cat#554413, BD Pharmingen), and interleukin (IL)-17A (PE; Cat#559502, BD Pharmingen). Appropriate isotype controls were used for each antibody. The cell surface markers were stained in the dark at 4°C for 30 min. For intracellular detection of IFN-γ and IL17, cells were stimulated with phorbol-myristate-acetate (25 ng/mL; Cat# P1585, Sigma–Aldrich) and ionomycin (1 μg/mL; Cat#407951, Sigma–Aldrich) in the presence of GolgiStop (containing monensin, Cat#554724, BD Pharmingen) for 4 h at 37°C in 5% CO2 before labeling. After surface staining, cells were fixed/infiltrated in a fixation/infiltration solution (Cytofix/Cytoperm™; Cat#554722, BD Pharmingen) according to the manufacturer’s instructions and dyed with APC-IFN-γ and PE-IL-17A (4°C, 30 min). Cells were then cleaned with buffer (Cat#554723, BD Pharmingen) and resuspended in PBS for flow cytometry analysis. To detect the transcription factor FOXP3, cells were fixed/infiltrated using the Foxp3/Transcription Factor Staining Buffer Set Kit (Cat#00-5523-00, eBioscience) after staining with PerCP-CD4 and PE-CD25. The cells were then stained with APC-FOXP3 for 30 min at 4°C. Flow cytometry was performed using a BD FACS Canto II (BD Biosciences, Franklin Lakes, NJ, USA). Data were analyzed using FlowJo 7.6 software.
Real-Time Quantitative Polymerase Chain Reaction (PCR)
Total RNA was obtained from lung tissue using TRIzol reagent according to the manufacturer’s instructions (Cat#9108, Takara, Dalian, China). The quality and quantity of total RNA were analyzed using a spectrophotometer (NanoDrop2000, Thermo Fisher Scientific, Waltham, MA, USA). Complementary DNA (cDNA) was transcribed from total RNA using a PrimeScript RT reagent kit with gDNA Eraser following the manufacturer’s protocol (Cat#RR047Q, Takara, Dalian, China). Real-time PCR was conducted using SYBR® Premix Ex Taq™ II (Cat#RR820A, Takara) in a typical 20-μL PCR mixture that included 10 μL of SYBR® Premix Ex Taq™ II, 0.8 μL of each PCR primer, 2 μL of template cDNA, 0.4 μL of ROX reference dye, and 6 μL of ddH2O. The cycling conditions were as follows: 95°C for 30s, followed by 40 cycles at 95°C for 5 s and 60°C for 30s. All samples were run on the ABI 7500 Real-Time PCR System (Applied Biosystems, Foster City, CA, USA). The following primers were used: β-actin forward, 5ʹ-CATCCGTAAAGACCTCTATGCCAAC-3ʹ; β-actin reverse, 5ʹ-ATGGAGCCACCGATCCACA-3ʹ; Rorγt forward, 5ʹ-GCTCCATATTTGACTTTTCCCACT-3ʹ; Rorγt reverse, 5ʹ-GATGTTCCACTCTCCTCTTCTCTTG-3ʹ; Ifn-γ forward, 5ʹ-AGGAACTGGCAAAAGGATGGT-3ʹ; Ifn-γ reverse, 5ʹ-ACGCTTATGTTGTTGCTGATGG-3ʹ; Foxp3 forward, 5ʹ-AGTGCCTGTGTCCTCAATGGT-3ʹ, Foxp3 reverse, 5ʹ-AGGGCCAGCATAGGTGCAAG-3ʹ. Relative gene expression was calculated using the 2−∆∆Ct method.
Cytokine Detection
For cytokine detection, the concentrations of IFN-γ, IL-17, IL-6, and transforming growth factor (TGF)-β in BALF were measured using ELISA kits (Cat#CSB-E04578m, Cat#CSB-E04608m, Cat#CSB-E04639m, Cat#CSB-E04726m, Cusabio, Wuhan, China) according to the manufacturer’s protocols. All samples were assayed in duplicate.
Statistical Analysis
Data are expressed as the mean ± standard deviation. According to whether the data were normally distributed, differences between groups were compared using parametric one-way ANOVA or nonparametric Kruskal–Wallis tests with Bonferroni adjustment. All statistical analyses were performed using SPSS version 16.0 Statistical Software (Chicago, IL, USA); P < 0.05 was considered statistically significant.
Results
Blocking CD40 Attenuates EP-Induced Emphysema in Mice
We have shown that EP promotes emphysema in a previous study.17 In this study, the MLI value was significantly higher in the EP group than in the control group (26.26 ± 2.49 μm vs 18.58 ± 1.66 μm, P < 0.001; Figure 1A and B). The DI in the EP group was also significantly higher than that in the control group (75.88 ± 4.93% vs 51.88 ± 7.68%, P < 0.001; Figure 1C). In contrast, after anti-CD40 treatment, the MLI in the EP+anti-CD40 group was lower than that in the EP group (18.69 ± 1.66 μm vs 26.26 ± 2.49 μm, P < 0.001; Figure 1A and B). Compared with the EP group, the DI in the EP+anti-CD40 group was also lower (63.95 ± 6.51% vs 75.88 ± 4.93%, P =0.004; Figure 1C).
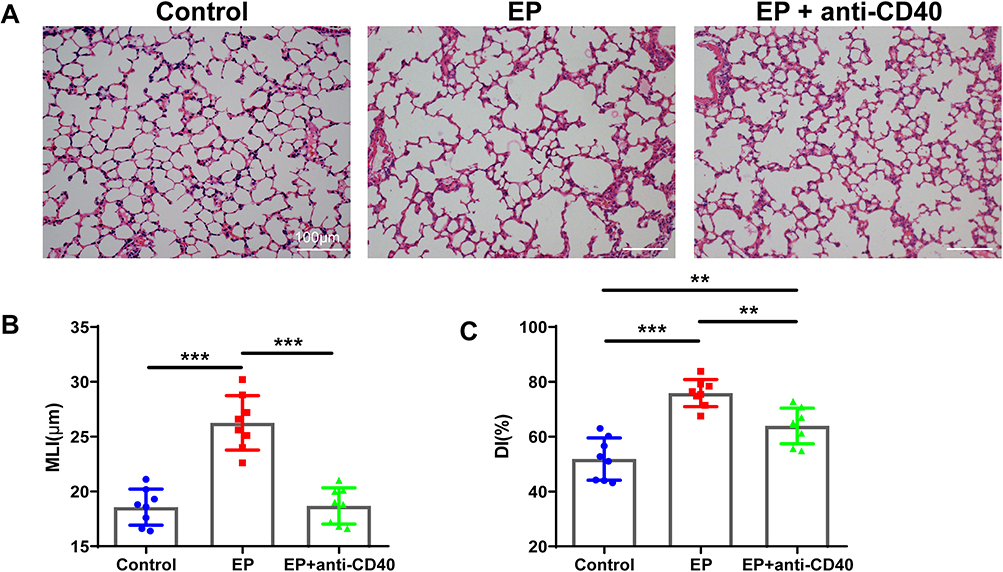 |
Figure 1 EP induces emphysema, and blocking the CD40-CD40L pathway reduces emphysema in mice. (A) Representative images of lung sections showing the pulmonary pathology of the control group, EP group, and EP+anti-CD40 group. Comparison of the lung (B) MLI and (C) DI among the three groups. Data are expressed as the mean ± standard deviation (n = 8 in each group). **P < 0.01, ***P < 0.001. Abbreviations: EP, elastin peptide; MLI, mean linear intercept; DI, destructive index. |
Anti-CD40 Antibody Decreases the Expression of CD40 on mDCs in EP-Treated Mice
Compared with the control group, the EP group showed increased proportions of mDCs in the spleen (0.93 ± 0.28% vs 0.46 ± 0.13%, P < 0.001) and lung (0.92 ± 0.38% vs 0.40 ± 0.14%, P = 0.021) (Figure 2A and B). The proportion of mDCs in the spleen of mice in the EP+anti-CD40 group showed no significant differences from that in the EP group (0.99 ± 0.21% vs 0.93 ± 0.28%, P > 0.999). The proportion of mDCs in the lungs of the EP+anti-CD40 group (3.70 ± 0.44%) was higher than that of the control group (0.40 ± 0.14%, P < 0.001) and EP group (0.92 ± 0.38%, P < 0.001) (Figure 2B).
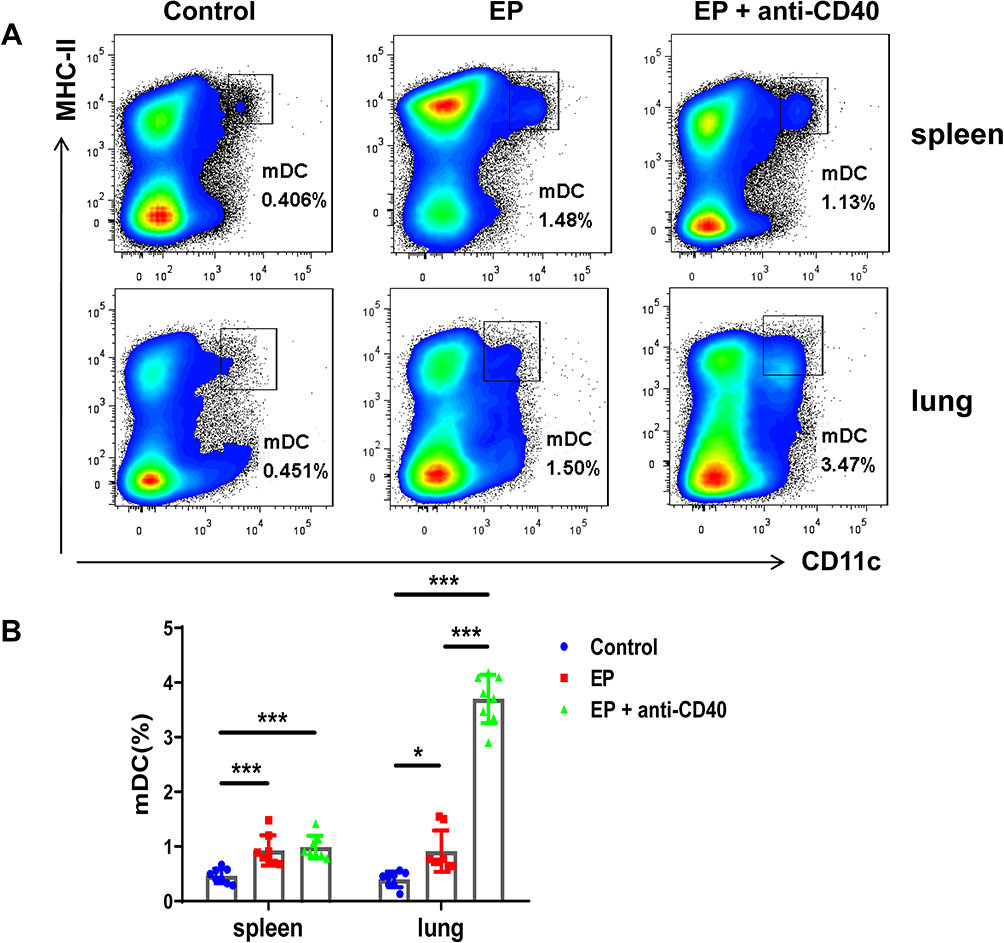 |
Figure 2 EP induces mDC expansion in mice. (A) Representative flow cytometric scatter plot of the proportions of mDCs in the spleen and lungs in the control group, EP group, and EP+anti-CD40 group. (B) Comparisons of the proportions of mDCs in the spleen and lungs among the three groups. Data are expressed as the mean ± standard deviation (n = 8 in each group). *P < 0.05, ***P < 0.001. Abbreviation: mDCs, myeloid dendritic cells. |
Next, we evaluated the expression of CD40 on mDCs in the lungs and spleen. The proportion of CD40 in the EP group was significantly higher than that in the spleens and lungs of the control group (26.24 ± 5.71% vs 10.21 ± 5.05%, 30.15 ± 7.33% vs 14.27 ± 2.63%, both P < 0.001; Figure 3A and B). After blocking with anti-CD40 antibody, the proportion of CD40 decreased in the spleens and lungs of mice in the EP+anti-CD40 group (11.75 ± 6.64% vs 26.24 ± 5.71%, 14.55 ± 3.90% vs 30.15 ± 7.33%, both P < 0.001; Figure 3A and B). The results of the geometric fluorescence intensity of CD40 showed the same trend as those of CD40 proportions (Figure 3C).
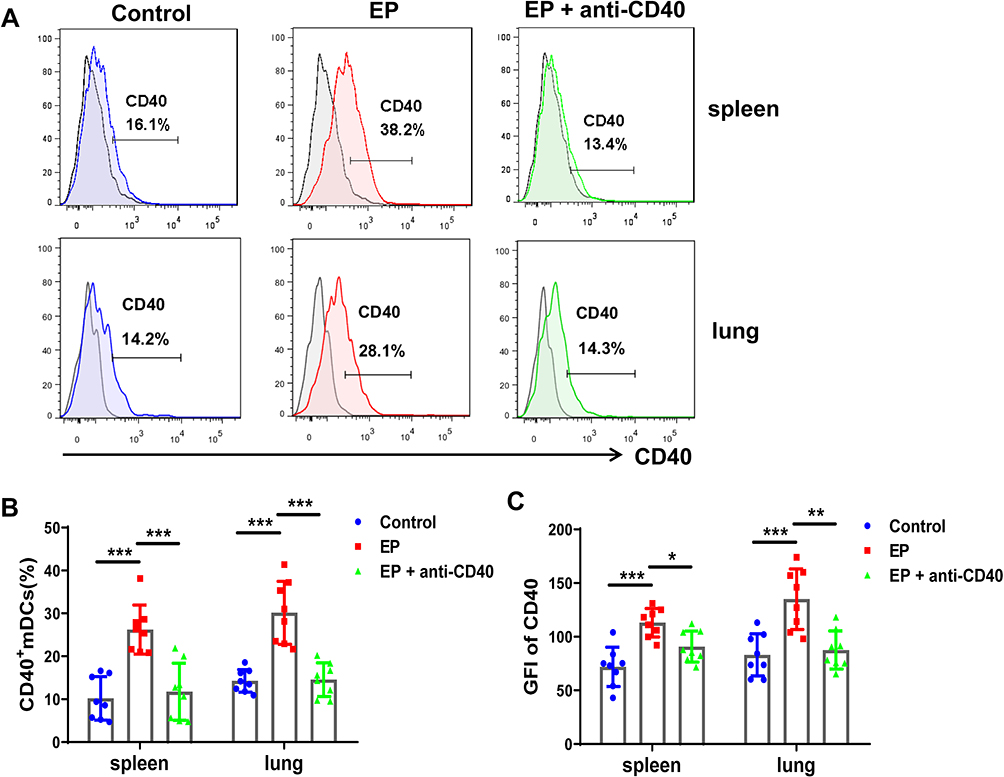 |
Figure 3 Effect of anti-CD40 antibody on CD40+ mDC expression. (A) Representative flow cytometric histogram of mDCs in the spleens and lungs of the control group, EP group, and EP+anti-CD40 group. (B) Comparisons of the proportions of CD40+ mDCs in the spleen and lungs among the three groups (n = 8 mice/group). (C) Comparisons of the GFI of CD40 in mDCs among the three groups. Data are expressed as the mean ± standard deviation (n = 8 in each group). *P < 0.05, **P < 0.01, ***P < 0.001. Abbreviation: GFI, geometric fluorescence intensity. |
Blocking CD40 Decreases EP-Induced Pulmonary and Systemic Th1 and Th17 Cell Responses and Promotes Treg Cell Expansion
We previously found that EP triggers Th1 and Th17 cell responses and suppresses Treg cells.17 To further investigate whether the CD40-CD40L pathway plays a critical role in Th17/Treg- and Th1-related cell responses in EP-treated mice, we measured the proportions of Th1, Th17, and Treg cells in EP-treated mice injected with the anti-CD40 antibody. As expected, Th1 and Th17 cell proportions in the blood, spleen, and lungs were significantly lower in the EP+anti-CD40 group than in the EP group (all P < 0.05; Figure 4A–D). In addition, Treg cells were significantly more abundant in the blood, spleen, and lungs of mice in the EP+anti-CD40 group than in the EP group (all P < 0.001; Figure 5A and B). The mRNA levels of Ifn-γ, Rorγt, and Foxp3 were in line with the trends of Th1, Th17, and Treg proportions, respectively (Figure 6A–C).
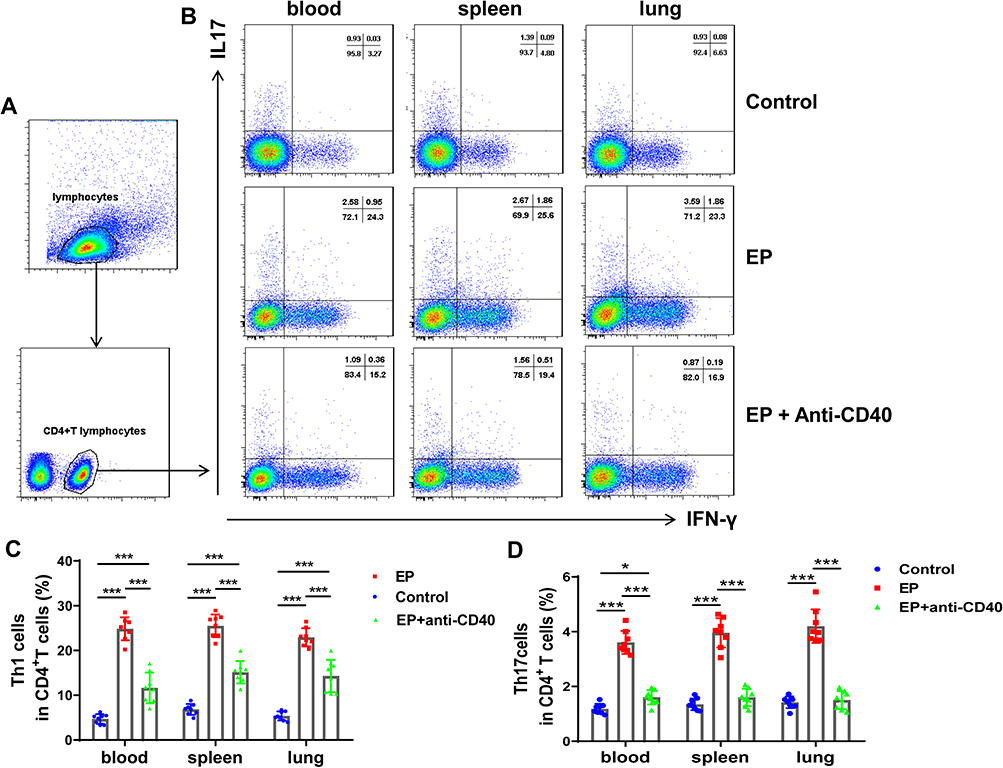 |
Figure 4 Blocking the CD40-CD40L pathway reduces EP-induced Th17 and Th1 cell responses in mice. (A) Gating strategy for Th1 and Th17 cells. Lymphocytes were identified based on FSC and SSC. Th1 cells were identified as CD4+IFN-γ+ cells, whereas Th17 cells were identified as CD4+IL-17+ cells. (B) Representative flow cytometry scatter plot of Th1 and Th17 cells in the spleens and lungs of the control group, EP group, and EP+anti-CD40 group. Comparisons of the proportions of (C) Th1 and (D) Th17 cells in the spleen and lungs among the three groups. Data are expressed as the mean ± standard deviation (n = 8 in each group). *P < 0.05, ***P < 0.001. Abbreviations: FSC, forward scatter; SSC, side scatter. |
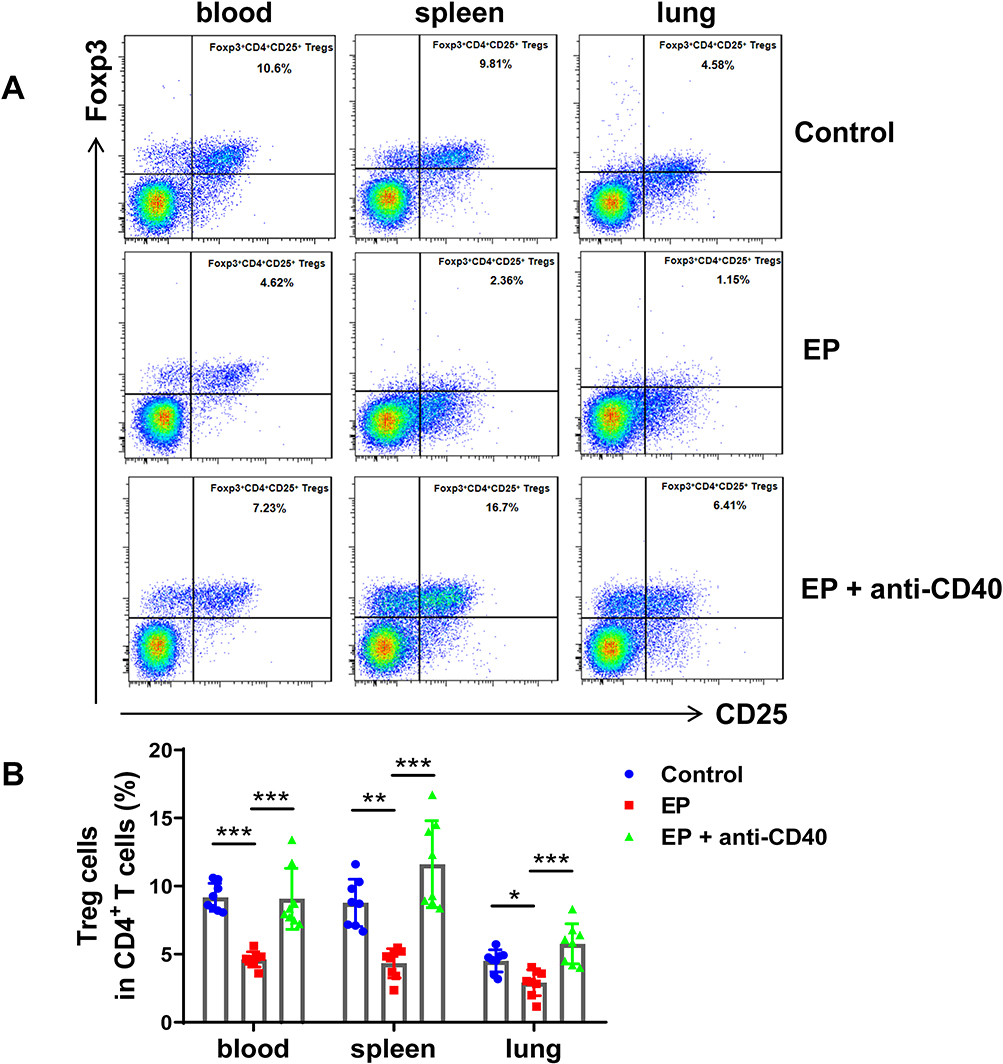 |
Figure 5 Blocking the CD40-CD40L pathway recovers Treg cells, which are depressed in EP-induced mice. (A) Representative flow cytometry scatter plot of Treg cells in the spleens and lungs of the control group, EP group, and EP+anti-CD40 group. (B) Comparisons of the proportions of CD4+CD25+FOXP3+ Treg cells in the spleen and lungs of the three groups. Data are expressed as the mean ± standard deviation (n = 8 in each group). *P < 0.05, **P < 0.01, ***P < 0.001. |
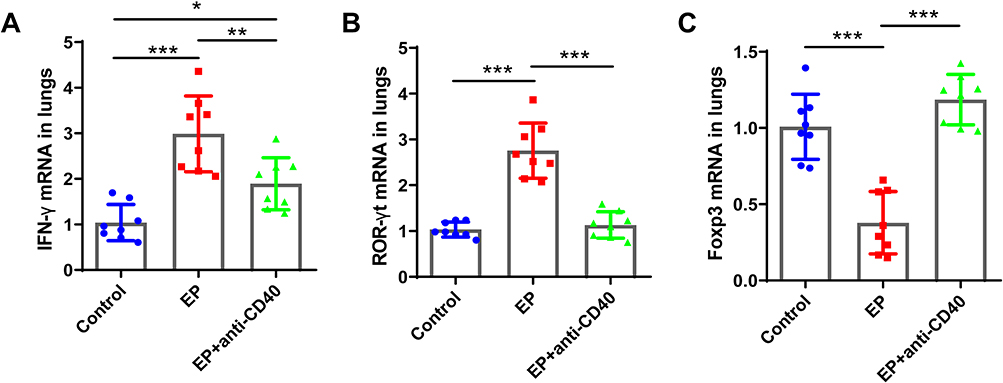 |
Figure 6 Comparisons of the relative mRNA expression levels of (A) Ifn-γ, (B) Rorγt, and (C) Foxp3 in the spleens and lungs of the control group, EP group, and EP+anti-CD40 group. Data are expressed as the mean ± standard deviation (n = 8 in each group). *P < 0.05, **P < 0.01, ***P < 0.001. |
Additionally, the levels of IL-6, IL-17, and IFN-γ were increased in the bronchoalveolar lavage fluid (BALF) of EP-exposed mice (Figure 7A–D). Compared with the EP group, the EP+anti-CD40 group showed significantly decreased levels of IL-6, IL-17, and IFN-γ (all P < 0.01; Figure 7A–C). In contrast, the level of TGF-β was markedly higher in the EP+anti-CD40 group than in the control and EP groups (all P < 0.001, Figure 7D).
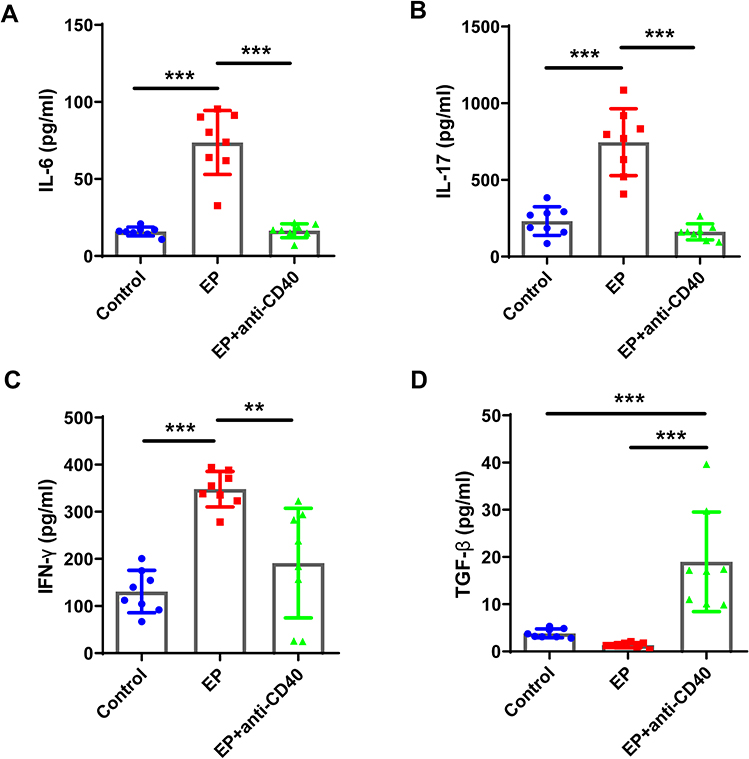 |
Figure 7 Comparisons of the levels of IL-6, IL-17, IFN-γ, and TGF-β in BALF. BALF was obtained from mice in the three groups and examined using ELISA. Comparison of the concentrations of (A) IL-6, (B) IL-17, (C) IFN-γ, and (D) TGF-β in BALF among the three groups. Data are expressed as the mean ± standard deviation (n = 8 in each group). **P < 0.01, ***P < 0.001. Abbreviations: BALF, bronchoalveolar lavage fluid; ELISA, enzyme-linked immunosorbent assay. |
Discussion
Adaptive immune dysregulation is one of the manifestations and mechanisms of COPD progression. Persistent immune inflammation leads to the remodeling and obstruction of small airways.27 The immune dysregulation of T cells in COPD is characterized by remarkable Th1-, Th17- and Tc1-cell responses and impaired function of Treg cells.28,29 CD4+ T-cell-mediated immune inflammation induced by EP synergistically strengthens the airway inflammatory response caused by smoking.17 We found that a single dose of 20 µg of VGVAPG promoted inflammatory Th1 and Th17 cell responses and reduced the number of Treg cells in the spleens and lungs of mice, leading to a systematic inflammatory response and immune dysregulation.17 The effect of EP is consistent with the immunologic changes associated with smoking-induced emphysema, such as an increased MLI; activation of DCs, Th1 cells, and Th17 cells; and suppression of Treg cells.18,30,31
Moreover, the proinflammatory effect of EP can be independent of cigarette smoke exposure.17,18 Herein, to rule out the effects of cigarette smoke on pulmonary immune cells, we used EP alone to induce the emphysema model. We found that EP exposure led to an accumulation of mDCs in the lungs with the upregulation of CD40 on mDCs, as well as an increase in Th1/Th17 cell responses and a decrease in Treg cells in mice. Blocking the CD40-CD40L pathway may alleviate Th1 and Th17 cell responses, increase Treg cell proportions, and alleviate the severity of emphysema. Our findings indicate that CD40-CD40L signaling could be one of the major pathways in the EP-triggered Th-cell response involved in the pathogenesis of EP-mediated emphysema.
CD40 expression is critical for the function of DCs, B cells, and monocytes. CD40 is upregulated when DCs are stimulated by microorganisms,21,32 Toll-like receptor ligands (eg, LPS),33 neutrophil extracellular traps,34 etc. DC activation plays a critical role in the initiation of the Th cell response.21 Herein, intravenous injection of an anti-CD40 antibody blocked the interaction between CD40 and CD40L, resulting in a decrease in the proportions of Th1 and Th17 cells and an increase in the proportion of Treg cells. Interestingly, blocking CD40 could promote the accumulation of mDCs in the lungs. A previous study showed that CD40 blockade could upregulate IL-4 produced by DCs.35 IL-4 can promote the expression of MHC-II and lead to an increasing number of CD11c+MHC-II+ mDCs. This may be the reason for the increased proportion of mDCs in the EP+anti-CD40 group compared with the EP group in our study. Notably, in the absence of analyzing CD40 on other immune cells together, we cannot fully attribute the change in Th cells to CD40 on DCs in our study, but we did not find any published studies that reported that CD40 on macrophages, monocytes or neutrophils could regulate Th1 or Th17 cells in COPD/emphysema.
Th1 and Th17 cells are two important proinflammatory cell types involved in airway inflammation in COPD.7 It was reported that CD40-CD40L influences Th17 differentiation more than Th1 cell differentiation.21,23 However, herein, we found that CD40 blockade also reduced the proportions of Th1 cells and Th17 cells. Indeed, it is clear that CD40L crosslinking can activate T cells and induce the production of IFN-γ.21 Blocking the CD40-CD40L interaction between DCs and T cells can also prevent the priming of IFN-γ production by CD8+ cells.36 IFN-γ can activate macrophages and strongly upregulate matrix metalloproteinase (MMP)12, a potent elastolytic proteinase that functions in emphysema development.7 In addition, IFN-γ and tumor necrosis factor (TNF)-α secreted by Th1 cells can promote B-cell differentiation into plasma cells, which express autoreactive antibodies.7 Th17 cells have attracted large amount of attention in recent years. Activated Th17 cells mainly function by producing the inflammatory cytokine IL-17, which is regulated by the transcription factor peroxisome proliferator-activated receptor-γ (RORγt).37 IL-17 was found to be critical for neutrophil and macrophage accumulation in a COPD mouse model.38 In addition, IL-17 can regulate the expression of receptor activator of nuclear factor-kB ligand (RANKL) on B cells and DCs and is involved in lymphoid neogenesis in COPD.39 Undoubtedly, Th17 cells play a critical role in the pathogenesis of COPD. Nevertheless, anti-IL17A monoclonal antibodies failed to show good efficacy in a Phase II clinical trial of COPD.40 This suggests that we need to search for more potentially effective targets.
In this study, we found that the Treg cell proportion was decreased when lung inflammation persisted in mice with EP-induced emphysema. Treg cells are subsets of CD4+ T cells with immunoregulatory functions that can inhibit autoimmunity and inflammation.29 Treg cells can inhibit other T cells or antigen-presenting DCs via a contact-dependent mechanism, such as CTLA-4 and CD80 and/or CD86 coinhibitory function, or by producing anti-inflammatory cytokines, such as IL-10 and TGF-β.29 CD40 inhibition was found to attenuate allergic symptoms and increase the proportion of Foxp3+ Tregs in mice.41 In our previous study, an increase in the proportion of Treg cells was observed when treatment for emphysema was effective.17 Here, after blocking CD40, Treg accumulation was increased, which was in line with that but may have different mechanisms. Adoptive Treg cell transfer treatment is successful in some organ transplant rejection and autoimmune diseases but is seldom reported in studies on COPD/emphysema.42 We believe that CD40 blockade may be an effective treatment to promote Foxp3+ Tregs and contribute to immune homeostasis.
This study has some limitations, including the lack of in vitro observations. Furthermore, we did not assess the proportions of other immune cells that expressed CD40. Further studies are needed to evaluate the effect of EP-induced emphysema on other immune cells (especially macrophages, monocytes and neutrophils). Despite these limitations, our study is valuable because, to our knowledge, this is the first report to identify CD40-CD40L as a regulator of the Th cell response in EP-induced emphysema. Moreover, our findings help provide a foundation for developing novel therapeutic modalities to regulate EP-triggered Th cell immune responses and airway inflammation in COPD.
Conclusion
In conclusion, blocking the CD40-CD40L pathway in vivo alleviated EP-triggered Th1 and Th17 cell responses, increased the proportion of Treg cells, and ultimately attenuated EP-induced emphysema. The levels of proinflammatory cytokines, such as IFN-γ, IL-6, and IL-17, were decreased in EP-exposed mice treated with the anti-CD40 antibody. Our findings prove that CD40-CD40L signaling is involved in EP-triggered CD4+ T-cell dysregulation and could serve as a therapeutic target in EP-mediated autoimmune emphysema.
Acknowledgments
This work was funded by grants from the National Natural Science Foundation of China (grant numbers 81460009 and 81760010), the Zhuhai People’s Hospital Cultivation Project (grant number 2019PY-18) and the Medical Science and Technology Research Fund Project of Guangdong Province (C2019042).
Disclosure
The authors declare that they have no conflicts of interest in this work.
References
1. Ananth S, Hurst JR. ERJ advances: state of the art in definitions and diagnosis of COPD. Eur Respir J. 2023;61(4):2202318. doi:10.1183/13993003.02318-2022
2. Stolz D, Mkorombindo T, Schumann DM, et al. Towards the elimination of chronic obstructive pulmonary disease: a lancet commission. Lancet. 2022;400(10356):921–972. doi:10.1016/s0140-6736(22)01273-9
3. Rosenwasser Y, Berger I, Loewy ZG. Therapeutic approaches for Chronic Obstructive Pulmonary Disease (COPD) exacerbations. Pathogens. 2022;11(12):1513. doi:10.3390/pathogens11121513
4. Milara J, Peiró T, Serrano A, et al. Roflumilast N-oxide inhibits bronchial epithelial to mesenchymal transition induced by cigarette smoke in smokers with COPD. Pulm Pharmacol Ther. 2014;28(2):138–148. doi:10.1016/j.pupt.2014.02.001
5. Wen L, Krauss-Etschmann S, Petersen F, Yu X. Autoantibodies in chronic obstructive pulmonary disease. Front Immunol. 2018;9:66. doi:10.3389/fimmu.2018.00066
6. Jin Y, Wan Y, Chen G, et al. Treg/IL-17 ratio and treg differentiation in patients with COPD. PLoS One. 2014;9(10):e111044. doi:10.1371/journal.pone.0111044
7. Kheradmand F, Zhang Y, Corry DB. Contribution of adaptive immunity to human COPD and experimental models of emphysema. Physiol Rev. 2023;103(2):1059–1093. doi:10.1152/physrev.00036.2021
8. Pouwels SD, Hesse L, Faiz A, et al. Susceptibility for cigarette smoke-induced DAMP release and DAMP-induced inflammation in COPD. Am J Physiol Lung Cell Mol Physiol. 2016;311(5):L881–L892. doi:10.1152/ajplung.00135.2016
9. Pouwels SD, Zijlstra GJ, van der Toorn M, et al. Cigarette smoke-induced necroptosis and DAMP release trigger neutrophilic airway inflammation in mice. Am J Physiol Lung Cell Mol Physiol. 2016;310(4):L377–L386. doi:10.1152/ajplung.00174.2015
10. Lee SH, Goswami S, Grudo A, et al. Antielastin autoimmunity in tobacco smoking-induced emphysema. Nat Med. 2007;13(5):567–569. doi:10.1038/nm1583
11. Zhou JS, Li ZY, Xu XC, et al. Cigarette smoke-initiated autoimmunity facilitates sensitisation to elastin-induced COPD-like pathologies in mice. Eur Respir J. 2020;56(3). doi:10.1183/13993003.00404-2020
12. He J, Turino GM, Lin YY. Characterization of peptide fragments from lung elastin degradation in chronic obstructive pulmonary disease. Exp Lung Res. 2010;36(9):548–557. doi:10.3109/01902148.2010.489143
13. Luisetti M, Ma S, Iadarola P, et al. Desmosine as a biomarker of elastin degradation in COPD: current status and future directions. Eur Respir J. 2008;32(5):1146–1157. doi:10.1183/09031936.00174807
14. Sellami M, Meghraoui-Kheddar A, Terryn C, et al. Induction and regulation of murine emphysema by elastin peptides. Am J Physiol Lung Cell Mol Physiol. 2016;310(1):L8–L23. doi:10.1152/ajplung.00068.2015
15. Rønnow SR, Langholm LL, Sand JMB, et al. Specific elastin degradation products are associated with poor outcome in the ECLIPSE COPD cohort. Sci Rep. 2019;9(1):4064. doi:10.1038/s41598-019-40785-2
16. Pierre A, Lemaire F, Meghraoui-Kheddar A, Audonnet S, Héry-Huynh S, Le Naour R. Impact of aging on inflammatory and immune responses during elastin peptide-induced murine emphysema. Am J Physiol Lung Cell Mol Physiol. 2019;316(4):L608–L620. doi:10.1152/ajplung.00402.2018
17. Tang S, Ma T, Zhang H, et al. Erythromycin prevents elastin peptide-induced emphysema and modulates CD4(+)T cell responses in mice. Int J Chron Obstruct Pulmon Dis. 2019;14:2697–2709. doi:10.2147/copd.S222195
18. Meghraoui-Kheddar A, Pierre A, Sellami M, Audonnet S, Lemaire F, Le Naour R. Elastin receptor (S-gal) occupancy by elastin peptides modulates T-cell response during murine emphysema. Am J Physiol Lung Cell Mol Physiol. 2017;313(3):L534–L547. doi:10.1152/ajplung.00465.2016
19. Liang Y, Shen Y, Kuang L, et al. Cigarette smoke exposure promotes differentiation of CD4(+) T cells toward Th17 cells by CD40-CD40L costimulatory pathway in mice. Int J Chron Obstruct Pulmon Dis. 2018;13:959–968. doi:10.2147/copd.S155754
20. Harding SA, Sarma J, Josephs DH, et al. Upregulation of the CD40/CD40 ligand dyad and platelet-monocyte aggregation in cigarette smokers. Circulation. 2004;109(16):1926–1929. doi:10.1161/01.Cir.0000127128.52679.E4
21. Ma DY, Clark EA. The role of CD40 and CD154/CD40L in dendritic cells. Semin Immunol. 2009;21(5):265–272. doi:10.1016/j.smim.2009.05.010
22. Sia JK, Bizzell E, Madan-Lala R, Rengarajan J, Salgame P. Engaging the CD40-CD40L pathway augments T-helper cell responses and improves control of Mycobacterium tuberculosis infection. PLoS Pathog. 2017;13(8):e1006530. doi:10.1371/journal.ppat.1006530
23. Iezzi G, Sonderegger I, Ampenberger F, Schmitz N, Marsland BJ, Kopf M. CD40-CD40L cross-talk integrates strong antigenic signals and microbial stimuli to induce development of IL-17-producing CD4+ T cells. Proc Natl Acad Sci U S A. 2009;106(3):876–881. doi:10.1073/pnas.0810769106
24. Bruder D, Westendorf AM, Geffers R, et al. CD4 T lymphocyte-mediated lung disease: steady state between pathological and tolerogenic immune reactions. Am J Respir Crit Care Med. 2004;170(11):1145–1152. doi:10.1164/rccm.200404-464OC
25. Thurlbeck WM. Internal surface area and other measurements in emphysema. Thorax. 1967;22(6):483–496. doi:10.1136/thx.22.6.483
26. Saetta M, Shiner RJ, Angus GE, et al. Destructive index: a measurement of lung parenchymal destruction in smokers. Am Rev Respir Dis. 1985;131(5):764–769. doi:10.1164/arrd.1985.131.5.764
27. Wang Y, Xu J, Meng Y, Adcock IM, Yao X. Role of inflammatory cells in airway remodeling in COPD. Int J Chron Obstruct Pulmon Dis. 2018;13:3341–3348. doi:10.2147/copd.S176122
28. Barnes PJ. Inflammatory mechanisms in patients with chronic obstructive pulmonary disease. J Allergy Clin Immunol. 2016;138(1):16–27. doi:10.1016/j.jaci.2016.05.011
29. Hou J, Sun Y. Role of regulatory T cells in disturbed immune homeostasis in patients with chronic obstructive pulmonary disease. Front Immunol. 2020;11:723. doi:10.3389/fimmu.2020.00723
30. Brusselle GG, Joos GF, Bracke KR. New insights into the immunology of chronic obstructive pulmonary disease. Lancet. 2011;378(9795):1015–1026. doi:10.1016/s0140-6736(11)60988-4
31. Yang L, Ma QL, Yao W, et al. Relationship between the anti-inflammatory properties of salmeterol/fluticasone and the expression of CD4⁺CD25⁺Foxp3⁺ regulatory T cells in COPD. Respir Res. 2011;12(1):142. doi:10.1186/1465-9921-12-142
32. Tang Y, Zhang H, Xu H, et al. Dendritic cells promote treg expansion but Not Th17 generation in response to talaromyces marneffei yeast cells. Infect Drug Resist. 2020;13:805–813. doi:10.2147/idr.S239906
33. Yin Y, Xu N, Shi Y, et al. Astaxanthin protects dendritic cells from lipopolysaccharide-induced immune dysfunction. Mar Drugs. 2021;19(6):346. doi:10.3390/md19060346
34. Zhang H, Qiu SL, Tang QY, et al. Erythromycin suppresses neutrophil extracellular traps in smoking-related chronic pulmonary inflammation. Cell Death Dis. 2019;10(9):678. doi:10.1038/s41419-019-1909-2
35. Karimi MH, Ebadi P, Pourfathollah AA, Moazzeni SM. Tolerance induction by CD40 blocking through specific antibody in dendritic cells. Iran J Allergy Asthma Immunol. 2010;9(3):141–147.
36. Kuang LJ, Deng TT, Wang Q, et al. Dendritic cells induce Tc1 cell differentiation via the CD40/CD40L pathway in mice after exposure to cigarette smoke. Am J Physiol Lung Cell Mol Physiol. 2016;311(3):L581–L589. doi:10.1152/ajplung.00002.2016
37. Ma R, Su H, Jiao K, Liu J. Role of Th17 cells, Treg cells, and Th17/Treg imbalance in immune homeostasis disorders in patients with chronic obstructive pulmonary disease. Immun Inflamm Dis. 2023;11(2):e784. doi:10.1002/iid3.784
38. Bozinovski S, Seow HJ, Chan SP, et al. Innate cellular sources of interleukin-17A regulate macrophage accumulation in cigarette- smoke-induced lung inflammation in mice. Clin Sci. 2015;129(9):785–796. doi:10.1042/cs20140703
39. Xiong J, Zhou L, Tian J, et al. Cigarette smoke-induced lymphoid neogenesis in COPD involves IL-17/RANKL pathway. Front Immunol. 2020;11:588522. doi:10.3389/fimmu.2020.588522
40. Eich A, Urban V, Jutel M, et al. A randomized, placebo-controlled Phase 2 Trial of CNTO 6785 in chronic obstructive pulmonary disease. Copd. 2017;14(5):476–483. doi:10.1080/15412555.2017.1335697
41. Cheng KJ, Zhou ML, Liu YC, Wang C, Xu YY. The role of CD40 in allergic rhinitis and airway remodelling. Mediators Inflamm. 2021;2021:6694109. doi:10.1155/2021/6694109
42. Raffin C, Vo LT, Bluestone JA. T(reg) cell-based therapies: challenges and perspectives. Nat Rev Immunol. 2020;20(3):158–172. doi:10.1038/s41577-019-0232-6
 © 2023 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2023 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.