Back to Journals » Blood and Lymphatic Cancer: Targets and Therapy » Volume 7
Blastic plasmacytoid dendritic cell neoplasm: challenges and future prospects
Authors Trottier AM, Cerquozzi S, Owen CJ
Received 24 August 2017
Accepted for publication 3 November 2017
Published 11 December 2017 Volume 2017:7 Pages 85—93
DOI https://doi.org/10.2147/BLCTT.S132060
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Professor David Dingli
Amy M Trottier, Sonia Cerquozzi, Carolyn J Owen
Division of Hematology and Hematological Malignancies, University of Calgary, Foothills Medical Centre, Calgary, AB, Canada
Abstract: Blastic plasmacytoid dendritic cell neoplasm (BPDCN) is a rare CD4+ CD56+ myeloid malignancy that is challenging to diagnose and treat. BPDCN typically presents with nonspecific cutaneous lesions with or without extra-cutaneous manifestations before progressing to leukemia. Currently, there is no standard of care for the treatment of BPDCN and various approaches have been used including acute myeloid leukemia, acute lymphoblastic leukemia, and lymphoma-based regimens with or without stem cell transplantation. Despite these treatment approaches, the prognosis of BPDCN remains poor and there is a lack of prospective data upon which to base treatment decisions. Recent work examining the mutational landscape and gene expression profiles of BPDCN has identified a number of potential therapeutic targets. One such target is CD123, the α subunit of the human interleukin-3 receptor, which is the subject of intervention studies using the novel agent SL-401. Other investigational therapies include UCART123, T-cell immunotherapy, and venetoclax. Prospective trials are needed to determine the best treatment for this uncommon and aggressive neoplasm.
Keywords: BPDCN, myeloid, neoplasm, cutaneous, dendritic cell
Introduction
Blastic plasmacytoid dendritic cell neoplasm (BPDCN) is a rare and clinically aggressive CD4+ CD56+ hematopoietic malignancy with no lineage-specific markers that has no defined standard of care. Prior to the discovery of its cell of origin, BPDCN was formerly named blastic natural killer (NK) cell lymphoma, agranular CD4+ NK cell leukemia, blastic NK leukemia/lymphoma, and agranular CD4+ CD56+ hematodermic neoplasm/tumor. The first clinical cases were described in the mid-1990s of uncommon, morphologically agranular CD4+ CD56+ hematopoietic malignancies with an aggressive clinical course and a propensity for cutaneous involvement.1,2 In the 2008 World Health Organization (WHO) Classification of Tumours of Haematopoietic and Lymphoid Tissues, this entity was renamed BPDCN and classified as myeloid lineage based on newer understanding of its plasmacytoid dendritic cell (pDC) origin.3 This nomenclature has remained in the 2016 WHO revision.4
BPDCN arises from precursors of pDCs (type 2 dendritic cells) but its underlying pathogenesis remains unknown.5,6 pDCs are also known as professional type 1 interferon producing cells or plasmacytoid monocytes because they are known to produce high amounts of interferon-α in response to viruses or viral nucleic acids and play a role in the innate immune system.7,8 Though this may hint at a role of viral exposure in the pathogenesis of BPDCN, there is no known association to any virus, including Epstein–Barr virus. BPDCN can occur at any age but is most common in the elderly, with a median age at diagnosis in the mid to late 60s.9–11 It has no known ethnic predisposition with a male to female ratio of 3:1.9 The true incidence of BPDCN is unknown, partly due to its changing nomenclature; however, it is very rare, estimated to be <1% of hematologic neoplasms.12 The prognosis of BPDCN is also very poor with a median overall survival (OS) of ~12–14 months.5
This review addresses the clinical and pathologic presentation of BPDCN, diagnostic challenges, treatment approaches, and outcomes with a focus on the adult population.
Clinical presentation
Patients with BPDCN frequently present with asymptomatic cutaneous manifestations with or without extra-cutaneous involvement, which can include lymphadenopathy, splenomegaly, cytopenias, and bone marrow involvement with progression to leukemia. Rarely, BPDCN can present as overt leukemia without cutaneous involvement.13 BPDCN cutaneous lesions can be variable and typically are either nodular, “bruise-like” patches, or disseminated and mixed lesions (macules and nodules).10 Figure 1 shows the images of mixed cutaneous lesions in a patient prior to treatment. In a large retrospective series, violaceous nodular lesions represented 73% of the cutaneous presentations,10 while another smaller series showed mixed lesions as more common than nodular lesions.11 Lesions can involve the extremities, trunk, face, and even mucous membranes. No prognostic significance has been noted for the different cutaneous presentations.10
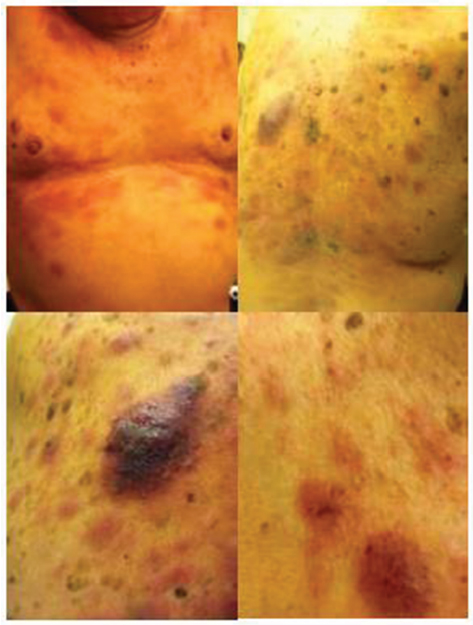 | Figure 1 Characteristic violaceous nodules of cutaneous involvement by blastic plasmacytoid dendritic cell neoplasm. |
Extra-cutaneous involvement at the time of diagnosis occurs in approximately two-thirds of patients.10 Bone marrow involvement is a common extra-cutaneous manifestation, ranging from minimal to extensive infiltration.9–11 Lymphadenopathy, splenomegaly, and hepatomegaly have also been observed. Central nervous system (CNS) involvement, primarily leptomeningeal, is less frequently reported at ~10%.10
BPDCN more rarely presents in a symptomatic leukemic state. An Italian multicenter retrospective study of 43 patients with leukemic BPDCN found that 77% of patients had concurrent cutaneous involvement, 56% had lymphadenopathy, 44% had splenomegaly, 42% had hepatomegaly, and 21% had other sites of disease including 9% with CNS disease.14 Interestingly, 23% of these cases were secondary leukemia (6 therapy-related cases and 4 patients with preexisting myelodysplastic syndrome). The median OS in this retrospective cohort was shorter at 8.7 months. Leukemic BPDCN without cutaneous lesions is also reported with similarly poor outcomes and no other distinguishing features.13
The diagnosis of BPDCN is frequently delayed due to its rarity, and is often a cutaneous-only presentation as well as overlapping immunophenotype with other benign and malignant disorders. Suspicion for BPDCN should arise when an older patient presents with persistent cutaneous lesions that do not respond to conventional dermatologic therapies with or without extra-cutaneous features such as lymphadenopathy, splenomegaly, and cytopenias. Cutaneous lesions require biopsy for histological and immunophenotypic evaluation in order to differentiate BPDCN from an array of other conditions with similar cutaneous involvement (Table 1).
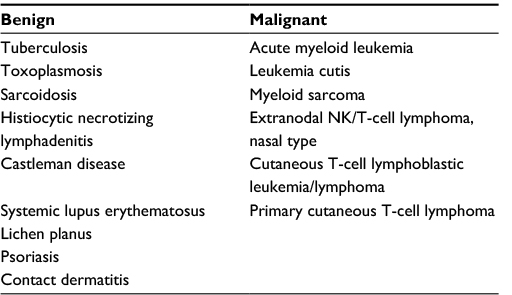 | Table 1 Differential diagnosis of blastic plasmacytoid dendritic cell neoplasm Abbreviation: NK, natural killer. |
Pathology
BPDCN can have similar immunohistochemistry patterns and morphologic appearance to benign states or other hematologic neoplasms such as acute myeloid leukemia (AML) and NK or T-cell lymphomas; thus, expert hematopathology review is recommended for diagnosis.
Morphology
On histology, cutaneous biopsies are characterized by the presence of a diffuse monomorphic infiltrate of medium-sized blast cells with fine chromatin, irregular nuclei, and one or more small nucleoli.11,15,16 The cells have only small amounts of agranular cytoplasm.15 Skin biopsies show a diffuse infiltration of these cells in the dermis with sparing of the epidermis but can have extension into the subcutaneous fat.11 There is no angioinvasion or coagulative necrosis. Lymph nodes are characterized by diffuse involvement of similar appearing malignant cells in interfollicular areas and medulla.15 Bone marrow involvement is variable with minimal to extensive infiltration and dysplastic features can be seen, particularly in the megakaryocytes. None of these features are diagnostic for BPDCN and further examination with extensive immunophenotyping is necessary.
Immunophenotype
The immunophenotype obtained by immunohistochemistry or flow cytometry can be used to identify BPDCN. BPDCN is characterized by the expression of both CD4 and CD56 in addition to one or more antigens specific to pDCs including CD123, BDCA-2 (also known as CD303), TCL1, and CD2AP as well as the absence of lineage-specific markers for B cells (CD19−, CD20−, CD79a−), T-cells (CD3− and CD5−), and myeloid and monocytic cells (MPO−, lysozyme−, CD13−, and CD117−);15,17–20 Figure 2 shows the morphology and immunohistochemistry images. CD34 is also negative and EBV-encoded RNA (EBER) is not present.15 Terminal deoxynucleotidyl transferase (TdT) can be positive or negative.15 CD68, which is found on normal pDCs, can be positive in 50% of BPDCN cases.21 CD7 and CD33 are also commonly positive.15
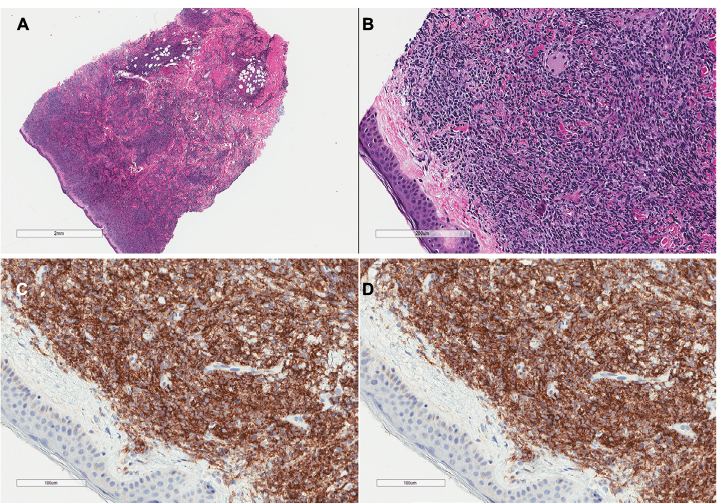 | Figure 2 H&E stains of a skin biopsy from a patient with blastic plasmacytoid dendritic cell neoplasm showing a dense infiltrate of medium-sized atypical cells with sparing of the epidermis (A, B). Immunohistochemical stains are positive for CD4 (C) and CD56 (D). Abbreviation: H&E, hematoxylin and eosin. |
The pathologic diagnosis of BPCDN can be very challenging due to atypical immunophenotyping. In rare cases, BPDCN can lack expression of CD56 and/or CD4.11,18,22 Also contributing to the diagnostic dilemma is the partially overlapping immunophenotype with benign pDCs in reactive lymph nodes, with the exception of CD56, which is negative in reactive cells.23 pDCs can accumulate in a variety of infections and inflammatory conditions, as listed in Table 1, and the cutaneous lesions can resemble those of BPDCN and have a similar immunophenotype with expression of pDC-specific antigens; however, reactive pDCs can usually be differentiated based on the lack of CD56 expression and TdT negativity, which is positive in about 30% of BPDCN cases.20
Other hematologic neoplasms that may express both CD56 and CD4 such as AML, AML-associated leukemia cutis, extranodal NK/T-cell lymphoma, nasal-type and some T-cell lymphomas must be differentiated from BPDCN by testing for the aforementioned pDC-specific antigens. Several myeloid antigens can help in differentiation including MPO, CD117, CD34, and lysozyme which are typically expressed by myeloid sarcoma, AML, and leukemia cutis but not in BPDCN.18,24 The WHO classifies tumors that share some but not all of the typical immunophenotypic characteristics of BPDCN as “acute leukaemia of ambiguous lineage.”15 Extranodal NK/T-cell, nasal-type lymphoma similarly are CD4+CD56+; however, unlike BPDCN, they are associated with EBV positivity (EBER positive).24–26 The expression of T-cell antigens helps rule out T-cell lymphoblastic leukemia/lymphoma and primary cutaneous T-cell lymphoma as do clonal TCR gene rearrangements.24,27 Table 2 outlines the immunophenotypic differences between BPDCN and other hematologic neoplasms that may express CD4 and CD56 as well as differences in sites of involvement.
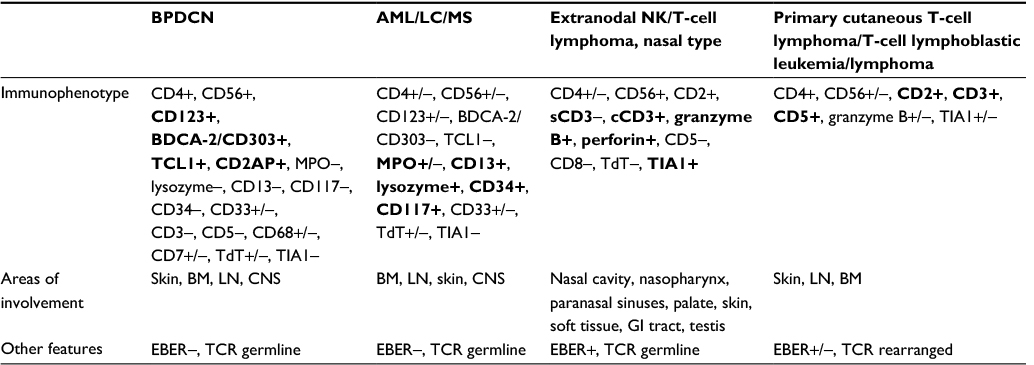 | Table 2 Comparison of BPDCN to other CD4/CD56 expressing hematologic neoplasms Note: Bold entries indicate the significant differences in immunophenotype between the various neoplasms. Abbreviations: AML, acute myeloid leukemia; BM, bone marrow; CNS, central nervous system; EBER, Epstein–Barr virus-encoded RNA; GI, gastrointestinal; LC, leukemia cutis; LN, lymph node; MS, myeloid sarcoma; TCR, T-cell receptor gene; BPDCN, blastic plasmacytoid dendritic cell neoplasm; NK, natural killer. |
Genetics
Cytogenetic abnormalities are found in up to two-thirds of patients with BPDCN and a complex karyotype is common.28 Recurrent abnormalities can be found in 5q, 12p, 13q, 6q, 15q, and 9, with 5q being the most common aberration, representing 72% of cases in one series.28 Using array-based comparative genomic hybridization, Lucioni et al found that genetic loss was more common than genetic gain mutations, with common deletions involving 9p21.3 (CDKN2A/CDKN2B), 13q13.1-q14.3 (RB1), 12p13.2-p13.1 (CDKN1B), 13q11-q12 (LATS2), and 7p12.2 (IKZF1).29 Biallelic loss of 9p21.3, which encodes the cyclin-dependent kinase inhibitors CDKN2A/CDKN2B, was associated with poorer OS.29 Wiesner et al similarly found common losses of cell cycle inhibitors including p16, encoded by CDKN2A, and loss of the RB1 gene on chromosome 13 in 43% of cases.30
A number of techniques including gene expression profiling, next-generation sequencing, and whole exome analysis have been used to study the mutational landscape of BPDCN.7 These studies have found recurrent mutations in TET2 (in 36% of cases), ASXL1, NRAS, NPM1, IKAROS, ZEB2, and TP53.31,32 The high frequency of TET2 mutations reinforces the myeloid origin of this malignancy. Using gene expression profiling, Dijkman et al found decreased expression of RB1 and LATS2 while expression of the oncogenes HES6, RUNX2, and FLT3 was found to be elevated.33 Among 33 BPDCN cases, Stenzinger et al detected point mutations in NRAS (27.3%), ATM (21.2%), MET, KRAS, IDH2, KIT, APC, and RB1, as well as in VHL, BRAF, MLH1, TP53, and RET (3% each). NRAS, KRAS, and ATM mutations were found to be mutually exclusive, which may suggest that different subgroups of BPDCN exist.34 Ultimately, none of these chromosomal abnormalities or genetic aberrations have been found to be pathognomonic of BPDCN and like in AML, it is likely that multiple driver mutations are contributory.
Treatment
There is currently no standard of care for BPDCN and due to the rarity of the disease, there is a lack of prospective data.
Pediatric BPDCN
BPDCN is exceptionally rare in children and data on treatment and outcomes are based primarily on case reports and series. The largest study of pediatric BPDCN was a case report and retrospective review of 33 patients.35 Various treatment approaches have been attempted including aggressive acute lymphoblastic leukemia (ALL), non-Hodgkin’s lymphoma (NHL), and AML-type regimens with or without allogeneic hematopoietic stem cell transplantation (HSCT). ALL regimens have been the most commonly used. Pediatric patients who received ALL-type regimens or NHL regimens with or without transplant had better outcomes compared to those receiving AML-based regimens.36
Pediatric patients tend to fare better than adult patients.36 Additionally, children with BPDCN who lack cutaneous symptoms appear to have better survival.35 As children can tolerate intensive induction therapy better than adults, most series recommend multi-agent intensive chemotherapy following an ALL protocol. The use of HSCT is a source of controversy. Some small series have described similar survival with and without consolidation HSCT for primary therapy such that there is currently no clear indication for HSCT in children who achieve complete remission (CR) with induction therapy.35,36
Adult BPDCN
Treatment of BPDCN in adults has been similarly heterogeneous including ALL, AML, and aggressive NHL-like regimens with or without autologous or allogeneic HSCT. BPDCN is usually responsive to initial chemotherapy with high CR rates; however, remissions are short and salvage therapy responses are poor leading to short OS.9,17
Induction therapy
Historical therapies for BPDCN include those used for aggressive NHL such as cyclophosphamide, doxorubicin, vincristine, prednisone (CHOP) or CHOP-like regimens or more intensive therapies such as hyperfractionated cyclophosphamide, vincristine, doxorubicin, and dexamethasone (hyper-CVAD) alternating with methotrexate and cytarabine or AML induction regimens (eg, mitoxantrone, idarubicin, cytarabine, and etoposide; cytarabine and an anthracycline [7+3]; fludarabine, cytarabine, granulocyte colony stimulating factor, and idarubicin), or other ALL regimens. Table 3 summarizes the outcomes of the reported chemotherapy regimens for adult BPDCN. With the above regimens, CR rates range between 40% and 100%.17 In 2002, Feuillard et al reviewed 23 patients, most treated with CHOP-like chemotherapy, and reported a CR rate of 86% but with short remission duration of 9 months and low OS (53% at 1 year and 25% at 2 years).9 CNS relapses were also common.
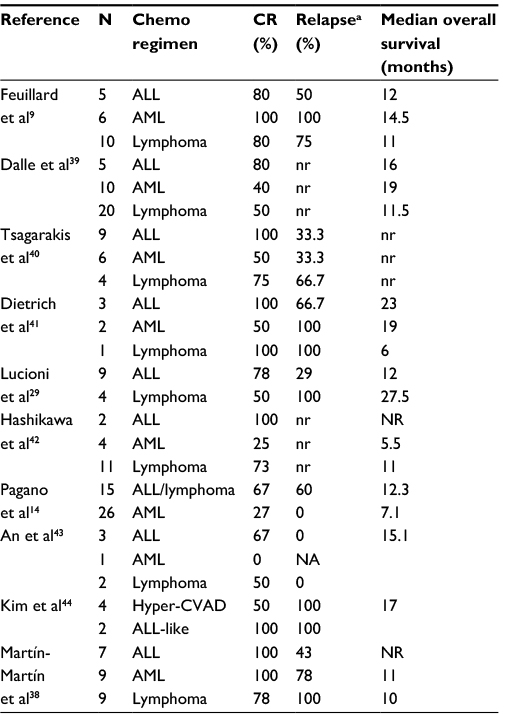 | Table 3 Outcomes of various induction regimens in blastic plasmacytoid dendritic cell neoplasm Notes: Lymphoma (CHOP/CHOP-like). a% relapse out of those achieving a CR with induction. Some studies include patients who received HSCT in consolidation or in first relapse. Abbreviations: nr, not reported; NR, not reached; CHOP, cyclophosphamide, doxorubicin, vincristine, prednisone; ALL, acute lymphoblastic leukemia; AML, acute myeloid leukemia; CVAD, cyclophosphamide, vincristine, doxorubicin, and dexamethasone; CR, complete remission; NA, not applicable; HSCT, hematopoietic stem cell transplantation. |
In 2003, Reimer et al published data on 93 patients compiled from publications on BPDCN and found that more aggressive approaches (acute leukemia protocols followed by HSCT) had superior sustained CR and OS compared to CHOP and CHOP-like regimens.37 The best outcomes were seen for those who underwent myeloablative HSCT in first CR with a median OS of 31.5 months.
In one of the largest retrospective multicenter studies, outcomes of treatment in 41 patients with BPDCN, of whom 26 were treated with an AML-type regimen and 15 with ALL/lymphoma-type, were 27% and 67%, respectively, for CR.14 Relapse occurred in only 35% of patients achieving CR but the median OS was 8.7 months. Median OS, though still short, was higher for patients treated with ALL/lymphoma-type chemotherapy at 12.3 months versus 7.1 months for AML-like therapy. Patients who received allogeneic HSCT in first CR (6 patients) had better outcomes with a median OS of 22.7 months.
The most recent report of 46 patients, 32 of whom received treatment with curative intent (including 4 children), confirmed an improved outcome with ALL-type regimens with CNS prophylaxis compared to CHOP or CHOP-like lymphoma protocols.38
Thus, the current evidence suggests no clear gold standard therapy for BPDCN but favors intensive multi-agent chemotherapy with an ALL-type protocol. Given the propensity for CNS relapse, CNS prophylaxis or CNS penetrating chemotherapy is recommended. Unfortunately, the majority of BPDCN patients are elderly and not fit for such intensive therapy and the optimal therapy for such patients is unclear. Given short remissions with all reported induction therapies, consolidation with HSCT or maintenance therapies should be investigated.
HSCT
The evidence for autologous and allogeneic HSCT is also limited to case series and retrospective reviews.
Autologous HSCT
The largest analyzed cohort of patients treated with autologous HSCT comes from a retrospective analysis of a Japanese transplant registry.45 In this study, 11 patients in first CR received an autologous HSCT. The OS and progression-free survival at 4 years were 82% and 73%, respectively. This study suggests that for patients achieving a CR, high-dose chemotherapy and autologous HSCT may be valuable. However, other smaller series have reported conflicting results with no clear advantage to the use of autologous HSCT.37,46 All of these reviews are confounded by varying induction and conditioning regimens and given the heterogeneity of results, the role of autologous HSCT in BPDCN remains uncertain.
Allogeneic HSCT
The largest retrospective study of patients receiving allogeneic HSCT for BPDCN was conducted using the European Group for Blood and Marrow Transplantation registry and included 34 patients with a relatively young median age of 41 years.47 The majority of these patients were induced with acute leukemia-type regimens. Fifty-six percent of patients were transplanted in first CR and 75% received myeloablative conditioning. Forty-seven percent of patients remained alive with a median survival of 28 months. However, 32% of patients had relapsed by 3 years and an additional 30% experienced non-relapse mortality. Three-year OS was 41%. Patients transplanted in first CR had significantly better 3-year OS at 52% compared to those transplanted beyond first CR. Several smaller series confirm the benefit of allogeneic HSCT and demonstrate improved outcomes for patient transplanted in first CR.37,45
Dalle et al used the French Study Group on Cutaneous Lymphoma database to examine the outcomes of various BPDCN treatment approaches (9 out of 10 received allogeneic HSCT).39 They found a significant survival advantage for those who underwent transplant compared to any other treatment and 6 of the 10 patients remained alive at the end of the study with a mean survival of 31.3 months. Allogeneic HSCT in the elderly is limited. Dietrich et al reported outcomes of 4 older BPDCN patients with a median age of 67 (range 55–80) who were treated with reduced intensity conditioning (RIC) HSCT.41 Two of the 4 patients remained in CR at the time of reporting while the other 2 patients relapsed and died (both had active disease at the time of transplant). This case series shows that RIC allogeneic HSCT may be successful in older BPDCN patients.
In summary, current studies suggest that treatment with high-dose chemotherapy followed by allogeneic HSCT in CR1 may result in improved OS for BPDCN. However, there is insufficient evidence to guide physicians in their choice of conditioning regimens and there is no clear evidence for the existence of a strong graft-versus-leukemia effect in this disease.
Relapsed and refractory disease
There is no established standard treatment for relapsed or refractory BPDCN. For patients who are fit for HSCT, salvage chemotherapy followed by allogeneic HSCT has been successful in some cases.47 For those who relapse after transplantation or are unfit for transplantation, there is little evidence to guide the choice of next therapy.
A single case report described a reasonable remission (1 year) and tolerability of oral etoposide in an older patient with relapsed BPDCN and another described partial disease control of cutaneous lesions with weekly infusions of pralatrexate.48,49 A case series supporting the use of azacytidine has also been reported as has the use of repeat courses of donor lymphocyte infusions after allogeneic HSCT relapse.50,51
Novel therapies and future prospects
Given the poor prognosis associated with BPDCN and high relapse rates with conventional treatments including CHOP/CHOP-like, AML, and ALL-type regimens, there has been interest in new approaches to therapy including targeted therapies. Examples include SL-401, UCART123, T-cell immunotherapy, and venetoclax. Though some of these agents show early promise, none are currently approved for use in BPDCN.
Montero et al investigated the use of venetoclax, a BCL2 inhibitor, in BPDCN based on high expression of BCL2 in BPDCN cells but not in normal pDCs.52 In vitro studies and a xenograft mouse model suggested efficacy leading to the off-label treatment of 2 patients with relapsed/refractory BPDCN who achieved rapid partial responses with no significant toxicity.
After gene expression profiling demonstrated aberrantly activated NF-kB in BPDCN cells, another group investigated the use of bortezomib in in vitro studies showing that the BPDCN cell line CAL-1 is sensitive to bortezomib resulting in inhibition of cellular proliferation and induction of cell death.32
The α subunit of the human interleukin-3 receptor (IL-3R), CD123, is ubiquitously and highly expressed by BPDCN making it another potential target for therapeutic intervention.53 One of the more promising novel targeted therapies for BPDCN is SL-401, an IL-3R-targeted drug.54 SL-401 exhibited high levels of cytotoxicity to CD123+ BPDCN cells, resulting in a 75% reduction in the viability of previously untreated BPDCN cells at 18 hours.55 In a mouse model, treatment with SL-401 significantly improved the survival of mice inoculated with a BPDCN cell line compared to untreated controls. These early data paved the way for the first prospective therapeutic study in BPDCN. The Phase I/expanded Phase II study of SL-401 for BPDCN included 11 patients with relapsed/refractory (7 patients) or previously untreated (4 patients) BPDCN.56 These patients received SL-401 at a dose of 12.5 μg/kg intravenously daily for up to 5 days. Seventy-eight percent of the patients achieved a CR or partial remission with a median duration of response of 5 months. These encouraging early-phase data have led to a larger open-label, non-randomized, multicenter trial (NCT02113982).
Finally, T-cell immunotherapy using chimeric antigen receptor T-cells has been investigated in a preclinical study targeting CD123 (UCART123).57 The study found a specific cytotoxic effect on BPDCN cells as well as prolonged survival in mouse xenograft models of BPDCN. UCART123 is being further evaluated for safety and clinical activity in patients with relapsed or refractory BPDCN in a Phase I clinical trial (NCT03203369). Another group is currently investigating genetically modified T-cell immunotherapy in persistent/recurrent BPDCN as well as relapsed/refractory AML in a Phase I trial (NCT02159495).
Conclusion
BPDCN is a rare and aggressive hematologic malignancy with a poor prognosis and no standard treatment. It is derived from pDCs and classified as a myeloid neoplasm. BPDCN typically expresses both CD4 and CD56 as well as one or more markers specific to pDCs including CD123, BDCA-2 (also known as CD303), TCL1, and CD2AP. The diagnosis can be challenging based on its varying presentations and clinically diverse array of cutaneous lesions. Prompt and accurate diagnosis requires appropriate clinical suspicion and review by an experienced hematopathologist using extensive immunophenotyping. Despite good understanding of the origin and lineage of this malignancy, the underlying pathobiology remains unknown and the prognosis is poor with a median OS of ~1 year.
There remains no standard first-line treatment and although the disease typically responds well to AML, ALL, and CHOP or CHOP-like chemotherapy with high CR rates, relapses are frequent and remissions are short. In the absence of prospective trials, treatment decisions are currently based only on case series and multicenter retrospective reviews. From these limited data, the most accepted therapy is intensive ALL-like induction therapy followed by allogeneic HSCT in first CR for those who are transplant eligible. The exception to this is pediatric patients, who tend to have better outcomes and for whom transplant is not typically recommended until the relapsed setting. Due to relatively high rates of CNS involvement or relapse, induction regimens that incorporate CNS prophylaxis or achieve CNS penetration are recommended. Despite these recommendations, the majority of patients with BPDCN are not eligible for allogenic HSCT or intensive multi-agent chemotherapy due to age and/or comorbidities and the best therapy for these patients remains unclear. Although no single driver mutation has been identified in BPDCN, gene expression profiles have identified a number of potential therapeutic targets. One of the most promising targeted therapies is SL-401, a conjugate of recombinant human IL-3 and a diphtheria toxin, which has shown encouraging results in early-phase studies and is the subject of further prospective evaluation. Prospective trials are needed to determine optimal therapy for this rare and aggressive neoplasm.
Acknowledgment
The authors thank Dr Etienne Mahe for providing the morphology and immunohistochemistry images.
Disclosure
The authors report no conflicts of interest in this work.
References
Adachi M, Maeda K, Takekawa M, et al. High expression of CD56 (N-CAM) in a patient with cutaneous CD4-positive lymphoma. Am J Hematol. 1994;47(4):278–282. | ||
Brody JP, Allen S, Schulman P, et al. Acute agranular CD4-positive natural killer cell leukemia. Comprehensive clinicopathologic studies including virologic and in vitro culture with inducing agents. Cancer. 1995;75(10):2474–2483. | ||
Vardiman JW, Thiele J, Arber DA, et al. The 2008 revision of the World Health Organization (WHO) classification of myeloid neoplasms and acute leukemia: rationale and important changes. Blood. 2009;114(5):937–951. | ||
Arber DA, Orazi A, Hasserjian R, et al. The 2016 revision to the World Health Organization classification of myeloid neoplasms and acute leukemia. Blood. 2016;127(20):2391–2405. | ||
Herling M, Jones D. CD4+/CD56+ hematodermic tumor: the features of an evolving entity and its relationship to dendritic cells. Am J Clin Pathol. 2007;127:687–700. | ||
Petrella T, Comeau MR, Maynadié M, et al. “Agranular CD4+ CD56+ hematodermic neoplasm” (blastic NK-cell lymphoma) originates from a population of CD56+ precursor cells related to plasmacytoid monocytes. Am J Surg Pathol. 2002;26(7):852–862. | ||
Reizis B, Bunin A, Ghosh HS, Lewis KL, Sisirak V. Plasmacytoid dendritic cells: recent progress and open questions. Annu Rev Immunol. 2011;29:163–183. | ||
Chaperot L, Bendriss N, Manches O, et al. Identification of a leukemic counterpart of the plasmacytoid dendritic cells. Blood. 2001;97(10):3210–3217. | ||
Feuillard J, Jacob M-C, Valensi F, et al. Clinical and biologic features of CD4(+)CD56(+) malignancies. Blood. 2002;99(5):1556–1563. | ||
Julia F, Petrella T, Beylot-Barry M, et al. Blastic plasmacytoid dendritic cell neoplasm: clinical features in 90 patients. Br J Dermatol. 2013;169(3):579–586. | ||
Cota C, Vale E, Viana I, et al. Cutaneous manifestations of blastic plasmacytoid dendritic cell neoplasm-morphologic and phenotypic variability in a series of 33 patients. Am J Surg Pathol. 2010;34(1):75–87. | ||
Bueno C, Almeida J, Lucio P, et al. Incidence and characteristics of CD4(+)/HLA DRhi dendritic cell malignancies. Haematologica. 2004;89(1):58–69. | ||
Rauh MJ, Rahman F, Good D, et al. Blastic plasmacytoid dendritic cell neoplasm with leukemic presentation, lacking cutaneous involvement: case series and literature review. Leuk Res. 2012;36(1):81–86. | ||
Pagano L, Valentini CG, Pulsoni A, et al. Blastic plasmacytoid dendritic cell neoplasm with leukemic presentation: an Italian multicenter study. Haematologica. 2013;98(2):239–246. | ||
Facchetti F, Jones D, Petrella T. Blastic plasmacytoid dendritic cell neoplasm. In: Swerdlow S, Campo E, Harris N, et al, editors. WHO Classification of Tumours of Haematopoietic and Lymphoid Tissues. 4th ed. Lyon: IARC; 2008:145–147. | ||
Riaz W, Zhang L, Horna P, Sokol L. Blastic plasmacytoid dendritic cell neoplasm: update on molecular biology, diagnosis, and therapy. Cancer Control. 2014;21(4):279–289. | ||
Pagano L, Valentini CG, Grammatico S, Pulsoni A. Blastic plasmacytoid dendritic cell neoplasm: diagnostic criteria and therapeutical approaches. Br J Haematol. 2016;174(2):188–202. | ||
Sullivan JM, Rizzieri DA. Treatment of blastic plasmacytoid dendritic cell neoplasm. Hematology. 2016;2016(1):16–23. | ||
Marafioti T, Paterson JC, Ballabio E, et al. Novel markers of normal and neoplastic human plasmacytoid dendritic cells. Blood. 2008;111(7):3778–3792. | ||
Facchetti F, Cigognetti M, Fisogni S, Rossi G, Lonardi S, Vermi W. Neoplasms derived from plasmacytoid dendritic cells. Mod Pathol. 2016;29(2):98–111. | ||
Petrella T, Bagot M, Willemze R, et al. Blastic NK-cell lymphomas (agranular CD4+CD56+ hematodermic neoplasms): a review. Am J Clin Pathol. 2005;123(5):662–675. | ||
Ascani S, Massone C, Ferrara G, et al. CD4-negative variant of CD4+/CD56+ hematodermic neoplasm: description of three cases. J Cutan Pathol. 2008;35(10):911–915. | ||
Facchetti F, Vermi W, Mason D, Colonna M. The plasmacytoid monocyte/interferon producing cells. Virchows Arch. 2003;443(6):703–717. | ||
Shi Y, Wang E. Blastic plasmacytoid dendritic cell neoplasm: a clinicopathologic review. Arch Pathol Lab Med. 2014;138(4):564–569. | ||
Gera S, Dekmezian MS, Duvic M, Tschen JA, Vega F, Cho-Vega JH. Blastic plasmacytoid dendritic cell neoplasm. Am J Dermatopathol. 2014;36(3):244–251. | ||
Assaf C, Gellrich S, Whittaker S, et al. CD56-positive haematological neoplasms of the skin: a multicentre study of the Cutaneous Lymphoma Project Group of the European Organisation for Research and Treatment of Cancer. J Clin Pathol. 2007;60(9):981–989. | ||
Laribi K, Denizon N, Besançon A, et al. Blastic plasmacytoid dendritic cell neoplasm: from origin of the cell to targeted therapies. Biol Blood Marrow Transplant. 2016;22(8):1357–1367. | ||
Leroux D, Mugneret F, Callanan M, et al. CD4+, CD56+ DC2 acute leukemia is characterized by recurrent clonal chromosomal changes affecting 6 major targets: a study of 21 cases by the Groupe Français de Cytogénétique Hématologique. Blood. 2002;99(11):4154–4159. | ||
Lucioni M, Novara F, Fiandrino G, et al. Twenty-one cases of blastic plasmacytoid dendritic cell neoplasm: focus on biallelic locus 9p21.3 deletion. Blood. 2011;118(17):4591–4594. | ||
Wiesner T, Obenauf AC, Cota C, Fried I, Speicher MR, Cerroni L. Alterations of the cell-cycle inhibitors p27(KIP1) and p16(INK4a) are frequent in blastic plasmacytoid dendritic cell neoplasms. J Invest Dermatol. 2010;130(4):1152–1157. | ||
Jardin F, Ruminy P, Parmentier F, et al. TET2 and TP53 mutations are frequently observed in blastic plasmacytoid dendritic cell neoplasm. Br J Haematol. 2011;153(3):413–416. | ||
Sapienza MR, Fuligni F, Agostinelli C, et al. Molecular profiling of blastic plasmacytoid dendritic cell neoplasm reveals a unique pattern and suggests selective sensitivity to NF-kB pathway inhibition. Leukemia. 2014;28(8):1606–1616. | ||
Dijkman R, van Doorn R, Szuhai K, Willemze R, Vermeer MH, Tensen CP. Gene-expression profiling and array-based CGH classify CD4+CD56+ hematodermic neoplasm and cutaneous myelomonocytic leukemia as distinct disease entities. Blood. 2007;109(4):1720–1727. | ||
Stenzinger A, Endris V, Pfarr N, et al. Targeted ultra-deep sequencing reveals recurrent and mutually exclusive mutations of cancer genes in blastic plasmacytoid dendritic cell neoplasm. Oncotarget. 2014;5(15):6404–6413. | ||
Sakashita K, Saito S, Yanagisawa R, et al. Usefulness of allogeneic hematopoietic stem cell transplantation in first complete remission for pediatric blastic plasmacytoid dendritic cell neoplasm with skin involvement: a case report and review of literature. Pediatr Blood Cancer. 2013;60(11):E140–E142. | ||
Jegalian AG, Buxbaum NP, Facchetti F, et al. Blastic plasmacytoid dendritic cell neoplasm in children: diagnostic features and clinical implications. Haematologica. 2010;95(11):1873–1879. | ||
Reimer P, Rüdiger T, Kraemer D, et al. What is CD4+CD56+ malignancy and how should it be treated? Bone Marrow Transplant. 2003;32(7):637–646. | ||
Martín-Martín L, López A, Vidriales B, et al. Classification and clinical behavior of blastic plasmacytoid dendritic cell neoplasms according to their maturation-associated immunophenotypic profile. Oncotarget. 2015;6(22):19204–19216. | ||
Dalle S, Beylot-Barry M, Bagot M, et al. Blastic plasmacytoid dendritic cell neoplasm: is transplantation the treatment of choice? Br J Dermatol. 2010;162(1):74–79. | ||
Tsagarakis NJ, Kentrou NA, Papadimitriou KA, et al. Acute lymphoplasmacytoid dendritic cell (DC2) leukemia: results from the Hellenic Dendritic Cell Leukemia Study Group. Leuk Res. 2010;34(4):438–446. | ||
Dietrich S, Andrulis M, Hegenbart U, et al. Blastic plasmacytoid dendritic cell neoplasia (BPDC) in elderly patients: results of a treatment algorithm employing allogeneic stem cell transplantation with moderately reduced conditioning intensity. Biol Blood Marrow Transplant. 2011;17(8):1250–1254. | ||
Hashikawa K, Niino D, Yasumoto S, et al. Clinicopathological features and prognostic significance of CXCL12 in blastic plasmacytoid dendritic cell neoplasm. J Am Acad Dermatol. 2012;66(2):278–291. | ||
An HJ, Yoon DH, Kim S, et al. Blastic plasmacytoid dendritic cell neoplasm: a single-center experience. Ann Hematol. 2013;92(3):351–356. | ||
Kim J-H, Park H-Y, Lee J-H, et al. Blastic plasmacytoid dendritic cell neoplasm: analysis of clinicopathological feature and treatment outcome of seven cases. Ann Dermatol. 2015;27(6):727. | ||
Aoki T, Suzuki R, Kuwatsuka Y, et al. Long-term survival following autologous and allogeneic stem cell transplantation for blastic plasmacytoid dendritic cell neoplasm. Blood. 2015;125(23):3559–3562. | ||
Suzuki R, Suzumiya J, Nakamura S, et al. Hematopoietic stem cell transplantation for natural killer-cell lineage neoplasms. Bone Marrow Transplant. 2006;37(4):425–431. | ||
Roos-Weil D, Dietrich S, Boumendil A, et al. Stem cell transplantation can provide durable disease control in blastic plasmacytoid dendritic cell neoplasm: a retrospective study from the European Group for Blood and Marrow Transplantation. Blood. 2013;121(3):440–446. | ||
Leitenberger JJ, Berthelot CN, Polder KD, et al. CD4+ CD56+ hematodermic/plasmacytoid dendritic cell tumor with response to pralatrexate. J Am Acad Dermatol. 2008;58(3):480–484. | ||
Hatano Y, Ogata M, Ohishi M, et al. Maintenance of long-term remission using oral administration of low-dose etoposide in a patient demonstrating a relapse of blastic natural killer-cell lymphoma. Clin Exp Dermatol. 2007;32(1):96–97. | ||
Khwaja R, Daly A, Wong M, Mahé E, Cerquozzi S, Owen C. Azacitidine in the treatment of blastic plasmacytoid dendritic cell neoplasm: a report of 3 cases. Leuk Lymphoma. 2016;57(11):2720–2722. | ||
Steinberg A, Kansal R, Wong M, et al. Good clinical response in a rare aggressive hematopoietic neoplasm: plasmacytoid dendritic cell leukemia with no cutaneous lesions responding to 4 donor lymphocyte infusions following transplant. Case Rep Transplant. 2011;2011:651906. | ||
Montero J, Stephansky J, Cai T, et al. Blastic plasmacytoid dendritic cell neoplasm is dependent on BCL2 and sensitive to venetoclax. Cancer Discov. 2017;7(2):156–164. | ||
Garnache-Ottou F, Feuillard J, Ferrand C, et al. Extended diagnostic criteria for plasmacytoid dendritic cell leukaemia. Br J Haematol. 2009;145(5):624–636. | ||
Kreitman RJ. Recombinant immunotoxins containing truncated bacterial toxins for the treatment of hematologic malignancies. BioDrugs. 2009;23(1):1–13. | ||
Angelot-Delettre F, Roggy A, Frankel AE, et al. In vivo and in vitro sensitivity of blastic plasmacytoid dendritic cell neoplasm to SL-401, an interleukin-3 receptor targeted biologic agent. Haematologica. 2015;100(2):223–230. | ||
Frankel AE, Woo JH, Ahn C, et al. Activity of SL-401, a targeted therapy directed to interleukin-3 receptor, in blastic plasmacytoid dendritic cell neoplasm patients. Blood. 2014;124(3):385–392. | ||
Cai T, Galetto R, Gouble A, et al. Pre-clinical studies of anti-CD123 CAR-T cells for the treatment of blastic plasmacytoid dendritic cell neoplasm (BPDCN). Blood. 2016;128(22):4039. |
 © 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.