Back to Journals » Pediatric Health, Medicine and Therapeutics » Volume 11
Biotinidase Deficiency: Prevalence, Impact And Management Strategies
Authors Canda E, Kalkan Uçar S, Çoker M ![]()
Received 30 September 2019
Accepted for publication 5 February 2020
Published 4 May 2020 Volume 2020:11 Pages 127—133
DOI https://doi.org/10.2147/PHMT.S198656
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Roosy Aulakh
Ebru Canda, Sema Kalkan Uçar, Mahmut Çoker
Department of Pediatrics, Faculty of Medicine, Ege University, Izmir, Turkey
Correspondence: Ebru Canda
Department of Pediatrics, Faculty of Medicine, Ege University, Izmir 35040, Turkey
Tel + 90 232 390 1293
Email [email protected]
Abstract: Biotinidase deficiency is an autosomal recessive inherited neurocutaneous disorder. Clinically untreated patients with BD can present with variable neurological and dermatological signs, such as seizures, hypotonia, feeding problems, developmental delay, hearing loss, optic atrophy ataxia, alopecia, and skin rash. Clinical findings of patients with partial BD reported in the literature show that it can occur from infancy to adulthood. Outcomes of newborn screening programs support the fact that biotin treatment started after birth prevents patients with biotinidase deficiency from developing symptoms. Presence of late-onset cases with different clinical findings indicates that there is still much to learn about BD.
Keywords: biotinidase, biotin, newborn screening
Introduction
Biotinidase (EC 3.5.1.12) is the enzyme that cleaves the vitamin, biotin, from the biocytin and from the dietary protein-bound sources, thereby recycling the biotin.1 Free biotin can directly enter the biotin pool and is used by four carboxylases to convert their active forms. Biotin is the coenzyme for four carboxylases that have roles in gluconeogenesis, the catabolism of several branch-chain amino acids and fatty acid synthesis.2,3 Biotinidase deficiency (BTD) is an autosomal recessive inherited neurocutaneous disorder.1,4 In BTD all the mitochondrial carboxylase activities become deficient. Deficient activity of pyruvate carboxylase results in the accumulation of lactic acid and alanine. Deficient activity in propionyl-CoA carboxylase results in the accumulation of propionate, 3-OH propionate, and methyl citrate. The accumulation of 3-methylcrotonylglycine and 3-hydoxyisolerate detected is due to the deficient 3-methylcrotonyl-CoA carboxylase enzyme activity. The fourth deficient carboxylase is acetyl CoA carboxylase.5 Since 1982 almost all individuals with juvenile or late-onset form of multiple carboxylase deficiency have been described to have biotinidase deficiency.1 Patients with biotinidase deficiency with less than 10% mean normal serum enzyme activity are classified as having profound deficiency and those with 10–30% mean normal serum activity are classified as having partial deficiency.6
Incidence
The incidence of BTD varies from 1:40,000 to 1:60,000 births in the world. In some countries such as Turkey and Saudi Arabia the prevalence is higher due to the high consanguinity rates.6–9 Based on the newborn screening outcome data from 2006, the incidence of profound BTD in United States is 1:80,000 births and partial deficiency between 1:31,000 and 1:40,000.10 The disease incidence varies between countries; BTD incidence is high in Brazil (1:9000).11 The national newborn screening program in Turkey revealed high incidence. Based on published data from Ministry of Health, the incidence is approximately 1:7,116.12
Clinical Findings
Clinically untreated patients with BTD can present with variable neurological, dermatological signs; such as seizures, hypotonia, feeding problems, developmental delay, hearing loss, optic atrophy ataxia, alopecia, skin rash.13 Cutaneous manifestations include skin rash, eczema, alopecia, conjunctivitis, viral and fungal infections due to immunological disfunction.14 Scaly erythematous plaques over the flexors and perioral areas and seborrheic dermatitis-like eruptions can be detected. In severe cases crusting, lichenification, and open lesions that may become secondary infections can be seen. Abnormalities in lipid metabolism and alterations in the skin’s composition may play a role in cutaneous manifestations in BTD patients.15 Figure 1 shows seborrheic dermatitis-like eruptions in a newborn with BTD.
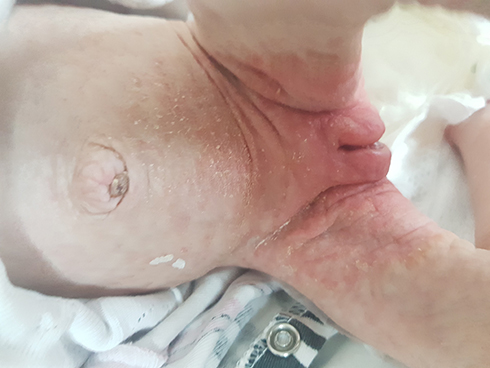 |
Figure 1 Seborrheic dermatitis-like eruptions in a newborn with biotinidase deficiency. |
Sometimes symptoms can mimic primary immune deficiencies.16 Due to immunologic dysfunction, recurrent viral or fungal infections can be seen. Respiratory problems such as hyperventilation, stridor, and apnea can be detected. Some children with BD manifest a single symptom, sometimes present with multiple neurological and cutaneous findings.17 Symptoms of BTD can be improved or prevented with pharmacological doses of oral biotin. However, if auditory and visual defects occur, they usually persist despite oral biotin therapy.18 If children with profound BTD detected with newborn screening are treated with biotin soon after birth, hearing loss can be prevented.6,10 The onset of symptoms varies from 2 weeks to 2 years of age but some patients develop symptoms of the disease much later in life.17,19 Approximately 76% of untreated patients with profound deficiency have sensorineural hearing loss.20
Patients with profound BTD develop symptoms of the disease at an early age.18 If affected patients are not treated with biotin, metabolic decompensation, coma or death can occur.21 The most common neurological symptoms of patients with untreated profound deficiency are seizures and hypotonia.17 Patients with partial BTD usually have milder symptoms. The disease can be misdiagnosed in children with developmental delay and autism.22 In rare cases that are not under treatment, stress might trigger developmental symptoms such as hypotonia, skin rash, and hair loss.14,17,23
Clinical findings of patients with partial BTD reported in the literature show that it can occur from infancy to adulthood. The symptoms range from minor symptoms such as rash and alopecia to hypotonia to seizures and developmental delay. Some symptoms may be irreversible even under biotin treatment, as we see in profound BTD.24
Seizure is a frequent symptom in patients with profound BTD. Venkatarman et al reported seizure as the presenting symptom of their case series and clonic seizure was predominant.25 Canda et al detected seizure in three of twelve symptomatic patients with BD, however most of their patients were diagnosed by newborn screening. Today patients are diagnosed early by newborn screening. They are treated with oral biotin. Due to this fact we do not see symptomatic patients frequently.26 Salbert et al detected that forty-three of 78 (55%) symptomatic children had seizures, and seizures were the presenting symptom in 38% of the enzyme-deficient patients in their study.27 BTD results in an absence of recycling of biotin with subsequent impairments of the main biotin-dependent carboxylases and accumulation of potentially neurotoxic and epileptogenic metabolites.28 In vitro studies in BTD fibroblast revealed that low biotin impairs the function of holocarboxylase synthase (HCS) and the transcription of the HCS gene.29 Low biotin levels, toxic metabolites such as organic acids, lactic acid, biocytin, and hyperammonemia may be responsible for different neurological abnormalities in BTD patients.5
Salbert et al found ophthalmic abnormalities in 51% of 78 symptomatic patients with BTD. These findings included 30% infections, 13% optic neuropathies, 13% motility disturbances, 4% retinal pigment changes, and 1% pupillary findings.27 Hayati et al summarized ocular signs of patients published form 1997 to 2011 in PubMed and reported that 6 patients presented with optic atrophy in both eyes.30
Neuromyelitis optica spectrum disorders (NMOSDs) are a group of demyelinating diseases of the central nervous system which essentially involve the optic nerves and spinal cord. In the literature, NMOSD cases were diagnosed with biotinidase deficiency.31–33 In the last several years, older adolescents and adults have been identified with profound deficiency who exhibited symptoms of myelitis, spastic paresis, paraplegia with or without retinal abnormalities. Most BTD cases are misdiagnosed as multiple sclerosis, transverse myelitis, myasthenia gravis, neuromyelitis optica, and brainstem encephalitis.34 The late onset of BTD should be kept in mind in patients with myelopathy with or without vision loss even if they had partial response to steroid treatment.33
Also, we know that there are several adult patients with profound BTD who remain asymptomatic without treatment. There is no explanation why these patients remain asymptomatic.10 Similarly, Canda et al reported 15 parents diagnosed with BD (two of them with profound BD) by family screening who were all asymptomatic and they did not have increased intake of dietary biotin. These adults may still be at risk for developing symptoms. It is possible that asymptomatic patients can become symptomatic at any time. Also, presence of asymptomatic patients with profound BTD suggests that there may be epigenetic factors that protect patients from developing symptoms.6,26,35 Another explanation for clinical heterogeneity is differences in biotinidase Km variants.36,37 The relationship between clinical symptoms and the severity of biotinidase activity is currently unknown.8,38
Diagnosis
Biotinidase activity can be measured in serum, plasma, and also in fibroblasts and leukocytes and other tissue extracts by radioassay.39 The measurement of biotinidase activity in plasma or serum by colorimetric assay is the most frequently used method for the diagnosis of BTD.1 Normal serum biotinidase activity in humans ranges from 4.4 to 10 nmol/min/mL with a mean activity and standard deviation of 7.1 ± 1.2 nmol/min/mL.4
Serum biotinidase activity of carriers may be similar to those with partial BTD, confounding diagnosis based on enzyme analysis.40 Wolf suggested that evaluation of parental biotinidase activity may be helpful.14 Mutation in the BTD gene results in deficient levels of enzyme activity.41 In some cases the enzyme activity does not differentiate partial deficiency from heterozygosity for profound deficiency, and genetic analysis is necessary.6
Biochemically, in untreated patients, metabolic ketoacidosis, lactic acidosis, and/or hyperammonemia can occur. Elevation of 3-hydroxyisovaleric, 3-hydroxypropionic, lactic acid, and 3-methylcrotonylglycine can be detected in urine organic acid analysis.10,42 In previous reports, it showed that urinary excretion of 3-hydroxyisovaleric acid was an indicator of biotin status.43
Differential Diagnosis
Clinical features including vomiting, hypotonia, and seizures accompanied by metabolic ketolactic acidosis or mild hyperammonemia can be detected in many inherited metabolic diseases.17 A diet containing raw eggs or protracted parenteral hyperalimentation without biotin supplementation may cause similar clinical findings and should be kept in mind in the differential diagnosis.14 Both biotinidase deficiency and holocarboxylase synthetase deficiency are characterized by deficient activities of the three mitochondrial carboxylases in peripheral blood leukocytes and clinical differentiation is often difficult. Biotinidase activity is normal in holocarboxylase synthetase deficiency.17
Sensorineural hearing loss has many causes. Biotinidase activity should be measured specifically in children with hearing loss who are exhibiting other clinical features consistent with biotinidase deficiency.17
Genetic Analysis
BTD gene is located on chromosome 3p25 and consists of four exons, with a total length of 1629 base pairs.44,45 More than 200 mutations have been recognized and it is likely that the list of mutations causing biotinidase deficiency will continue to increase.46 All types of mutations have been found in BTD patients; missense, nonsense, single and multiple nucleotide deletions, single and multiple nucleotide insertions, deletion/insertions, cryptic splice site mutations, and compound allelic mutations.24 Four mutations, c.98_104delinsTCC, p.Q456H, p.R538C, and complex mutation p.D444H/A171T are the most common mutations with BTD, as reported by Hymes et al.41 The first microdeletion of BTD that involves three exons of the gene was reported by Wolf and this deletion results in complete loss of biotinidase enzyme activity. The possibility of a deletion in BTD gene must be considered in patients with low enzyme activity which is inconsistent with BTD gene analysis.47
It is well known that the spectrum of mutations varies in different countries. A large group of patients with BTD ascertained by newborn and family screening from Turkey showed that common mutations are p.R157H, p.D444H, c.98-104del7ins3, and p.T532M. In this study, nine of the patients who were diagnosed by newborn screening had initial symptoms such as seizures, encephalopathy, and lactic acidemia and a common mutation in this group was c.98_104delinsTCC.8 Similar findings were observed about common mutations in different studies from Turkey.26,48
The p.A171T:D444H mutation is a common cause of profound BTD in children. This mutation is one of the most common mutations in children diagnosed by newborn screening program in United States.49 Patients carrying homozygote p.D444H mutations have nearly 50% mean serum biotinidase activity in serum.35,49 Although p.D444H mutation causes a considerable decrease in biotinidase enzyme activity alone, in combination with p. A171H mutations, it results in profound BTD.49 The p.D444H mutation alone causes milder mutation, but in combination with mutation for profound BTD on the other allele, it usually results in partial deficiency.6 There have been several polymorphisms detected, and most are single nucleotide alterations. One variant c.117C>T results in P391S with normal enzyme activity.41
Although genotype-phenotype correlations are not well established in BD, Sivri et al reported that children with profound BD with null mutations were more likely to develop hearing loss than those with missense mutations.50
After detecting a patient with BD, genetic counseling should be done. To evaluate the relatives at risk, measurement of biotinidase enzyme activity and molecular genetic testing should be performed.
Radiological Findings
Cerebral atrophy, ventriculomegaly, widened extra cerebral spaces, subdural effusions, basal ganglion calcifications and T2 hyperintensities were the findings of brain imaging in BTD patients. Bhat et al reported T2 and FLAIR hyperintensities in bilateral hippocampi and para hippocampal gyri as novel observation in BTD patients.51,52 Spinal cord involvement was described especially in older patients with late onset BTD.53
Treatment and Management
Patients with profound BTD are treated with 5–20 mg/day biotin. All symptomatic individuals improve clinically with biotin therapy. Seizures usually resolve within hours to days and cutaneous manifestations usually resolve within weeks.6 Oral biotin therapy provides improvement in symptoms such as alopecia, skin rash, ataxia, and developmental delay.13
As the child grows the quantity of biotin administration decreases. Some have monitored urine organic acid to control whether the dosage of biotin is enough.6 Treatment of partial BTD was initially controversial but over the years, due to the presence of symptomatic partial BTD patients who were not screened and presence of untreated patients who developed symptoms, treatment of partial BTD patients is also recommended.26
Symptomatic children often have developmental delay and are at risk of irreversible damage to auditory, visual, or central nervous system functions. Children with profound BTD who were diagnosed pre-symptomatically following newborn screening and treated with biotin supplementation, do not experience these effects.54 Based on mutation analysis and biotinidase enzyme activity, it is currently not clear whether an untreated patient will develop symptoms or not. The patients with activities lower than 1% are at a high risk for developing symptoms of the disease.55
Wolf at el recommend that the children with both profound and partial BTD should be monitored for hearing loss periodically (yearly in profound BD, every two years in partial BD).6 Also, children with BD should be evaluated for psychomotor deficit, and have ophthalmologic examination (yearly in profound BD, every two years in partial BD) and physical examination for neurological abnormalities, cutaneous findings and infections.29 In the literature, two girls with profound BD developed hair-loss during adolescence that was resolved by increasing the dose of biotin.29 We need more data to determine the dosage of biotin that is necessary for children with BD. Measurement of blood or urine biotin levels is not useful. Also, it can determine compliance with treatment. Evaluation of urine organic acid analysis can be performed if the symptoms return after treatment.29
Biotin is considered safe and non-toxic even in high concentrations. This fact brought a specific problem to medical practitioners' attention.56 Most clinical immunoassays that measure analytics in small concentrations, such as hormones, use specific and sensitive biotin (avidin) technologies.57
High concentrations of biotin in the samples caused incorrect abnormal results. Biotin use can result in false high levels of T3 and T4 and false low TSH levels leading to wrong diagnosis of hyperthyroidism. It should be kept in mind during follow-up.58
Newborn- Screening
The first newborn screening program for BTD was initiated in Virginia in 1984.59 Determination of biotinidase activity on dried blood samples was first used in a pilot study program in Virginia. BTD screening is performed by direct immune assay and results are not related to dietary protein intake. There has been a high rate of false positive results due to prematurity and mishandling of samples.10,60 Wolf at el showed that biotinidase activity is between 50–70% of adult serum biotinidase activity in term infants.6,61 The enzyme activity reaches adult levels in days to weeks.22 Neonatal screening for BTD is conducted in many countries. In Turkey BD screening has been included in neonatal screening program since 2008.8
False positive results can be detected if the patient has above normal albumin levels or if the patient is being treated with valproic acid or has high triglyceride levels. If an individual is being treated with sulfa medications, false negative results can occur when biotinyl-6-aminoquinoline was used for substrate, fortunately sulfa medications are contraindicated in pregnant women and infants.14
Outcomes of newborn screening programs support the fact that biotin treatment started after birth prevents patients with BTD from developing symptoms. There has not been much time between the first description of the disease and newborn BTD screening programs and due to this fact there is a lack of information about long-term natural history of the disorder.62,63
Conclusion
Biotinidase deficiency is an excellent example of early detection and successful treatment of an inherited metabolic disorder. Symptomatic patients with BD can be easily treated with biotin. Today most patients diagnosed by newborn screening are treated with biotin at early ages and due to this fact the number of asymptomatic patients is increasing. Presence of late-onset cases with different clinical findings indicates that there is still much to learn about BD. By the increasing number of patients with BD and detection of many mutations in BTD gene we expect to have more information about genotype and phenotype correlation.
Disclosure
The authors report no conflicts of interest in this work.
References
1. Wolf B, Grier RE, Allen RJ, Goodman SI, Kien CL. Biotinidase deficiency: the enzymatic defect in late-onset multiple carboxylase deficiency. Clin Chim Acta. 1983;131(3):273–281. doi:10.1016/0009-8981(83)90096-7
2. Moss J, Lane MD.The biotin-dependent enzymes. Adv Enzymol Relat Areas Mol Biol.1971;35:321–442. doi:10.1002/9780470122808.ch7
3. Wolf B, Hsia YE, Sweetman L, et al. Multiple carboxylase deficiency: clinical and biochemical improvement following neonatal biotin treatment. Pediatrics. 1981;68(1):113–118.
4. Wolf B, Grier RE, Secor McVoy JR, Heard GS. Biotinidase deficiency: a novel vitamin recycling defect. J Inherit Metab Dis. 1985;8(Suppl 1):53–58. doi:10.1007/BF01800660
5. Wolf B. The neurology of biotinidase deficiency. Mol Genet Metab. 2011;104(1–2):27–34.Review. doi:10.1016/j.ymgme.2011.06.001
6. Wolf B. Clinical issues and frequent questions about biotinidase deficiency. Mol Genet Metab. 2010;100(1):6–13. doi:10.1016/j.ymgme.2010.01.003
7. Baumgardner MR, Suormala T. Biotin-Responsive Disorders. In: Saudubray JM, Baumgardner MR, Walter J, editors. Inborn Metabolic Diseases Diagnosis and Treatment. Heidelberg: Springer-Verlag Berlin Heidelberg; 2016:375–383.
8. Karaca M, Özgül RK, Ö Ü, et al. Detection of biotinidase gene mutations in Turkish patients ascertained by newborn and family screening. Eur J Pediatr. 2015;174(8):1077–1084. doi:10.1007/s00431-015-2509-5
9. Cowan TM, Blitzer MG, Wolf B, Working Group of the American College of Medical Genetics Laboratory Quality Assurance Committee. Technical standards and guidelines for the diagnosis of biotinidase deficiency. Genet Med. 2010;12(7):464–470. doi:10.1097/GIM.0b013e3181e4cc0f
10. Strovel ET, Cowan TM, Scott AI, Wolf B. Laboratory diagnosis of biotinidase deficiency, 2017 update: a technical standard and guideline of the American College of Medical Genetics and Genomics. Genet Med. 2017;19(10):Oct. doi:10.1038/gim.2017.84
11. Neto EC, Schulte J, Rubim R, et al. Newborn screening for biotinidase deficiency in Brazil: biochemical and molecular characterizations. Braz J Med Biol Res. 2004;37(3):295–299. doi:10.1590/S0100-879X2004000300001
12. Baykal T, Hüner G, Sarbat G, Demirkol M. Incidence of biotinidase deficiency in Turkish newborns. Acta Paediatr. 1998;87:1102–1103. doi:10.1080/080352598750031518
13. Wolf B, Heard GS, Weissbecker KA, McVoy JR, Grier RE, Leshner RT. Biotinidase deficiency: initial clinical features and rapid diagnosis. Ann Neurol. 1985;18(5):614–617. doi:10.1002/ana.410180517
14. Wolf B. Biotinidase deficiency: “if you have to have an inherited metabolic disease, this is the one to have”. Genet Med. 2012;14(6):565–575. doi:10.1038/gim.2011.6
15. Mock DM. Skin manifestations of biotin deficiency. Semin Dermatol. 1991;10(4):296–302.
16. Kiykim E, Kiykim A, Cansever MS, Zeybek CA. Biotinidase deficiency mimicking primary immune deficiencies. BMJ Case Rep. 2015;8:2015.
17. Wolf B. Biotinidase Deficiency. In: Adam MP, Ardinger HH, Pagon RA, et al. editors. GeneReviews® [Internet]. Seattle: University of Washington; 2000 March 24:1993–2019. Available from http://www.ncbi.nlm.nih.gov/books/NBK1322/.
18. Wolf B, Spencer R, Gleason T. Hearing loss is a common feature of symptomatic children with profound biotinidase deficiency. J Pediatr. 2002;140(2):242–246. doi:10.1067/mpd.2002.121938
19. Ferreira P, Chan A, Wolf B. Irreversibility of symptoms with biotin therapy in an adult with profound biotinidase deficiency. JIMD Rep. 2017;36:117–120.
20. Wolf B. Children with profound biotinidase deficiency should be treated with biotin regardless of their residual enzyme activity or genotype. Eur J Pediatr. 2002;161(3):167–168. doi:10.1007/s00431-001-0902-8
21. Wolf B, Grier RE, Allen RJ, et al. Phenotypic variation in biotinidase deficiency. J Pediatr. 1983;103(2):233–237. doi:10.1016/S0022-3476(83)80351-5
22. Manzi B, Loizzo AL, Giana G, Curatolo P. Autism and metabolic diseases. J Child Neurol. 2008;23(3):307–314. doi:10.1177/0883073807308698
23. Küry S, Ramaekers V, Bézieau S, Wolf B. Clinical utility gene card for: biotinidase deficiency-update 2015. Eur J Hum Genet. 2016;24(7):3–5.
24. Wolf B. Why screen newborns for profound and partial biotinidase deficiency? Mol Genet Metab. 2015;114(3):382–387. doi:10.1016/j.ymgme.2015.01.003
25. Venkataraman V, Balaji P, Panigrahi D, Jamal R. Biotinidase deficiency in childhood. Neurol India. 2013;61(4):411–413. doi:10.4103/0028-3886.117614
26. Canda E, Yazici H, Er E, et al. Single center experience of biotinidase deficiency: 259 patients and six novel mutations. J Pediatr Endocrinol Metab. 2018;31(8):917–926. doi:10.1515/jpem-2018-0148
27. Salbert BA, Astruc J, Wolf B. Ophthalmologic findings in biotinidase deficiency. Ophthalmologica. 1993;206(4):177–181. doi:10.1159/000310387
28. Mastrangelo M. Actual insights into treatable inborn errors of metabolism causing epilepsy. J Pediatr Neurosci. 2018;13(1):13–23. doi:10.4103/JPN.JPN_160_16
29. León-Del-Río A. Biotin in metabolism, gene expression, and human disease. J Inherit Metab Dis. 2019;42(4):647–654. doi:10.1002/jimd.12073
30. Hayati AA, Wan-Hitam WH, Cheong MT, Yunus R, Shatriah I. Optic neuritis in a child with biotinidase deficiency: case report and literature review. Clin Ophthalmol. 2012;6:389–395. doi:10.2147/OPTH.S29048
31. Girard B, Bonnemains C, Schmitt E, Raffo E, Bilbault C. Biotinidase deficiency mimicking neuromyelitis optica beginning at the age of 4: a treatable disease. Mult Scler. 2017;23(1):119–122. doi:10.1177/1352458516646087
32. Bottin L, Prud’hon S, Guey S, et al. Biotinidase deficiency mimicking neuromyelitis optica: initially exhibiting symptoms in adulthood. Mult Scler. 2015;21(12):1604–1607. doi:10.1177/1352458515596457
33. Yilmaz S, Serin M, Canda E, et al. A treatable cause of myelopathy and vision loss mimicking neuromyelitis optica spectrum disorder: late-onset biotinidase deficiency. Metab Brain Dis. 2017;32(3):675–678. doi:10.1007/s11011-017-9984-5
34. Wolf B. “Think metabolic” in adults with diagnostic challenges: biotinidase deficiency as a paradigm disorder. Neurol Clin Pract. 2017;7(6):518–522. doi:10.1212/CPJ.0000000000000379
35. Swango KL, Demirkol M, Hüner G, et al. Partial biotinidase deficiency is usually due to the D444H mutation in the biotinidase gene. Hum Genet. 1998;102(5):571–575. doi:10.1007/s004390050742
36. Baykal T, Gokcay G, Gokdemir Y, et al. Asymptomatic adults and older siblings with biotinidase deficiency ascertained by family studies of index cases. J Inherit Metab Dis. 2005;28(6):903–912. doi:10.1007/s10545-005-0161-3
37. Suormala T, Ramaekers VT, Schweitzer S, et al. Biotinidase Km-variants: detection and detailed biochemical investigations. J Inherit Metab Dis. 1995;18(6):689–700. doi:10.1007/BF02436758
38. Ye J, Wang T, Han LS, et al. Diagnosis, treatment, follow-up and gene mutation analysis in four Chinese children with biotinidase deficiency. J Inherit Metab Dis. 2009;32(Suppl1):S295–302. doi:10.1007/s10545-009-1238-1
39. Wolf B, Secor McVoy J. A sensitive radioassay for biotinidase activity: deficient activity in tissues of serum biotinidase-deficient individuals. Clin Chim Acta. 1983;135(3):275–281. doi:10.1016/0009-8981(83)90286-3
40. Gannavarapu S, Prasad C, DiRaimo J, et al. Biotinidase deficiency: spectrum of molecular, enzymatic and clinical information from newborn screening Ontario, Canada (2007–2014). Mol Genet Metab. 2015;116(3):146–151. doi:10.1016/j.ymgme.2015.08.010
41. Hymes J, Stanley CM, Wolf B. Mutations in BTD causing biotinidase deficiency. Hum Mutat. 2001;18(5):375–381. doi:10.1002/(ISSN)1098-1004
42. Schubiger G, Caflisch U, Baumgartner R, Suormala T, Bachmann C. Biotinidase deficiency: clinical course and biochemical findings. J Inherit Metab Dis. 1984;7(3):129–130. doi:10.1007/BF01801771
43. Horvath TD, Matthews NI, Stratton SL, Mock DM, Boysen G. Measurement of 3-hydroxyisovaleric acid in urine from marginally biotin-deficient humans by UPLC-MS/MS. Anal Bioanal Chem. 2011;401(9):2805–2810. doi:10.1007/s00216-011-5356-x
44. Cole H, Reynolds TR, Lockyer JM, et al. Human serum biotinidase. cDNA cloning, sequence, and characterization. J Biol Chem. 1994;269(9):6566–6570.
45. Knight HC, Reynolds TR, Meyers GA, Pomponio RJ, Buck GA, Wolf B. Structure of the human biotinidase gene. Mamm Genome. 1998;9(4):327–330. doi:10.1007/s003359900760
46. Procter M, Wolf B, Mao R. Forty-eight novel mutations causing biotinidase deficiency. Mol Genet Metab. 2016;117(3):369–372. doi:10.1016/j.ymgme.2016.01.002
47. Wolf B. First microdeletion involving only the biotinidase gene that can cause biotinidase deficiency: a lesson for clinical practice. Mol Genet Metab Rep. 2016;2(6):74–76. doi:10.1016/j.ymgmr.2016.02.006
48. Seker Yilmaz B, Mungan NO, Kor D, et al. Twenty-seven mutations with three novel pathologenic variants causing biotinidase deficiency: a report of 203 patients from the southeastern part of Turkey. J Pediatr Endocrinol Metab. 2018;31(3):339–343. doi:10.1515/jpem-2017-0406
49. Norrgard KJ, Pomponio RJ, Swango KL, et al. Double mutation (A171T and D444H) is a common cause of profound biotinidase deficiency in children ascertained by newborn screening the the United States. Hum Mutat. 1998;11(5):410. doi:10.1002/(SICI)1098-1004(1998)11:5<410::AID-HUMU10>3.0.CO;2-8
50. Sivri HS, Genç GA, Tokatli A, et al. Hearing loss in biotinidase deficiency: genotype-phenotype correlation. J Pediatr. 2007;150(4):439–442. doi:10.1016/j.jpeds.2007.01.036
51. Desai S, Ganesan K, Hegde A. Biotinidase deficiency: a reversible metabolic encephalopathy. Neuroimaging and MR spectroscopic findings in a series of four patients. Pediatric Radiology. 2008;38(8):848–856. doi:10.1007/s00247-008-0904-z
52. Bhat MD, Bindu PS, Christopher R, Prasad C, Verma A. Novel imaging findings in two cases of biotinidase deficiency-a treatable metabolic disorder. Metab Brain Dis. 2015;30(5):1291–1294. doi:10.1007/s11011-015-9690-0
53. Raha S, Udani V. Biotinidase deficiency presenting as recurrent myelopathy in a 7-year-old boy and a review of the literature. Pediatr Neurol. 2011;45(4):261–264. doi:10.1016/j.pediatrneurol.2011.06.010
54. Weber P, Scholl S, Baumgartner ER. Outcome in patients with profound biotinidase deficiency: relevance of newborn screening. Dev Med Child Neurol. 2004;46(7):481–484. doi:10.1111/j.1469-8749.2004.tb00509.x
55. Möslinger D, Stöckler-Ipsiroglu S, Scheibenreiter S, et al. Clinical and neuropsychological outcome in 33 patients with biotinidase deficiency ascertained by nationwide newborn screening and family studies in Austria. Eur J Pediatr. 2001;160(5):277–282. doi:10.1007/s004310100740
56. Wolf B. High doses of biotin can interfere with immunoassays that use biotin-strept(avidin) technologies: implications for individuals with biotin-responsive inherited metabolic disorders. Mol Genet Metab. 2019;127(4):321–324. doi:10.1016/j.ymgme.2019.07.003
57. Luong JHT, Male KB, Glennon JD. Biotin interference in immunoassays based on biotin-strept(avidin) chemistry: an emerging threat. Biotechnol Adv. 2019;37(5):634–641. doi:10.1016/j.biotechadv.2019.03.007
58. Odhaib SA, Mansour AA, Haddad NS. How biotin induces misleading results in thyroid bioassays: case series. Cureus. 2019;11(5):e4727.
59. Wolf B, Heard GS, Jefferson LG, Proud VK, Nance WE, Weissbecker KA. Clinical findings in four children with biotinidase deficiency detected through a statewide neonatal screening program. N Engl J Med. 1985;313(1):16–19. doi:10.1056/NEJM198507043130104
60. Suormala T, Wick H, Baumgartner ER. Low biotinidase activity in plasma of some preterm infants: possible source of false-positive screening results. Eur J Pediatr. 1988;147(5):478–480. doi:10.1007/BF00441970
61. Heard GS, Wolf B, Jefferson LG, et al. Neonatal screening for biotinidase deficiency: results of a 1-year pilot study. J Pediatr. 1986;108(1):40–46. doi:10.1016/S0022-3476(86)80766-1
62. Wolf B. Biotinidase deficiency should be considered in individuals thought to have multiple sclerosis and related disorders. Mult Scler Relat Disord. 2019;28:26–30. doi:10.1016/j.msard.2018.11.030
63. Jay AM, Conway RL, Feldman GL, Nahhas F, Spencer L, Wolf B. Outcomes of individuals with profound and partial biotinidase deficiency ascertained by newborn screening in Michigan over 25 years. Genet Med. 2015;17(3):205–209. doi:10.1038/gim.2014.104
 © 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.