Back to Journals » ClinicoEconomics and Outcomes Research » Volume 15
Biosimilars Adoption: Recognizing and Removing the RoadBlocks
Authors Niazi SK ![]()
Received 9 January 2023
Accepted for publication 29 March 2023
Published 12 April 2023 Volume 2023:15 Pages 281—294
DOI https://doi.org/10.2147/CEOR.S404175
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Prof. Dr. Dean Smith
Sarfaraz K Niazi
College of Pharmacy, University of Illinois, Chicago, IL, 60612, USA
Correspondence: Sarfaraz K Niazi, Tel +1-312-297-0000, Email [email protected]
Abstract: Almost two decades since biosimilars arrived, we still await their broader adoption, as anticipated. The roadblocks to this adoption include the high amortized cost of goods due to regulatory burden, hurdles created by the distribution system, perception of safety and efficacy, and lack of focus by stakeholders on removing these roadblocks. In this paper, I analyze the source of these roadblocks and offer practical solutions to remove them. These efforts are needed to maximize the adoption of biosimilars to encourage the entry of 100+ biological molecules that can bring affordable healthcare direly missing today across the globe.
Keywords: biosimilars, biosimilarity, development cost, regulatory planning, accessibility, affordability, FDA, EMA, WHO, MHRA
Introduction
The per capita cost of prescription drugs is the highest in the US, with an expenditure of over USD 600 billion in 2021, rising more than 7% annually.1 Of this cost, a majority goes to biological drugs that are given a monopoly for 12 years2,3 to recoup their investment of billions of dollars.4,5 Since US sales bring more than 60% of the global revenue for new drugs and there are no price controls in the US, it is understandable why the entry of new drugs is motivated in the US. From 2002 to 2021, 883 new molecular entities have been introduced globally, of which 779 are available in the US.6
The success of generic drugs in reducing the cost to patients led to legislation7 allowing copies of biological drugs, biosimilars, to be approved using an abbreviated path, though still more complex and expensive than chemical drugs, requiring comparison of product- and process-related attributes, clinical pharmacology and clinical efficacy. The US legislation (BPCIA) has been amended recently to replace “animal toxicology” testing requirements with “nonclinical”, and other changes are imminent that might reduce the cost burden of the development of biosimilars in the US.
Biosimilars were supposed to be adopted quickly to bring the promised savings of billions of dollars. A global trend of biosimilar adoption is shown in Figure 1, comparing market share as a percentage of units contributed by biosimilars with the discount compared with the price of the reference product in 24 European countries and the US. Nine European countries showed 90% or better adoption, all related to the price drop and the lowest price reductions yielding the highest market adoption in some instances.
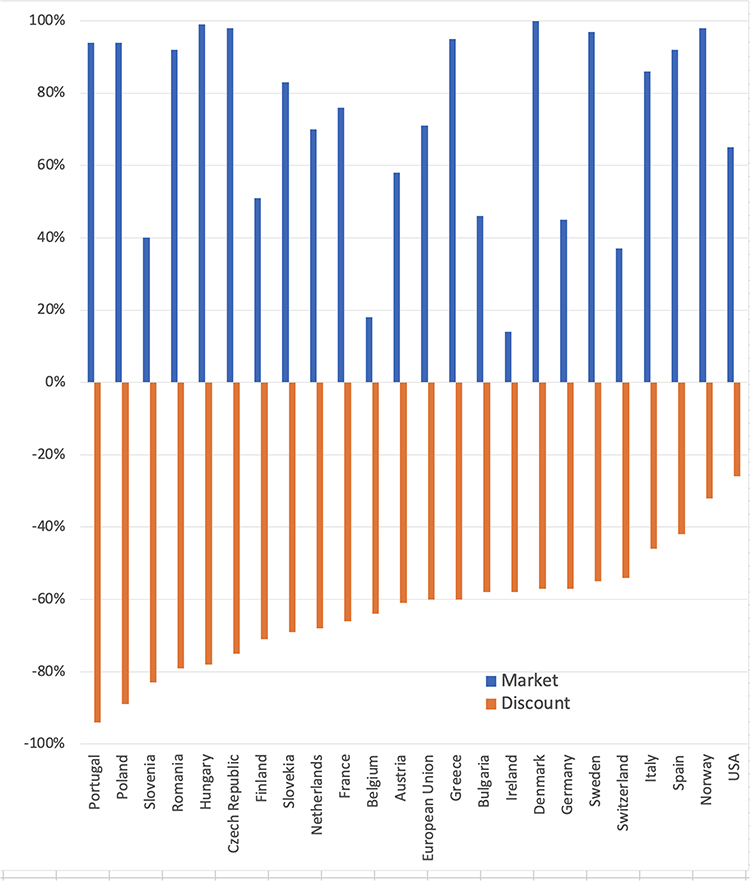 |
Figure 1 Comparison of market share as a percentage of units contributed by biosimilars with the discount from the price of the reference product in 24 European countries and the US. European data [https://www.iqvia.com/-/media/iqvia/pdfs/library/white-papers/the-impact-of-biosimilar-competition-in-Europe-2021.pdf]. US data [Barclay Global Pharmaceuticals; US Specialty Pharma. September 2022.] The US data are based on filgrastim, pegfilgrastim, trastuzumab, infliximab, and bevacizumab products till the third quarter of 2022. |
The US adoption was unpredictable (Figure 2); infliximab and rituximab showed the highest price drop, both products introduced by Amgen, the largest biotechnology company. However, the price reduction (as percent units) did not correlate with the percent price reduction in the US (Figure 3). Filgrastim, the lowest-priced product, gained the largest market share. In contrast, pegfilgrastim, the highest-priced product, gained only a smaller market share as biosimilars, despite offering a higher discount of 37% than the discount for filgrastim biosimilars. One lesson learned from these data is that unless the price drop is more than 80%, the market adoption is not correlated; it can only be contributed to distribution systems in Europe where these products are purchased under a tender system; in the US, it is due to the interference by the pharmacy benefit managers (PBMs) and the ill-conceived perception of the safety and efficacy taught by the reference product companies. To improve the adoption of biosimilars, we need to identify the foundations of these roadblocks and make a plan to remove them.
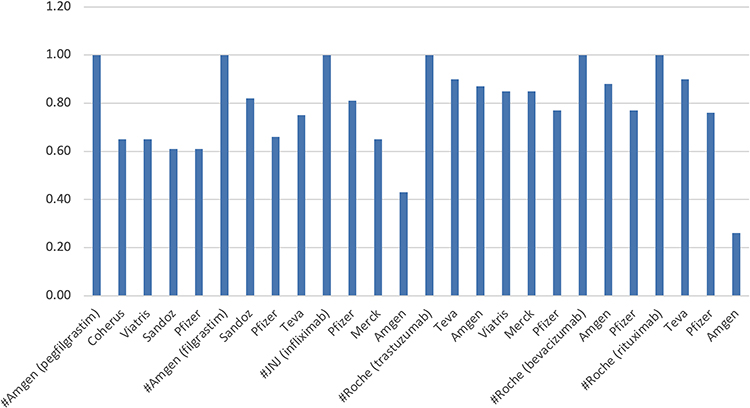 |
Figure 2 WAC Percent reduction of the biosimilars in the US. The WAC does not include gross-net sale discounts. [Source: https://www.iqvia.com; https://www.cms.gov]. |
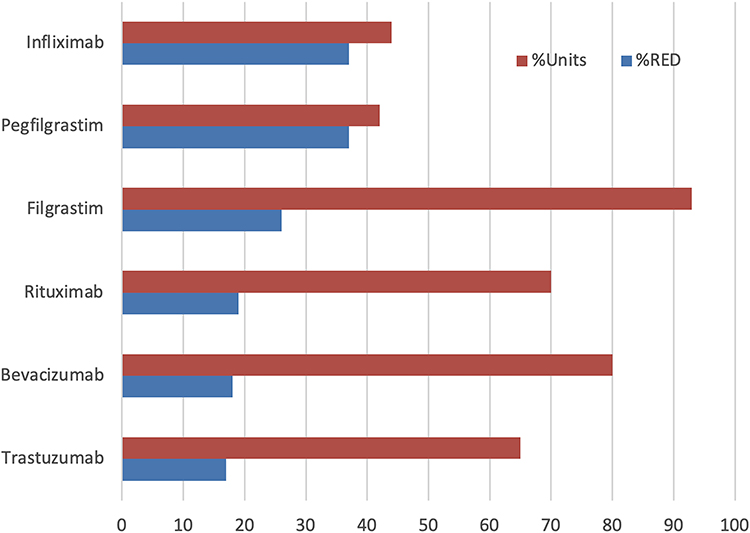 |
Figure 3 Comparison of percent reduction of WAC price and percent adoption of biosimilars in the US. WAC Price does not include GTN discounts. The reference product is scored as 1.0. |
 |
Figure 4 The four reins are pulling back the adoption of biosimilars and artwork created by the Open AI platform, DALLE-2. (https://openai.com/product/dall-e-2). |
Cost Containment
Implementing plans to overcome the hurdles above starts with a clear understanding of the reasonable cost of biological drugs based on the costs involved in the development and distribution cycle, keeping in mind that introducing biosimilars is to reduce the cost of biological drugs to patients.
Table 1 shows the cost that the CMS reimburses for monoclonal antibodies or drug-antibody conjugates, ranging from about $25 million to about $4000 per gram;8 the public prices are even higher. Most reference product sell for reference products at $10–12,000 per gram. Lower-dose cytokines like filgrastim are charged from $650,000 to $1.3 million per gram, compared to their production cost of $1000-$3000.9
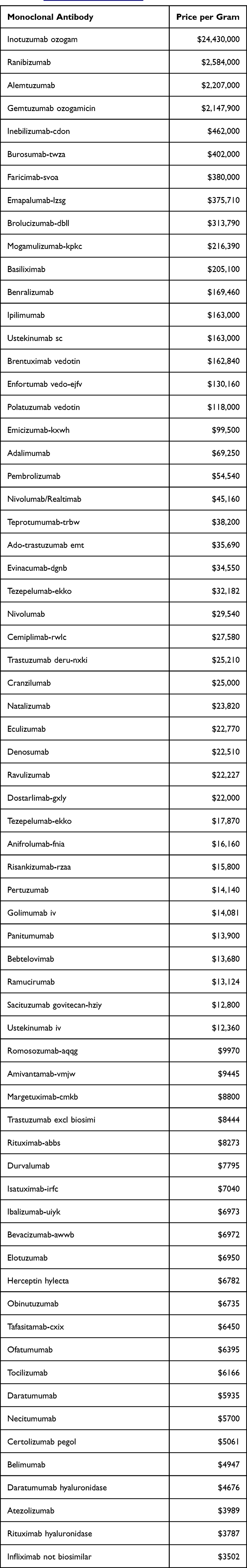 |
Table 1 Cost per Gram of Monoclonal Antibodies in the Finished Product, as Reimbursed by CMS (Source: https://www.cms.gov] |
For example, in the data presented in Figures 2 and 3, the infliximab reference product cost was $11,680; for trastuzumab, $10,386; for bevacizumab, $7970 and for rituximab, $9400 per gram. A similar analysis of part D and part B drugs reimbursement costs shows that the highest cost is in millions of dollars for the reference product (Table 1; data for monoclonal antibodies only). These costs are likely the lowest as it is based on CMS reimbursement; it will be higher for other patients. Given that a new biological drug will amortize its investment of approximately a billion dollars for at least 12 years, the price of 35 to 24,000 times the cost can still be questioned.
A new US law can now force price reductions for biological drugs that have a monopoly on the market for 12 years if no biosimilar is available or anticipated over the next two years and if it falls in the top reimbursement scale. Based on current data, none of the existing biological drugs will fall into this category.
Regulatory
The material cost of biological drugs is relatively small. For example, the well-established cost of goods of monoclonal antibodies is between $95 and $200 per gram, as calculated by the WHO,10 and even lower, as suggested by others.11 For example, Humira (adalimumab) sells at over $90,000 per gram.12 These price structures partly contributed to the amortization of the development cost of around two billion dollars for a new biological drug. In addition, the biosimilar development cost is $100–300 million,13 which also significantly adds to the amortized cost.
Reducing the cost of regulatory approval has been a focus of the FDA and has brought many changes in the approval guidelines. Yet, it remains the responsibility of the developers to make ingenious use of these suggestions, as suggested below, derived from the scientific advances and the novel techniques focussed by the FDA in its new research program.14
- The analytical assessment provides the foundation to establish biosimilarity; over the past decade, the testing technologies have improved significantly to a point where even the slightest differences in the structure can be demonstrated. As a result, developers should use more innovative methods, particularly analytical assessment, beyond what is recommended in related guidelines.15
- New technologies that can assist in establishing molecular similarity now focus on the relationship between the primary amino acid sequence that is readily analyzed with the 3D structure responsible for the pharmacology and toxicology of the molecule. For example, the artificial intelligence (AI) based AlphaFold16,17 (Google’s DeepMind) and ESMFold 15 16 (Facebook’s Meta) predict the 3D structure, as well as its reliability that can be compared with expected variability in protein structure between the batches. Proteins with high reproducibility may not require extensive testing; argument developers can present to the FDA.18
- Another test assuring high similarity of secondary and tertiary structure is the fluorescent spectroscopy under different stress conditions to stretch the bonds and demonstrate equivalence of bonding to assure similar domain structure.19
- Also significant is the correlation of glycosylation microheterogeneity with Fc-receptor binding and ADCC for mAbs, and the examination of initial levels of aggregates (non-covalent and covalent) and correlation with clinical immunogenicity and differences found in the structure.20
- The FDA has recently disclosed a plan to ensure that protein aggregation that contributes to immunogenicity can be monitored using flow imaging microscopy (FIM), and to ensure that the images are correctly interpreted, using an artificial intelligence/machine learning approach (AI/ML) of convolutional neural networks (CNNs or ConvNets).21 The FDA has also disclosed a suitable software, ParticleSentry AI,22 to analyze these data during development and commercial manufacturing.23 Developers are strongly urged to adopt this technology to reduce the burden of safety testing.
- Analytical assessment begins with selecting critical quality attributes to characterize the product expression-dependent (product-related) or manufacturing process-dependent (process-related); selecting only the relevant attributes reduces redundant testing (Table 1).
- Biosimilar products can have different formulations than the reference product, and it is often necessary to avoid patent infringement; however, this can result in serious safety issues if any novel inactive component is used. It is best to use the same formulation if available and use inactive ingredients already used for the given molecule.
- The process-related attributes are tested when the product is released; to establish release specifications, multiple kinds of reference products must be tested, except for those whose limits fall under legacy attributes such as sterility, protein content, and potency to reduce the testing burden. (Table 2) Developers can only use one testing method that provides the same property evaluation; these are not considered orthogonal methods.
- Since the key concern of the FDA is assuring analytical similarity, delays in approvals often come from audit findings of analytical data, particularly Part 11 compliance; one way to avoid such delays is to outsource the final analytical assessment to cGMP-compliant CROs. The cost of outsourcing will be well outweighed by shorter approval time.
- Until the beginning of 2023, animal toxicology testing was part of the BPCIA, resulting in thousands of such studies submitted with regulatory applications, frequently using these data to justify the differences in the analytical profile,24 and most were rejected.25 However, more than 130 products have received FDA and EMA approval, and none of the animal studies failed because they cannot fail. The FDA Modernization 2.0 that became law in 2023 removes “animal” and “toxicology” from the abovementioned legislation and replaces them with “nonclinical”, which may include animal testing that can not be justified for testing of biological drugs in species that do not have the receptors to show pharmacological response. Now FDA recommends not conducting animal studies for new biological drugs.26
- The FDA has begun approving biosimilars based on pharmacokinetic and pharmacodynamic data derived from healthy individuals. These data are labeled as clinical efficacy testing in healthy subjects. Developers should question the need for patient testing that adds to more than 70% of the development cost. Here are some suggestions that may hold. Of the hundreds of clinical efficacy trials conducted on biosimilars, all have succeeded compared to their reference products. The portal clinicaltrials.gov27 reports successful comparisons for about 150 trials with final results. The research database, PubMed, lists over 400 trials over the past 20 years that were also successful.28 Clear scientific reasoning states that the study power cannot be achieved to show the difference between the biosimilar candidate and its reference product,29 as confirmed by the MHRA.30
- The FDA has taken several measures to expedite the development of biosimilars at a much-reduced cost, for example, by suggesting to replace comparative clinical efficacy testing, which adds to almost 70% or more of the development cost, with in silico models.31
- Unnecessary testing in humans is highly discouraged as codified in the US 21 CFR 320.25(a)(13).32 Therefore, if patient efficacy testing cannot fail, it is tantamount to human abuse.
- An argument is that patients may have different disposition kinetics, receptor binding, and immunogenic profile, so it is better to test in patients. If the disposition kinetics are different in patients, then these should be the same for the biosimilar product; any difference in the structure that might affect disposition kinetics is already studied in clinical pharmacology testing, allowing a similar robustness of comparison in healthy subjects. This is more relevant in testing immunogenicity responses that can be muted in patients due to exposure to drugs or the disease, making healthy subject testing more relevant.
- The FDA has declared that any product with a PD marker will not need to demonstrate efficacy in patients, even if the PD markers do not correlate with clinical response.31 However, antibodies do not demonstrate a PD response excluding them from this category. Since the PD response results from receptor binding, should functional studies suffice to qualify for the waiver of Phase 3 studies?
- Post-market surveillance, as currently required, is sufficient to demonstrate any residual risks with biosimilars.
- Biosimilars are expected to share the filing submissions to FDA with the reference product company within 30 days of filing.33 The language of the law states that the biosimilar developers “shall” share what has been interpreted by the Supreme Court as optional. The current advice is to refrain from sharing if other biosimilars are already available; otherwise, sharing is preferred to avoid longer litigations. Recently, the US Congress passed a bill to restrict this practice.34
- Manufacturing costs can be significantly reduced by introducing newer technologies, using a smaller-scale lot as an “at-scale” lot and scaling up after approval under ICHQ5E,35 using higher-yielding cell lines, and considering continuous manufacturing are some emerging ideas that should be evaluated.
- Capital expenditure can be minimized by outsourcing the development and transferring manufacturing in-house after approval using the ICHQ5E to avoid regulatory inquiries that can delay the approval. Outsourcing the development work to CDMOs also opens up the possibility of many products arriving sooner since every developer comes to bottlenecks, not if dozens of CDMOs are available.
- The FDA allows developers to hold as many meetings as requested once they have enrolled in the plan and paid the fee. However, each meeting has a 4–6 month toll; initially, when developers needed clarification, they demanded multiple meetings. The most important meeting is after the completion of clinical pharmacology or what the FDA now calls clinical efficacy in healthy subjects. At this stage, the developers should ask if the FDA can identify data uncertainties requiring more clinical testing. Any residual uncertainty may require additional clinical studies; shifting the onus to regulatory agencies makes it possible to avoid many such studies; however, if developers offer extensive testing on patients or other healthy subject studies, the agencies will not refuse.
- The FTC and the FDA are tracking down the “anti-competitive practices, such as making false or misleading statements comparing biological reference products and biosimilars, may be slowing progress and hampering the uptake of these important therapies”.36
- The FDA also agreed to “take appropriate steps to address companies making false or misleading communications about biologics, including biosimilars and interchangeable products, which will help deter anti-competitive behavior in the biologics market and lead to the use of all available biological product” when the FDA said it in a press release in 2021, blaming Amgen for false claims that the is more effective due to its delivery system.37
- The Executive Order “Promoting Competition in the American Economy”38 directs the FTC to stop “unfair anticompetitive conduct or agreements in the prescription drug industries, such as agreements to delay the market entry of generic drugs or biosimilars.”
- The “Advancing Education on Biosimilars Act”39 calls for the government to “provide educational materials to healthcare providers, patients, and the general public to increase awareness, knowledge, and confidence in the safety and efficacy of approved biosimilars.”
- The “Star Rating for Biosimilars Act”40 introduced a method of qualifying Medicare plans.
- The “Bolstering Innovative Options to Save Immediately on Medicines” (BIOSIM) Act41 suggests increasing, albeit temporarily, by 2% when biosimilars are used.
- The “Preserve Access to Affordable Generics and Biosimilars Act”42 requires the FTC to end “pay-for-delay” tactics to delay the entry of generics or biosimilars.
- The “Inflation Reduction Act” introduces price reduction of Medicare part B and later part D drugs that exceed a billion dollars of reimbursement, have had a monopoly for more than 12 years for biological and 7 years for chemical drugs, and for which no biosimilar or generic product is available or imminent to arrive.43
- The “FDA Modernization Act 2.0” became law in 2023, amending the BPCIA to remove “animal testing” with “nonclinical testing”.44
- The FDA withdrew a pivotal guideline for the analytical assessment and replaced it with a more rational approach that reduces the cost of establishing analytical similarity of biosimilar candidates with the reference product.45,46
- The FDA removed the requirement of immunogenicity testing if it meets other analytical similarity requirements.47 This suggestion applies to other immunogenic biological drugs where immunogenicity is not known to alter the PK profile.
- The FDA began approving biosimilars without testing in patients and listing testing in healthy subjects as “clinical efficacy in healthy subjects”, where the molecule has a pharmacodynamic response to compare the two products.48
- The FDA has started several innovation directives on demonstrating efficacy, and the developers need to catch up to their expertise here. As a result, the old mindset that patient data is golden no longer holds for biosimilars.49
- The FDA has taken several significant scientific innovation and regulatory modification steps, including funding research, both in-house and by others, to develop innovative approaches that would give more confidence in the testing and reduce or eliminate irrelevant studies, including this publication.50
- The FDA has introduced a new AI-driven method to assure the safety of biotherapeutics that can be used to support the claim of biosimilarity.23
- The FDA has made over 200 funding opportunities available for biological drugs and biosimilars51 to innovate their development.
- A new Bill in the Senate is removing “interchangeable” biosimilars.52 The issue of interchangeability in the US53 is a unique situation with no scientific rationale; other agencies like the EMA54 and MHRA55 consider biosimilars interchangeable, even with other biosimilars. In the EU, where biosimilars are part of the negotiated purchase, the patients have no choice but to use whatever product is made available. Historically, insulin was interchanged without question for decades as a chemical drug. Now that it is part of the biological drugs program, the interchangeability has risen again, which is redundant since the CMS has already put a monthly ceiling cost to Medicare patients. Most big insulin producers have begun to reduce the cost.56 One major misunderstanding about interchangeability comes from the limited relevance to the biological drugs dispensed at the pharmacy level; this excludes all intravenously injected products and most others requiring clinician supervision. More than 90% of biological drugs are purchased through institutional plans that decide which brand to dispense when a reference product is prescribed. The only use left for this category of a biosimilar is to gain added reputation by declaring an interchangeable as a better product, even if it is not dispensed at the pharmacy level; this option then goes only to larger companies that will spend scores of additional millions to gain this reputation.
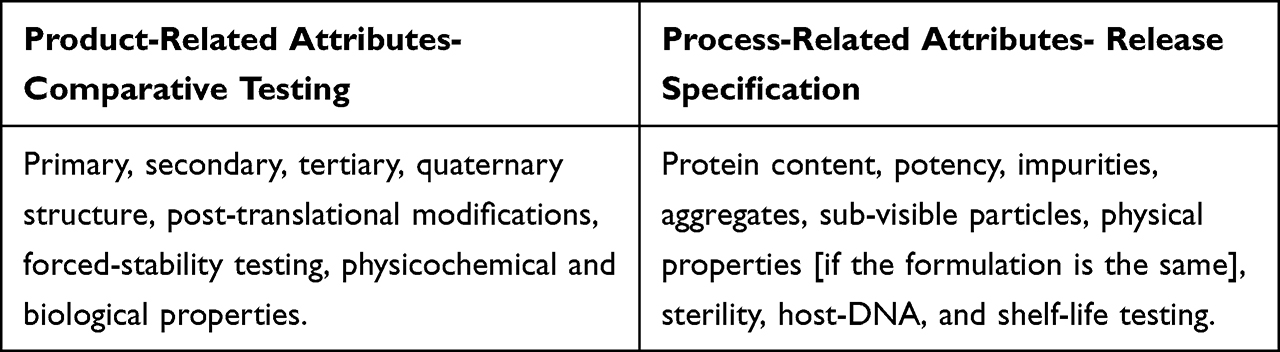 |
Table 2 Critical Quality Attributes Classification |
Distribution
This per capita cost of pharmaceuticals at $1400 is the highest in the US,57 partly attributed to the complexity of the distribution system that allows manipulations by the PBMs. On 26 January 2023, a bill was introduced in the Senate, Pharmacy Benefit Manager Transparency Act of 2023, “to prevent unfair and deceptive acts or practices and the dissemination of false information related to pharmacy benefit management services for prescription drugs and for other purposes”.58 In addition, 42 Committee Reports59 and 100 Bills in the US Congress identify and criticize the role of PBMs.60 However, the political standing of PBMs keeps them in play and remains a significant hurdle in allowing the adoption of biosimilars.
To overcome these constraints, several efforts have been made by not-for-profit organizations such as CivicaRx61 and the state governments such as California,62 planning their supply of biological drugs at the lowest price, going around the distribution channels. On a broader scale, Mark Cuban’s efforts63 to eliminate all distribution channels are beginning to show great promise and stand the best chance to lower biosimilars’ prices significantly.
In the EU, where most countries have socialized medicine, the costs are much lower, but they, too, have issues with fair bidding and pricing of pharmaceutical products.64
Stakeholders
As biosimilars entered the US market in 2015, many stakeholders began promoting biosimilars to improve their adoption. These efforts include educating the prescribers and patients that may have backfired. The FDA declares that “biosimilars have no clinically meaningful difference with their reference products.” Pushing this statement raises the question about the credibility of the FDA; why is there a need to clarify if this is a fact? The FDA has developed several education platforms, and the claims of safety and efficacy should be left to the FDA, not the marketing teams or industry associations.
The savings claims are also redundant since this is the main reason for bringing biosimilars to the market. A recent RAND Corporation study estimates that biosimilars could save $38.4 billion over five years if their cost is optimized.65 It should not be news; instead, it should raise a question, why so little? It almost sounds like promoting biosimilar use to claim more savings; it should be evident to payors. Yet, there is rarely a talk about chemical generics savings billions of dollars.
Competition
The expectations of an 80–90% discount on biosimilars now seem far-fetched,66 as it has come out in several European countries. There was high anticipation of the Indian and Chinese companies entering the race, which has now cooled off67 mainly due to poor cGMP compliance of the most qualified Indian companies, including Biocon, which has secured FDA approval for its insulin product.68–71 What is keeping the Indian companies away is the reputation of their biosimilar products if judged based on the regulatory guidelines in India that are highly lax and often meaningless.72 Thus, based on these guidelines, the 100+ biosimilars marketed in India will require a complete rework, a cost burden that the companies will need more money to afford.
Another source of lower-cost biosimilars could come from China, which is fast expanding its biotechnology base. However, the need for a sound regulatory path for hundreds of biosimilars developed in China73 will keep them from entry into the US.74 Until China adopts globally harmonizing guidelines for biosimilars. The FDA has also struck other non-US competitors like Alvotech,75 reducing the number of potential competitors. For now, the major pharma that controls the US biosimilar will continue to add more biosimilars, and the discounts will likely be at most 50% for many years.
Conclusion
Adopting biosimilars requires that they be affordable, the primary purpose mandated by the EMA76 and the FDA.77 Both agencies have made significant changes and revisions to their guidelines, but many more are needed to remove proven redundant studies. The impeccable safety and efficacy record of approved biosimilars since their approval began 18 years ago is the best evidence that the regulatory agencies will moderate the approval guidelines to a point where small and medium-sized companies will enter the market. The development costs are expected to drop to less than $30 million for products where Phase III studies are not required to $50 million where they are, with a wide margin based on the indications; the oncology drugs with efficacy testing form the highest cost development. In addition, the availability of newer technologies that can readily detect meaningful differences makes it plausible that the development cost will drop significantly.
The developers are also responsible for reducing the cost of development by adopting better science, rational and systematic planning, and taking advantage of outsourcing that is now readily available for biosimilar development and commercial supply.
Industry associations such as the Biosimilars Council,78 Association for Affordable Medicines,79 International Generics and Biosimilars Association,80 Biologics and Biosimilars Collective Intelligence Consortium,81 British Biosimilar Council,82 Generic and Biosimilars Association,83 Medicines for Europe,84 Danish Generic and Biosimilars Medicines Industry Association,85 Biosimilars Canada,86 and other consortiums of stakeholders are wrongfully focussed on promoting biosimilars to prescribers and patients. This role should be left to the FDA.87 Over time, all concerns will wither away if they start educating developers and working with regulatory agencies to rationalize the approval guidelines. Instead, millions of dollars spent in promotional advertising that biosimilars will save money should be spent on improving the science, creating collaborative projects, and streamlining partnerships. The FDA has given millions of dollars in grants to improve the testing of biosimilars and create proof that clinical efficacy testing may not be required; the stakeholders should be funding such research.
The distribution systems in the US and Europe continue to be a significant impediment that should also be a priority for the stakeholders to overcome. There are multiple bills in the Senate to control the power of the Pharmacy Benefits Managers (PBM) who are primarily responsible for keeping the pricing non-competitive; the holds in the EU, where the bureaucracy of tender business has yet to yield a consistent result.
However, despite all hurdles, constraints, politics, and misgivings, the inflection points for biosimilars are imminent, and a significant shift will occur soon88 as the prices drop and the markets expand. It is only apparent and anticipated.
All concerns and payors’ hesitations will evaporate when the prices drop by 70 to 80%. Gary Kasparov said, “at the end of the day, it is all about money”, which applies to biosimilars most suitably.89 But that is happening differently than expected. Figure 4 summarizes the relative importance of all impediments, surprisingly, the most commonly blamed regulatory hurdles are of the least conern.
Disclosure
The author declares no conflict of interest.
References
1. Tichy EM, Hoffman JM, Suda KJ, et al. National trends in prescription drug expenditures and projections for 2022. Am J Health Syst Pharm. 2022;79(14):1158–1172. PMID: 35385103; PMCID: PMC9383648. doi:10.1093/ajhp/zxac102
2. Cicchiello A, Gustafsson L. Brand-Name Drug Prices: The Key Driver of High Pharmaceutical Spending in the US. - An International Comparison of Prescription Drug Spending and Costs. New York, NY: Commonwealth Fund; 2021.
3. Conti RM, Berndt ER. Four facts concerning competition in US. generic prescription drug markets. Int J Econ Bus. 2020;27(1):27–48. doi:10.1080/13571516.2019.1654324
4. Congressional Budget Office. Research and development in the pharmaceutical industry. Available from: https://www.cbo.gov/publication/57126.
5. Wouters OJ, McKee M, Luyten J. Estimated research and development investment needed to bring a new medicine to market, 2009–2018. J Am Med Assoc. 2020;323(9):844–853. doi:10.1001/jama.2020.1166
6. IQVIA Global Trends in R&D. Available from: https://www.iqvia.com/insights/the-iqvia-institute/reports/global-trends-in-r-and-d-2023.
7. Title VII—improving access to innovative medical therapies subtitle a—biologics price competition and innovation. Available from: https://www.fda.gov/media/78946/download.
8. CMS. 2023 ASP Drug Pricing Files Available from: https://www.cms.gov/medicare/medicare-part-b-drug-average-sales-price/2023-asp-drug-pricing-files.
9. Babaeipour V, Khanchezar S, Mofid MR, et al. Efficient process development of recombinant human granulocyte colony-stimulating factor (rh-GCSF) production in Escherichia coli. Iran Biomed J. 2015;19(2):102–110. PMID: 25864815; PMCID: PMC4412921. doi:10.6091/ibj.1338.2015
10. WHO. Call for consultant on monoclonal antibodies for infectious diseases. Available from: https://www.who.int/news-room/articles-detail/call-for-consultant-on-monoclonal-antibodies-for-infectious-diseases.
11. Eri Amasawa, Hiroaki Kuroda, Kozue Okamura, Sara Badr, and Hirokazu Sugiyama, Cost–Benefit Analysis of Monoclonal Antibody Cultivation Scenarios in Terms of Life Cycle Environmental Impact and Operating Cost. ACS Sustainable Chemistry & Engineering 2021 9 (42), 14012-14021. DOI: 10.1021/acssuschemeng.1c01435 1. .
12. Humira Prices, Coupons and Patient Assistance Programs Available from: https://www.drugs.com/price-guide/humira.
13. McKinsey Three imperatives for research in biosimilars. Available from: https://www.mckinsey.com/industries/life-sciences/our-insights/three-imperatives-for-r-and-d-in-biosimilars.
14. FDA. Biosimilar science and research. Available from: https://www.fda.gov/drugs/biosimilars/biosimilars-science-and-research.
15. FDA. Development of therapeutic protein biosimilars: comparative analytical assessment and other quality-related considerations. Guidance for Industry. Available from: https://www.fda.gov/media/159261/download.
16. Jumper J, Evans R, Pritzel A, et al. Highly accurate protein structure prediction with AlphaFold. Nature. 2021;596:583–589. doi:10.1038/s41586-021-03819-2
17. Callaway, E. News July 2022 ‘The entire protein universe’: AI predicts shape of nearly every known protein DeepMind’s AlphaFold tool has determined the structures of around 200 million proteins.. . Available from: https://www.nature.com/articles/d41586-022-02083-2.
18. AlphaFoldAphaFold Protein Structure Database.. Available from: https://alphafold.ebi.ac.uk/.
19. Niazi SK. US patent application. Available from: https://patents.google.com/patent/US20180024137A1/en?oq=US20180024137A1.
20. Coghlan J, Benet A, Kumaran P, et al. Streamlining the characterization of disulfide bond shuffling and protein degradation in IgG1 biopharmaceuticals under native and stressed conditions. Front Bioeng Biotechnol. 2022;10:862456. PMID: 35360407; PMCID: PMC8963993. doi:10.3389/fbioe.2022.862456
21. Stanford University Course. Completed biosimilar trials with results. Available from: https://clinicaltrials.gov/ct2/results?term=biosimilar&age_v=&gndr=&type=&rslt=With&Search=Apply.
22. Particle Sentry AI: Quality Control in Drug Product Manufacturing Available from: https://www.semitorr.com/specialties/particle-sentry-ai-quality-control-in-drug-product-manufacturing/.
23. FDA: Artificial Intelligence/Machine Learning Assisted Image Analysis for Characterizing Biotherapeutics Available from: https://www.fda.gov/drugs/regulatory-science-action/artificial-intelligencemachine-learning-assisted-image-analysis-characterizing-biotherapeutics?utm_medium=email&utm_source=govdelivery.
24. Moore TJ, Mouslim MC, Blunt JL, Alexander GC, Shermock KM. Assessment of availability, clinical testing, and US food and drug administration review of biosimilar biologic products. JAMA Intern Med. 2020. doi:10.1001/jamainternmed.2020.3997
25. Niazi SK. Biosimilars: a futuristic fast-to-market advice to developers. Expert Opin Biol Ther. 2022;22(2):149–155. PMID: 34913776. doi:10.1080/14712598.2022.2020241
26. Li J, Florian J, Campbell E, et al. Advancing biosimilar development using pharmacodynamic biomarkers in clinical pharmacology trials. Clin Pharmacol Ther. 2020;107:40–42. doi:10.1002/cpt.1653
27. Completed biosimilar trials with results. https://clinicaltrials.gov/ct2/results?term=biosimilar&age_v=&gndr=&type=&rslt=With&Search=Apply. Available from: https://clinicaltrials.gov/ct2/results?term=biosimilar&age_v=&gndr=&type=&rslt=With&Search=Apply.
28. Biosimilar Clinical Trials in peer-reviewed articles as reported in PubMed. Available from: https://pubmed.ncbi.nlm.nih.gov/?term=biosimilar+clinical+trial.
29. Niazi S. Scientific rationale for waiving clinical efficacy testing of biosimilars. Drug Des Devel Ther. 2022;16:2803–2815. PMID: 36043044; PMCID: PMC9420434. doi:10.2147/DDDT.S378813
30. MHRA. Biosimilar guidance. Available from: https://www.gov.uk/government/publications/guidance-on-The-licensing-of-biosimilar-products/guidance-on-The-licensing-of-biosimilar-products.
31. Chiu K, Racz R, Burkhart K, et al. New science, drug regulation, and emergent public health issues: the work of FDA’s division of applied regulatory science. Front Med. 2023;9:1109541. doi:10.3389/fmed.2022.1109541
32. US Congress. BPCIA. Available from: https://www.congress.gov/bill/111th-congress/house-bill/3590.
33. Van de Wiele VL, Kesselheim AS, Sarpatwari A. Barriers to US biosimilar market growth: lessons from biosimilar patent litigation. Health Aff. 2021;40(8):1198–1205. Erratum in: Health Aff (Millwood). 2021 Oct;40(10):1671.PMID: 34339242. doi:10.1377/hlthaff.2020.02484
34. US Congress H.R.2884 - affordable prescriptions for patients through improvements to patent litigation act. Available from: https://www.congress.gov/bill/117th-congress/house-bill/2884.
35. Niazi SK. Reinventing commercial biomanufacturing. Available from: https://www.europeanpharmaceuticalreview.com/article/77395/reinventing-commercial-biomanufacturing/.
36. FDA Press Announcement. FDA and FTC announce new efforts to further deter anticompetitive business practices. Available from: https://www.fda.gov/news-events/press-announcements/fda-and-ftc-announce-new-efforts-further-deter-anti-competitive-business-practices-support.
37. FDA. Notifies amgen of branding its product neulasta. Available from: https://www.fda.gov/drugs/drug-safety-and-availability/fda-notifies-amgen-misbranding-its-biological-product-neulasta-due-false-or-misleading-promotional.
38. Whitehouse Announcement Presidential order on promoting competition in the American economy. Available from: https://www.whitehouse.gov/briefing-room/presidential-actions/2021/07/09/executive-order-on-promoting-competition-in-The-american-economy/.
39. US Senate. Advancing education on biosimilars act. Available from: https://www.cassidy.senate.gov/imo/media/doc/AdvancingEducationonBiosimilarsAct.pdf. (
40. US Congress. Star rating for biosimilars act. Available from: https://www.congress.gov/bill/117th-congress/house-bill/2855?q=%7B%22search%22%3A%5B%22Star+Rating+for+Biosimilars+Act%22%2C%22Star%22%2C%22Rating%22%2C%22for%22%2C%22Biosimilars%22%2C%22Act%22%5D%7D&s=1&r=1. (
41. US Congress. H.R.2815—BIOSIM act. Available from: https://www.congress.gov/bill/117th-congress/house-bill/2815/text?r=1&s=1.
42. US Congress. Preserve access to Af3 fordable generics and biosimilars act. Available from: https://www.congress.gov/117/bills/s1428/BILLS-117s1428rs.pdf.
43. Niazi SK. The inflation reduction act: a boon for the generic and biosimilar industry. J Clin Pharm Ther. 2022;47(11):1738–1751. PMID: 36207987. doi:10.1111/jcpt.13783
44. US Congress S.5002 - FDA modernization act 2.0. 117th Congress (2021–2022). Available from: https://www.congress.gov/bill/117th-congress/senate-bill/5002/text.
45. FDA News Release. Withdraws draft guidance for industry: statistical approaches to evaluate analytical similarity. Available from: https://www.fda.gov/drugs/drug-safety-and-availability/fda-withdraws-draft-guidance-industry-statistical-approaches-evaluate-analytical-similarity.
46. Forbes Magazine. Scientist invented a new pathway to approve biosimilars, and the FDA is listening. Available from: https://www.forbes.com/sites/nicolefisher/2018/07/25/one-mans-mission-to-fix-The-fdas-biosimilar-problem/?sh=67e656742380.
47. FDA. Clinical immunogenicity considerations for biosimilar and interchangeable insulin products. Available from: https://www.fda.gov/regulatory-information/search-fda-guidance-documents/clinical-immunogenicity-considerations-biosimilar-and-interchangeable-insulin-products.
48. FDA. Theragrastim/Releuko approval package. Available from: https://www.accessdata.fda.gov/drugsatfda_docs/nda/2022/761082Orig1s000MedR.pdf.
49. FDA: Innovation and Regulatory Science Available from: https://www.fda.gov/vaccines-blood-biologics/science-research-biologics/innovation-and-regulatory-science.
50. FDA Innovation and Regulatory Incentives. Available from: https://www.fda.gov/vaccines-blood-biologics/science-research-biologics/innovation-and-regulatory-science.
51. FDA Grant awarded under BsuFA. FDA biosimilars. Available from: https://www.grants.gov/web/grants/search-grants.html?keywords=fda%20biosimilar.
52. Press Release US Senate. Senator Lee introduces bill to remove interchangeability of biosimilars.Available from: https://www.lee.senate.gov/2022/11/sen-lee-introduces-biosimilar-red-tape-elimination-act.
53. Niazi SK. No two classes of biosimilars: urgent advice to the US congress and the FDA. J Clin Pharm Ther. 2022;47(9):1352–1361. PMID: 35869625. doi:10.1111/jcpt.13743
54. EMA. Biosimilar Medicines Can be Changed Available from: https://www.ema.europa.eu/en/news/biosimilar-medicines-can-be-interchanged.
55. MHRA, Guidance on the licensing of biosimilar products. https://www.gov.uk/government/publications/guidance-on-the-licensing-of-biosimilar-products/guidance-on-the-licensing-of-biosimilar-products Available from: https://www.gov.uk/government/publications/guidance-on-The-licensing-of-biosimilar-products/guidance-on-The-licensing-of-biosimilar-products.
56. News Release. Eli LIlly Company. Lilly Cuts Insulin Prices by 70% and Caps Patient Insulin Out-of-Pocket Costs at $35 Per Month Available from: https://investor.lilly.com/news-releases/news-release-details/lilly-cuts-insulin-prices-70-and-caps-patient-insulin-out-pocket.
57. OECD Data. Pharmaceutical Spending Available from: https://data.oecd.org/healthres/pharmaceutical-spending.htm.
58. US Congress. Congressional Committee Reports mentioning PBMs. Available from: https://www.congress.gov/bill/118th-congress/senate-bill/127/all-info.
59. US Congress. Pharmacy Benefit Manager Transparency Act of 2023 Available from: https://www.congress.gov/search?q=%7B%22source%22%3A%5B%22comreports%22%5D%2C%22search%22%3A%22PBM%22%7D.
60. US Congress. Pharmacy Benefit Manager Transparency Act of 2023 Available from: https://www.congress.gov/search?q=%7B%22source%22%3A%5B%22legislation%22%2C%22comreports%22%5D%2C%22search%22%3A%22PBM%22%7D.
61. News Release. Civica to Manufacture and Distribute Affordable Insulin Available from: https://civicarx.org/.
62. News release. Insulin is way too expensive. California has a solution: Make its own. Available from: https://www.vox.com/policy-and-politics/23574178/insulin-cost-california-biden-medicare-coverage.
63. Mark Cuban Cost-plus Drug Company Available from: https://costplusdrugs.com/.
64. Ferreri, D. Policy Recommendations for Improving Biosimilar Uptake in Belgium Mar 11, 2023 Available from: https://www.centerforbiosimilars.com/view/policy-recommendations-for-improving-biosimilar-uptake-in-belgium.
65. Andrew W. Mulcahy, “Biosimilar Drugs Could Generate $38.4 Billion in Savings Over Five Years. RAND Corporation; 2022.
66. Press Release. Organon Foresees US Adalimumab Discounts Hitting 80%-90% CEO Suggests Heavy Competition Will Lead To Steep Price Cuts By 2025-2026 Available from: https://generics.pharmaintelligence.informa.com/GB152419/Organon-Foresees-US-Adalimumab-Discounts-Hitting-8090.
67. Barclays Bank. U.S. Specialty pharmaceuticals, Sunday soliloquies in spec pharma - can biosimilars pricing head the generics way? Equity research healthcare. One of the more distinctive; 2023.
68. Podcast: Panelists Suggest More Groundwork Is Needed for Indian Biologics to Podcast. Achieve Global Market Access Available from: https://www.centerforbiosimilars.com/view/part-1-panelists-suggest-more-groundwork-is-needed-for-indian-biologics-to-achieve-global-market-access.
69. Indian Biosimilar Standards: A Work in Progress Apr 5, 2021
Deana Ferreri, PhD
Paul Cornes, BM, BCH, MA, MRCP, FRCR Available from: https://www.centerforbiosimilars.com/view/part-2-panelists-advise-patterning-guidelines-on-international-standards.
70. Column: India Struggles to Meet International Biologics Standards Apr 10, 2021
Paul Cornes, BM, BCH, MA, MRCP, FRCR Available from: https://www.centerforbiosimilars.com/view/column-india-struggles-to-meet-international-biologics-standards.
71. Indian Biosimilar Standards: A Work in Progress Apr 5, 2021
Deana Ferreri, PhD Available from: https://www.centerforbiosimilars.com/view/indian-biosimilar-standards-a-work-in-progress.
72. CDSCO India. GUIDELINES ON SIMILAR BIOLOGICS: Regulatory Requirements for Marketing Authorization in India, 2016 Available from: https://cdsco.gov.in/opencms/resources/UploadCDSCOWeb/2018/UploadAlertsFiles/BiosimilarGuideline2016.pdf.
73. Yang J. Study on the purple book system in the United States and discussion on the necessity and feasibility of building the “biologics listed product catalogue collection” in China. Chin Pharm Aff. 2019;33:966–971.
74. Yang J, Zhao X, Li J, et al. Creating China’s biosimilar drugs regulatory system: a calculated approach. Front Pharmacol. 2022;13:815074. PMID: 35185570; PMCID: PMC8848751. doi:10.3389/fphar.2022.815074
75. Eye on Pharma: Good News, Bad News for Alvotech; EpiVax Secures FDA Grant for Biosimilar Testing Sep 12, 2022
Skylar Jeremias
https://www.centerforbiosimilars.com/view/eye-on-pharma-good-news-bad-news-for-alvotech-epivax-secures-fda-grant-for-biosimilar-testing Available from: https://www.centerforbiosimilars.com/view/eye-on-pharma-good-news-bad-news-for-alvotech-epivax-secures-fda-grant-for-biosimilar-testing.
76. EMA. Centrally approved biosimilars. Available from: https://www.ema.europa.eu/en/medicines/field_ema_web_categories%253Aname_field/Human/ema_group_types/ema_medicine/field_ema_med_status/authorised-36/ema_medicine_types/field_ema_med_biosimilar/search_api_aggregation_ema_medicine_types/field_ema_med_biosimilar.
77. US Congress. Title VII—improving access to innovative medical therapies subtitle a—biologics price competition and innovation. US Congress. Available from: https://www.fda.gov/media/78946/download.
78. Industry Association: Biosimilars Council. Available from: https://biosimilarscouncil.org/.
79. Industry Association: Association for affordable medicines. Available from: https://accessiblemeds.org/.
80. Industry Association: International Generics and Biosimilars Association. Available from: https://www.igbamedicines.org/.
81. Industry Association: Biologics and biosimilars collective intelligence consortium. Available from: https://www.bbcic.org/.
82. Industry Association: British Biosimilar Council. Available from: https://britishbiosimilars.co.uk/.
83. Industry Association: Generic and Biosimilars Association. Available from: https://www.gbma.com.au/.
84. Industry Association: Medicines for Europe. Available from: https://www.medicinesforeurope.com/biosimilar-medicines/.
85. Industry Association: Danish Generic and Biosimilars Medicines Industry Association Available from: http://www.igldk.dk/english-about.
86. Industry Association: Biosimilars Canada. Available from: https://biosimilarscanada.ca.
87. Industry Association: Biotechnology Innovation Organization. Available from: http://www.bio.org.
88. McKinsey. Inflection point for biosimilars. Available from: https://www.mckinsey.com/industries/life-sciences/our-insights/an-inflection-point-for-biosimilars.
89. Niazi SK. Obstacles to success for biosimilars in the US market. Available from: https://www.europeanpharmaceuticalreview.com/article/70987/obstacles-success-biosimilars-us-market/.
 © 2023 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2023 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
Recommended articles
Scientific Rationale for Waiving Clinical Efficacy Testing of Biosimilars
Niazi S
Drug Design, Development and Therapy 2022, 16:2803-2815
Published Date: 24 August 2022