")
Back to Journals » Open Access Rheumatology: Research and Reviews » Volume 11
Biomarkers of subclinical atherosclerosis in patients with psoriatic arthritis
Authors Peluso R , Caso F , Tasso M, Sabbatino V, Lupoli R, Di Minno MND, Ursini F, Costa L, Scarpa R
Received 27 February 2019
Accepted for publication 15 April 2019
Published 28 June 2019 Volume 2019:11 Pages 143—156
DOI https://doi.org/10.2147/OARRR.S206931
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Professor Chuan-Ju Liu
Rosario Peluso,1 Francesco Caso,1 Marco Tasso,1 Vincenzo Sabbatino,1 Roberta Lupoli,2 Matteo Nicola Dario Di Minno,3 Francesco Ursini,4 Luisa Costa,1 Raffaele Scarpa1, On behalf of CaRRDs study group
1Department of Clinical Medicine and Surgery, Rheumatology Research Unit, Federico II University, Naples, Italy; 2Department of Clinical Medicine and Surgery, Division of Internal Medicine, Federico II University, Naples, Italy; 3Department of Advanced Biomedical Sciences, Division of Cardiology, Federico II University, Naples, Italy; 4Internal Medicine Unit, Department of Medical Sciences, University of Ferrara, Ferrara, Italy
Background: Psoriatic arthritis (PsA) is a chronic immune-mediated disease. It is associated with an increase in cardiovascular risk factors (obesity, hypertension, diabetes, and dyslipidemia), giving a higher risk of major adverse cardiovascular events. Patients with PsA have an increased incidence of subclinical atherosclerosis and endothelial dysfunction. The aim of this study is to perform a review of the biomarkers of subclinical atherosclerosis in patients with PsA.
Methods: A search was performed in the electronic databases (PubMed, Web of Science, Scopus, and Embase) up until July 2017. Studies were considered if they included data on biomarkers of subclinical atherosclerosis in PsA, and each article was then reviewed for quality and clinical relevance. After completing the literature search, all screened literature was summarized and discussed in our study group (CaRRDs study group).
Results: The initial search produced 532 abstracts, which were limited to 258 potentially relevant articles by preliminary review of the titles and by excluding review articles and case reports (n=274). A further 102 articles were deemed ineligible after examining the abstracts. Full texts of the remaining 156 articles were retrieved. Most articles were excluded because they were not relevant to the biomarkers of subclinical atherosclerosis in psoriasis and/or PsA. In the end, 54 articles were deemed eligible for this review.
Conclusion: Patients with PsA showed more severe atherosclerotic disease compared with patients with only psoriasis. This may have been due to the higher systemic inflammatory burden from the combination of both diseases. In patients with PsA some molecules may be considered as markers of atherosclerotic disease, and their detection may be a prognostic marker, in addition to imaging procedures, for the development of atherosclerotic disease, and could be suitable for the management of patients with PsA.
Keywords: psoriatic disease, insulin resistance, lipid profile, serum uric acid, complement C3, primary and secondary hemostasis
Introduction
Psoriatic arthritis (PsA) is a chronic immune-mediated disease. One-third of patients with skin and/or nail psoriasis will develop an inflammatory arthritis leading to severe physical limitations and disability.1,2 In addition to skin and joint involvement, PsA has a high prevalence of extra-articular manifestations3 and comorbidities.4–6 The literature reports an increased cardiovascular risk in patients with PsA.7,8 PsA patients show a higher prevalence of metabolic syndrome (MetS), hypertension, hyperlipidemia, obesity, and diabetes compared with those who have only psoriasis.4 An additional vascular risk factor is hyperhomocysteinemia, which may be determined by medications used for the treatment of PsA9,10 as much as by genetic and/or nutritional variations. PsA subjects may have increased fibrinogen, a major predictor of stroke and myocardial infarction,11 and C-reactive protein (CRP) levels.12 Moreover, a higher incidence of arterial thrombosis is related to platelet hyperreactivity, so the inflammation influences platelet reactivity and the achievement of minimal disease activity (MDA) may normalize platelet hyperreactivity, thus reducing thrombotic events.13
The aim of this manuscript is to perform a review on the biomarkers of subclinical atherosclerosis in patients with PsA.
Methods
A search was performed in the electronic databases (PubMed, Web of Science, Scopus, and Embase) up until July 2017. In MEDLINE, we used the search terms “psoriatic arthritis” AND “cardiovascular risk”, OR “oxidized low-density lipoproteins”, OR “nitric oxide”, OR “-nitrotyrosine”, OR “vitamin A”, OR “vitamin E”, OR “beta-carotene”, OR “homocysteine”, OR “fibrinogen”, OR “increased platelets”, OR “hypercoagulability”, OR “complement C3”, OR “C-reactive protein”, OR “uric acid”. Search limits included links to full text only, humans, English language articles, males and females, and all adult ages (Table S1). The “Related Articles” function of PubMed was used to cross-check for any additional relevant studies and the references of the reviewed articles were manually scanned for other relevant studies. Studies were included if they contained data on biomarkers of subclinical atherosclerosis in PsA, and each article was then reviewed for quality and clinical relevance. After completing the literature search, all screened literature was summarized and discussed in our study group (Cardiovascular Risk in Rheumatic Diseases [CaRRDs] study group). All literature and comments were included in the systematic review.
Results
The initial search produced 532 abstracts, which were limited to 258 potentially relevant articles by preliminary review of the titles and by excluding review articles and case reports (n=274). A further 102 articles were considered ineligible after examining the abstracts. Full texts of the remaining 156 articles were retrieved. The majority of articles were excluded because they were not relevant to the biomarkers of subclinical atherosclerosis in psoriasis and/or PsA. In the end, 54 articles were considered eligible for this review (Figure 1).
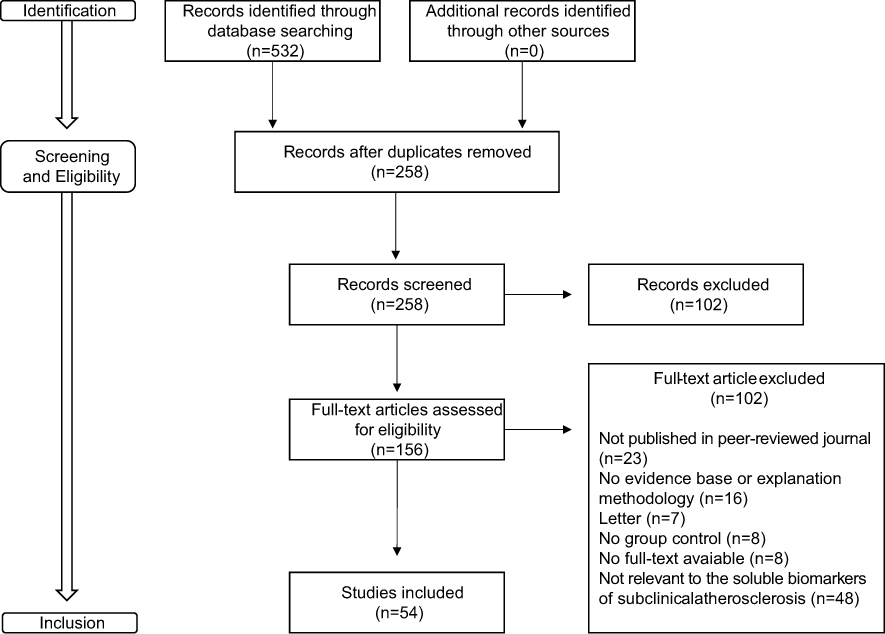 |
Figure 1 Flowchart of the literature review process (adapted from the PRISMA flow diagram). |
PsA and psoriasis are associated with a significantly increased risk of cardiovascular risk factors and major adverse cardiovascular events (MACEs). Ogdie et al showed a higher risk of developing MACEs in patients with PsA who were not using disease-modifying anti-rheumatic drugs (DMARDs) and a similar risk to that in patients with psoriasis and rheumatoid arthritis (RA).14 However, irrespective of classical cardiovascular risk factors, systematic inflammation in PsA patients plays an important role in increasing cardiovascular diseases.
A high body mass index (BMI) and obesity are both associated with an increased risk of cardiovascular mortality and morbidity.15 It has been widely demonstrated that obesity is more common in patients with psoriasis, and it is notably more common in PsA than in RA patients (28% vs 15% with BMI >27 kg/m2).16 A meta-analysis by Armstrong et al showed that there is a connection between psoriasis and obesity. In particular, psoriasis patients have 45% increased odds of being obese compared with the general population.17
Data from the literature prove that nutritional assessment in psoriatic patients may improve both the disease severity and the obesity-related comorbidities. In particular, some studies show that body weight and diet may trigger the psoriatic disease, or even exacerbate the clinical manifestations.18,19 PsA patients present a higher BMI than patients without joint involvement and a higher prevalence of obesity than in the general population.18 Obesity is also associated with a higher disease activity in PsA patients, but few studies have evaluated the relationship between joint disease severity and obesity in PsA patients. Similarly, in patients without joint involvement, the severity of the skin disease has been associated with BMI,20 and the prevalence of obesity is higher in those with severe compared with mild psoriasis, with an OR of 1.47 (95% CI 1.32–1.63).21 Di Minno et al showed, in a prospective study, that increased BMI predicted lower response to tumor necrosis factor-α (TNF-α) blockers in PsA patients.22 In this study, it was also reported that weight reduction was linked with greater response to treatment with TNF-α blockers, probably due to the underdosing of medications, which may explain the lower response of the obese patients to the treatment.23
The exact mechanism underlying the association between psoriatic disease and obesity is still unidentified. Obesity is considered a chronic, low-grade inflammatory condition which causes changes in levels of cytokines (TNF-α and IL-6) and “adipokines” (leptin and adiponectin).24–26 Adiponectin has anti-inflammatory and insulin-sensitizing properties as it inhibits TNF-α, IL-6, and interferon-γ production; conversely, it also has proinflammatory effects. It leads monocytes and macrophages to produce proinflammatory IL-6, TNF-α, and IL-12. In addition, leptin is involved in the production of proinflammatory Th1 cytokines and in the inhibition of anti-inflammatory Th2 cytokines. Chen et al demonstrated higher leptin levels in psoriatic patients compared with healthy controls, and they considered psoriasis an independent risk factor for hyperleptinemia.27 Similarly, resistin concentration was increased in patients with psoriasis and correlated with disease severity. Differently, in these patients, there is a decrement in plasma levels of cytokines with anti-inflammatory properties such as adiponectin compared with healthy controls, and this is inversely related to psoriasis severity and TNF-α level.28,29
Moreover, monocytes, CD4 T lymphocytes, and most proinflammatory cytokines (TNF-α, IL-1β, IL-6, and IL-18) involved in the pathophysiology of major arthritides30-32 play a role in the induction and maintenance of the atherosclerotic process.33–35 Thus, in obese patients with PsA, there is a synergic action between the obesity-related inflammatory status and the immunity-related inflammation.36,37 In addition to this hypothesis, it was demonstrated that obesity represents a negative predictor of success of treatment with TNF-α blockers in patients with PsA.22
Insulin resistance and diabetes
The relationship between diabetes and RA is interesting for its association with a well-documented increased risk of cardiovascular disease.38 Whereas several studies support the relationship between insulin resistance and rheumatic diseases, there are few data about rheumatic diseases and diabetes. Han et al, in a cross-sectional comparative study through a large insurance database, documented an increased risk of diabetes in RA, ankylosing spondylitis, and PsA patients (prevalence ratios 1.4, 1.2, and 1.5, respectively).39 In the Rochester Epidemiology Project, the authors described no increase in the risk of new-onset diabetes (relative risk [RR]=0.978) in RA patients, with an incidence rate of 7.9 per 1,000 person-years.40 Salomon et al studied the incidence rate of diabetes among subjects with RA or psoriatic disease, and established a higher RR for incident diabetes among those with psoriatic disease, in comparison with healthy subjects.41 Similarly, RA patients showed a higher RR for diabetes in both sexes, however, this risk is reduced with age. The elevated adjusted HRs seen among subjects without glucocorticoid therapy imply that this risk is not primarily an adverse effect of this treatment.41
While several cross-sectional studies reported a higher prevalence of diabetes in PsA patients, fewer studies have assessed the risk of developing incident diabetes in patients with PsA.16,19,42–44 Diabetes and other metabolic diseases were reported to be at increased prevalence in many studies on PsA patients, with an OR of 2.18 (95% CI 1.36–3.50) of diabetes in PsA, and patients with severe psoriasis having a higher risk.45–47 Several mechanisms could explain the association between PsA and diabetes, such as patients' unhealthy lifestyles, the inflammatory cytokine setting that drives insulin resistance,48–50 and shared genetic loci for susceptibility to psoriasis and diabetes.51,52
Lipid profile
The lipid profile in psoriatic patients has been studied for more than 50 years; nevertheless, there are still controversial results on this association. Many authors report reduced levels of high-density lipoprotein (HDL) and/or augmented levels of low-density lipoprotein (LDL), very-low-density lipoprotein (VLDL), and triglycerides (TGs).53–56 Other findings suggest no significant association between lipid serum levels and psoriasis.57–60 These studies are heterogeneous; they include patients with different disease durations, comorbidities (such as diabetes and obesity), and systemic treatments, factors that surely influence the lipid metabolism. Mallbris et al report a higher cholesterol concentration in VLDL and HDL in 200 patients studied at the onset of skin disease.61 Another important issue concerns the quality of HDL in psoriatic patients, which reflects their metabolic function. Mehta et al found a pro-atherogenic HDL profile in 112 psoriasis patients compared to controls.62
Rheumatic diseases, such as RA and PsA, are associated with alterations in lipid metabolism.63 It is widely recognized that an increase in TGs and a reduction in high-density lipoprotein-cholesterol (HDL-C) concentrations occur in acute-phase responses. These changes are accompanied by alterations in total cholesterol (TC) and LDL-cholesterol (LDL-C). The lipid levels, which are mediated by cytokines, are linked to host defense and tissue repair,64 but a chronic inflammation can be involved in the development of cardiovascular disease.39,50 It has been hypothesized that apolipoproteins and lipoproteins contribute toward both acute and chronic inflammation.65 HDL-C, in particular, plays an important anti-inflammatory role because it inhibits production of proinflammatory cytokines induced by T-cell contact.66 In PsA, the data on serum lipid profiles are controversial. Indeed, a study examining the lipid profile in PsA patients67 did not find a relationship between high levels of LDL-C and PsA. Nevertheless, increased levels of TC68 and TGs50,69 were found to be associated with subclinical atherosclerosis in patients with PsA. Jones et al, in a study on serum lipid profile in 50 PsA patients, found an important shift in the distribution of LDL in patients with active joint disease: there was a reduction in LDL1 and LDL2 levels and an increase in LDL3 levels.70 They also found a significant reduction in HDL-C levels, particularly subclass HDL3, which is important as HDL3-C protects less against atherosclerosis than other HDL subclasses.70 In relation to the control group, patients with psoriasis presented a lower ratio of TC contained in HDL2 to its total content (0.05 vs 0.08) and a lower ratio in HDL3 to its total content (0.18 vs 0.25). These modifications were not linked with the significant changes in the level serum of TGs. Moreover, the reduction in HDL-C was not connected to significant changes in LDL-C in serum.70 Significantly, Skoczyňska et al also found lower levels of HDL-C and its subclasses (both HDL3 and HDL2) in PsA patients.71 The serum levels of TC and TG were normal, whereas the plasma levels of HDL2 and HDL3 were lower than those in the control group (p<0.001 and p<0.05, respectively).71 More recently, Tam et al, in a case–control study, showed that patients with PsA had higher HDL-C levels, lower TC and LDL-C levels, and a lower TC/HDL-C ratio.50 Although all these studies investigated the lipid profile in PsA patients, a possible relationship between high levels of small dense (sd)-LDL and PsA has not been demonstrated. Only the study by Jones et al looked at the presence of sd-LDL in PsA patients, but there were only 13 patients and LDL size analysis was performed by a different method (ultracentrifugation).70 Gentile et al established that PsA patients have increased serum levels of sd-LDL independently of the presence of MetS. These findings show a possible association between PsA and the development of atherosclerosis mediated by sd-LDL. LDL size measurement can be useful both in the risk assessment for atherosclerotic disease and in identifying a subsample of high-risk PsA patients with lipoprotein alteration, who could benefit from lipid-lowering intervention.72 Finally, there seems to be a potential relationship between the level of inflammation and the lipid profile; indeed, dyslipidemia is more important in PsA patients with active disease.50,70
Primary hemostasis (platelet aggregation)
Platelet hyperreactivity is a major predictor of arterial thrombosis and, in turn, of cardiovascular events.73,74 Platelets produce inflammatory mediators and mediate leukocyte incorporation into plaques through platelet-mediated leukocyte adhesion. Pathomechanisms of psoriasis imply platelet activation, mediated by chronic inflammation.75–78 Psoriatic patients show high titers of serum platelet-derived microparticles and P-selectin, which are both markers of platelet hyperreactivity, as reported by several studies.76–78
On the other hand, several cytokines/chemokines involved in PsA, by interacting with specific platelet receptors, cause intracellular calcium mobilization, nucleotide secretion, and platelet activation.79,80 These data suggest a synergism between inflammation and atherothrombosis.81 However, little is known about the association of disease activity and platelet reactivity in PsA subjects. Di Minno et al evaluated platelet aggregability in 114 PsA patients by assessing the maximal light transmittance (max-A%) achieved within 5 minutes after the addition of very low concentrations of pro-aggregating agents.13 The authors found that max-A% values of PsA patients who achieved MDA, during treatment with TNF-α inhibitors, were comparable to those of controls and significantly lower than those of individuals with active disease. CRP values were lower in subjects with MDA than in those with active disease, and directly correlated with max-A%. Platelet hyperreactivity is a major predictor of cardiovascular events and of arterial thrombosis,74 and these findings strongly support a synergism between inflammation and pathobiology of atherothrombosis.78 Moreover, the study by Di Minno et al showed that platelet function is increased in patients with PsA, especially in those with poorly controlled disease.13,82 The correlation of CRP with max-A% and the decreasing prevalence of MDA for increasing quartiles of max-A% argue for a link between inflammation and platelet reactivity. By interacting with specific platelet receptors, cytokines/chemokines involved in PsA80 cause intracellular calcium mobilization, nucleotide secretion, and platelet activation.81 Hyperreactivity to ADP has been reported in rheumatic diseases.83 However, almost 50% of patients in that sample were receiving NSAIDs84 and only 17% had PsA. Platelet hyperreactivity was correlated with an elevated incidence of arterial thrombosis,73,74 and the effect of antiplatelet agents in the vascular risk profile of subjects with PsA requires investigation.83 These data suggest that inflammation influences platelet reactivity and that achievement of MDA may normalize platelet hyperreactivity.
Secondary hemostasis (coagulation and fibrinolysis)
Novel evidence suggests an important role for changes in hemostatic system parameters in determining the cardiovascular risk in patients with RA.84,85 In addition to primary hemostasis (platelet reactivity), changes in fibrinolytic (tissue plasminogen activator [t-PA] and plasminogen activator inhibitor-1 [PAI-1]) and secondary hemostasis variables (coagulation proteins and natural anticoagulants) play a relevant role in the cardiovascular disease risk. Impaired fibrinolysis and/or raised levels of coagulation factors and/or reduced levels of natural anticoagulants (protein C, protein S, and antithrombin) have been recognized as major determinants of both arterial and venous thrombosis.86 Marongiu et al described increased plasma levels of prothrombin fragment 1+2, thrombin–antithrombin complexes, and D-dimer in a cohort of 48 psoriatic patients, indicating a pro-coagulant state.87
By enhancing platelet reactivity and affecting a series of coagulation and fibrinolytic variables, proinflammatory cytokines (ie, TNF-α and IL-6) may trigger the thrombotic risk in rheumatic patients.13,88 In a prospective study, Di Minno et al evaluated the changes in hemostatic and fibrinolytic variables in PsA patients starting treatment with TNF-α inhibitors.89 They also compared changes in these variables with those found in subjects who had achieved MDA with traditional DMARDs and were on continuous treatment with such drugs. The analysis of the data on patients receiving a 6-month treatment showed that, with the exception of antithrombin, all the other hemostatic and fibrinolytic variables changed significantly.89 In addition, the reduction in protein S, one of the major natural anticoagulants, is likely to mirror the progressive reduction in the hypercoagulative state due to treatment with TNF-α inhibitors. Moreover, the results of this prospective study provide further evidence about the link between inflammation and thrombotic risk. In particular, the authors documented that the control of the inflammatory process induced by treatment with TNF-α inhibitors is associated with a significant improvement in hemostatic and fibrinolytic parameters in PsA patients, most changes being documented in patients achieving MDA. These variables have been found to predict arterial and venous thrombosis, which are major complications in PsA.90,91 Previous studies have already shown that the overproduction of proinflammatory cytokines (TNF-α and IL-6), besides playing a crucial role in the inflammatory process correlated with rheumatic disease activity,92 are also involved in the modulation of the fibrinolytic system.93 The balance between plasminogen activators (eg, t-PA) and plasminogen activator inhibitors (eg, PAI-1) determines the total fibrinolytic potential of human blood. TNF-α has proved to be a strong agonist of PAI-1 expression and regulation.94 In addition, high plasma levels of prothrombin fragment 1+2 and D-dimer (markers of thrombin activation and fibrinolysis, respectively) have been found in RA patients.95 Thus, by inducing a procoagulant shift in the hemostatic balance, chronic inflammation contributes to fibrin generation and, in turn, thrombosis.96,97 Protein C and protein S are natural anticoagulant proteins that work in opposing hypercoagulable states.98 In accordance with the association between natural anticoagulants and variables involved in hypercoagulable states, the changes we have reported in protein S levels are likely to be related to the changes that occur in PAI-1 and t-PA levels. In the study by Di Minno et al, besides the control of inflammation, TNF-α inhibitors have been found to downregulate fibrinolytic and hemostatic parameters and to normalize platelet hyperreactivity, thus leading to a reduction in the cardiovascular risk.98,99 In addition, maximal changes in coagulation variables were found in patients achieving the MDA during treatment with TNF-α inhibitors.
High-sensitivity C-reactive protein
High-sensitivity C-reactive protein (hsCRP) was suggested as a risk marker for potential cardiovascular events100 Some cytokines, such as IL-6, induce its production from the liver101 A wide literature has shown that an elevated value of hsCRP, when added to traditional cardiovascular risk factors, can be used to support the hypothesis that atherosclerosis is first and foremost an inflammatory disease.102–104 Indeed, abdominal obesity and insulin resistance are predictors of an elevated hsCRP, and the presence of MetS correlates strongly with an elevated hsCRP level.100,101 Conversely, patients with fewer than two risk factors for MetS have low hsCRP levels.100 Even in patients with mild or inactive PsA disease, low-grade inflammation as reflected by the hsCRP level was associated with obesity, insulin resistance, hypertension, and dyslipidemia, as reported by Tam et al.50 In the same study, the authors showed that hsCRP was also associated with an increased thrombotic tendency, as demonstrated by the increased platelet count.50 Moreover, no association was found between hsCRP and patient’s age, TC, LDL-C, TGs, apolipoprotein B, insulin, urate, or serum creatinine levels. The results were similar when only PsA patients were included in the analysis, except that the hsCRP level correlated inversely with TC.50 Therefore, the hsCRP level in PsA patients would make the differences in the prevalence of most cardiovascular risk factors non-significant. This suggests that elevated hsCRP may not be a universal feature of chronic inflammation and may mistakenly predict coronary artery disease in PsA patients, indicating a need for alternative cardiovascular biomarkers in these at-risk populations.
Vitamins A, E, and C, and β-carotene
The increased production of reactive oxygen species (ROS) induced by atherosclerotic risk factors accelerates the disease progression in patients with rheumatic disease. This development is neutralized by natural antioxidants, such as vitamins C and E and carotenoids.105 These antioxidants are scavengers of free radicals, which reduce the oxidative damage and protect LDL against oxidation.106 Moreover, vitamins and carotenoids, by combating oxidative stress, may protect patients against the development of rheumatic diseases.107 In patients with rheumatic diseases, although an inverse relationship between systemic inflammation and antioxidant blood levels has been reported in the literature, there is a lack of information about the relationship between antioxidants and accelerated atherosclerosis.108,109
A clinical study by Profumo et al contributed toward clarifying the association between serum levels of natural antioxidants and atherosclerotic disease in RA and PsA patients.109 The authors described that in PsA patients, there was a significantly higher level of vitamin A only in those with intima–media thickness (IMT) ≤1 mm, suggesting a possible protective action of vitamin A on the cardiovascular system in these patients. However, further studies are needed to confirm this hypothesis. Moreover, Profumo et al observed that β-carotene levels were significantly lower in RA and PsA patients than in healthy subjects, but they did not differ in accordance with the presence of subclinical atherosclerosis.109 These results were in line with previous findings by De Pablo et al, showing that plasma levels of carotenoid were significantly lower in RA patients, having modified potential confounders such as smoking (which affects the serum concentrations of antioxidants).110 In addition, a reduced antioxidant concentration among RA patients results from increased metabolism of antioxidants. Profumo et al did not find any associations between antioxidant levels and potential confounders among the clinical and serological variables of patients. An association of β-carotene levels in autoimmune disease but not with IMT values is suggested by lower concentrations of β-carotene in RA and PsA patients in comparison with healthy subjects, and by the inverse correlation between β-carotene content and the duration of RA. This finding further suggests the occurrence of a redox imbalance in this type of pathology, and could be linked to the higher levels of oxidized LDLs present in these patients. A recent report found that plasma concentrations of β-carotene can cause some oxidative modification of LDL in vivo.111 In this report, the authors also demonstrated that vitamin E does not influence the occurrence of oxidized LDLs.111
Serum uric acid
Hyperuricemia is a common characteristic of PsA patients,112 and it has been associated with an increased incidence of cardiovascular disease and MACEs in the general population.112,113 Hyperuricemia is often associated with other classic cardiovascular risk factors. Fukui et al described a positive correlation between serum uric acid concentration and atherosclerosis through the carotid intima–media thickness (c-IMT) in men with diabetes.114 Moreover, it has been shown that an increased serum uric acid level represents a significant and independent risk factor for cardiovascular mortality in women,115 and an independent association has been found between serum uric acid concentration and c-IMT in Japanese men without MetS.116 In RA, there is an association between c-IMT >0.60 mm and carotid plaques, markers of subclinical atherosclerosis, and serum uric acid concentrations,117 and serum uric acid levels are significantly higher in RA patients with cardiovascular disease.118 A cross-sectional study shows that this association between serum uric acid levels and cardiovascular disease in RA patients is independent of other traditional cardiovascular risk factors.118 Gonzalez-Gay et al reported a significant correlation between serum uric acid concentration and subclinical atherosclerosis in PsA patients without clinically evident cardiovascular risk factors.119 In this study in PsA patients, there were significantly higher serum uric acid levels when c-IMT was >0.90 mm than when c-IMT was <0.60 mm. Moreover, the serum uric acid concentration had a high predictive power for the presence of severe subclinical atherosclerosis in PsA patients without clinically evident cardiovascular disease. In addition, in PsA patients with hyperuricemia, severe subclinical atherosclerosis was found through c-IMT values >0.90 mm or the presence of carotid plaques in the ultrasonographic assessment of the common carotid artery.119
Complement C3
Complement C3 (C3) is widely accepted as an emerging risk factor for cardiovascular diseases.120 Serum C3 has been demonstrated as a reliable marker of insulin resistance in different populations.121–122 C3 is involved in complement system activation because all the main activation pathways lead to the generation of C3 products (C3a and C3b). Thus, from an immunological perspective C3b is the main effector of the complement system, while from a metabolic perspective C3a appears to be more important.123 Proinflammatory cytokines (TNF-α, IL-1, IL-6, and interferon-γ) augment the production of C3.124,125 In PsA patients, inflammatory cytokines may play a role in the increased production of C3 in adipose tissue, where gene expression of C3 is high.126–128 This could cause an increased insulin resistance which, in turn, determines fat storage; thus, inflammation and visceral obesity lead to the maintenance of elevated levels of insulin resistance. Ursini et al, in a cross-sectional study, suggested that serum C3 is associated with estimated insulin resistance in PsA patients.125 Early evidence suggests that elevated C3 and C4 levels can be reduced by anti-TNF-α treatment.129
Vascular endothelial growth factor
Vascular endothelial growth factor (VEGF) plays an important role in angiogenesis, which occurs in PsA and in atherosclerosis.130,131 It is possible that the increased levels of VEGF observed in PsA patients has a balancing effect regarding the endothelial progenitor cells (EPCs). EPCs are a population of bone-marrow derived cells, which have the ability to migrate into areas of tissue ischemia and possess reparative qualities. They have been shown to be decreased in level and function in various inflammatory disorders, such as RA and inflammatory bowel disease.132–134 Some studies have demonstrated defects in the levels or function of EPCs among RA and inflammatory bowel disease patients, with an increased risk regarding cardiovascular morbidity and mortality.135–137 In view of these results, defective EPC function may play a role in the increased cardiovascular morbidity and mortality in these conditions. The levels and function of EPCs can improve in treated RA patients in association with clinical improvements.19,39,134,135,138 Nevertheless, Patschan et al showed that neither EPC colonies nor percentages of circulating cells differed between controls and PsA patients.139 Ablin et al presented equivalent data, with no significant differences in EPC levels among healthy controls, patients with psoriasis, and PsA patients.140 This may be due to the possibility that the EPC system does not serve as a ubiquitous surrogate marker of higher cardiovascular risk in subjects with autoimmune-mediated inflammatory diseases.139
Conclusion
Cardiovascular disease is the major cause of morbidity and mortality among PsA patients.81,141,142 PsA patients have an increased risk of MACEs, specifically myocardial infarction, stroke, and cardiovascular death. Ahlehoff et al showed that PsA is directly related to composite myocardial infarction, stroke, or cardiovascular death, with a rate ratio of 1.79 (95% CI 1.31–2.45).121 Ogidie et al, moreover, showed that PsA confers a fully adjusted composite cardiovascular risk among PsA patients.14 In addition, in PsA patients the prevalence of MetS and its components is higher in comparison to the general population and to other types of rheumatic disease.16,142 Boehncke et al described the evolution of atherosclerosis in psoriatic disease with the term “psoriatic march”.143 In particular, the authors described that the chronic systemic inflammation in PsA patients leads to insulin resistance, with endothelial dysfunction and atherosclerosis. Patients with PsA were affected by more severe atherosclerotic disease compared with patients with only psoriasis, maybe because of the higher systemic inflammatory burden due to the combination of both diseases. Finally, the increase in cardiovascular risk in patients with PsA compared with both healthy populations and subjects matched for vascular risk factors highlights that systemic inflammation is an independent cardiovascular risk factor.144 Indeed, this hypothesis is supported by the improvement in the cardiovascular risk profile following the control of systemic inflammation with anti-inflammatory treatments.145–147 Some molecules may be useful, as adjuncts to imaging procedures, as markers of atherosclerotic disease in PsA patients, and could be used for the management of PsA patients. To this end, an interaction among medical specialists, general practitioners, and educational programs is needed to achieve adequate cardiovascular preventive strategies in rheumatic patients.
Abbreviation list
PsA, psoriatic arthritis; MetS, metabolic syndrome; CRP, C-reactive protein; MDA, minimal disease activity; MACE, major adverse cardiovascular event; DMARD, disease-modifying anti-rheumatic drug; RA, rheumatoid arthritis; BMI, body mass index; TNF, tumor necrosis factor; RR, relative risk; HDL, high-density lipoprotein; LDL, low-density lipoprotein; VLDL, very-low density lipoprotein; TG, triglyceride; sd, small dense; max-A%, maximal light transmittance; HDL-C, HDL-cholesterol; TC, total cholesterol; t-PA, tissue plasminogen activator; PAI-1, plasminogen activator inhibitor-1; ROS, reactive oxygen species; IMT, intima–media thickness; c-IMT, carotid intima–media thickness; C3, complement C3.
Acknowledgments
The authors want to thank the members of the CaRRDs (Cardiovascular Risk in Rheumatic Diseases) study group: Matteo Nicola Dario Di Minno, Roberta Lupoli, Antonella Scalera, Alessandro Di Minno, Pasquale Ambrosino, Giovanni Tarantino, and Giovanni Di Minno (Department of Clinical Medicine and Surgery, Regional Reference Centre for Coagulation Disorders, Federico II University, Naples, Italy); Rosario Peluso and Raffaele Scarpa (Department of Clinical Medicine and Surgery, Rheumatology Research Unit, Psoriatic Arthritis Clinic, Federico II University, Naples, Italy); and Salvatore Iervolino and Nicola Pappone (Rheumatology and Rehabilitation Research Unit “Salvatore Maugeri” Foundation, Telese Terme [BN], Italy). The authors declare that they did not receive any external funding.
Author contributions
All authors contributed to data analysis, drafting and revising the article, gave final approval of the version to be published, and agree to be accountable for all aspects of the work.
Disclosure
RS, RP, LC, FC, and MNDDM have acted as paid lecturers or board members and received grants and honoraria in the past 36 months for research unrelated to the present study. RP is a member of the Editorial Board for BMC Muscoskeletal Disorders. The authors report no other conflicts of interest in this work.
References
1. Gladman DD, Antoni C, Mease P, et al. Psoriatic arthritis: epidemiology, clinical features, course, and outcome. Ann Rheum Dis. 2005;64(Suppl 2):ii14–17. doi: 10.1136/ard.2004.032482
2. Ibrahim G, Waxman R, Helliwell PS. The prevalence of psoriatic arthritis in people with psoriasis. Arthritis Rheum. 2009;61:1373–1378. doi:10.1002/art.v61:10
3. Peluso R, Iervolino S, Vitiello M, Bruner V, Lupoli G, Di Minno MN. Extra-articular manifestations in psoriatic arthritis patients. Clin Rheumatol. 2015;34(4):745–753. doi:10.1007/s10067-014-2652-9
4. Husted JA, Thavaneswaran A, Chandran V, et al. Cardiovascular and other comorbidities in patients with psoriatic arthritis: a comparison. Arthritis Care Res (Hoboken). 2011;63:1729–1735. doi:10.1002/acr.20373
5. Edson-Heredia E, Zhu B, Lefevre C, et al. Prevalence and incidence rates of cardiovascular, autoimmune, and other diseases in patients with psoriatic or psoriatic arthritis: a retrospective study using clinical practice research datalink. J Eur Acad Dermatol Venereol. 2015;29(5):955–963. doi:10.1111/jdv.12669
6. Costa L, Caso F, Del Puente A, Di Minno MN, Peluso R, Scarpa R. Incidence of malignancies in a cohort of psoriatic arthritis patients taking traditional disease modifying antirheumatic drug and tumor necrosis factor inhibitor therapy: an observational study. J Rheumatol. 2016;43(12):2149–2154. doi:10.3899/jrheum.160542
7. Tobin AM, Veale DJ, Fitzgerald O, et al. Cardiovascular disease and risk factors in patients with psoriasis and psoriatic arthritis. J Rheumatol. 2010;37:1386–94. 7. doi:10.3899/jrheum.090822
8. Jamnitski A, Symmons D, Peters MJ, Sattar N, McInnes I, Nurmohamed MT. Cardiovascular comorbidities in patients with psoriatic arthritis: a systematic review. Ann Rheum Dis. 2013;72:211–216. doi:10.1136/annrheumdis-2011-201194
9. Di Minno MN, Tremoli E, Coppola A, Lupoli R, Di Minno G. Homocysteine and arterial thrombosis: challenge and opportunity. Thromb Haemost. 2010;103(5):942–961. doi:10.1160/TH09-06-0393
10. Slot O. Changes in plasma homocysteine in arthritis patients starting treatment with low-dose methotrexate subsequently supplemented with folic acid. Scand J Rheumatol. 2001;30(5):305–307. doi:10.1080/030097401753180408
11. Maresca G, Di Blasio A, Marchioli R, Di Minno G. Measuring plasma fibrinogen to predict stroke and myocardial infarction: an update. Arterioscler Thromb Vasc Biol. 1999;19(6):1368–1377. doi:10.1161/01.ATV.19.6.1368
12. Laurent MR, Panayi GS, Shepherd P. Circulating immune complexes, serum immunoglobulins, and acute phase proteins in psoriasis and psoriatic arthritis. Ann Rheum Dis. 1981;40(1):66–69. doi:10.1136/ard.40.1.66
13. Di Minno MN, Iervolino S, Peluso R, Scarpa R, Di Minno G. Platelet reactivity and disease activity in subjects with psoriatic arthritis. J Rheumatol. 2012;39:334–336. doi:10.3899/jrheum.110741
14. Ogidie A, Yu Y, Haynes K, et al. Risk of major cardiovascular events in patients with psoriatic arthritis, psoriasis and rheumatoid arthritis: a population-based cohort study. Ann Rheum Dis. 2015;74(2):326–332. doi:10.1136/annrheumdis-2014-205675
15. Calle EE, Thun MJ, Petrelli JM, Rodriguez C, Heath CW. Body-mass index and mortality in a prospective cohort of U.S. N Engl J Med. 1999;341:1097–1105. doi:10.1056/NEJM199910073411501
16. Mok CC, Ko GT, Ho LY, Yu KL, Chan PT, To CH. Prevalence of atherosclerotic risk factors and the metabolic syndrome in patients with chronic inflammatory arthritis. Arthritis Care Res (Hoboken). 2011;63(2):195–202. doi:10.1002/acr.20373
17. Armstrong AW, Harskamp CT, Armstrong EJ. The association between psoriasis and obesity: a systematic review and meta-analysis of observational studies. Nutr Diabetes. 2012;3(2):e54. doi:10.1038/nutd.2012.26
18. Bhole VM, Choi HK, Burns LC, et al. Differences in body mass index among individuals with PsA, psoriasis, RA and the general population. Rheumatology (Oxford). 2012;51:610–618. doi:10.1093/rheumatology/ker154
19. Kimhi O, Caspi D, Bornstein MN, et al. Prevalence and risk factors of atherosclerosis in patients with psoriatic arthritis. Semin Arthritis Rheum. 2007;36(4):203–9.73. doi:10.1016/j.semarthrit.2006.09.001
20. Bardazzi F, Balestri R, Baldi E, et al. Correlation between BMI and PASI in patients affected by moderate to severe psoriasis undergoing biological therapy. Dermatol Ther. 2010;23(Suppl 1):S14–9. doi:10.1111/j.1529-8019.2009.01281.x
21. Neimann AL, Shin DB, Wang X, et al. Prevalence of cardiovascular risk factors in patients with psoriasis. J Am Acad Dermatol. 2006;55:829–835. doi:10.1016/j.jaad.2006.03.021
22. Di Minno MN, Peluso R, Iervolino S, et al. Obesity and the prediction of minimal disease activity: a prospective study in psoriatic arthritis. Arthritis Care Res (Hoboken). 2013;65:141–147. doi:10.1002/acr.21711
23. Di Minno MN, Peluso R, Iervolino S, et al. Weight loss and achievement of minimal disease activity in patients with psoriatic arthritis starting treatment with tumour necrosis factor alpha blockers. Ann Rheum Dis. 2014;73:1157–1162. doi:10.1136/annrheumdis-2014-205310
24. Barrea L, Macchia PE, Di Somma C, et al. Bioelectrical phase angle and psoriasis: a novel association with psoriasis severity, quality of life and metabolic syndrome. J Transl Med. 2016;14(1):130. doi:10.1186/s12967-016-0867-z
25. Marini E, Buffa R, Saragat B, et al. The potential of classic and specific bioelectrical impedance vector analysis for the assessment of sarcopenia and sarcopenic obesity. Clin Interv Aging. 2012;7:585–591. doi:10.2147/CIA.S38488
26. Lowes MA, Bowcock AM, Krueger JG. Pathogenesis and therapy of psoriasis. Nature. 2007;445(7130):866–873. doi:10.1038/nature05663
27. Chen YJ, Wu CY, Shen JL, et al. Psoriasis independently associated with hyperleptinemia contributing to metabolic syndrome. Arch Dermatol. 2008;144:1571–1575. doi:10.1001/archderm.144.12.1571
28. Peluso I, Palmery M. The relationship between body weight and inflammation: lesson from anti-TNF-α antibody therapy. Hum Immunol. 2016;77:47–53. doi:10.1016/j.humimm.2015.10.008
29. Barrea L, Nappi F, Di Somma C, et al. Environmental risk factors in psoriasis: the point of view of the nutritionist. Int J Environ Res Public Health. 2016;13(5):E743. doi:10.3390/ijerph13121252
30. Sattar N, Dw M, Capell H, et al. Explaining how “high-grade” systemic inflammation accelerates vascular risk in rheumatoid arthritis. Circulation. 2003;108:2957–2963. doi:10.1161/01.CIR.0000099844.31524.05
31. Del Rincon ID, Williams K, Stern MP, et al. High incidence of cardiovascular events in a rheumatoid arthritis cohort not explained by traditional cardiac risk factors. Arthritis Rheum. 2001;44:2737–2745.
32. van Kuijk AW, Reinders-Blankert P, Smeets TJ, et al. Detailed analysis of the cell infiltrate and the expression of mediators of synovial inflammation and joint destruction in the synovium of patients with psoriatic arthritis: implications for treatment. Ann Rheum Dis. 2006;65:1551–1557. doi:10.1136/ard.2005.050963
33. Dixon WG, Symmons DP. What effects might anti-TNF-alpha treatment be expected to have on cardiovascular morbidity and mortality in rheumatoid arthritis? A review of the role of TNF-alpha in cardiovascular pathophysiology. Ann Rheum Dis. 2007;66:1132–1136. doi:10.1136/ard.2006.063867
34. Popa C, van Den Hoogen FH, Radstake TR, et al. Modulation of lipoprotein plasma concentrations during long-term anti-TNF therapy in patients with active rheumatoid arthritis. Ann Rheum Dis. 2007;66:1503–1507. doi:10.1136/ard.2006.066191
35. Libby P. Changing concepts of atherogenesis. J Intern Med. 2000;247:349–358. doi:10.1046/j.1365-2796.2000.00654.x
36. Russolillo A, Iervolino S, Peluso R, et al. Obesity and psoriatic arthritis: from pathogenesis to clinical outcome and management. Rheumatology (Oxford). 2013;52:62–67. doi:10.1093/rheumatology/kes242
37. Rondinone CM. Adipocyte-derived hormones, cytokines, and mediators. Endocrine. 2006;29:81–90. doi:10.1385/ENDO:29:1
38. Solomon DH, Karlson EW, Rimm EB, et al. Cardiovascular morbidity and mortality in women diagnosed with rheumatoid arthritis. Circulation. 2003;107:1303–1307. doi:10.1161/01.CIR.0000054612.26458.B2
39. Han C, Robinson DW
40. Gabriel SE, Crowson CS, O’Fallon WM. The epidemiology of rheumatoid arthritis in Rochester, Minnesota, 1955–1985. Arthritis Rheum. 1999;42:415–420. doi:10.1002/1529-0131(199904)42:3<415::AID-ANR4>3.0.CO;2-Z
41. Solomon DH, Love TJ, Canning C, Schneeweiss S. Risk of diabetes among patients with rheumatoid arthritis, psoriatic arthritis and psoriasis. Ann Rheum Dis. 2010;69(12):2114–2117. doi:10.1136/ard.2009.125476
42. Dubreuil M, Rho YH, Man A, et al. Diabetes incidence in psoriatic arthritis, psoriasis and rheumatoid arthritis: a UK population-based cohort study. Rheumatology (Oxford). 2014;72. doi:10.1093/rheumatology/ket343
43. Dreiher J, Freud T, Cohen AD. Psoriatic arthritis and diabetes: a population-based cross-sectional study. Dermatol Res Pract. 2013;2013:580404. doi:10.1155/2013/580404
44. Di Minno MN, Iervolino S, Lupoli R, et al. Cardiovascular risk in rheumatic patients: the link between inflammation and atherothrombosis. Semin Thromb Hemost. 2012;38(497–505):15.
45. Labitigan M, Bahče-Altuntas A, Kremer JM, et al. Higher rates and clustering of abnormal lipids, obesity, and diabetes mellitus in psoriatic arthritis com¬pared with rheumatoid arthritis. Arthritis Care Res (Hoboken). 2014;66:600–7.74. doi:10.1002/acr.22185
46. Johnsson H, McInnes IB, Sattar N. Cardiovascular and metabolic risks in psoriasis and psoriatic arthritis: pragmatic clinical management based on available evidence. Ann Rheum Dis. 2012;71:480–483. doi:10.1136/annrheumdis-2011-200567
47. Coto-Segura P, Eiris-Salvado N, Gonzalez-Lara L, et al. Psoriasis, psoriatic arthritis and type 2 diabetes mellitus: a systematic review and meta-analysis. Br J Dermatol. 2013;169:783–793. doi:10.1111/bjd.12473
48. Gottlieb AB, Dann F, Menter A. Psoriasis and the metabolic syndrome. J Drugs Dermatol. 2008;7:563–572.
49. Sonnenberg GE, Krakower GR, Kissebah AH. A novel pathway to the manifestations of metabolic syndrome. Obes Res. 2004;12:180–186. doi:10.1038/oby.2004.24
50. Tam LS, Tomlinson B, Chu TTW, et al. Cardiovascular risk profile of patients with psoriatic arthritis compared to controls-the role of inflammation. Rheumatology. 2008;47(5):718–723. doi:10.1093/rheumatology/ken090
51. Das SK, Elbein SC. The search for type 2 diabetes susceptibility loci: the chromosome 1q story. Curr Diab Rep. 2007;7:154–164. doi:10.1007/s11892-007-0025-3
52. Wolf N, Quaranta M, Prescott NJ, et al. Psoriasis is associated with pleiotropic susceptibility loci identified in type II diabetes and Crohn disease. J Med Genet. 2008;45:114–116. doi:10.1136/jmg.2007.053595
53. Ma C, Harskamp CT, Armstrong EJ, Armstrong AW. The association between psoriasis and dyslipidaemia: a systematic review. Br J Dermatol. 2013;168:486–495. doi:10.1111/bjd.12101
54. Langan SM, Seminara NM, Shin DB, et al. Prevalence of metabolic syndrome in patients with psoriasis: a population-based study in the United Kingdom. J Invest Dermatol. 2012;132:556–562. doi:10.1038/jid.2011.365
55. Kimball AB, Szapary P, Mrowietz U, et al. Underdiagnosis and undertreatment of cardiovascular risk factors in patients with moderate to severe psoriasis. J Am Acad Dermatol. 2012;67:76–85. doi:10.1016/j.jaad.2011.06.035
56. Pietrzak A, Michalak-Stoma A, Chodorowska G, Szepietowski JC. Lipid disturbances in psoriasis: an update. Mediat Inflamm. 2010;2010:1–13. doi:10.1155/2010/535612
57. Farshchian M, Zamanian A, Farshchian M, Monsef A-R MH. Serum lipid level in Iranian patients with psoriasis. J Eur Acad Dermatol Venereol. 2007;21:802–805. doi:10.1111/j.1468-3083.2006.02099.x
58. Toker A, Kadi M, Yildirim AK, Aksoy H, Akçay F. Serum lipid profile paraoxonase and arylesterase activities in psoriasis. Cell Biochem Funct. 2009;27:176–180. doi:10.1002/cbf.1553
59. Ma C, Schupp CW, Armstrong EJ, Armstrong AW. Psoriasis and dyslipidemia: a population-based study analyzing the National Health and Nutrition Examination Survey (NHANES). J Eur Acad Dermatol Venereol. 2014;28:1109–1112. doi:10.1111/jdv.2014.28.issue-8
60. Caso F, Del Puente A, Oliviero F, et al. Metabolic syndrome in psoriatic arthritis: the interplay with cutaneous involvement. Clin Rheumatol. 2018;37(3):579–586.
61. Mallbris L, Granath F, Hamsten A, Ståhle M. Psoriasis is associated with lipid abnormalities at the onset of skin disease. J Am Acad Dermatol. 2006;54:614–621. doi:10.1016/j.jaad.2005.11.1079
62. Mehta NN, Li R, Krishnamoorthy P, et al. Abnormal lipoprotein particles and cholesterol efflux capacity in patients with psoriasis. Atherosclerosis. 2012;224:218–221. doi:10.1016/j.atherosclerosis.2012.06.068
63. Toms TE, Symmons DP, Kitas GD. Dyslipidaemia in rheumatoid arthritis: the role of inflammation, drugs, lifestyle and genetic factors. Curr Vasc Pharmacol. 2010;8:301–326. doi:10.2174/157016110791112269
64. Esteve E, Ricart W, Fernández-Real JM. Dyslipidemia and inflammation: an evolutionary conserved mechanism. Clin Nutr. 2005;24:16–31. doi:10.1016/j.clnu.2004.08.004
65. Bresnihan B, Gogarty M, FitzGerald O, Dayer JM, Burger D. Apolipoprotein A-I infiltration in rheumatoid arthritis synovial tissue: a control mechanism of cytokine production? Arthritis Res Ther. 2004;6(6):R563–6. doi:10.1186/ar1443
66. Gruaz L, Delucinge-Vivier C, Descombes P, Dayer JM, Burger D. Blockade of T cell contact-activation of human monocytes by high-density lipoproteins reveals a new pattern of cytokine and inflammatory genes. PLoS One. 2010;5:e9418. doi:10.1371/journal.pone.0009418
67. Di Minno MN, Ambrosino P, Peluso R, Di Minno A, Lupoli R, Dentali F, et al. Lipid profile changes in patients with rheumatic diseases receiving a treatment with TNF-α blockers: a meta-analysis of prospective studies. Ann Med. 2014;46:73–83. doi:10.3109/07853890.2013.874661
68. Gonzalez-Juanatey C, Llorca J, Amigo-Diaz E, Dierssen T, Martin J, Gonzalez-Gay MA. High prevalence of subclinical atherosclerosis in psoriatic arthritis patients without clinically evident cardiovascular disease or classic atherosclerosis risk factors. Arthritis Rheum. 2007;57(6):1074–1080. doi:10.1002/(ISSN)1529-0131
69. Eder L, Zisman D, Barzilai M, et al. Subclinical atherosclerosis in psoriatic arthritis: a case-control study. J Rheumatol. 2008;35(5):877–882.
70. Jones SM, Harris CPD, Lloyd J, Stirling CA, Jpd R, McHugh NJ. Lipoproteins and their sub-fractions in psoriatic arthritis: identification of an atherogenic profile with active joint disease. Ann Rheum Dis. 2000;59(904–9):91. doi:10.1136/ard.59.11.904
71. Skoczyňska AH, Turczyn B, Barancewicz-Losek M, Martynowicz H. High-dansity lipoprotein cholesterol in patients with psoriatic arthritis. J Eur Acad Dermatol Venereol. 2003;17:362. doi:10.1046/j.1468-3083.2003.00792_11.x
72. Gentile M, Peluso R, Di Minno MN, et al. Association between small dense LDL and sub-clinical atherosclerosis in patients with psoriatic arthritis. Clin Rheumatol. 2016;35(8):2023–2029. doi:10.1007/s10067-016-3344-4
73. Di Minno MN, Guida A, Camera M, et al. Overcoming limitations of current antiplatelet drugs. A concerted effort for more profitable strategies of intervention. Ann Med. 2011;43:531–544. doi:10.3109/07853890.2011.582137
74. Bray PF. Platelet hyperreactivity: predictive and intrinsic properties. Hematol Oncol Clin North Am. 2007;21:633–645. doi:10.1016/j.hoc.2007.06.002
75. Ludwig RJ, Schultz JE, Boehncke WH, et al. Activated, not resting, platelets increase leukocyte rolling in murine skin utilizing a distinct set of adhesion molecules. J Invest Dermatol. 2004;122:830–836. doi:10.1111/j.0022-202X.2004.22318.x
76. Tamagawa-Mineoka R, Katoh N, Kishimoto S. Platelet activation in patients with psoriasis: increased plasma levels of platelet-derived microparticles and soluble P-selectin. J Am Acad Dermatol. 2010;62:621–626. doi:10.1016/j.jaad.2009.06.053
77. Pelletier F, Garnache-Ottou F, Angelot F, et al. Increased levels of circulating endothelial-derived microparticles and small-size platelet-derived microparticles in psoriasis. J Invest Dermatol. 2011;131:1573–1576. doi:10.1038/jid.2011.57
78. Papadavid E, Diamanti K, Spathis A, et al. Increased levels of circulating platelet-derived microparticles in psoriasis: possible implications for the associated cardiovascular risk. World J Cardiol. 2016;8(11):667–675. doi:10.4330/wjc.v8.i11.667
79. Loffredo S, Ayala F, Marone GC, et al. Immunopathogenesis of psoriasis and psoriatic arthritis and pharmacological perspectives. Reumatismo. 2007;59:28–39.
80. Barrett NE, Holbrook L, Jones S, et al. Future innovations in anti-platelet therapies. Br J Pharmacol. 2008;154:918–939.
81. Libby P, Ridker PM, Hansson GK. Leducq transatlantic network on atherothrombosis. Inflammation in atherosclerosis: from pathophysiology to practice. J Am Coll Cardiol. 2009;54:2129–2138. doi:10.1016/j.jacc.2009.09.009
82. Di Minno MN, Iervolino S, Zincarelli C, Lupoli R, Ambrosino P, Pizzicato P, DI Minno A et al. Cardiovascular effects of etanercept in patients with psoriatic arthritis: evidence from the cardiovascular risk in rheumatic disease databese. Expert Opin Drug Safety. 2015;14(12):1905–1912.
83. MacMullan PA, Peace AJ, Madigan AM, Tedesco AF, Kenny D, McCarthy GM. Platelet hyper-reactivity in active inflammatory arthritis is unique to the adenosine diphosphate pathway: a novel finding and potential therapeutic target. Rheumatology. 2010;49:240–245. doi:10.1093/rheumatology/kep377
84. Galliard-Grigioni KS, Reinhart WH. A randomized, controlled study on the influence of acetaminophen, diclofenac, or naproxen on aspirin-induced inhibition of platelet aggregation. Eur J Pharmacol. 2009;609:96–99. doi:10.1016/j.ejphar.2009.02.042
85. Busso N, Hamilton JA. Extravascular coagulation and the plasminogen activator/plasmin system in rheumatoid arthritis. Arthritis Rheum. 2002;46:2268–2279. doi:10.1002/art.10498
86. Ikuta T, Naruko T, Ikura Y, et al. Immunolocalization of platelet glycoprotein IIb/IIIa and P-selectin, and neutrophil-platelet interaction in human coronary unstable plaques. Int J Mol Med. 2005;15:573–577.
87. Marongiu F, Sorano GG, Bibbò C, et al. Abnormalities of blood coagulation and fibrinolysis in psoriasis. Dermatology. 1994;189(1):32–37. doi:10.1159/000246755
88. Ingegnoli F, Fantini F, Favalli EG, et al. Inflammatory and prothrombotic biomarkersin patients with rheumatoid arthritis: effects of tumor necrosis factor-alpha blockade. J Autoimmun. 2008;31:175–179. doi:10.1016/j.jaut.2008.07.002
89. Di Minno MN, Iervolino S, Peluso R, et al. Hemostatic and fibrinolytic changes are related to inflammatory conditions in patients with psoriatic arthritis: effect of different treatments. J Rheumatol. 2014;41:714–722. doi:10.3899/jrheum.140167
90. Di Minno MN, Iervolino S, Peluso R, et al., CaRRDS Study Group. Hepatic steatosis and disease activity in subjects with psoriatic arthritis receiving tumor necrosis factor-α blockers. J Rheumatol. 2012;39(5):1042–6.10. doi:10.3899/jrheum.111391
91. Di Minno MN, Peluso R, Iervolino S, et al. Hepatic steatosis, carotid plaques and achieving MDA in psoriatic arthritis patients starting TNF-α blockers treatment: a prospective study. Arthritis Res Ther. 2012;14(5):R211. doi:10.1186/ar4049
92. Felson DT, Anderson JJ, Boers M, et al. The American College of Rheumatology preliminary core set of disease activity measures for rheumatoid arthritis clinical trials. Arthritis Rheum. 1993;36:729–740.
93. Agirbasli M, Inanc N, Baykan OA, et al. The effects of TNF alpha inhibition on plasma fibrinolytic balance in patients with chronic inflammatory rheumatical disorders. Clin Exp Rheumatol. 2006;24:580–583.
94. Hou B, Eren M, Painter CA, et al. Tumor necrosis factor α activates the human plasminogen activator inhibitor-1 gene through a distal nuclear factor kappa B. J Biol Chemistry. 2004;279:18127–18136. doi:10.1074/jbc.M310438200
95. McEntegart A, Capell HA, Creran D, et al. Cardiovascular risk factors, including thrombotic variables, in a population with rheumatoid arthritis. Rheumatology. 2001;40:640–644. doi:10.1093/rheumatology/40.6.640
96. Cugno M, Ingegnoli F, Gualtierotti R, et al. Potential effect of anti-tumour necrosis factor-alpha treatment on reducing the cardiovascular risk related to rheumatoid arthritis. Curr Vasc Pharmacol. 2010;8:285–292. doi:10.2174/157016110790886965
97. Medcalf RL. Fibrinolysis, inflammation, and regulation of the plasminogen activating system. J Thromb Haemost. 2007;5:132–142. doi:10.1111/j.1538-7836.2007.02464.x
98. Di Minno MN, Pezzullo S, Palmieri V, et al. Protein C and protein S changes in GH-deficient adults on r-HGH replacement therapy: correlations with PAI-1 and t-PA plasma levels. Thromb Res. 2010;126:e434–438. doi:10.1016/j.thromres.2010.08.028
99. Ingegnoli F, Fantini F, Griffini S, et al. Anti-tumor necrosis factor alpha therapy normalizes fibrinolysis impairment in patients with active rheumatoid arthritis. Clin Exp Rheumatol. 2010;28:254–257.
100. Ridker P. C-reactive protein and the prediction of cardiovascular events among those at intermediate risk: moving an inflammatory hypothesis toward consensus. J Am Coll Cardiol. 2007;49:2129–2138. doi:10.1016/j.jacc.2007.02.052
101. Lemieux I, Pascot A, Prud’homme D, et al. Elevated C-reactive protein: another component of the atherothrombotic profile of abdominal obesity. Arterioscler Thromb Vasc Biol. 2001;21:961–967. doi:10.1161/01.ATV.21.6.961
102. Wang TJ, Gona P, Larson MG, et al. Multiple biomarkers for the prediction of first major cardiovascular events and death. N Engl J Med. 2006;355:2631–2639. doi:10.1056/NEJMoa055373
103. Sattar N, Murray HM, McConnachie A, et al. C-reactive protein and prediction of coronary heart disease and global vascular events in the prospective study of pravastatin in the elderly at risk (PROSPER). Circulation. 2007;115:981–989. doi:10.1161/CIRCULATIONAHA.106.643114
104. Khera A, deLemos JA, Peshock RM. Relationship between C-reactive protein and subclinical atherosclerosis. The Dallas heart study. Circulation. 2006;113:38–43. doi:10.1161/CIRCULATIONAHA.105.575241
105. Zadák Z, Hyspler R, Tichá A, et al. Antioxidants and vitamins in clinical conditions. Physiol Res. 2009;58(1):S13–S17.
106. Brady WE, Mares-Perlman JA, Bowen P, Stacewicz-Sapuntzakis M. Human serum carotenoid concentrations are related to physiologic and lifestyle factors. J Nutr. 1996;126(1):129–137. doi:10.1093/jn/126.1.129
107. Costenbader KH, Kang JH, Karlson EW. Antioxidant intake and risks of rheumatoid arthritis and systemic lupus erythematosus in women. Am J Epidemiol. 2010;172(2):205–216. doi:10.1093/aje/kwq089
108. Profumo E, Buttari B, Tosti ME, et al. Subclinical Atherosclerosis in Patients with Rheumatoid and Psoriatic Arthritis; 2008. Autoimmunity: Role, Regulation and Disorders by Vogel and Zimmermann, Eds. New York: Nova Science. ISBN:978-1-60456-833-2.
109. Profumo E, Di Franco M, Buttari B, et al. Biomarkers of subclinical atherosclerosis in patients with autoimmune disorders. Mediators Inflamm. 2012;2012:503942. doi:10.1155/2012/503942
110. De Pablo P, Dietrich T, Karlson EW. Antioxidants and other novel cardiovascular risk factors in subjects with rheumatoid arthritis in a large population sample. Arthritis Rheum. 2007;57(6):953–962. doi:10.1002/art.22912
111. Steinberg D. The LDL modification hypothesis of atherogenesis: an update. J Lipid Res. 2009;50:S376–S381. doi:10.1194/jlr.R800087-JLR200
112. Lambert JR, Wright V. Serum uric acid levels in psoriatic arthritis. Ann Rheum Dis. 1977;36:264–267. doi:10.1136/ard.36.3.264
113. Johnson RJ, Kang DH, Feig D, et al. Is there a pathogenetic role for uric acid in hypertension and cardiovascular and renal disease? Hypertension. 2003;41:1183–1190. doi:10.1161/01.HYP.0000069700.62727.C5
114. Fukui M, Tanaka M, Shiraishi E, et al. Serum uric acid is associated with microalbuminuria and subclinical atherosclerosis in men with type 2 diabetes mellitus. Metabolism. 2008;57:625–629. doi:10.1016/j.metabol.2007.12.005
115. Hakoda M, Masunari N, Yamada M, et al. Serum uric acid concentration as a risk factor for cardiovascular mortality: a longterm cohort study of atomic bomb survivors. J Rheumatol. 2005;32:906–912.
116. Kawamoto R, Tomita H, Oka Y, Ohtsuka N. Relationship between serum uric acid concentration, metabolic syndrome and carotid atherosclerosis. Intern Med. 2006;45:605–614.
117. Dessein PH, Joffe BI, Veller MG, et al. Traditional and nontraditional cardiovascular risk factors are associated with atherosclerosis in rheumatoid arthritis. J Rheumatol. 2005;32:435–442.
118. Panoulas VF, Milionis HJ, Douglas KM, et al. Association of serum uric acid with cardiovascular disease in rheumatoid arthritis. Rheumatology (Oxford). 2007;46:1466–1470. doi:10.1093/rheumatology/kem159
119. Gonzalez-Gay MA, Gonzalez-Juanatey C, Vazquez-Rodriguez TR, et al. Asymptomatic hyperuricemia and serum uric acid concentration correlate with subclinical atherosclerosis in psoriatic arthritis patients without clinically evident cardiovascular disease. Semin Arthritis Rheum. 2009;39(3):157–162. doi:10.1016/j.semarthrit.2008.06.001
120. Hertle E, van Greevenbroek MM, Stehouwer CD. Complement C3: an emerging risk factor in cardiometabolic disease. Diabetologia. 2012;55(4):881–884. doi:10.1007/s00125-012-2462-z
121. Ahlehoff O, Gislason GH, Charlot M, et al. Psoriasis is associated with clinically significant cardiovascular risk: a Danish nationwide cohort study. J Intern Med. 2011;270(2):147–157. doi:10.1111/j.1365-2796.2010.02310.x
122. Yang S, Li Q, Song Y, et al. Serum complement C3 has a stronger association with insulin resistance than high-sensitivity C-reactive protein in women with polycystic ovary syndrome. Fertil Steril. 2011;95(5):1749–53. 29. doi:10.1016/j.fertnstert.2011.01.136
123. Wang B, Li Q, Jiang Y, et al. Serum complement C3 has a stronger association with insulin resistance than high sensitive C-reactive protein in non-diabetic Chinese. Inflamm Res. 2011;60(1):63–68. doi:10.1007/s00011-010-0236-y
124. Muscari A, Antonelli S, Bianchi G, et al., Pianoro Study Group. Serum C3 is a stronger inflammatory marker of insulin resistance than C-reactive protein, leukocyte count, and erythrocyte sedimentation rate: comparison study in an elderly population. Diabetes Care. 2007;30(9):2362–2368. doi:10.2337/dc07-0637
125. Ursini F, Grembiale A, Naty S, Grembiale RD. Serum complement C3 correlates with insulin resistance in never treated psoriatic arthritis patients. Clin Rheumatol. 2014;33(12):1759–1764. doi:10.1007/s10067-013-2366-4
126. Volanakis JE. Transcriptional regulation of complement genes. Annu Rev Immunol. 1995;13:277–305. doi:10.1146/annurev.iy.13.040195.001425
127. Peake PW, O’Grady S, Pussell BA, Charlesworth JA. Detection and quantification of the control proteins of the alternative pathway of complement in 3T3-L1 adipocytes. Eur J Clin Invest. 1997;27(11):922–927. doi:10.1046/j.1365-2362.1997.2090759.x
128. Gabrielsson BG, Johansson JM, Lönn M, et al. High expression of complement components in omental adipose tissue in obese men. Obes Res. 2003;11(6):699–708. doi:10.1038/oby.2003.100
129. Chimenti MS, Perricone C, Graceffa D, et al. Complement system in psoriatic arthritis: a useful marker in response prediction and monitoring of anti-TNF treatment. Clin Exp Rheumatol. 2012;30(1):23–30.
130. Leong TT, Fearon U, Veale DJ. Angiogenesis in psoriasis and psoriatic arthritis: clues to disease pathogenesis. Curr Rheumatol Rep. 2005;7:325–329.
131. Herrmann J, Lerman LO, Mukhopadhyay D, Napoli C, Lerman A. Angiogenesis in atherogenesis. Arterioscler Thromb Vasc Biol. 2006;26:1948–1957. doi:10.1161/01.ATV.0000233387.90257.9b
132. Masuda J, Mitsuyama K, Yamasaki H, et al. Depletion of endothelial progenitor cells in the peripheral blood of patients with ulcerative colitis. Int J Mol Med. 2007;19(2):221–228.
133. Grisar J, Aletaha D, Steiner CW, et al. Depletion of endothelial progenitor cells in the peripheral blood of patients with rheumatoid arthritis. Circulation. 2005;111(2):204–211. doi:10.1161/01.CIR.0000151875.21836.AE
134. Ablin JN, Boguslavski V, Aloush V, et al. Effect of anti-TNFalpha treatment on circulating endothelial progenitor cells (EPCs) in rheumatoid arthritis. Life Sci. 2006;75(25):2364–2369. doi:10.1016/j.lfs.2006.07.035
135. Peters MJ, van der Horst-Bruinsma IE, Dijkmans BA, Nurmohamed MT. Cardiovascular risk profile of patients with spondylarthropathies, particularly ankylosing spondylitis and psoriatic arthritis. Semin Arthritis Rheum. 2004;34(3):585–592. doi:10.1016/j.semarthrit.2004.07.010
136. Westerweel PE, Luijten RK, Hoefer IE, Koomans HA, Derksen RH, Verhaar MC. Haematopoietic and endothelial progenitor cells are deWcient in quiescent systemic lupus erythematosus. Ann Rheum Dis. 2007;66(7):865–870. doi:10.1136/ard.2006.065631
137. Fadini GP, Agostini C, Sartore S, Avogaro A. Endothelial progenitor cells in the natural history of atherosclerosis. Atherosclerosis. 2007;194(1):46–54. doi:10.1016/j.atherosclerosis.2007.03.046
138. Gonzalez-Juanatey C, Llorca J, Miranda-Filloy JA, et al. Endothelial dysfunction in psoriatic arthritis patients without clinically evident cardiovascular disease or classic atherosclerosis risk factors. Arthritis Rheum. 2007;15(57):287–293. doi:10.1002/art.22530
139. Patschan D, Sugiarto N, Henze E, et al. Early endothelial progenitor cells and vascular stiffness in psoriasis and psoriatic arthritis. Eur J Med Res. 2018;23(1):56. doi:10.1186/s40001-018-0306-0
140. Ablin JN, Goldstein Z, Aloush V, et al. Normal levels and function of endothelial progenitor cells in patients with psoriatic arthritis. Rheumatol Int. 2009;29(3):257–262. doi:10.1007/s00296-008-0676-7
141. Horreau C, Pouplard C, Brenaut E, et al. Cardiovascular morbidity and mortality in psoriasis and psoriatic arthritis: a systematic literature review. J Eur Acad Dermatol Venereol. 2013;27(Suppl 3):12–29. doi:10.1111/jdv.12163
142. Peluso R, Caso F, Tasso M, et al. Cardiovascular Risk Markers and Major Adverse Cardiovascular Events in Psoriatic Arthritis Patients. Rev Recent Clin Trials. 2018;13(3):199–209.
143. Boehncke WH, Boehncke S, Tobin AM, Kirby B. The ‘psoriatic march’: a concept of how severe psoriasis may drive cardiovascular comorbidity. Exp Dermatol. 2011;20(4):303–307. doi:10.1111/j.1600-0625.2011.01261.x
144. Kitas GD, Gabriel SE. Cardiovascular disease in rheumatoid arthritis: state of the art and future perspectives. Ann Rheum Dis. 2011;70(1):8–14. doi:10.1136/ard.2010.142133
145. Jacobsson LT, Turesson C, Gülfe A, et al. Treatment with tumor necrosis factor blockers is associated with a lower incidence of first cardiovascular events in patients with rheumatoid arthritis. J Rheumatol. 2005;32(7):1213–1218.
146. Caso F, Del Puente A, Peluso R, et al. Emerging drugs for psoriatic arthritis. Expert Opin Emerg Drugs. 2016;21(1):69–79. doi:10.1517/14728214.2016.1146679
147. Dixon WG, Watson KD, Lunt M, Hyrich KL, Silman AJ, Symmons DP. Reduction in the incidence of myocardial infarction in patients with rheumatoid arthritis who respond to anti-tumor necrosis factor alpha therapy: results from the British Society for Rheumatology Biologics Register. Arthritis Rheum. 2007;56(9):2905–2912. doi:10.1002/art.22862
Supplementary material
 |
Table S1 Literature search strategy |
 © 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.