Back to Journals » International Journal of Chronic Obstructive Pulmonary Disease » Volume 9 » Issue 1
Biomarkers in chronic obstructive pulmonary disease: confusing or useful?
Authors Stockley R ![]()
Received 1 November 2013
Accepted for publication 2 December 2013
Published 7 February 2014 Volume 2014:9(1) Pages 163—177
DOI https://doi.org/10.2147/COPD.S42362
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Robert A Stockley
Queen Elizabeth Hospital Birmingham, Lung Function and Sleep Department, Birmingham, UK
Abstract: The field of biomarker research has almost reached unmanageable proportions in chronic obstructive pulmonary disease (COPD). The developments of new technology platforms have generated a huge information data base, both cross sectionally and increasingly, longitudinally. The knowledge emerging provides an enormous potential for understanding the disease pathophysiology, for developing markers specific for long-term outcomes, and for developing new therapeutic strategies. However, the excitement must be tempered with an understanding of the limitations of the data collection techniques, and of the variations in disease state, activity, impact, and progression. Nevertheless, the most crucial aspect in interpreting the current literature is the recognition of the relatively superficial characterization of what is a complex group of pathological processes with a common end point of airflow limitation. The current review explores some of these issues together with those areas where real progress appears to have been made, and provides caution on interpretation.
Keyword: emphysema, inflammation, secretions, technology platforms
Introduction
“Biomarkers” have become a hot topic in the study and treatment of chronic obstructive pulmonary disease (COPD). In simple terms, a biomarker is a measurable characteristic that reflects the presence, severity, or state of a disease. For instance, whilst spirometry reflects the presence and dyspnea the severity or impact of COPD, temperature may reflect an infectious state. Furthermore, a change in the expression of a biomarker may reflect progression, the risk of progression, or the response to treatment, whilst genes and their products reflect the likelihood of disease emergence. Thus biomarkers can be specific cells, molecules, genes, gene products, organ functions, or general clinical characteristics that reflect the damage done, or the process that the damage sets in motion or the process that leads to the damage, or can indicate the prognosis and/or response to therapy. More recently, the use of general technology platforms has led to an exponential increase in information, accompanied by uncertainty as to whether this reflects cause or effect.
The current article attempts to review (at least in part) this enormous field together with some guidelines, based on potential pitfalls and on what we need to know. In general the “hot” questions we need answers to are:
- Can we identify the susceptible individual before the disease state develops?
- Can we identify “early” disease whilst it is still relatively asymptomatic?
- Can we predict the rate of progression of the disease?
- Can we identify or confirm the key pathophysiological processes, leading to new drug discovery?
- Can we identify factors that predict the response to current treatment or short Phase II clinical trials that provide confidence to undertake more extensive and definitive Phase III studies?
- Can we be sure of the impact of our new strategies on the patient, in the present or future?
Defining COPD status
COPD is a slowly progressing disease, although the rate of progression varies. This is an important concept as it impacts upon three features of COPD, namely, the disease activity, its severity, and the impact on the patient. The activity of the disease process will influence the current or future severity in a time dependent manner, ie, a highly active disease process will produce more severe disease at an earlier age. Similarly, the severity of the disease will have a variable impact on the patient, dependent upon the rate at which it developed. Small changes that develop rapidly are more likely to have a greater impact on the younger patient than the same change developing slowly will have on a more elderly patient (see Figure 1).
 | Figure 1 Line graph representation of disease progression in 3 idealised patients. Patient (Pt) A has slow disease progression and with age may notice or report few symptoms (ie low impact/low disease activity/mild disease). Patient B has greater progression and symptoms become noticeable in the 60s (moderate impact/moderate disease activity/moderate disease). Patient C has rapid disease progression leading to symptoms in the 30s (major impact/high disease activity/mild disease). |
In addition, it is increasingly recognized that COPD, although defined by major changes in spirometry, has several distinct phenotypes (clinically and pathologically) as well as additional features, such as the exacerbation complex (also consisting of many pathophysiological processes) and the recognized comorbidities, which may have some common physiological yet distinct pathological mechanisms.1 “Omics” is a neologism that aims at the collective characterization and quantification of pools of biological molecules that translate into the structure, function, and dynamics of an organism. Trying to understand the complexities of the enormous data generated by the “omics” platforms in COPD (proteomics, genomics, metabolomics, transcriptomics etc) is almost impossible without a similar high-quality and detailed patient characterization, and all biomarker studies need to be interpreted in this light.
Clinical biomarkers
Chest hyperinflation, low body mass index (BMI), the use of accessory muscles of respiration, and prolonged expiration have always been the physical markers of the presence of airflow obstruction, especially that occurring with a predominant emphysema phenotype. However the objective measures of lung physiology have been the “gold standard” biomarkers to date. In particular, not only has spirometry been the lynchpin of diagnosis but forced expired volume in 1 second (FEV1) has been central to our description of severity. It is a marker of disease progression and response to therapy, as well as a sound predictor of mortality2 and a weak indicator of future exacerbations.3 Nevertheless, it only measures one aspect of COPD and relates poorly to the presence of emphysema4 or to patient-reported quality of life,5 and its deterioration is not linear with time.6
However spirometry remains central to our thinking of COPD, requires little equipment, is relatively simple to perform and interpret (with appropriate training), and hence is a useful tool to detect “early” disease (at least when sufficiently advanced to fulfill the cross-sectional diagnostic criterion). Yet, even the term “early disease” is interpreted in different ways. To a patient, this may be understood as the stage in the disease when the symptoms or their impact is mild, whereas to physiologists, it is understood to occur when the spirometry is abnormal and yet is relatively close to the normal range. This concept is summarized in Figure 1, showing that the speed with which airflow obstruction develops will influence the symptoms perceived by the patient, which will clearly be dependent upon the activity of their lifestyle and expectations.
Furthermore, the presence of comorbidity and other physiological impairment (gas trapping and defects of gas transfer) will also influence the overall impact on the patient. Indeed, the impact, as determined by health status tools, is also a fair guide to long-term mortality.7 However, the poor correlation of health status with physiology suggests such tools only partly measure the same thing. Mortality is related to many factors, including the degree of breathlessness,8 exercise limitation,9 and low BMI.10 These markers predict the same end point but do not necessarily reflect the same thing. For this reason, composite scores, such as the BODE index (Body mass index, airflow Obstruction, Dyspnoea and Exercise), may be better predictors of long-term mortality.11 Because these relatively insensitive tests have been studied for many years, their use in assessing patients’ prognosis, progression, and response to treatment has been well documented. However, although useful as cross-sectional measures of cohort risks, these composite markers are less robust in assessing individual patients,12 and a progression of health status can occur independent of spirometry, even though progression of both also predict mortality.12 However, the composite markers do fulfill two other criteria, namely, that changes, both by decline or improvement, also relate to mortality,13 although may not be predictive of progression per se.
Patient-reported outcomes
Symptoms are included in many patient-reported outcomes and can be scored, in their own right, to provide both an indicator of the disease impact in individual patients but also, of progression and, importantly, the response to treatment. This alone consists of a large literature of assessment and validation. A good start for the interested reader is the validation of the COPD Assessment Test and its comparison with other patient-reported outcomes (which again highlights the weak relationship to spirometry).14 Nevertheless, these subjective tests do act as a biomarker of the impact of the disease on the patient, even if this is not entirely (or even only partly) explained by the current objective measures (a disjunction between disease severity and impact).
Radiology
More recently, the widespread use of high-resolution computed tomography (CT) scans and other radiological techniques has been applied to COPD populations. The CT scan is now the marker of choice to identify and quantify the presence and type of emphysema. This patient phenotype also relates to health status (though also weakly) as shown in Figure 2. Emphysema is a good predictor of mortality,15,16 can be present with normal spirometry,17 and is the outcome of choice in treatments aimed at improving18 or slowing down the progression19 of emphysema. CT scanning is recognized as the most sensitive test of progression in patients with emphysema,20 being significantly more sensitive than spirometry and even gas transfer, in such patients. More recently, further refinements of CT analysis have enabled the relationship between small airways disease and emphysema to be characterized in more detail,21 and the preliminary data suggest that small airways disease may precede the development of radiological emphysema. The implications of this for our understanding of pathophysiology clearly requires further study.
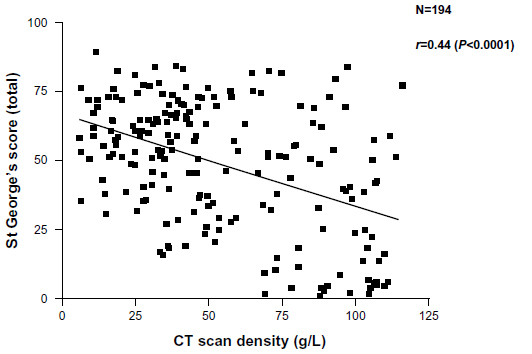 | Figure 2 Relationship between health status recorded as the SGRQ score and decreasing lung density as a marker of emphysema. Individual patient data points are shown and the significance of the correlation is given. |
Radiology can also study the airways, indicating responses to bronchodilator therapy22 and the presence and distribution of accompanying bronchiectasis, which is also a predictor of mortality23 (although the reasons are currently unclear).
Chronic bronchitis
The chronic bronchitis phenotype received little attention as a marker until recently and is now recognized as a predictor of disease progression24 and recurrent exacerbations.25 The latter defines a further clinical phenotype (and hence marker) likely to respond to anti-inflammatory therapy, both by the inhaled route26 and oral administration.27
Despite the well-tried and accepted roles of these “clinical” biomarkers, in more recent research, the term has come to mean a fluid sample test that is specific, particularly in terms of the pathophysiological processes taking place. Such research was initially based on a hypothesis-testing strategy, but the development and ready availability of technology platforms has caused this to become a data-collecting and possibly, a hypothesis-generating strategy. The latter relies on extensive data collection, which is then probed for patterns (generally the “omics” strategy) that may become informative about the potential process(es) taking place.
Biomarkers based on pathophysiology
The generally accepted concept is that the development of COPD occurs in susceptible individuals as a result of an enhanced inflammatory response to inhaled substances, like cigarette smoke. However, not only is COPD a generically defined syndrome but, inflammation itself is a highly complex, interrelated network of cells, cytokines, and other proteins that both amplify and dampen down the process – in part, this represents a balance between the destructive elements and the reparative ones. However, once damage has occurred to the delicate structures of the lung, repair cannot completely restore function, especially in the alveolar regions. This has been best demonstrated in animal models of emphysema, where a single challenge has been shown to result in the loss of lung elastin and, although it reaccumulates, the development of emphysema.28 To date, the majority of biomarker studies in COPD have concentrated on the damage process.
Our understanding of the mechanisms involved in the pathophysiology of COPD relates back to the single observation that alpha-1-antitrypsin (AAT)-deficiency (AATD) resulted in an increased susceptibility to the development of early-onset basal emphysema.29 The cascade of events that followed this observation resulted in many studies of AAT phenotypes in COPD, but apart from the recognition of the Pi ZZ severe form of deficiency and some very rare null genes, little was found. However, even the deficiency itself is not a highly sensitive marker since many affected patients remain healthy and with normal lung function. The true incidence of the deficiency and disease and hence, the sensitivity of even this genetic/biochemical marker remains unknown.
The mechanism involved became relatively easy to define. AAT is a specific inhibitor of serine proteinases, and elastase30 and proteinase 3,31 released by activated neutrophils, have both been shown to produce emphysematous lesions in experimental animals. Since neutrophil recruitment to the lung results in the release of these enzymes, elastin digestion during transit should be excessive in the presence of low concentrations of AAT. Completion of the pathophysiological axis required a chemoattractant signal and cell activation, adhesion, and migration, as summarized in Figure 3. The figure also summarizes the relevance of fluid sampling related to the parts of the process being studied.
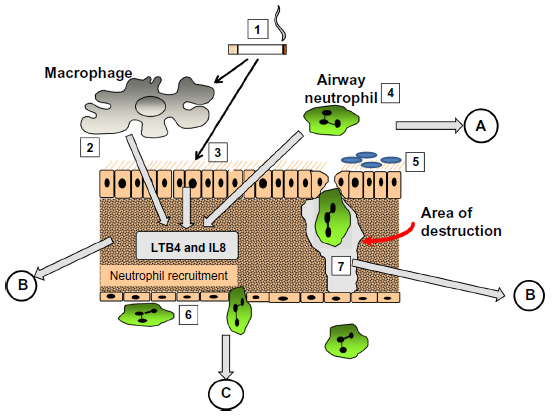 | Figure 3 The pathological process involved in emphysema. (1) cigarette smoke activates macrophages (2), epithelial cells (3) and airway neutrophils (4) to release pro inflammatory cytokines and neutrophil chemoattractants. At the same time oxidant stress in smoke damages local airway proteins (3). Harvesting airway secretions (A) detects markers of these effects including the influence of airway colonisation (5) and local mucus over production. The chemokines activate endothelial cells and circulating neutrophils (6) leading to adhesion and migration. Blood biomarkers (C) reflect these events. Migrating neutrophils ± local activated macrophages destroy connective tissue releasing specific fragments into the lymph and together with locally produced chemokines circulate into the circulation where they can be detected (B). |
It was strongly believed that monitoring the destructive process would identify a biomarker that would be increased with progression and decreased by effective therapy. Assays were developed to detect elastin-breakdown products/peptides and the cross-linking peptides, desmosine and isodesmosine. Lung elastin is a very long-lived connective tissue,32 and hence, breakdown products should not be a feature of health. Unfortunately elastin is not confined to the lung, and these elastin-breakdown products, despite being elevated in patients with neutrophilic lung diseases, do not help with the management of even AATD. There is some evidence that these elastin-breakdown products increase with disease progression33 but do not mark any therapeutic benefit.34 Furthermore a variety of other inflammatory lung diseases and variable clinical states also affect the levels of those markers,35 and for many reasons, their validity remains controversial. It is possible that only specific elastase-generated fragments would be useful and hence results may depend on both enzyme-cleavage preparation of antigen and monoclonal antibody.36 More recently, renewed interest in the role of desmosine (an elastin cross-linking peptide) has occurred, reviewed extensively by Luisetti et al34 and shown to relate to lung physiology.37 The relationship with spirometry was weak but that to gas transfer was stronger, consistent with elastin breakdown being more a feature of emphysema. However, studies of elastin breakdown may be more relevant if assessed in airways secretions.38 So despite the studies that evaluated lung elastin-breakdown products as far back as the 1970s and strong evidence that this is a key process in the development of emphysema, its validation as a biomarker remains an unresolved problem.34
Elastase and other proteinases
The release of elastase from activated neutrophils within the lung tissues is necessary to cause the proteolytic damage implicated, at least, in emphysema. This would seem, therefore, to be a much more relevant and direct biomarker of this clinical/pathological phenotype. The problem is that although elastase release in the lung interstitium is likely to be critical for matrix degradation at this site, the enzyme is rapidly inactivated (even in AATD), especially by local inhibitors, even if already bound to elastin.39 An alternative approach has been to measure the enzyme activity in the lung secretions. Sputum elastase activity is detectable in usual COPD but only during neutrophilic exacerbations,40 again reflecting the capacity of the local inhibitors. However, although elastase activity is more easily detectable in AATD patients, even in the stable state,41 it is also present in the secretions of bronchiectasis patients with42 and without43 cystic fibrosis. Since these latter diseases do not develop emphysema, the data suggest neutrophil recruitment more locally (bronchial circulation), sparing the interstitium, in response to bacterial load,44 and overwhelming local- and serum-derived inhibitors play a role, potentially damaging the airway epithelium and local host defenses.45
Indeed, other enzymes are often detected in these same secretions, including proteinase 3,46 metalloproteinases,47 and cysteine proteinases,48 all of which have been implicated in emphysema but which also form part of an independent proteinase cascade.49 Whatever the final pathway, all these enzymes and their inhibitors may become both specific and nonspecific biomarkers. Thus, whether these markers provide insight into airways disease above and beyond simple associations or merely reflect sputum purulence (see Exacerbations section) remains to be determined.
For the emphysema process, bronchoalveolar lavage (BAL) would be the only viable option for the study of markers in the airway. Although this approach has been used for more general inflammation markers (see General inflammation markers section), it has added little to the data because of the difficulty in obtaining a true alveolar sampling. However, in AATD, lavage, AAT, and elastase inhibitory capacity have been used to support the benefit of AAT augmentation therapy,50 which has also been reflected in sputum studies.51
More recently, an alternative approach to the detection of lung elastase activity has been developed and at least partly validated. As enzyme is released from neutrophils, there is a finite area and time within which the enzyme remains active. Specific cleavage of the surrounding substrate can produce a biochemical footprint of enzyme activity. A specific cleavage product of fibrinogen (Aα-Val360) has been shown to relate to lung physiology, be reproducible, increase during exacerbations, and to decrease with augmentation therapy, in AATD.52 Concentrations were also found to be higher than normal in usual COPD and to be related to physiological and radiological measure of emphysema as well as radiological progression of emphysema in the lower zone;53 this suggests a similar elastolytic process is at least partly responsible even in the presence of normal AAT. These latter findings may well relate to the abnormal neutrophil function seen in usual COPD (see The neutrophil section). Whether studies of similar markers of other elastolytic enzymes will also be informative remains to be seen. However, of interest, it should be noted that as part of the proteinase cascade, neutrophil elastase (NE) is responsible for the activation of the cysteine proteinase cathepsin B.54 Although this enzyme is not inhibited by AAT, the inhalation of AAT, which would inactivate NE, was also shown to result in the inactivation of cathepsin B,55 suggesting the latter is also a downstream biomarker of local NE activity. In the same vein NE activates matrix metalloproteinases (MMPs),56 and these enzymes are responsible for tumor necrosis factor (TNF)α processing,57 which, in its own right, drives neutrophilic inflammation. Thus, understanding these parts of the inflammatory cascade provides, not only potential therapeutic targets but also, potential biomarker readouts of efficacy (Figure 4).
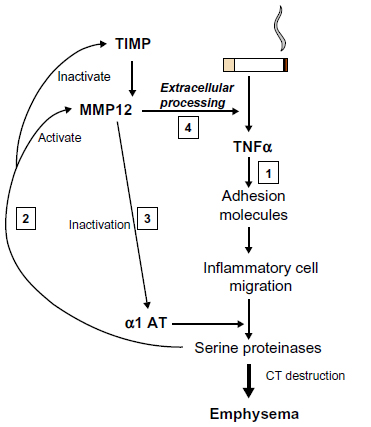 | Figure 4 Interrelation of proteinases and TNF. Cigarette smoking leads to TNF release and sequential events leading to emphysema (1). Serine proteinases released by recruited neutrophils activate MMP12 and inactivate its’ cognate inhibitor/s (2). MMP12 inactivates α1AT facilitating its own activation by serine proteinases (3). MMP12 then leads to extracellular processing of the interaction of TNF with its receptor (4) facilitating the main pathway (1). |
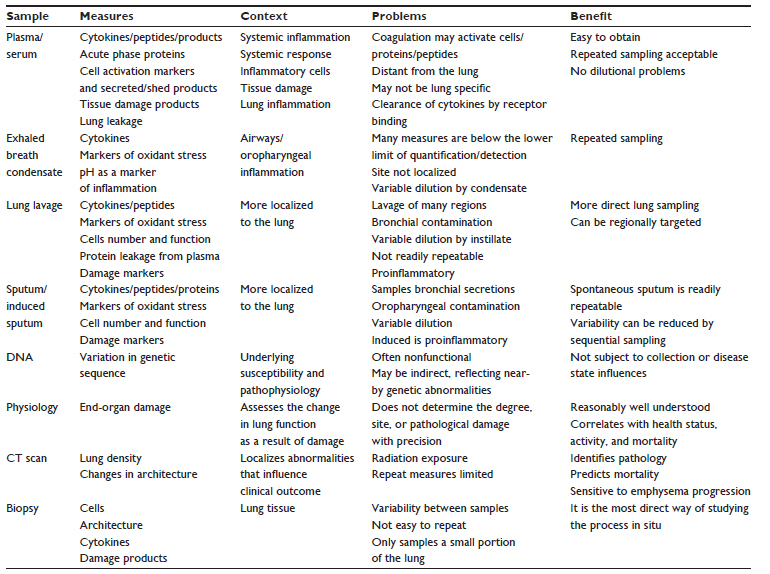 | Table 1 Biomarkers in context |
The neutrophil
Since the identification of NE as a potential mediator of emphysema, studies of this cell have become central to our understanding of the end point of tissue destruction in this subtype of COPD. There is extensive literature documenting evidence of the activation of neutrophils and their adhesion molecules in COPD. However, few of these studies have determined whether this is COPD-specific or reflects lung neutrophilic disease states in general. However, airway neutrophils have been shown to relate to the decline in lung function58 and emphysema quantified by CT scan.59 Nevertheless, the findings of such studies (as with all inflammatory mediators) beg the question of whether the observations are physiological, caused by the “severity” of the disease, or pathological, causing the severity of the disease itself. The data implicating the neutrophil is extensive but remains circumstantial. Although it is recognized that the degree of emphysema relates to the amount of NE in the lung60 and that NE destroys elastin, for this to be pathological would require that there be an abnormal response but not in those not demonstrating the phenotype (ie, healthy age-matched smokers). AATD provides another control since the pathological mechanism (low AAT) is well established, and disease severity (to obviate any physiological response) and treatment (to overcome any influences of drugs) can be matched to patients with usual COPD.
It was recognized in 1987 that the polymorphonuclear leukocytes (PMN) from emphysema patients with COPD behaved abnormally. The cells demonstrated a rapid motility and an excessive ability to destroy connective tissue.61 This was related to increased endothelial adhesion underflow62 and reflects a defective chemotactic response to routine chemoattractants.63 These features are not found in healthy smokers and matched patients with AATD. The defect has been linked to the abnormal expression of phosphoinositide 3 (PI3) kinase and may reflect excessive production by phosphatidylinositol (3,4,5)-triphosphate (PIP3), a genetic defect of regulation, or defective breakdown by the phosphates, phosphatase and tensin homolog (PTEN) and SH2 domain-containing inositol 5′-phosphatase 1 (SHIP1). Thus further studies are required to determine the exact mechanism, but it does reflect a pathologically relevant trait that needs further validation, including determining whether this is a feature of susceptible smokers alone and relates to the rate of progression. Nevertheless, recent data has shown that activated neutrophils in usual COPD colocalize to the area of the lung where the emphysema develops.59 The signal is also clearly different to healthy controls and AATD patients and provides a platform for early Phase II studies of neutrophil-modifying therapies.
This whole pathophysiological hypothesis of the role of the neutrophil in the development and progression of disease (at least of emphysema) has therefore a long and well-established history based on a human genetic model. With all this knowledge, the potential for biomarker discovery would seem relatively straightforward, and yet, no such validated biomarker that fulfils all the necessary requirements (marks the disease process, reflects the activity of the disease process, is stable, predicts progression, and identifies patients amenable to therapy) has emerged. With this lesson as an example, all other markers need similar understanding and background before true advances in specific disease management can occur.
The eosinophil
The eosinophil is more classically associated with asthma. However, one study in COPD has shown that an increase in the proportion of eosinophils in induced (or spontaneous) sputum is indicative of a good spirometric response to inhaled corticosteroids.64 Furthermore a recent publication has also indicated that this marker (reflected in blood eosinophils) also identifies patients who are likely to benefit from oral steroid therapy for exacerbations.65 Importantly, the study also suggested that the noneosinophilic group faired less well with steroid therapy. These studies would be consistent with an asthma/COPD overlap syndrome and provide guidance for a more steroid-orientated treatment strategy. Hence, eosinophils can be seen to fulfill one of the requirements for a biomarker. Although bronchodilator reversibility, per se, influences spirometric decline,66 it remains unknown whether it is the eosinophilic subset that marks the susceptible, yet steroid-responsive patient or predicts future progression.
Oxidative stress
The other key process implicated in the pathophysiology of COPD is oxidative stress. Cigarette smoke itself contains high concentrations of oxygen-free radicals with the potential to damage tissues and proteins, and stimulate inflammation. Despite an efficient antioxidant defense, there has been clear and repeated evidence of oxidant stress in COPD. Again, whether this is pathological or physiological (a general response-to-smoking) remains unclear. This topic was extensively reviewed in 200867 and more recently in 2013,68 and the oxidant and proteinase story has been brought together in a further recent review.69 However, to date, the markers of oxidative stress have not been widely used other than to reflect the process itself. Validation of these markers and their use in supporting effective antioxidant therapy is awaited.
Genetic studies have, however, cast some light on the potential pathological, rather than physiological, nature of the ability to deal with oxidative stress (see Technology platforms section).
Inflammation markers
Although the preceding sections addressed relatively specific marker studies in COPD that were based on clinical and pathophysiological processes, the rapidly expanding literature on biomarkers has taken a more generic stance. It is widely accepted that the majority of diseases in the COPD syndrome have inflammation as the key underlying mechanism, and studies of a vast array of mediators have been undertaken, addressing three distinct questions.
- Is it/are they elevated in COPD?
- Does it/do they relate to the physiological markers (usually spirometric) of severity?
- Does it/do they reflect a specific patient phenotype or a therapeutic target?
Using the search terms “biomarkers and COPD”, in excess of 1,000 papers can be found that have been published since 2008. It is clearly beyond the scope of the current review to discuss such a large body of literature, so selective aspects have been chosen to illustrate the general principles required for interpretation (as few have emerged to answer most of the questions outlined in the introduction). C-reactive protein (CRP) is probably the most nonspecific marker of inflammation and yet, has been widely studied in COPD. At present, over 300 articles can be identified via PubMed, using the search words of “CRP and COPD”. CRP levels, even those within the accepted normal range, predict future cardiac events in a general population,70 and they are implicated in the pathophysiology of vascular disease.1 There is an inverse relationship between plasma CRP levels and lung function, even in subjects assumed to be healthy.71 The levels are elevated in COPD patients without evidence of ischemic heart disease; these are stable and are reduced with inhaled corticosteroids,72 although this does not reflect a disease-modifying effect.
There has been no clear mechanism to implicate CRP in the pathophysiology of COPD, nor does it reflect known genetic polymorphisms, suggesting it is merely a general marker of the underlying inflammatory process associated with COPD.73 However, the data did suggest that high CRP was associated with an increased odds ratio for hospitalization, although probably reflecting disease severity. The conclusions were based on a large population cohort and as such, only indicate trends and will have little impact on managing individual patients.73
CRP levels rise during exacerbations particularly when there is an increased neutrophilic influx due to a bacterial cause.74 In addition, a raised CRP in the stable state predicts recurrent exacerbations due either to a failure to completely resolve the first episode or a persistent underlying airway colonization that predisposes to further episodes.75
Fibrinogen has also been widely studied in COPD and has been well covered in a recent review.76 Fibrinogen is also an acute-phase protein and is raised in COPD (as part of the systemic effects of the disease), increases during exacerbations, and is higher in those with frequent exacerbations and those with more severe airflow obstruction. It may also play a role in comorbidities, especially vascular disease and potentially cardiovascular death due to an increased clotting tendency. Again, no genetic polymorphisms have been identified, indicating fibrinogen is also more likely to be a reflective feature of COPD. Although studies indicate that therapy can reduce fibrinogen levels, it remains unknown whether this affects progression or other outcomes. If so, fibrinogen could act as an ideal biomarker at least for some aspects of COPD, and its management and further studies are awaited. The references related to these data are included in the review article.76
Many other inflammatory markers have been measured in COPD, and some seem to be persistently raised. The nonspecific nature of these changes provides little insight into management or pathophysiology. Surfactant protein D (SPD) is a lung-derived protein. SPD is elevated in the plasma in COPD and falls with corticosteroid therapy,77 suggesting that steroids reduce systemic leak from the lungs as part of a general anti-inflammatory effect. However, in one study, the changes in SPD following inhaled therapy were also reflected in the degree of improvement of health status and FEV1, in the short term,78 suggesting that lung inflammation itself affects these features. Of interest, the CRP and interleukin (IL)-6 did not change, suggesting that the effects were acting locally in the lung and not influencing other aspects of generic systemic inflammation (although the SPD effect could be measured systemically).
Studies are beginning to address the requirements of an ideal biomarker, with therapeutic interventions for Phase II and Phase III studies in mind. For example, fibrinogen was used as a marker in a Phase II study of p38 mitogen-activated protein kinase (MAPK) inhibitors,79 which effectively block the proinflammatory pathway. The treatment clearly had an effect, as determined by reducing fibrinogen levels. However, whether this pathway is merely physiological, rather than pathological, remains unknown, and hence, fibrinogen still remains uncertain as a therapeutic marker option in the long-term or short-term management of COPD. Nevertheless, this study represents a first step toward the development and assessment of new therapies using a biomarker, perhaps with a cardiovascular benefit (in those with this comorbidity), rather than a pulmonary one.
Other inflammatory markers are measurable and elevated in the plasma, and although none have proven to be specific, an alternative has been to measure several to develop a multicomponent approach.80 Using the Evaluation of COPD Longitudinally to Identify Predictive Surrogate Endpoints (ECLIPSE) cohort,80 the authors measured several blood biomarkers that were increased in COPD patients compared with healthy smokers and nonsmokers. The white blood cell (WBC) count, CRP, and fibrinogen levels fitted this criterion, as did IL-6, IL-8, and TNFα. The proportion outside the 95th percentile of nonsmokers was networked at baseline and at 1-year follow-up to determine consistency. A subgroup (16% of the patients) with persistently elevated inflammatory markers (two or more) was identified who had a higher incidence of exacerbations (cause or effect?) and worse survival.
This approach of selecting certain markers or traits to identify patient groups with common features is the basis for “cluster analysis”, which may be informative but clearly depends on the traits or factors selected and always needs validation in a second patient cohort; such studies are awaited.
The ECLIPSE cohort has been used to study a large number of other potential biomarkers. In 2011 Dickens et al reported on the stability of 34 blood biomarkers over 3 months.81 The importance of stability is that single measurements could be informative of prognosis and particularly, may reflect the underlying disease activity that leads to progressive deterioration. Fifteen of the markers were different in COPD compared with the controls, and four reflected patients who had exacerbations between baseline and follow-up (CRP, fibrinogen, IL-6, and SPD), but few were classified as stable and hence useful as markers of baseline disease activity (fibrinogen, Clara cell protein [CC]16, and SPD). Although there were weak correlations of the other markers with spirometry, fibrinogen, which did not correlate, emerged as the most promising as it was stable and was a marker of a frequent exacerbation phenotype, exercise impairment, the severity of the BODE index, and dyspnea.
However, of these “stable” markers, only CC16 showed both a relationship to baseline spirometry and the subsequent annual decline.82 At present, only this protein and the Aα-Val360 (see Elastase and other proteinases section) biomarker have shown such characteristics. CC16 is an immunosuppressant and antioxidant produced in the airways. The fact that it is elevated in COPD suggests it is reflective of the disease, rather than causative; however, it is possible that its measurement in serum (like SPD) reflects leakage from the lung due to local inflammation, which may be the generic process associated with, rather than causing, decline. Nevertheless, this series of studies demonstrates several of the steps needed to validate a biomarker, which if subsequently implicated in the pathophysiology, will also direct thinking to new specific therapeutic targets.
SPD levels in blood are also raised in COPD and respond to steroid therapy as indicated above. Since low SPD levels in the lung have been implicated in the pathophysiology of COPD,83 are reduced in BAL in COPD,84 and relate to spirometry,85 it fulfils more of the pathophysiological role than does CC16. However, although SPD responds to oral steroids with a decrease in plasma levels, it is unknown what happens in the lung in the same patients. Nevertheless, in a separate study, COPD was associated with low lung levels of SPD, and inhaled steroid therapy was associated with higher levels,84 suggesting that SPD is a good marker of disease and response to treatment. Subsequently, a polymorphism of the SPD gene was identified that also relates to COPD86 and that is reflected in the level of SPD − at least in the serum (transcription and lung levels are currently unknown). This latter point is of importance:whereas this appears to “close the loop”, it should be noted that a functional polymorphism of SPD that is of pathophysiological importance should result in low lung levels of SPD, but the presence of airway inflammation and leak may, counterintuitively, lead to high blood levels. Nevertheless, the studies do suggest the potential for local therapy with SPD in COPD and that it is certainly worth pursuing.
Sampling issues
Studies of the pathophysiology of COPD are made all the more complex because of the different pathological phenotypes and the fact that the site of damage/repair is in the local tissues. Clearly this is not an issue if the biomarker is either genetic (as in AATD), or systemically reflects the disease process or specifically samples the disease area. To date, biomarkers have been studied in exhaled breath condensate, spontaneous or induced sputum, BAL, or serum/plasma. All of these have inherent problems in interpretation, especially when using high-throughput methodology.
Firstly, many commercially available assays have not been validated using the biological medium collected and processed for the measurement (especially airway secretions) and can produce inaccurate results.87 Secondly, the mediator measurement (especially in exhaled breath condensate) may be detectable but often below the lower limit of quantification.88 Thirdly, the biological medium may not specifically sample the area where the disease process is occurring. For instance, sputum or induced sputum is unlikely to represent samples derived from the small airways or alveoli, and BAL does not exclusively sample the small airways or alveolar region. In addition, the variability between samples from the lung is often wider than the differences between subjects or clinical states. For instance, markers in spontaneous sputum may have a day to day variability of up to 200%, for many reasons.89 Obtaining samples sequentially and averaging the results drastically reduces this variability, if the patient is stable;89 however, this approach cannot easily be applied to invasive procedures, such as BAL or induced sputum, since the introduction of saline or hypertonic saline itself is proinflammatory, and several days are needed between sampling to enable a return to the stable state. However, serum or plasma stability is much more reliable, at least for some biomarkers.81 Pathological specimens provide an opportunity to sample individual cells and their nature/metabolic activity, by laser capture methodology, but other sampling techniques provide varying information from normal and abnormal tissue and may not reflect the pathological area unless performed on lung resection material within defined pathological areas. Finally, mediators measured in blood samples may reflect processes occurring in the interstitium, recirculated from lymph but not necessarily the airways. The subsequent volume of distribution, not only dilutes mediators but also, clears them by appropriate receptor binding.90 Even then, studies like proteomics and metabolomics will differ in plasma compared with serum, due to coagulation and also, by sample collection, distribution, processing, and storage. Careful matching with appropriate controls and interpretation of data is therefore essential in all studies but is rarely done or reported, making the comparison between studies relatively tenuous.
Exacerbations
Exacerbations are an important contributor to health status, health care utilization, and death, particularly if leading to hospitalization. The definition of this symptom-led event, its complexity, and the broad approach to treatment and prevention has generated a search for an objective measure of an exacerbation and therapeutic guidance.
In general, exacerbations are events that are associated with an increase in inflammation, both in the lung and systematically.91 However, in general, the events may reflect a bacterial or viral cause or be related to other causes of increased airway obstruction or may even reflect the destabilization of other organ systems, such as in associated cardiovascular decompensation.92 The main decision required for such events is whether to increase bronchodilator therapy alone, to treat with corticosteroids or antibiotics, or both. Of these, the use of antibiotics has received most attention because overuse is likely to lead to the continued development of bacterial resistance.
Serum procalcitonin is normally produced by neuroendocrine cells of the thyroid and lung but is also produced elsewhere in the body, in response to bacterial infection. For this reason, it has been used to predict bacterial infections and outcome in patients with sepsis.93 However in general this approach has been unsuccessful in COPD.94.95 Serum amyloid A has also been suggested for this as it is increased during exacerbations with a viral or bacteriological aetiology;96 however, it failed to differentiate them, despite reflecting severity of the episode, and hence would not facilitate treatment guidance.
To date, the best two markers that influence therapy are symptom deterioration with known or current eosinophilia, which likely benefit from steroid therapy,65 or sputum color. The latter reflects the presence or absence of a large neutrophilic load, imparting various shades of green. A change in color reflects an increase or decrease in sputum neutrophilia. The color can be characterized at least semiobjectively by both the patient and health care worker, using a standard color chart. In the initial study using this approach in primary care, there was high sensitivity to bacteria isolated from sputum and in particular, an increase relating to the colonizing microbial load, even in the stable clinical state.44 This approach, using sputum color, enabled antibiotics to be withheld without detriment from patients with white or colorless sputum74 and also enabled monitoring of the antibiotic response by the patients.97 Subsequent studies have confirmed the robust nature of this approach in identifying patients with significant colonization98 and also, that withholding antibiotics from those without yellow or green sputum had no adverse effects.94 Interestingly the authors found that CRP and not procalcitonin was increased in patients with purulent sputum, suggesting that CRP may be a better discriminator for bacterial infection. Clearly, further clinical trials are indicated to consolidate this approach.
Technology platforms and the “omics” revolution
In recent years, the ability to obtain multiple assay kits commercially and the development of the proteomics, metabolomics, transcriptomics, and genomics approaches to patient assessment has led to an overwhelming plethora of data. This does lead to some technical issues:the assay may not have been validated for the biological sample or the processing methodology used may lead to spurious and unrecognized effects on the result, both within and between patients. In addition, the data generated has a sensitivity that far outweighs our current ability to phenotype patients sufficiently from a clinical, physiological, and radiological viewpoint.It may be that this approach will lead to a pathophysiological classification of COPD that negates clinical phenotyping, although this remains to be determined and would have to be validated against the clinical “gold standard”.
Proteomics (with 75 citations in the past year) is based on the identification of COPD proteins in biological fluids by a multitude of techniques, which produce a two-dimensional spectrum of data (Figure 5) to determine the similarities or differences between subjects. This demonstrates the amount of data that can be generated by the “omics” approach. The benefit is that this is not a “hypothesis-led” approach, and new data can emerge as “hypothesis generating”. Nevertheless, such an approach can lead to false-positive and false-negative results, and new statistical approaches to the data are needed.99
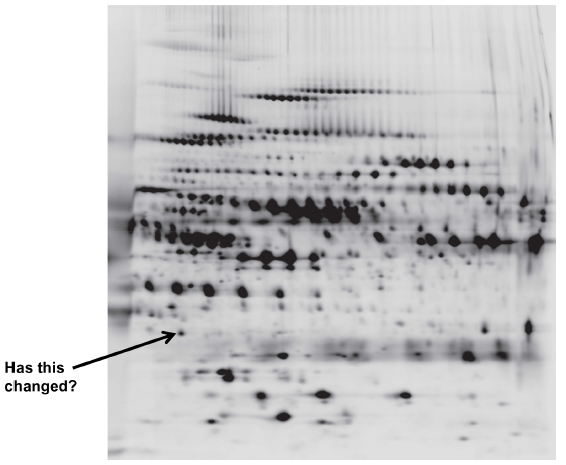 | Figure 5 A two dimensional proteomics read out. Each dot/blot represents a single peptide. Changes in density/presence are considered as biomarkers of the disease progress or activity. |
Indeed it has even been suggested that combining the data from different “omics” platforms, as part of an integrated system biological approach to understanding lung diseases, may be necessary to make progress.100 The methodology is advancing at great pace. The review by Nicholas et al101 defines the principles and aims of the methodology and is worth reading, for those interested.
Less has been published on metabolomics, but in a small study of smokers with emphysema, the assessment of 3,534 metabolite markers was able to separate smokers with and without emphysema,102 and the top 12 were used for predictive models, some of which produced clear separation (as good as CT scan?).
Genetic studies have dominated the COPD world since the early 2000s, with both a candidate gene approach and genome-wide association studies. The data have aimed at detecting the genetic markers of COPD predisposition, thereby leading to new therapies based on gene function (over- or underactive) or a preventative approach to the disease(s). Many of the genetic markers failed to identify polymorphisms that influence the targeted gene function, but associations with COPD or its features suggested the marker was in linkage disequilibrium, probably with a nearby functional abnormality. Such susceptibility markers known to be associated with functional abnormalities include matrix metallopeptidase 9 (MMP9) and apical emphysema,103 TNFα and bronchitis,104 superoxide dismutase and emphysema,105 iron-responsive element binding protein 2 (IREB2) and COPD,106 and IL-13 promoter polymorphisms and an interaction with cigarette smoking on FEV1.107 Nevertheless, such associations represent only minor contributors to the COPD complex that could reflect either the superficial phenotyping of patients or indeed the complexity of the disease(s).108
Such studies do reflect potential susceptibility genes and hence, pathophysiological pathways, but how or whether they reflect the rate of progression or long-term outcomes has yet to be determined. Nevertheless, in other diseases, treatments have been influenced by genetic studies, suggesting the same may follow for COPD, at least for those associated with a functional contribution.109 It may be that a panel of genes or a panel of gene transcripts may provide further insight into progression or the response to treatment, although far from personalized medicine.110 Interestingly, a recent study111 has suggested that a polymorphism in the a disintegrin and metalloprotease domain 33 (ADAM33) gene is associated with all-cause and cardiovascular mortality in COPD. The group of proteins encoded by this gene domain play important roles in cell adhesion, migration, and proteolysis, and ADAM33 has also been implicated in the rapid decline in lung function and development of COPD.112,113 Whether this reflects a functional abnormality and hence, a treatment target needs to be determined.
Summary
Biomarker discovery has become a rapidly expanding field of research primarily to develop easy and early readouts for delivery of Phase II clinical studies and a shortening of pivotal Phase III studies that displace conventional markers like FEV1. A variety of methodologies and sample collections have been undertaken, measuring things as diverse as the mechanics of the lung, patient symptomatology, markers of exposure (such as inhaled oxidants from cigarette smoke), markers of tissue damage (connective tissue degradation products), number and function of implicated cells, generalized inflammatory markers, cell-activation products, underlying proteins (their function, metabolism, and source), and the genetic background of the patients. These have involved a variety of biological samples collected in different ways, all with both benefits and drawbacks and all requiring caution in interpretation, as summarized in Table 1. However, the identification and understanding of such markers, either in a hypothesis-testing or hypothesis-generating approach, may prove sufficiently informative for understanding the disease process and for developing new therapeutic strategies. Nevertheless, without accurate patient characterization, biomarker validation, and an understanding of the issues that likely influence the measurements (including current therapy and dissecting cause and effect), this process may prove to be both fruitless and confusing.
Disclosure
The author has lectured widely as part of pharmaceutical sponsored symposia, sat on numerous advisory boards for drug design and trial implementation, and received noncommercial grant funding. The author reports no other conflicts of interest in this work.
References
Sevenoaks MJ, Stockley RA. Chronic Obstructive Pulmonary Disease, inflammation and co-morbidity – a common inflammatory phenotype? Respir Res. 2006;7(70):1–9. | |
Cote CG. Surrogates of mortality in chronic obstructive pulmonary disease. Am J Med. 2006;119(10 Suppl 1):S54–S62. | |
Donaldson GC, Wedzicha JA. COPD exacerbations 1:Epidemiology. Thorax. 2006;61(2):164–168. | |
Clark KD, Wardrobe-Wong N, Elliott JJ, Gill PT, Tait NP, Snashall PD. Patterns of lung disease in a “normal” smoking population:are emphysema and airflow obstruction found together? Chest. 2001;120(3):743–747. | |
Jones PW. Health status and the spiral of decline. COPD. 2009;6(1):59–63. | |
Dawkins PA, Dawkins CL, Wood AM, Nightingale PG, Stockley JA, Stockley RA. Rate of progression of lung function impairment in alpha1-antitrypsin deficiency. Eur Respir J. 2009;33(6):1338–1344. | |
Domingo-Salvany A, Lamarca R, Ferrer M, et al. Health-related quality of life and mortality in male patients with chronic obstructive pulmonary disease. Am J Respir Crit Care Med. 2002;166(5):680–685. | |
Nishimura K, Izumi T, Tsukino M, Oga T. Dyspnea is a better predictor of 5-year survival than airway obstruction in patients with COPD. Chest. 2002;121(5):1434–1440. | |
Gerardi DA, Lovett L, Benoit-Connors ML, Reardon JZ, ZuWallack RL. Variables related to increased mortality following out-patient pulmonary rehabilitation. Eur Respir J. 1996;9(3):431–435. | |
Landbo C, Prescott E, Lange P, Vestbo J, Almdal TP. Prognostic value of nutritional status in chronic obstructive pulmonary disease. Am J Respir Crit Care Med. 1999;160(6):1856–1861. | |
Celli BR, Cote CG, Marin JM, et al. The body-mass index, airflow obstruction, dyspnea, and exercise capacity index in chronic obstructive pulmonary disease. N Engl J Med. 2004;350(10):1005–1012. | |
Casanova C, de Torres JP, Aguirre-Jaíme A, et al. The progression of chronic obstructive pulmonary disease is heterogeneous:the experience of the BODE cohort. Am J Respir Crit Care Med. 2011;184(9):1015–1021. | |
Martinez FJ, Han MK, Andrei AC, et al; National Emphysema Treatment Trial Research Group. Longitudinal change in the BODE index predicts mortality in severe emphysema. Am J Respir Crit Care Med. 2008;178(5):491–499. | |
Jones PW, Brusselle G, Dal Negro RW, et al. Properties of the COPD assessment test in a cross-sectional European study. Eur Respir J. 2011;38(1):29–35. | |
Parr DG, Stoel BC, Stolk J, Stockley RA. Pattern of emphysema distribution in alpha1-antitrypsin deficiency influences lung function impairment. Am J Respir Crit Care Med. 2004;170(11):1172–1178. | |
Haruna A, Muro S, Nakano Y, et al. CT scan findings of emphysema predict mortality in COPD. Chest. 2010;138(3):635–640. | |
Klein JS, Gamsu G, Webb WR, Golden JA, Müller NL. High-resolution CT diagnosis of emphysema in symptomatic patients with normal chest radiographs and isolated low diffusing capacity. Radiology. 1992;182(3):817–821. | |
Stolk J, Stockley RA, Stoel BC, et al. Randomised controlled trial for emphysema with a selective agonist of the γ-type retinoic acid receptor. Eur Respir J. 2012;40(2):306–312. | |
Dirksen A, Piitulainen E, Parr DG, et al. Exploring the role of CT densitometry:a randomised study of augmentation therapy in alpha1-antitrypsin deficiency. Eur Respir J. 2009;33(6):1345–1353. | |
Stolk J, Cooper BG, Stoel B, et al. Retinoid treatment of Emphysema in Patients on the Alpha-1 International Registry. The REPAIR study:study design, methodology and quality control of study assessments. Ther Adv Respir Dis. 2010;4(6):319–332. | |
Galbán CJ, Han MK, Boes JL, et al. Computed tomography-based biomarker provides unique signature for diagnosis of COPD phenotypes and disease progression. Nat Med. 2012;18(11):1711–1715. | |
Tanabe N, Muro S, Oguma T, et al. Computed tomography assessment of pharmacological lung volume reduction induced by bronchodilators in COPD. COPD. 2012;9(4):401–408. | |
Martínez-García MA, de la Rosa Carrillo D, Soler-Cataluña JJ, et al. Prognostic value of bronchiectasis in patients with moderate-to-severe chronic obstructive pulmonary disease. Am J Respir Crit Care Med. 2013;187(8):823–831. | |
Guerra S, Sherrill DL, Venker C, Ceccato CM, Halonen M, Martinez FD. Chronic bronchitis before age 50 years predicts incident airflow limitation and mortality risk. Thorax. 2009;64(10):894–900. | |
de Oca MM, Halbert RJ, Lopez MV, et al. The chronic bronchitis phenotype in subjects with and without COPD:the PLATINO study. Eur Respir J. 2012;40(1):28–36. | |
Calverley PM, Boonsawat W, Cseke Z, Zhong N, Peterson S, Olsson H. Maintenance therapy with budesonide and formoterol in chronic obstructive pulmonary disease. Eur Respir J. 2003;22(6):912–919. | |
Calverley PM, Rabe KF, Goehring UM, Kristiansen S, Fabbri LM, Martinez FJ; M2-124 and M2-125 study groups. Roflumilast in symptomatic chronic obstructive pulmonary disease:two randomised clinical trials. Lancet. 2009;374(9691):685–694. | |
Kuhn C, Yu SY, Chraplyvy M, Linder HE, Senior RM. The induction of emphysema with elastase. II. Changes in connective tissue. Lab Invest. 1976;34(4):372–380. | |
Laurell CB, Eriksson S. The electrophoretic alpha 1-globulin pattern of serum in alpha 1-antitrypsin deficiency. Scand J Clin Lab Invest. 1963;15:132–140. | |
Senior RM, Tegner H, Kuhn C, Ohlsson K, Starcher BC, Pierce JA. The induction of pulmonary emphysema with human leukocyte elastase. Am Rev Respir Dis. 1977;116(3):469–475. | |
Kao RC, Wehner NG, Skubitz KM, Gray BH, Hoidal JR. Proteinase 3. A distinct human polymorphonuclear leukocyte proteinase that produces emphysema in hamsters. J Clin Invest. 1988;82(6):1963–1973. | |
Shapiro SD, Endicott SK, Province MA, Pierce JA, Campbell EJ. Marked longevity of human lung parenchymal elastic fibers deduced from prevalence of D-aspartate and nuclear weapons-related radiocarbon. J Clin Invest. 1991;87(5):1828–1834. | |
Fregonese L, Ferrari F, Fumagalli M, Luisetti M, Stolk J, Iadarola P. Long-term variability of desmosine/isodesmosine as biomarker in alpha-1-antritrypsin deficiency-related COPD. COPD. 2011;8(5):329–333. | |
Luisetti M, Ma S, Iadarola P, et al. Desmosine as a biomarker of elastin degradation in COPD:current status and future directions. Eur Respir J. 2008;32(5):1146–1157. | |
Viglio S, Iadarola P, Lupi A, et al. MEKC of desmosine and isodesmosine in urine of chronic destructive lung disease patients. Eur Respir J. 2000;15(6):1039–1045. | |
Skjøt-Arkil H, Clausen RE, Rasmussen LM, et al. Acute Myocardial Infarction and Pulmonary Diseases Result in Two Different Degradation Profiles of Elastin as Quantified by Two Novel ELISAs. PLoS One. 2013;8(6):e60936. | |
Lindberg CA, Engström G, de Verdier MG, et al. Total desmosines in plasma and urine correlate with lung function. Eur Respir J. 2012;39(4):839–845. | |
Turino GM, Ma S, Lin YY, Cantor JO, Luisetti M. Matrix elastin:a promising biomarker for chronic obstructive pulmonary disease. Am J Respir Crit Care Med. 2011;184(6):637–641. | |
Morrison HM, Welgus HG, Stockley RA, Burnett D, Campbell EJ. Inhibition of human leukocyte elastase bound to elastin:relative ineffectiveness and two mechanisms of inhibitory activity. Am J Respir Cell Mol Biol. 1990;2(3):263–269. | |
Stockley RA, Burnett D. Alpha1-antitrypsin and leukocyte elastase in infected and non-infected sputum. Am Rev Respir Dis. 1979;120:1081–1086. | |
Hill AT, Campbell EJ, Bayley DL, Hill SL, Stockley RA. Evidence for excessive bronchial inflammation during an acute exacerbation of chronic obstructive pulmonary disease in patients with alpha(1)-antitrypsin deficiency (PiZ). Am J Respir Crit Care Med. 1999;160(6):1968–1975. | |
Stockley RA, Hill SL, Morrison HM, Starkie CM. Elastolytic activity of sputum and its relation to purulence and to lung function in patients with bronchiectasis. Thorax. 1984;39(6):408–413. | |
Jackson AH, Hill SL, Afford SC, Stockley RA. Sputum sol-phase proteins and elastase activity in patients with cystic fibrosis. Eur J Respir Dis. 1984;65(2):114–124. | |
Hill AT, Campbell EJ, Hill SL, Bayley DL, Stockley RA. Association between airway bacterial load and markers of airway inflammation in patients with stable chronic bronchitis. Am J Med. 2000;109(4):288–295. | |
Sapey E, Stockley RA. Neutrophils. In:Barnes PJ, editor. Chronic Obstructive Pulmonary Disease:Cellular and Molecular Mechanisms. Vol 198. New York, NY:Taylor and Francis Group; 2005:133–170. | |
Sinden NJ, Stockley RA. Proteinase 3 activity in sputum from subjects with alpha-1-antitrypsin deficiency and COPD. Eur Respir J. 2013; 41(5):1042–1050. | |
Burnett D, Reynolds JJ, Ward RV, Afford SC, Stockley RA. Tissue inhibitor of metalloproteinases and collagenase inhibitory activity in lung secretions from patients with chronic obstructive bronchitis:effect of corticosteroid treatment. Thorax. 1986;41(10):740–745. | |
Burnett D, Crocker J, Stockley RA. Cathepsin B-like cysteine proteinase activity in sputum and immunohistologic identification of cathepsin B in alveolar macrophages. Am Rev Respir Dis. 1983;128(5):915–919. | |
Stockley RA, Rennard SI, Rabe K, Celli B. Proteinases and COPD. In:Sullivan AL, Stockley RA, editors. Chronic Obstructive Pulmonary Disease. Oxford:Blackwell Publishing Ltd; 2008:349–366. | |
Gadek JE, Klein HG, Holland PV, Crystal RG. Replacement therapy of alpha 1-antitrypsin deficiency. Reversal of protease-antiprotease imbalance within the alveolar structures of PiZ subjects. J Clin Invest. 1981;68(5):1158–1165. | |
Stockley RA, Bayley DL, Unsal I, Dowson LJ. The effect of augmentation therapy on bronchial inflammation in alpha1-antitrypsin deficiency. Am J Respir Crit Care Med. 2002;165(11):1494–1498. | |
Carter RI, Mumford RA, Treonze KM, et al. The fibrinogen cleavage product Aα-Val360, a specific marker of neutrophil elastase activity in vivo. Thorax. 2011;66(8):686–691. | |
Carter RI, Ungurs MJ, Mumford RA, Stockley RA. Aα-Val360:a marker of neutrophil elastase and COPD disease activity. Eur Respir J. 2013;41(1):31–38. | |
Buttle DJ, Abrahamson M, Burnett D, et al. Human sputum cathepsin B degrades proteoglycan, is inhibited by alpha 2-macroglobulin and is modulated by neutrophil elastase cleavage of cathepsin B precursor and cystatin C. Biochem J. 1991;276(Pt 2):325–331. | |
Geraghty P, Rogan MP, Greene CM, et al. Alpha-1-antitrypsin aerosolised augmentation abrogates neutrophil elastase-induced expression of cathepsin B and matrix metalloprotease 2 in vivo and in vitro. Thorax. 2008;63(7):621–626. | |
Okada Y, Nakanishi I. Activation of matrix metalloproteinase 3 (stromelysin) and matrix metalloproteinase 2 (‘gelatinase’) by human neutrophil elastase and cathepsin G. FEBS Lett. 1989;249(2):353–356. | |
Churg A, Zhou S, Wright JL. Series “matrix metalloproteinases in lung health and disease”:Matrix metalloproteinases in COPD. Eur Respir J. 2012;39(1):197–209. | |
Stănescu D, Sanna A, Veriter C, et al. Airways obstruction, chronic expectoration, and rapid decline of FEV1 in smokers are associated with increased levels of sputum neutrophils. Thorax. 1996;51(3):267–271. | |
Subramanian DR, Jenkins L, Edgar R, Quraishi N, Stockley RA, Parr DG. Assessment of pulmonary neutrophilic inflammation in emphysema by quantitative positron emission tomography. Am J Respir Crit Care Med. 2012;186(11):1125–1132. | |
Damiano VV, Tsang A, Kucich U, et al. Immunolocalization of elastase in human emphysematous lungs. J Clin Invest. 1986;78(2):482–493. | |
Burnett D, Chamba A, Hill SL, Stockley RA. Neutrophils from subjects with chronic obstructive lung disease show enhanced chemotaxis and extracellular proteolysis. Lancet. 1987;2(8567):1043–1046. | |
Woolhouse IS, Bayley DL, Lalor P, Adams DH, Stockley RA. Endothelial interactions of neutrophils under flow in chronic obstructive pulmonary disease. Eur Respir J. 2005;25(4):612–617. | |
Sapey E, Stockley JA, Greenwood H, et al. Behavioral and structural differences in migrating peripheral neutrophils from patients with chronic obstructive pulmonary disease. Am J Respir Crit Care Med. 2011;183(9):1176–1186. | |
Brightling CE, Monteiro W, Ward R, et al. Sputum eosinophilia and short-term response to prednisolone in chronic obstructive pulmonary disease:a randomised controlled trial. Lancet. 2000;356(9240):1480–1485. | |
Bafadhel M, McKenna S, Terry S, et al. Blood eosinophils to direct corticosteroid treatment of exacerbations of chronic obstructive pulmonary disease:a randomized placebo-controlled trial. Am J Respir Crit Care Med. 2012;186(1):48–55. | |
Postma DS, de Vries K, Koëter GH, Sluiter HJ. Independent influence of reversibility of air-flow obstruction and nonspecific hyperreactivity on the long-term course of lung function in chronic air-flow obstruction. Am Rev Respir Dis. 1986;134(2):276–280. | |
Louhelainen N, Myllärniemi M, Rahman I, Kinnula VL. Airway biomarkers of the oxidant burden in asthma and chronic obstructive pulmonary disease:current and future perspectives. Int J Chron Obstruct Pulmon Dis. 2008;3(4):585–603. | |
Kirkham PA, Barnes PJ. Oxidative stress in COPD. Chest. 2013;144(1):266–273. | |
Fischer BM, Pavlisko E, Voynow JA. Pathogenic triad in COPD:oxidative stress, protease-antiprotease imbalance, and inflammation. Int J Chron Obstruct Pulmon Dis. 2011;6:413–421. | |
Ridker PM, Rifai N, Rose L, Buring JE, Cook NR. Comparison of C-reactive protein and low-density lipoprotein cholesterol levels in the prediction of first cardiovascular events. N Engl J Med. 2002;347(20):1557–1565. | |
Aronson D, Roterman I, Yigla M, et al. Inverse association between pulmonary function and C-reactive protein in apparently healthy subjects. Am J Respir Crit Care Med. 2006;174(6):626–632. | |
Pinto-Plata VM, Müllerova H, Toso JF, et al. C-reactive protein in patients with COPD, control smokers and non-smokers. Thorax. 2006;61(1):23–28. | |
Dahl M, Vestbo J, Zacho J, Lange P, Tybjærg-Hansen A, Nordestgaard BG. C reactive protein and chronic obstructive pulmonary disease:a Mendelian randomisation approach. Thorax. 2011;66(3):197–204. | |
Stockley RA, O’Brien C, Pye A, Hill SL. Relationship of sputum color to nature and outpatient management of acute exacerbations of COPD. Chest. 2000;117(6):1638–1645. | |
Perera WR, Hurst JR, Wilkinson TM, et al. Inflammatory changes, recovery and recurrence at COPD exacerbation. Eur Respir J. 2007;29(3):527–534. | |
Duvoix A, Dickens J, Haq I, et al. Blood fibrinogen as a biomarker of chronic obstructive pulmonary disease. Thorax. 2013;68(7):670–676. | |
Lomas DA, Silverman EK, Edwards LD, et al; Evaluation of COPD Longitudinally to Identify Predictive Surrogate Endpoints Study Investigators. Serum surfactant protein D is steroid sensitive and associated with exacerbations of COPD. Eur Respir J. 2009;34(1):95–102. | |
Sin DD, Man SF, Marciniuk DD, et al; ABC (Advair, Biomarkers in COPD) Investigators. The effects of fluticasone with or without salmeterol on systemic biomarkers of inflammation in chronic obstructive pulmonary disease. Am J Respir Crit Care Med. 2008;177(11):1207–1214. | |
Lomas DA, Lipson DA, Miller BE, et al; Losmapimod Study Investigators. An oral inhibitor of p38 MAP kinase reduces plasma fibrinogen in patients with chronic obstructive pulmonary disease. J Clin Pharmacol. 2012;52(3):416–424. | |
Agustí A, Edwards LD, Rennard SI, et al; Evaluation of COPD Longitudinally to Identify Predictive Surrogate Endpoints (ECLIPSE) Investigators. Persistent systemic inflammation is associated with poor clinical outcomes in COPD:a novel phenotype. PLoS One. 2012;7(5):e37483. | |
Dickens JA, Miller BE, Edwards LD, Silverman EK, Lomas DA, Tal-Singer R; Evaluation of COPD Longitudinally to Identify Surrogate Endpoints (ECLIPSE) study investigators. COPD association and repeatability of blood biomarkers in the ECLIPSE cohort. Respir Res. 2011;12:146. | |
Vestbo J, Edwards LD, Scanlon PD, et al; ECLIPSE Investigators. Changes in forced expiratory volume in 1 second over time in COPD. N Engl J Med. 2011;365(13):1184–1192. | |
Knudsen L, Ochs M, Mackay R, et al. Truncated recombinant human SP-D attenuates emphysema and type II cell changes in SP-D deficient mice. Respir Res. 2007;8:70. | |
Sims MW, Tal-Singer RM, Kierstein S, et al. Chronic obstructive pulmonary disease and inhaled steroids alter surfactant protein D (SP-D) levels:a cross-sectional study. Respir Res. 2008;9:13. | |
Sin DD, Leung R, Gan WQ, Man SP. Circulating surfactant protein D as a potential lung-specific biomarker of health outcomes in COPD:a pilot study. BMC Pulm Med. 2007;7:13. | |
Foreman MG, Kong X, DeMeo DL, et al. Polymorphisms in surfactant protein-D are associated with chronic obstructive pulmonary disease. Am J Respir Cell Mol Biol. 2011;44(3):316–322. | |
Woolhouse IS, Bayley DL, Stockley RA. Effect of sputum processing with dithiothreitol on the detection of inflammatory mediators in chronic bronchitis and bronchiectasis. Thorax. 2002;57(8):667–671. | |
Bayley DL, Abusriwil H, Ahmad A, Stockley RA. Validation of assays for inflammatory mediators in exhaled breath condensate. Eur Respir J. 2008;31(5):943–948. | |
Sapey E, Bayley D, Ahmad A, Newbold P, Snell N, Stockley RA. Inter-relationships between inflammatory markers in patients with stable COPD with bronchitis:intra-patient and inter-patient variability. Thorax. 2008;63(6):493–499. | |
Sinden NJ, Stockley RA. Systemic inflammation and comorbidity in COPD:a result of ‘overspill’ of inflammatory mediators from the lungs? Review of the evidence. Thorax. 2010;65(10):930–936. | |
Aaron SD, Angel JB, Lunau M, et al. Granulocyte inflammatory markers and airway infection during acute exacerbation of chronic obstructive pulmonary disease. Am J Respir Crit Care Med. 2001;163(2):349–355. | |
Beghé B, Verduri A, Roca M, Fabbri LM. Exacerbation of respiratory symptoms in COPD patients may not be exacerbations of COPD. Eur Respir J. 2013;41(4):993–995. | |
Becker KL, Snider R, Nylen ES. Procalcitonin in sepsis and systemic inflammation:a harmful biomarker and a therapeutic target. Br J Pharmacol. 2010;159(2):253–264. | |
Soler N, Esperatti M, Ewig S, Huerta A, Agustí C, Torres A. Sputum purulence-guided antibiotic use in hospitalised patients with exacerbations of COPD. Eur Respir J. 2012;40(6):1344–1353. | |
Falsey AR, Becker KL, Swinburne AJ, et al. Utility of serum procalcitonin values in patients with acute exacerbations of chronic obstructive pulmonary disease:a cautionary note. Int J Chron Obstruct Pulmon Dis. 2012;7:127–135. | |
Bozinovski S, Hutchinson A, Thompson M, et al. Serum amyloid a is a biomarker of acute exacerbations of chronic obstructive pulmonary disease. Am J Respir Crit Care Med. 2008;177(3):269–278. | |
Woolhouse IS, Hill SL, Stockley RA. Symptom resolution assessed using a patient directed diary card during treatment of acute exacerbations of chronic bronchitis. Thorax. 2001;56(12):947–953. | |
Miravitlles M, Marín A, Monsó E, et al. Colour of sputum is a marker for bacterial colonisation in chronic obstructive pulmonary disease. Respir Res. 2010;11:58. | |
Wheelock ÅM, Wheelock CE. Trials and tribulations of ‘omics data analysis:assessing quality of SIMCA-based multivariate models using examples from pulmonary medicine. Mol Biosyst. 2013;9(11):2589–2596. | |
Auffray C, Adcock IM, Chung KF, Djukanovic R, Pison C, Sterk PJ. An integrative systems biology approach to understanding pulmonary diseases. Chest. 2010;137(6):1410–1416. | |
Nicholas BL, O’Connor CD, Djukanovic R. From proteomics to prescription-the search for COPD biomarkers. COPD. 2009;6(4):298–303. | |
Paige M, Burdick MD, Kim S, Xu J, Lee JK, Michael Shim Y. Pilot analysis of the plasma metabolite profiles associated with emphysematous Chronic Obstructive Pulmonary Disease phenotype. Biochem Biophys Res Commun. 2011;413(4):588–593. | |
Ito I, Nagai S, Handa T, et al. Matrix metalloproteinase-9 promoter polymorphism associated with upper lung dominant emphysema. Am J Respir Crit Care Med. 2005;172(11):1378–1382. | |
Sapey E, Wood AM, Ahmad A, Stockley RA. Tumor necrosis factor-{alpha} rs361525 polymorphism is associated with increased local production and downstream inflammation in chronic obstructive pulmonary disease. Am J Respir Crit Care Med. 2010;182(2):192–199. | |
Sørheim IC, DeMeo DL, Washko G, et al; International COPD Genetics Network Investigators. Polymorphisms in the superoxide dismutase-3 gene are associated with emphysema in COPD. COPD. 2010;7(4):262–268. | |
DeMeo DL, Mariani T, Bhattacharya S, et al. Integration of genomic and genetic approaches implicates IREB2 as a COPD susceptibility gene. Am J Hum Genet. 2009;85(4):493–502. | |
Sadeghnejad A, Meyers DA, Bottai M, Sterling DA, Bleecker ER, Ohar JA. IL13 promoter polymorphism 1112C/T modulates the adverse effect of tobacco smoking on lung function. Am J Respir Crit Care Med. 2007;176(8):748–752. | |
Cookson WO, Moffatt MF. Genetics of complex airway disease. Proc Am Thorac Soc. 2011;8(2):149–153. | |
Hall IP. Stratified medicine:drugs meet genetics. Eur Respir Rev. 2013;22(127):53–57. | |
Mariani TJ, Martinez F. Chronic obstructive pulmonary disease genomics:yesterday, discovering population biomarkers; tomorrow, defining disease clusters. Am J Respir Crit Care Med. 2013;187(9):900–902. | |
Figarska SM, Vonk JM, van Diemen CC, Postma DS, Boezen HM. ADAM33 gene polymorphisms and mortality. A prospective cohort study. PLoS One. 2013;8(7):e67768. | |
van Diemen CC, Postma DS, Vonk JM, Bruinenberg M, Schouten JP, Boezen HM. A disintegrin and metalloprotease 33 polymorphisms and lung function decline in the general population. Am J Respir Crit Care Med. 2005;172(3):329–333. | |
Gosman MM, Boezen HM, van Diemen CC, et al. A disintegrin and metalloprotease 33 and chronic obstructive pulmonary disease pathophysiology. Thorax. 2007;62(3):242–247. |
 © 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.