Back to Journals » OncoTargets and Therapy » Volume 13
Bioinformatics Analysis and RNA-Sequencing of SCAMP3 Expression and Correlated Gene Regulation in Hepatocellular Carcinoma
Authors Han S, Feng Z, Liu R, Ye J, Cheng W, Bao J
Received 3 July 2019
Accepted for publication 17 December 2019
Published 4 February 2020 Volume 2020:13 Pages 1047—1057
DOI https://doi.org/10.2147/OTT.S221785
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Yao Dai
Shan-shan Han,1 Zhi-qiang Feng,1 Rui Liu,2 Jun Ye,2 Wei-wei Cheng,2 Jun-bo Bao3
1Beijing Chaoyang Emergency Medical Center, Department of General Surgery, Chaoyang, Beijing 100020, People’s Republic of China; 2Medical University of Anhui Air Force Clinical School, Department of Hepatobiliary Surgery, Beijing 100142, People’s Republic of China; 3First People’s Hospital of Suqian, Department of Medicine, Suqian, Jiangsu 223800, People’s Republic of China
Correspondence: Zhi-qiang Feng
Beijing Chaoyang Emergency Medical Center, Department of General Surgery, Chaoyang, Beijing 100020, People’s Republic of China
Email [email protected]
Background: Secretory Carrier Membrane Proteins 3 (SCAMP3) is a transmembrane protein that affects intracellular trafficking, protein sorting and vesicle formation. Overexpression of SCAMP3 correlates with poorly differentiated hepatocellular carcinoma (HCC). However, the expression and corresponding gene regulation of SCAMP3 in HCC remain unclear.
Methods: Bioinformatics analyses of clinical parameters and survival data were conducted to predict the prognostic value of SCAMP3 in HCC. RNA sequencing and real-time PCR were conducted to confirm the SCAMP3 expression in HCC tissue. Expression was analyzed using OncomineTM and UALCAN, while SCAMP3 alterations and survival analysis were identified by cBioPortal. Differential gene expression with SCAMP3 was analyzed by LinkedOmics and GEPIA. The target networks of enzymes and co-transcriptional factors were identified using Gene enrichment analysis. Expression of SCAMP3 in HCC tissue was detected by RNA-sequencing and Western-blotting.
Results: Based on bioinformatics analysis and detection of mRNA expression, SCAMP3 was over-expressed in numerous tumors, especially in HCC. SCAMP3 level was positively correlated with disease stages and tumor grades and negatively correlated with patient survival. Furthermore, functional network analysis indicated that SCAMP3 regulated metabolic process and DNA replication through oxidative phosphorylation and chromatin remodeling or Ribosome. SCAMP3 regulated a number of gene expressions including PPAP2B, SNRK, ARID4A, PRCC, VPS72 via protein binding and proteasome, which may affect cell adhesion, proliferation, transcription, cell cycle and metabolism. Further, Real-time PCR and Western-blotting showed that the SCAMP3 level was increased in HCC tissue.
Conclusion: The present data analysis efficiently reveals information about SCAMP3 expression and correlated function in HCC, laying a foundation for further study of SCAMP3 in the tumor.
Keywords: SCAMP3, hepatocellular carcinoma, bioinformatics analysis, RNA-sequencing, real-time PCR
Introduction
Hepatocellular carcinoma (HCC) is regarded as the majority of primary liver cancers. Liver cancer remains one of the most common malignant tumors and the fourth leading cause of cancer-related death globally.1 The incidence of HCC and related cancer death is increasing in developing countries.2 The World Health Organization reported that more than 1 million patients would die of HCC in 2030.1 Due to the complex pathogenesis of HCC such as hepatic inflammation, fibrosis and aberrant hepatocyte regeneration, identifying the key network of multiple genes at multiple steps is needed. Although progress in the treatment of HCC with various targeted drugs has been achieved, the 5-year survival rate of patients is less than 5%.3 Prognosis of HCC at early stages and grades is helpful to prolong the overall life expectancy of HCC patients.4 Therefore, exploring accurate biomarkers for early diagnosis and effective therapeutic strategies have crucial clinical significance.
Secretory Carrier Membrane Proteins (SCAMPs) are tetraspanin, integral membrane proteins that exist in endosomes, plasma membrane recycling and Trans-Golgi network.5,6 Through influencing cell recycling process such as trafficking of transporters and receptors, protein sorting and so on, SCAMPs play a key role in homeostasis and cellular responses to stress in the environment.5,6 In humans, the SCAMPs family consists of four ubiquitous isoforms, SCAMPs1-4, and a central isoform SCAMP5.7 SCAMP3 has been shown to regulate the size of early-endosomes and recruit the ESCRT-0 protein to early-endosomes. It is reported that SCAMP3 negatively modulated degradation of the epidermal growth factor receptor, leading to receptor recycling.7 However, very little is known about the role and mechanism of SCAMP3 in carcinogenesis. Until now, Wael found that overexpression of SCAMP3 was detected in 84 HCC patients, which is associated with hepatocellular carcinoma dedifferentiation and HCC progression.8
In the present study, bioinformatics analysis of several large online databases was mined to evaluate the expression profile of SCAMP3 and the relationship between SCAMP3 expression and clinical pathological parameters in HCC. Furthermore, genomic alterations and biological function related to SCAMP3 in HCC were also examined. This study may potentially supply a new target for HCC diagnosis and treatment.
Materials and Methods
Gene Expression Analysis Using Oncomine and HCCDB Databases
The gene expression and mRNA level of SCAMP3 were evaluated using Oncomine database (http://www.oncomine.org), which is the world’s largest oncogene database containing 715 data sets and 86,733 samples.9 We focused on several HCC studies such as Chen Liver, Mas Liver and so on.10 SCAMP3 gene was queried in the database and the data was shown by selecting HCC and cancer vs Normal tissue. The cut-off of P-value was 0.01 and fold change was defined as 2. HCCDB is a one-stop online resource for visualizing HCC gene expression from around 4000 clinical samples.11 We used HCCDB to analyze CAMP3 expression in HCC.
UALCAN Analysis
UALCAN database (http://ualcan.path.uab.edu), an interactive web-portal for investigating the relative expression of SCAMP3 across HCC and normal tissues, as well as in various sub-groups such as cancer stages, tumor grades, ages, gender and so on. UALCAN is based on the analyses of TCGA (The Cancer Genome Atlas) gene expression and clinical data from 31 cancer types.10
TIMER Analysis
TIMER (http://cistrome.Shinyapps.io/timer/) is a web server for exploring the correlation between abundance of tumor-infiltrating immune cells and expression of query gene, which uses the statistical method of deconvolution. It also supplies an analysis of relative expression of the goal gene across tumor and normal tissues, currently containing 10.897 samples from TCGA.12 SCAMP3 expression was assessed in cancers paired to normal tissues.
cBioportal Database Analysis
The altered expression of SCAMP3 in LIHC (Liver hepatocellular carcinoma) and relative overall survival in patients were analyzed by cBioportal database (http://cbioportal.org), which is an open access web-based resource for exploring the multidimensional cancer genomics data sets.13 It currently provides data from 225 cancer studies. The tab Oncoprint shows the overview of SCAMP3 alterations in LIHC patients.
LinkedOmics and GSE (Gene Set Enrichment) Analysis
LinkedOmics database (http://www.linkedomics.org/login.php) is a web tool for elucidating multi-omics data and clinical data from TCGA, currently containing 32 cancer types.14 It has three analysis modules in web application. We used Linked Finder module to analyze the correlation of SCAMP3 and differentially expressed genes in LIHC. The heat maps show the statistical plots for individual genes. GSEA is a powerful analytical method for interpreting gene expression data.15 We used GSEA to analyze the data from LinkFinder, obtaining the GO (cellular component, CC; biological process, BP; molecular function, MF) and KEGG (Kyoto Encyclopedia of Genes and Genomes) pathway of SCAMP3 co-expression genes in LIHC.
GEPIA Analysis
GEPIA (Gene Expression Profiling Interactive Analysis, http://gepia.cancer-pku.cn/) dataset, an interactive web server, uses a standard processing pipeline to analyze differential gene expression, similar gene detection and correlation, dimensionality reduction and so on.16 The correlation between SCAMP3 and the top three differentially expressed genes was evaluated by GEPIA.
Transcriptome Sequencing (mRNA-Seq) and Real-Time PCR
Total RNA from HCC and normal tissues was isolated with TRIzol (Invitrogen), which was sent to a sequencing company (Vazyme, Nanjing, China) for mRNA-seq. Written informed consent was provided by the patients, and the study was conducted in accordance with the Declaration of Helsinki and approved by the Air Force Medical Center of PLA Ethics committee. Briefly, the RNA quality was examined and sequencing libraries were generated using the VAHTS mRNA-seq v2 Library Prep Kit for Illumina® (Vazyme, NR601). After measuring the library concentration, the clustering of the index-coded samples was performed on a cBot Cluster Generation System (Illumina, USA) and the library preparations were sequenced on an Illumina Hiseq X Ten platform and 150-bp paired-end module. Gene differential expression was analyzed by Cuffdiff (v2.2.1), Genes with corrected p-value≤0.05 and the absolute value of log2 (fold change) ≥1 were assigned as significantly differentially expressed. For real-time PCR, 1 µg mRNA was reverse transcribed and amplified, which detected by Bio-Rad CFX Connect System (Bio-Rad). The primers were as follows: SCAMP3 from the Primer Bank (https://pga.mgh.harvard.edu/primerbank/), ACCGCCCCATGTATAAGGCT (forward), CATGAGCACGGATACTGCTGT (reverse) and GAPDH TGTTCGACAGTCAGCCGC (forward), GGTGTCTGAGCGATGTGGC (reverse).17 The changes in gene expressions were determined with the comparative CT (△△CT) method. The results were expressed as means±S.E.M. Data was analyzed through GraphPad Prism 5.0 software by One-way ANOVA with Turkey’s multiple comparison tests. P<0.05 was considered to be of statistical significance.
Western Blots
Total protein from HCC or Normal tissues was extracted using RIPA buffer (Kaiji, Beijing, China). Lysates were centrifuged and separated on the Lysates were centrifuged and separated on the gel, blotted onto a PVDF membrane (Bio Rad, USA), and analysed using SCAMP3 and GAPDH antibodies (Proteintech, Wuhan, China). Bands were visualized using enhanced chemiluminescence. Written informed consent was provided by the patients, and the study was conducted in accordance with the Declaration of Helsinki and approved by the Air Force Medical Center of PLA Ethics committee.
Results
Increased SCAMP3 Expression in HCC
We initially evaluated SCAMP3 transcription levels in multiple cancers from TCGA. Data from the Oncomine database showed that mRNA expression was significantly higher in breast cancer, leukemia and especially in Liver cancer (Figure 1A and B). To evaluate SCAMP3 gene expression, we analyzed SCAMP3 expression using RNA-seq data from TIMER. As shown in Figure 2A, compared with adjacent normal tissues, SCAMP3 transcription expression was significantly higher in LIHC (liver hepatocellular carcinoma), BLCA (bladder urothelial carcinoma), BRCA (breast invasive carcinoma) and so on. The 4D metric from HCCDB datasets (Figure 1C) revealed that SCAMP3 is not a liver-specific gene (logFC1=−0.38). While it was up-regulated in HCC compared with adjacent samples (logFC2=1.08), with other normal tissues (logFC3=1.19) and other kinds of tumors (logFC4=0.63). Furthermore, the sub-group evaluation of liver hepatocellular carcinoma (LIHC) samples with multiple clinic pathological features showed a high expression of SCAMP3 (UALCAN databases). The expression of SCAMP3 was significantly higher in LIHC than normal in sub-group analyses based on tumor grade and stages, patient’s age, gender, race and weight (Figure 2B–G). Taken together, SCAMP3 can be recognized as a potential diagnostic marker in HCC.
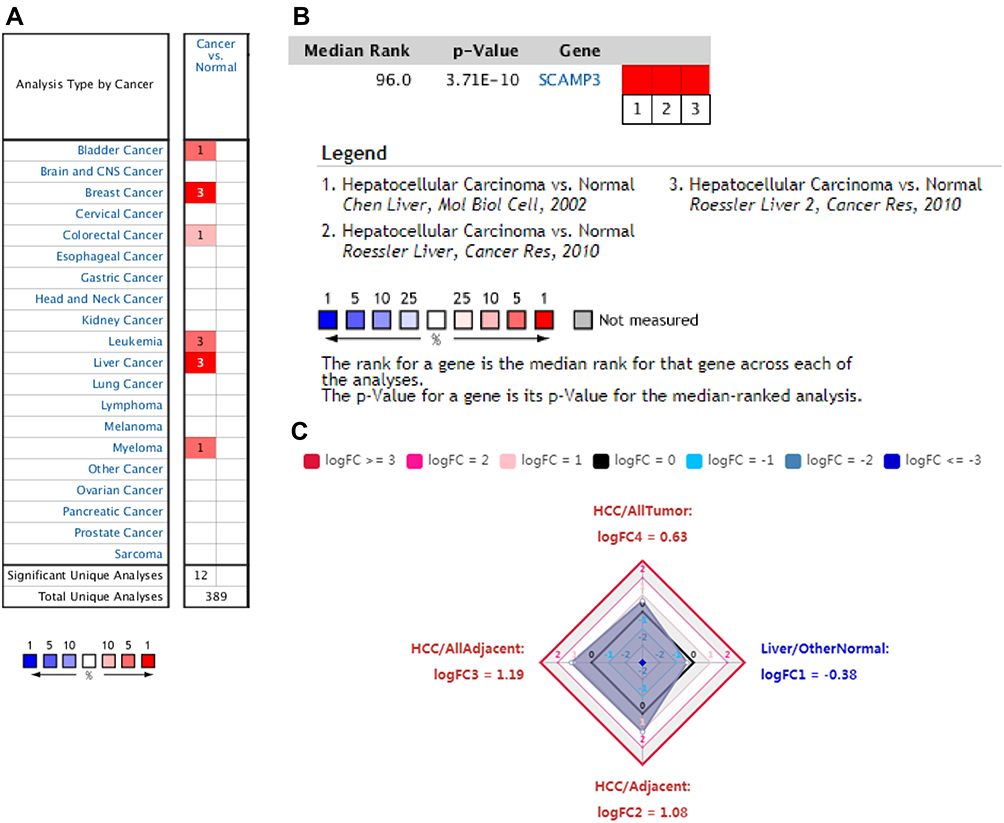 |
Figure 1 SCAMP3 transcription in hepatocellular carcinoma. (A) Expression of SCAMP3 gene in common cancers vs paired normal tissues using the Oncomine database. (B) The mRNA level of SCAMP3 was evaluated in HCC among three studies using ONCOMINE. (C) Expression summary of SCAMP3 in HCCDB database. |
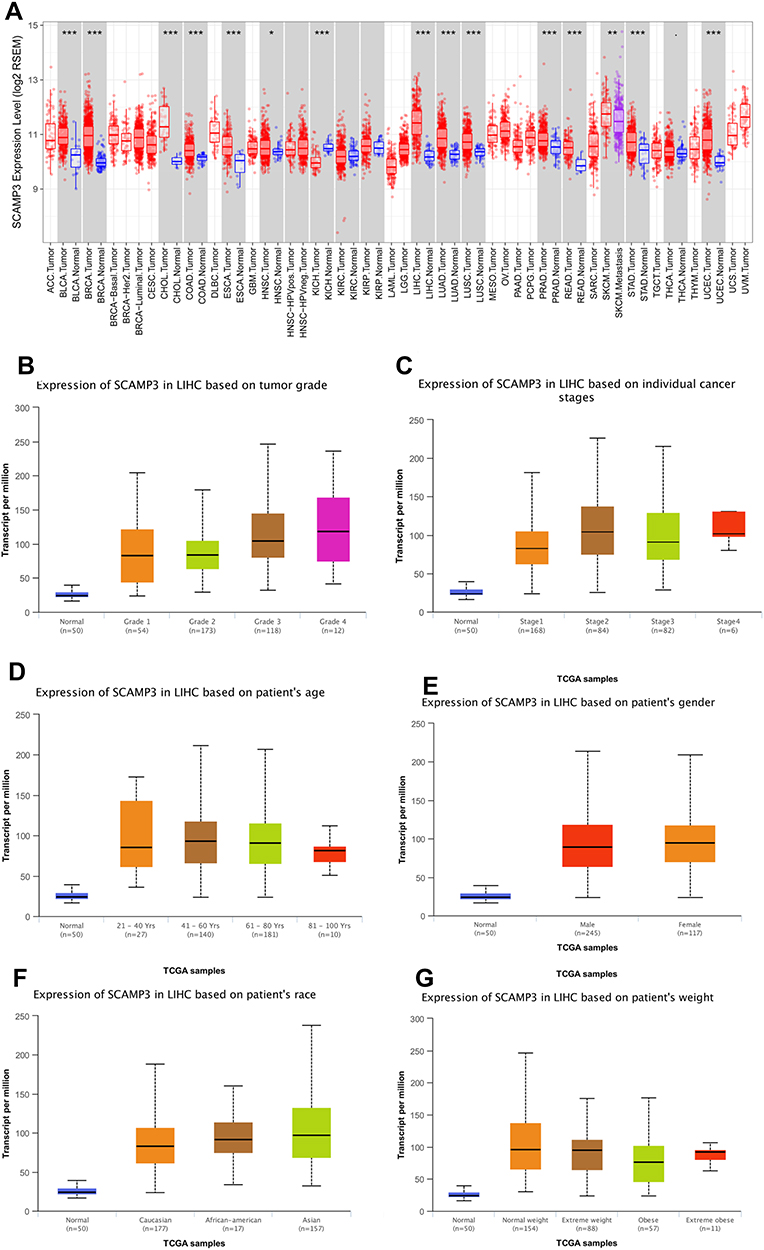 |
Figure 2 SCAMP3 transcription in hepatocellular carcinoma based on TCGA database (A) Expression of SCAMP3 gene in cancers vs paired normal tissues (TIMER). (B) Boxplot showing relative expression of SCAMP3 in normal individuals or LIHC patients with grade 1, 2, 3 or 4 tumors. (C) Boxplot showing relative expression of SCAMP3 in normal individuals or LIHC patients with stage 1, 2, 3 or 4 tumors. (D) Boxplot showing relative expression of SCAMP3 in normal individuals of any age or in LIHC patients aged 21–40, 41–60, 61–80, or 81–100 yr. (E) Boxplot showing relative expression of SCAMP3 in normal individuals of either gender or male or female LIHC patients. (F) Boxplot showing relative expression of SCAMP3 in normal individuals of any ethnicity or in LIHC patients of Caucasian, African-American or Asian ethnicity. (G) Boxplot showing relative expression of SCAMP3 in normal individuals or in LIHC patients in normal weight, extreme weight, obese, or extremely obese. Figure (B–G) were conducted using UALCAN. |
Increased SCAMP3 Expression Correlated with Patient Survival in HCC
Alterations in SCAMP3 mRNA expression were analyzed by the cBioPortal dataset. As shown in Figure 3A, the most frequent alteration type in LIHC was up-regulation of SCAMP3 and the highest alteration frequency got to 7%. In addition, patients with SCAMP3 amplification had significantly lower overall survival compared to that with no alteration (Figure 3C). UALCAN analysis showed that high SCAMP3 expression was associated with poor overall survival (Figure 3B). Therefore, these results suggest that SCAMP3 amplification is positively correlated with patient poor survival in HCC.
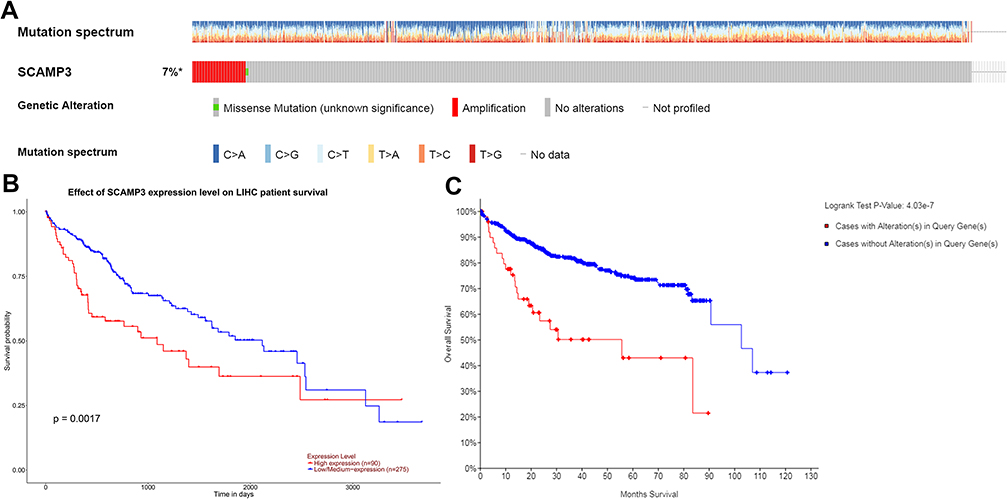 |
Figure 3 Visual summary of SCAMP3 alterations and survival analysis in hepatocellular carcinoma. (A) OncoPrint of SCAMP3 alterations in LIHC (cBioPortal). (B) High expression of SCAMP3 was associated with poor survive (UALCAN). (C) Patients with SCAMP3 alterations were associated with poor survive (cBioPortal). |
Co-Expression Genes of SCAMP3 in HCC
Genes co-expressed with SCAMP3 were analyzed by LInkedOmics, which analyzed mRNA sequencing data of 371 LIHC patients from TCGA project. Figure 4A and B showed the 50 significant genes positively and negatively correlated with SCAMP3. The volcano plot (Figure 4C) revealed all the genes associated with SCAMP3. PPAP2B, SNRK and ARID4A were the top three genes positively correlated with SCAMP3 expression (Figure 5A–C). While PRCC, VPS72 and FLAD1 were the ones negatively correlated with SCAMP3 expression (Figure 5D–F). These genes may play roles in cell adhesion, proliferation, transcription, cell cycle and metabolism. Significantly enriched GO annotations were analyzed by Gene set enrichment analysis (GSEA). The genes differentially expressed associated with SCAMP3 were located mainly in membrane, nucleus, membrane-enclosed lumen, protein-containing complex (Figure 6B), where they participate primarily in protein and ion binding, nucleic acid binding and hydrolase and transferase activity (Figure 6C), suggesting the effects on biological regulation, transcription and metabolic process (Figure 6A). KEGG pathway analysis found enrichment in the Ribosome, Proteasome, Spliceosome, DNA replication and Oxidative phosphorylation pathways (Figure 6D).
 |
Figure 4 Genes differentially expressed in correlation with SCAMP3 in hepatocellular carcinoma (LinkedOmics). (A–B) Heat maps showing genes negatively and positively correlated with SCAMP3 in LIHC (TOP 50). (C) A Pearson test was used to analyze correlations between SCAMP3 and genes differentially expressed in LIHC. |
 |
Figure 5 Verfication of the correlation of SCAMP3 and the top three differentially expressed genes using GEPIA. (A–C) The negative correlation between SCAMP3 and the top three genes (PPAP2B, SNRK, and ARID4A). (D–F) The positive correlation between SCAMP3 and the top three genes (PRCC, VPS72, and FLAD1). |
 |
Figure 6 Significantly enriched GO annotations and KEGG pathways of SCAMP3 in hepatocellular carcinoma (LinkedOmics). The significantly enriched GO annotations and KEGG pathways of SCAMP3 co-expression genes in LIHC were analyzed using GSEA. (A) Cellular components. (B) Biological processes. (C) Molecular functions. (D) KEGG pathway analysis. |
mRNA and Protein Expressions of SCAMP3 Were Increased in HCC
To identify mRNAs that were differentially expressed in HCC patients, the secondary sequencing was used to profile mRNA expression in tissue samples from three HCC patients and three matched normal patients. The heatmap and volcano plot showed the variation of mRNA expression level between HCC patients and normal. As shown in Figure 7A and B, the SCAMP3 expression was significantly up-regulated in HCC patients compared to that in normal. Further, we detected the mRNA and protein expressions of SCAMP3 from HCC tissues by real-time PCR and Western blotting. As shown in Figure 7C and D, SCAMP3 expression increased significantly in HCC tissues, consistent with the analysis of the databases. The experiments were done after the approval from a formally Ethics committee and informed consent by the patients.
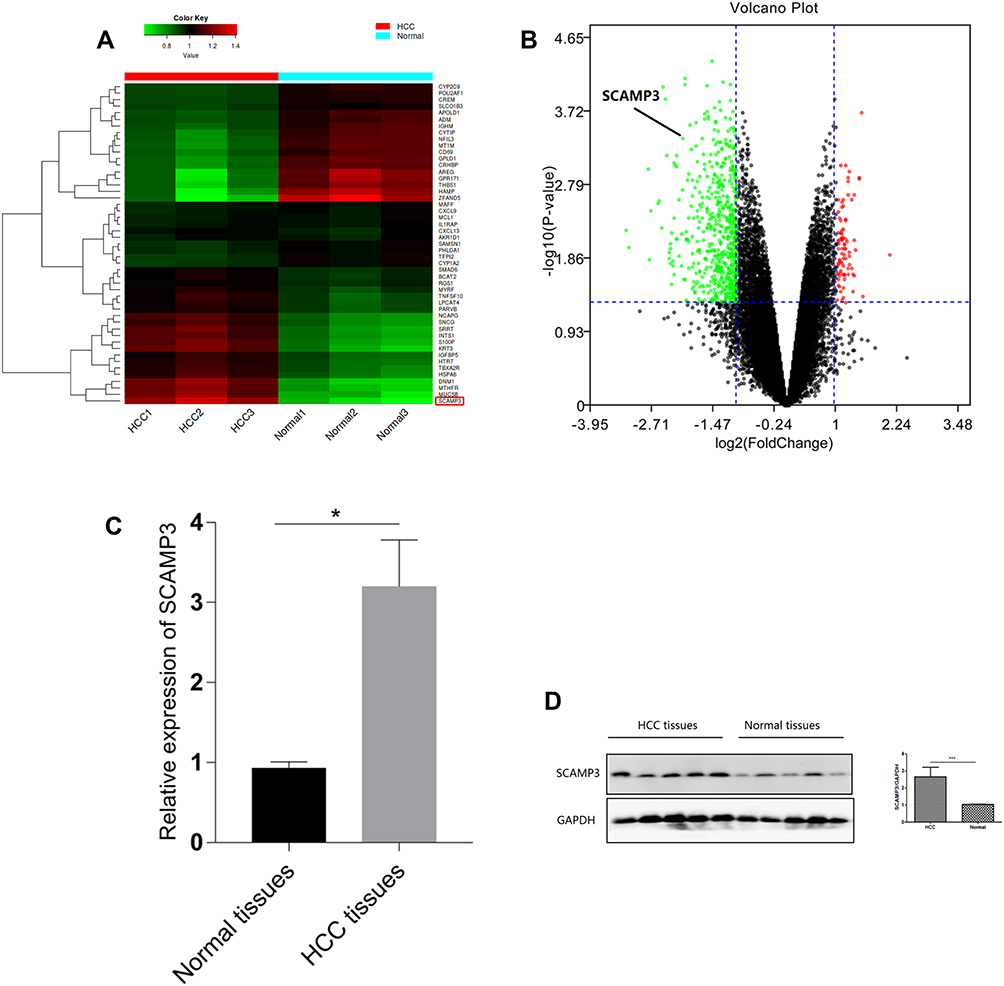 |
Figure 7 Prediction and identification of mRNAs expressed in hepatocellular carcinoma. (A) Expression profiles of the mRNAs are displayed in a heatmap. (B) Differentially expressed mRNAs are displayed in a volcano plot. (C,D) qRT-PCR and Western-blotting verification of SCAMP3 expression in HCC tissues (n=5) and normal tissues (n=5). * p < 0.05; *** p<0.001. |
Discussion
SCAMP3, a member of the Secretory Carrier Membrane Proteins, has been reported to have a high level in HCC tissue samples.18 Differentially regulated endocytosis-associated proteins such as SCAMP3 have been identified in HCC progression. Barbara found that SCAMP3 expression was associated with HCC differentiation grades and mostly SCAMP3 expression was increased in poorly differentiated tumors,8 suggesting that SCAMP3 is probably involved in Epidermal Growth Factor Receptor (EGFR) regulation on the cell membrane.7 To the best of our knowledge, there are few reports about SCAMP3 expression and corresponding mechanism in a tumor at present. And the significance of SCAMP3 expression in the development and prognosis of HCC remains largely unclear. The present studies were conducted based on a lot of databases and mRNA-sequencing profiling. Following analyzing the Oncomine, HCCDB and TIMER databases, SCAMP3 transcriptional level was significantly upregulated in HCC. Furthermore, the transcriptional level of SCAMP3 was significantly higher in HCC patients than healthy people in subgroup analyses based on disease stages, tumor grade, age, gender, race and weight. Subsequently, the frequencies of alterations and survival analysis related to SCAMP3 were analyzed through cBioPortal and UALCAN databases. The present results revealed that the amplification is the only type of mutation and higher SCAMP3 expression correlated with worse survival rates, which indicated that the mRNA level of SCAMP3 may be a valuable biomarker for the prognosis of patients with HCC.
By mining the related functional networks through the KEGG pathway, SCAMP3 participated primarily in the ribosome, proteasome, spliceosome, DNA replication and oxidative phosphorylation. As we have known, SCAMP3 is a secretory carrier membrane protein, which might contribute to sorting events in endosomes. It is reported that SCAMP3 modified ubiquitin, interacted with multivesicular bodies to the regulated endosomal pathway and inhibited degradation of Epidermal Growth Factor Receptor.7 As Ubiquitin-proteasome system-mediated ribosome abundance, which participated in the degradation of protein,19 we speculated that SCAMP3 plays an important role in the ubiquitin-proteasome system.
In addition, our study identified several genes correlated with SCAMP3. Expressions of PPAP2B, SNRK and ARID4A were negatively associated with SCAMP3 transcription. PPAP2B, also known as Phospholipid phosphatase 3, is reported to inhibit endothelial inflammation and promote monolayer integrity by hydrolyzing lysophosphatidic acid (LPA)20, which may be involved in repressing cell adhesion and atherosclerosis.21 SNRK, SNF-related serine/threonine-protein kinase, is an AMPK subfamily member,22 which plays a role in hematopoietic cell differentiation.23 Recently, Zou found SNRK activated NF-kB to induce inflammation.24 Whether SNRK is involved in SCAMP3-mediated HCC process needs to be studied. ARID4A, AT-rich interactive domain-containing protein 4A, modulates the activity of several transcription factors and expression of various genes involved in hematopoiesis.25 Exome sequencing revealed that frequent inactivating mutations in ARID4 existed in microsatellite unstable colorectal cancer.26 Therefore, our analyses suggest that SCAMP3 inhibits PPAP2B expression to induce metastasis, SNRK expression to suppress differentiation and ARID4A expression to modulate several transcriptions. PRCC, VPS72 and FLAD1 were co-upregulated with SCAMP3 in HCC. PRCC, Proline-rich protein PRCC, was a protein of 491 amino acids with a proline-, glycine- and leucine-rich N-terminal region.27 It regulated cell cycle progression through interaction with MAD2B in renal cell carcinomas.28 VPS72 (Vacuolar protein sorting-associated protein 72 homolog) induced the ATP-dependent exchange of histone H2AFZ/H2B dimers for nucleosomal H2A/H2B, resulting in transcriptional regulation of selected genes by chromatin remodeling.29 Thus, SCAMP3 may disturb the normal cell cycle to form tumors through PRCC. And VPS72 may devote to induce transcriptions of some oncogenes. FLAD1 is the FAD synthase that catalyzes the adenylation of flavin mononucleotide (FMN) to form flavin adenine dinucleotide (FAD) coenzyme.30,31 Mutated FLAD1 correlated with the patients with myopathy, scoliosis and cataracts.32 We suspected that FLAD1 may participate in the SCAMP3-mediated metabolic process. As these genes above are new and there are few works of literature, further studies should test this hypothesis.
In conclusion, the present study suggests that SCAMP3 is upper-expressed in HCC compared with normal tissues and is associated with several networks. SCAMP3 can be served as a promising predictive biomarker for the prognosis of HCC. More in-depth experiments and clinical trials are needed to validate the value of SCAMP3 in HCC and other cancers.
Disclosure
The authors have no conflicts of interest to disclose in this work.
References
1. Villanueva A. Hepatocellular carcinoma. N Engl J Med. 2019;380:1450–1462.
2. Chen W. Cancer statistics: updated cancer burden in China. Chin J Cancer Res. 2015;27:1.
3. Louafi S, Boige V, Ducreux M, et al. Gemcitabine plus oxaliplatin (GEMOX) in patients with advanced hepatocellular carcinoma (HCC): results of a Phase II study. Cancer. 2007;109:1384–1390. doi:10.1002/(ISSN)1097-0142
4. Bialecki ES, Di Bisceglie AM. Diagnosis of hepatocellular carcinoma. HPB. 2005;7:26–34. doi:10.1080/13651820410024049
5. Castle A, Castle D. Ubiquitously expressed secretory carrier membrane proteins (SCAMPs) 1-4 mark different pathways and exhibit limited constitutive trafficking to and from the cell surface. J Cell Sci. 2005;118:3769–3780. doi:10.1242/jcs.02503
6. Hubbard C, Singleton D, Rauch M, Jayasinghe S, Cafiso D, Castle D. The secretory carrier membrane protein family: structure and membrane topology. Mol Biol Cell. 2000;11:2933–2947. doi:10.1091/mbc.11.9.2933
7. Aoh QL, Castle AM, Hubbard CH, Katsumata O, Castle JD. SCAMP3 negatively regulates epidermal growth factor receptor degradation and promotes receptor recycling. Mol Biol Cell. 2009;20:1816–1832. doi:10.1091/mbc.e08-09-0894
8. Naboulsi W, Bracht T, Megger DA, et al. Quantitative proteome analysis reveals the correlation between endocytosis-associated proteins and hepatocellular carcinoma dedifferentiation. Biochim Biophys Acta. 2016;1864:1579–1585. doi:10.1016/j.bbapap.2016.08.005
9. Rhodes DR, Kalyana-Sundaram S, Mahavisno V, et al. Oncomine 3.0: genes, pathways, and networks in a collection of 18,000 cancer gene expression profiles. Neoplasia. 2007;9:166–180. doi:10.1593/neo.07112
10. Lin Y, Liang R, Qiu Y, et al. Expression and gene regulation network of RBM8A in hepatocellular carcinoma based on data mining. Aging. 2019;11:423–447. doi:10.18632/aging.101749
11. Lian Q, Wang S, Zhang G, et al. HCCDB: a database of hepatocellular carcinoma expression atlas. Genomics Proteomics Bioinformatics. 2018;16::269–275. doi:10.1016/j.gpb.2018.07.003
12. Pan JH, Zhou H, Cooper L, et al. LAYN is a prognostic biomarker and correlated with immune infiltrates in gastric and colon cancers. Front Immunol. 2019;10:6. doi:10.3389/fimmu.2019.00006
13. Gao J, Aksoy BA, Dogrusoz U, et al. Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci Signal. 2013;6:pl1. doi:10.1126/scisignal.2004088
14. Vasaikar SV, Straub P, Wang J, Zhang B. LinkedOmics: analyzing multi-omics data within and across 32 cancer types. Nucleic Acids Res. 2018;46:D956–D963. doi:10.1093/nar/gkx1090
15. Subramanian A, Tamayo P, Mootha VK, et al. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci U S A. 2005;102:15545–15550. doi:10.1073/pnas.0506580102
16. Huang C, Liu J, Xiong B, Yonemura Y, Yang X. Expression and prognosis analyses of forkhead box A (FOXA) family in human lung cancer. Gene. 2019;685:202–210. doi:10.1016/j.gene.2018.11.022
17. Jin L, Chen J, Li L, Li C, Chen C, Li S. CRH suppressed TGFbeta1-induced epithelial-mesenchymal transition via induction of E-cadherin in breast cancer cells. Cell Signal. 2014;26:757–765. doi:10.1016/j.cellsig.2013.12.017
18. Skawran B, Steinemann D, Weigmann A, et al. Gene expression profiling in hepatocellular carcinoma: upregulation of genes in amplified chromosome regions. Modern Pathol. 2008;21:505–516. doi:10.1038/modpathol.3800998
19. An H, Harper JW. Ribosome abundance control via the ubiquitin-proteasome system and autophagy. J Mol Biol. 2019. doi:10.1016/j.jmb.2019.06.001
20. Krause MD, Huang RT, Wu D, et al. Genetic variant at coronary artery disease and ischemic stroke locus 1p32.2 regulates endothelial responses to hemodynamics. Proc Natl Acad Sci U S A. 2018;115:E11349–E11358. doi:10.1073/pnas.1810568115
21. Aldi S, Matic LP, Hamm G, et al. Integrated human evaluation of the lysophosphatidic acid pathway as a novel therapeutic target in atherosclerosis. Mol Ther Meth Clin Develop. 2018;10:17–28.
22. Jaleel M, McBride A, Lizcano JM, et al. Identification of the sucrose non-fermenting related kinase SNRK, as a novel LKB1 substrate. FEBS Lett. 2005;579:1417–1423. doi:10.1016/j.febslet.2005.01.042
23. Kertesz N, Samson J, Debacker C, Wu H, Labastie MC. Cloning and characterization of human and mouse SNRK sucrose non-fermenting protein (SNF-1)-related kinases. Gene. 2002;294:13–24. doi:10.1016/S0378-1119(02)00829-6
24. Lu Q, Ma Z, Ding Y, et al. Circulating miR-103a-3p contributes to angiotensin II-induced renal inflammation and fibrosis via a SNRK/NF-kappaB/p65 regulatory axis. Nat Commun. 2019;10:2145. doi:10.1038/s41467-019-10116-0
25. Meehan WJ, Samant RS, Hopper JE, et al. Breast cancer metastasis suppressor 1 (BRMS1) forms complexes with retinoblastoma-binding protein 1 (RBP1) and the mSin3 histone deacetylase complex and represses transcription. J Biol Chem. 2004;279:1562–1569. doi:10.1074/jbc.M307969200
26. Cajuso T, Hanninen UA, Kondelin J, et al. Exome sequencing reveals frequent inactivating mutations in ARID1A, ARID1B, ARID2 and ARID4A in microsatellite unstable colorectal cancer. Int J Cancer. 2014;135:611–623. doi:10.1002/ijc.28705
27. Skalsky YM, Ajuh PM, Parker C, Lamond AI, Goodwin G, Cooper CS. PRCC, the commonest TFE3 fusion partner in papillary renal carcinoma is associated with pre-mRNA splicing factors. Oncogene. 2001;20:178–187. doi:10.1038/sj.onc.1204056
28. Weterman MA, van Groningen JJ, Tertoolen L, van Kessel AG. Impairment of MAD2B-PRCC interaction in mitotic checkpoint defective t(X;1)-positive renal cell carcinomas. Proc Natl Acad Sci U S A. 2001;98:13808–13813. doi:10.1073/pnas.241304198
29. Latrick CM, Marek M, Ouararhni K, et al. Molecular basis and specificity of H2A.Z-H2B recognition and deposition by the histone chaperone YL1. Nat Struct Mol Biol. 2016;23:309–316. doi:10.1038/nsmb.3189
30. Brizio C, Galluccio M, Wait R, et al. Over-expression in Escherichia coli and characterization of two recombinant isoforms of human FAD synthetase. Biochem Biophys Res Commun. 2006;344:1008–1016. doi:10.1016/j.bbrc.2006.04.003
31. Olsen RKJ, Konarikova E, Giancaspero TA, et al. Riboflavin-responsive and -non-responsive mutations in FAD synthase cause multiple Acyl-CoA dehydrogenase and combined respiratory-chain deficiency. Am J Hum Genet. 2016;98:1130–1145. doi:10.1016/j.ajhg.2016.04.006
32. Garcia-Villoria J, De Azua B, Tort F, et al. FLAD1, encoding FAD synthase, is mutated in a patient with myopathy, scoliosis and cataracts. Clin Genet. 2018;94:592–593. doi:10.1111/cge.13452
 © 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.