Back to Archived Journals » Advanced Health Care Technologies » Volume 4
Biofunctionalization of biomaterials for nitric oxide delivery: potential applications in regenerative medicine
Authors Tsai KJ, Rammou A, Gao C, de Mel A
Received 25 June 2017
Accepted for publication 10 April 2018
Published 27 August 2018 Volume 2018:4 Pages 25—36
DOI https://doi.org/10.2147/AHCT.S144816
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 4
Editor who approved publication: Dr Gozde Durmus
Kai Jen Tsai,1,* Anna Rammou,1,* Chuanyu Gao,1 Achala de Mel1,2
1UCL Division of Surgery and Interventional Sciences, University College London, London, UK; 2School of Chemistry, University of Nottingham, Nottingham, UK
*These authors contributed equally to this work
Introduction: Mimicking physiological functions of nitric oxide (NO) has applications in regenerative medicine. However, few NO delivery systems have progressed to clinical trials owing to limitations in delivery.
Materials and methods: A novel NO delivery system was explored by integrating S-nitro-N-acetylpenicillamine-functionalized long-chain aliphatic hydrocarbons (LCAHs) into a polyurethane-based polymer.
Results and discussion: Contact angle analysis determined the novel delivery system to be significantly more hydrophobic than control. Chemilluminscence showed a four-phase NO release profile of the delivery system with more stable and prolong NO release than control.
Conclusion: LCAHs can optimize the duration and rate of NO delivery and present a viable option for use in surgical implants and biomedical applications.
Keywords: nitric oxide, biofunctionalization, materials, aliphatic hydrocarbons
Introduction
Biomimetic materials are of interest to regenerative medicine endeavors as they present innovative solutions to repair or restore damaged or diseased organs.1 In particular, there is an interest to develop materials mimicking endogenous nitric oxide (NO), given its vital role in influencing biological systems.2 NO mainly acts via the Nitric oxide-souble guanylate cyclase-cyclic guanosine monophosphate (NO–sGC–cGMP) pathway to contribute to physiological homeostasis.3 NO has potent antithrombogenic effects and stimulates endothelial cell proliferation, mobilization, and adhesion and inhibits smooth muscle cell proliferation and adhesion.4–6,19 Therefore, NO shows great promise for applications in cardiovascular implants that can address the damage or the absence of an endothelium by restoring cardiovascular homeostasis.7 Other functions of NO include neurotransmission, antimicrobial, and anticancer properties (Figure 1).8–11
 | Figure 1 NO can influence a great many physiological systems through a range of bio-physiological mechanisms. Abbreviation: NO, nitric oxide. |
Gaseous NO is short-lived and highly reactive under physiological conditions; thus, a range of NO donor molecules capable of storing and enabling effective localized delivery of NO have been studied. Current strategies seek to optimize the delivery of NO from these donor molecules considering dose, duration, and rate of delivery. Owing to such considerations, few studies have progressed to an in vivo test stage. It is significant to establish NO delivery systems of greater storage stability with a low cost of synthesis and biomimetic levels of NO release over prolonged durations. One group of viable NO donors are the S-nitrosothiols (RSNOS), which are naturally present in the body and therefore infer negligible toxicity in comparison to other NO donors. Additionally, they do not spontaneously decompose but release NO by exposure to heat, UV ions, transition metal catalysis, and ascorbic acid, thus providing opportunity for tissue-specific release.12
In previous attempts to stabilize the release of NO, RSNOS have been blended with polymers, derivatized to fibrin-based hydrogels or covalently bound to nanoparticles.13–16 NO release can be further controlled by incorporating NO donors into polymers, considering the ability of polymers to form hydrophobic cores, hence shielding the NO donor from aqueous environments and prolonging NO release. There is an interest in stabilizing NO with N-diazeniumdiolates (NONOates)17 but RSNOS are preferred due to their greater biological compatibility. It has been previously presented that polyhedral oligomeric silsesquioxane poly(carbonate-urea) (PCU) urethane, a relatively hydrophobic polyurethane-based biocompatible polymer scaffold, proved to be effective for the integration of RSNOS, which were passively incorporated and fabricated into bypass grafts.18,19 This study aimed to exploit the inherent hydrophobic properties of long-chain aliphatic hydrocarbons (LCAHs) for NO delivery. We hypothesized that amine terminated LCAHs, dodecylamine (DD), and/or octadecylamine (OD) would form hydrophobic, spherical structures when conjugated to S-nitro-N-acetylpenicillamine (SNAP), an RSNOS, which has been previously determined to have antithrombogenic properties.20 We predicted that this delivery system would supply a more stable NO release profile within a polyurethane-based polymer poly(carbonate-urea) urethane (PCU)-based polymer system under physiological conditions, when compared with passively incorporated, unmodified SNAP. SNAP with DD conjugated (DD–SNAP) or with OD conjugated (OD–SNAP) was produced with simple amide chemistry and was incorporated within a biocompatible polyurethane-based polymer system and characterized. NO release profiles of the delivery system confirmed a steadier, sustained NO release, providing a novel NO release system with potential applications for surgical implants.
Materials and methods
All materials used in this study were obtained from Sigma-Aldrich Co. (St Louis, MO, USA).
Organic synthesis
DD–SNAP and OD–SNAP were synthesized always maintaining the samples in ice and in the dark to avoid SNAP decomposition. SNAP was dissolved into 1 mL of dimethylformamide (DMF)–methylmorpholine (MMP). The amide bond was formed using a two-step procedure involving activation and aminolysis steps using O-(1H-benzotriazol-1-yl)-N,N,N′,N′-tetramethyluronium hexoflurophosphate (HBTU) as a coupling agent. HBTU was added to the solution in a 1:1 ratio with SNAP, and the solution was left on the roller for 1 h to allow activation of the –OH group of the SNAP molecule (Figure 2A). DD is a white wax-like solid at room temperature that dissolves in DMF while OD is a white powder that only partially dissolves in DMF (resulting in both solid and liquid phases in the reaction chamber). DD or OD was added, respectively, in a 2.5:1 (SNAP:DD/OD) ratio and was allowed to react for 14 h. HBTU was precipitated out of the DD. The samples were centrifuged (at 3°C) and vacuum-dried to get rid of the solvents and stored at 3°C.
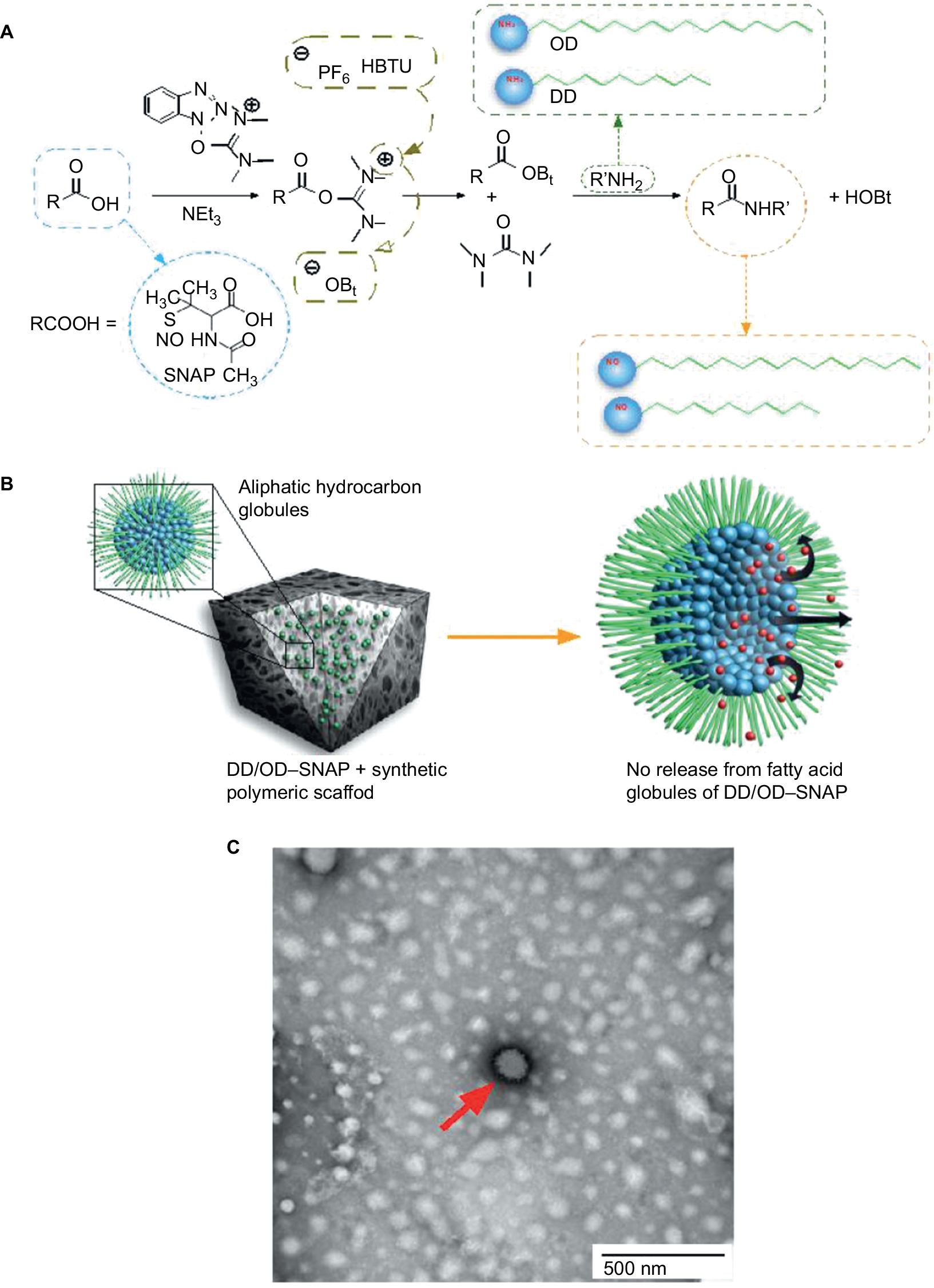 | Figure 2 Overview of globule formation and SNAP conjugation. Notes: (A) Chemical diagram of conjugation of long chain aliphatic hydrocarbons (OD/DD) with SNAP and formation of OD–SNAP and DD–SNAP. (B) Incorporation of DD–SNAP or OD–SNAP with a hydrophobic polymeric scaffold and the proposed distribution of SNAP-conjugated long-chain aliphatic hydrocarbons and the diagrammatic illustration of NO release. (C) Negatively stained TEM imaging of aliphatic hydrocarbon globules; the red arrow indicates a fatty acid globule. Abbreviations: DD, dodecylamine; HBTU, O-(1H-benzotriazol-1-yl)-N,N,N′,N′-tetramethyluronium hexoflurophosphate; HOBt, Hydroxybenzotriazole; NO, nitric oxide; OD, octadecylamine; SNAP, S-nitro-N-acetylpenicillamine. |
Fourier transform infrared spectrometry
Samples were analyzed using a JASCO FT/IR-420 spectrometer. Absorptions due to water and atmospheric CO2 were subtracted using the JASCO software. Spectra were obtained, after taking a background reading for every sample.
Thin layer chromatography (TLC)–column chromatography
TLC was performed to identify the components of the reaction mixture after the organic synthesis. Five microdrops of the DD-SNAP reaction mixture, SNAP, and DD dissolved in diethyl ether were placed on a silica gel glass sheet. An acetone:toluene (2:1) mixture was used as a mobile phase to separate the components by capillary action. The spots of the constituents were then visualized using iodine. The spot that believed to correspond to DD–SNAP was scraped off the plate, resuspended in DMF, and centrifuged to remove the silica, and the washed supernatant was put in the chemiluminescence. In an attempt to separate the components of the mixture, column chromatography was employed. A custom-made column using silica gel and a simple glass tube was manufactured. The column was thoroughly washed with acetone:toluene (2:1) mixture to achieve uniformity inside the column. Using the same conditions employed in the TLC, fractions were collected after calibrating the column with the pure constituents (DD and SNAP dissolved in diethyl ether). The column was loaded with the mixture, and the components were eluted using a stable flow rate of acetone:toluene (2:1).
Liquid chromatography–mass spectrometry (LC–MS)
Mass spectrometry (MS) analysis using electrospray-coupled LC–MS was performed in UCL School of Pharmacy and UCL Department of Chemistry and carried out by Ramesh and Dr Karu Kersti. LC–MS/MS analyses were carried out using an Agilent HP 6400 Series Triple Quadrupole LC/MS System (Agilent Technologies, Santa Clara, CA, USA).
Electrospray MS uses a soft ionization technique to determine the masses of molecular ions (molecular ion mass = 65) present in the solution, and this study allows us to determine the presence of the newly synthesized compounds OD–SNAP and DD–SNAP. Liquid chromatography was coupled with the electrospray technique in order to first separate the components of the samples chromatographically and then ionize them for MS, so that it is easier to separate masses in quite impure samples. Samples dissolved in 400 μL of methanol were analyzed considering the molecular weights of 220.25 for uncoupled SNAP, 185.35 for uncoupled DD, 387.58 for DD–SNAP, 269.51 for uncoupled OD, and 471.74 for OD–SNAP. Ionization and the handling of the samples can lead to possible NO breakdown, and therefore, masses of 357.58 and 441.74 were also searched to account for DD–SNAP and OD–SNAP, respectively, but without the NO attached to the thiol group. In the absence of NO attached to the thiol group, molecular mass is 30 less than the original molecule.
High-performance liquid chromatography (HPLC)
In an effort to purify DD–SNAP from unreacted SNAP and DD in the reaction mixture, HPLC was performed in methanol (1 mL)-dissolved samples using an acetonitrile/water gradient. HPLC pumps the liquid sample through a column with absorbent material that has different affinities for various components of the mixture and therefore creates different flow rates, which enables the separation of the components through elution at different time points. HPLC was performed using an Agilent 1200 Series instrument with UV detection (Agilent Technologies), and chromatography was carried out over Spherisorb ODS2 Prep Column (250 mm ×10 mm, pore size =10 μm; Waters, Milford, MA, USA).
Passive incorporation of –NH3 terminated aliphatic hydrocarbons into PCU and material characterization
Methods of incorporating –NH3 terminated aliphatic hydrocarbons within PCU and sample processing is as described in a previous study.22
Scanning electron microscopy (SEM)
Test samples were inserted into a Q150 Rotary-Pumped Sputter Coater (Quorum, Essex, UK) in order to gold-coat the samples. Images were taken after loading the samples into a Zeiss EVO HD15 Scanning Electron Microscope.
Atomic force microscopy (AFM)
AFM was used for sample surface analysis. The equipment used was a JPK Nanowizard microscope, with the samples imaged using MSNL-10 (Bruker, San Diego, CA, USA) cantilevers in contact mode.
Contact angle
An EasyDrop DSA20E drop-shape analysis system (Kruss) with analysis software (DSA1) was used to measure θ. Contact angles θ>90° and θ<90° are considered to indicate a hydrophobic surface and a hydrophilic surface, respectively. The measurements are then averaged for each sample.
Dielectric analysis
Samples were placed in a Dielectric Analyser (DS6000 Dielectric Thermal Analyser; Lacerta Technology, Leicestershire, UK) with controlled humidity unit. Dielectric measurements were made under conditions of controlled humidity in order to investigate how much water is absorbed on the polymer (water absorbed by the polymer increases its permittivity), ie, its wettability after the modifications. Samples (1 cm2) were placed between two electrodes, and dielectric properties were measured as the humidity was increased from 20% to 80% (at 1%/min). In dielectric analysis, an alternating voltage is placed across the sample (AC field). The alternating strain equivalent of the dynamic mechanical analysis becomes the stored charge (Q) in the sample on the Dielectric Thermal Analyser. The outputs are as follows: e′ (the dielectric constant or permittivity) and e″ the loss factor.
Chemiluminescence
NO release was measured with ozone-induced chemiluminescence. Copper(II) sulfate at 0.1 M (1.5 mL per sample and supplemented with 0.5 mL at 3 h) was used as a catalyst for the release, and the reaction chamber was always covered to eliminate break down by light. The mechanism of detection is based on the fact that NO reacts with ozone (O3) to form nitrogen dioxide (NO2) in the gas phase. The samples were analyzed using a 280i Nitric Oxide Analyzer (NOA™), which consists of an ozone generator from oxygen, a reaction chamber, the optical filter, and a photomultiplier tube (PMT) to detect the red light emission. A vacuum pump is used to maintain the reaction chamber at low pressure (4–7 Torr) to push the gas into the NOA through a frit restrictor to maintain a constant flow rate (200 mL/min). The signal from the PMT is processed, and results are received in a connected computer using the corresponding software.
Results and discussion
DD–SNAP and OD–SNAP were coupled by reacting with HBTU as a coupling agent (Figure 2A). The different degrees of solubility of the LCAH, with the use of more soluble DD in comparison to the relatively less soluble OD, lead to the use of different but complementary methods of characterization of the conjugation of DD or OD with SNAP. OD–SNAP was separated out from the reaction mixture in the presence of diethyl ether with relative ease.
TLC was used to confirm the presence of DD–SNAP. Solubilized DD–SNAP was purified from the reaction mixture using HPLC in a custom-made silica column, and this separation was re-confirmed with TLC.
DD–SNAP formed a solution with diethyl ether, and only HBTU and by-products were separated from the DD–SNAP–HBTU mixture. The amide bond between OD and SNAP was verified using the Fourier transform infrared spectroscopy (Figure S1).
LC–MS with positive electrospray ionization was used to identify the synthesis of both DD–SNAP and OD–SNAP (Table 1). Peaks at 358.5 and 442.7 u were also identified; as these values are ~30 u lower than those for DD–SNAP and OD–SNAP, they were recognized that they represent molecules of DD–SNAP and OD–SNAP from which NO had dissociated. However, this purification process was used solely for characterization purposes, as the product of HPLC would not be suitable for biological application considering the lysis of the S–NO bond of the NO donor by H+ during HPLC.
 | Table 1 Summary of the peaks obtained by LC–MS of SNAP, DD–SNAP, and OD–SNAP Abbreviations: DD, dodecylamine; LC–MS, liquid chromatography–mass spectrometry; NO, nitric oxide; OD, octadecylamine; PCU, poly(carbonate-urea); SNAP, S-nitro-N-acetylpenicillamine. |
DD–SNAP and OD–SNAP were passively incorporated into PCU in the presence of the solvent dimethylacetamide (DMAC) to form DD–SNAP-P or OD–SNAP-P (Figure 2B). The functionalized polymer solution was fabricated as vascular conduits of 5 cm in diameter considering the potential cardiovascular applications of the material, using previously established methods of a solvent exchange coagulating process.18 The integration of LCAH–SNAP within the polymer scaffold and its resulting surface topography were verified by SEM (Figure 3A) and AFM (Figure S2). The particles observed with SEM are clusters due to the hydrophobic environment of the surrounding hydrophobic polymer with relatively larger particle size, but examinations with transmission electron microscopy (TEM) in solution are not clustered and relatively small in size (Figures 2C and S3).
 | Figure 3 Characterization of LCAH–SNAP–PCU. Notes: (A) SEM images indicating porous structures of PCU and DD/OD–SNAP globular structures (pseudo colored green). (B) Mean contact angle measurements (θ) in mean ± SD and images of water drop on the surface of PCU (control) =107.8±1.9, DD–SNAP-P (2%) =109.0±5.1, and OD–SNAP-P (2%) =135.8±14.5. Abbreviations: DD, dodecylamine; LCAH, long-chain aliphatic hydrocarbon; OD, octadecylamine; PCU, poly(carbonate-urea); SEM, scanning electron microscopy; SNAP, S-nitro-N-acetylpenicillamine. |
The degree to which the hydrophobic properties of the LCAHs influence the overall hydrophobicity of the delivery system was confirmed with contact angle analysis (DSA100; Kruss, Hamburg, Germany) (Figure 3B).
On AFM (Figure S2), the control polyurethane had an amorphous and homogenous structure. The topography image showed a high range of height values, ranging from 0 to 780 nm. Some globular structures were visible on the surface in the DD–SNAP image. OD–SNAP particles had a cobblestone-like pattern when viewed in 2 dimension. The corresponding height image showed clearly defined domains of different dimensions but with a height of 450–780 nm. The domains could be as a result of the longer chain length when compared with DD–SNAP. There were clearly defined groves between the domains, which were deeper in the sample, and could correspond to the variation in height seen in the control PCU sample.
SEM indicates the presence of DD and OD in a form of spherical structures within the pores of relatively hydrophobic PCU (Figure 3A), while the surface of the polymer was free of such particles. Formation of a porous structure is well recognized with the solvent exchange coagulating process of PCU.19 Porosity is a significant feature of materials used in surgical implants to confer optimal mechanical properties and to facilitate cell–material interactions.29
Amphiphiles, such as LCAHs, with the amine groups such as those of OD and DD are expected to form micellar geometries with varying degrees of surface area to volume ratios. The range of structures that could form includes spheres, ellipsoids, or globular-bilobed distorted spheres, rods and cylinders, or bilayers. The polymorphisms of hydrated LCAH–SNAP particles within the synthetic polymer would be more precisely determined with recognized methods such as phosphorus nuclear magnetic resonance, X-ray diffraction, or differential scanning calorimetry. Hydrated lipid aggregates are known to entropically adopt a confirmation and become stabilized with hydrophobic force as the major thermodynamic force, with relatively minor influences of forces exerted by van der Waals and hydrogen bonds. However, considering standard lipid thermodynamics, we envisage that the nonpolar long chain lipid group of LCAHs might prefer interactions with porous hydrophobic polymer surface and therefore avert more polar SNAP groups away from the polymer pores, thus forming “reverse micelles” (Figure 2B). However, this theory requires further thorough investigations and techniques such as dynamic light scattering can be used to examine the size and consistency of the micelle and its structure. NO has previously been incorporated into reverse micelle of N-acryloylmorpholine and N-acryloyl2,5-dimethylpierazine to create 50 nm spherical micelles. It was shown that the hydrophobic core shielded NO from release and delayed its release to a half-life of 7 days.25 Using amine functionalized silica nanoparticles, NO was incorporated into the following three different diameters of reverse micelle: 50, 100, and 200 nm. It was shown that the smaller micelle of 50 nm had more significant antimicrobial activity and showed no toxic effect on fibroblasts at therapeutic dose.28
The chain length of the simple alkane chain amphiphiles OD and DD is predicted to influence their hydrophobicity. The effect of incorporating OD and DD into PCU was demonstrated by controlled humidity dielectric analysis, which was used to evaluate the effect of moisture regain on these materials. Figure S4 shows that the conductivity values of the modified materials (OD and DD) at 80% relative humidity (RH) are significantly lower in comparison with the unmodified material indicating more hydrophobic characteristics. The van der Waals radii reflect the proportionality between hydrophobicity and surface area of contact between water and the nonpolar solute. This suggests that larger molecules with longer chain alkanes cause a larger perturbation on the water structure due to a greater contact area. Moreover, biophysical factors, including hydrophobic transfer free energy, predict that greater hydrophobic molecules can aggregate at lower concentrations. Contact angle study showed that incorporation of OD of a concentration as low as 2% significantly increases the water contact angle of PCU to 135.8°, thereby increasing the hydrophobicity of the delivery system. No significant difference was observed with the shorter chain, smaller molecule DD–SNAP-P compared to the control. This result is in agreement with a previous study with graphite sheets.21
As suggested, chemiluminescence was used to determine NO release profiles of OD–SNAP-P and DD–SNAP-P with SNAP–PCU as a control of graft (2 cm length and 5 mm diameter; 120.95 cm2) with similar dimensions as tested in a previous study to determine NO release from SNAP (Figure 4A).22 All these three test forms used a similar starting concentration of 2% SNAP; a concentration that has been shown to be effective in previous study, with a theoretical amount of 4810 nmol of NO.18 The system was introduced with highest levels of catalytic conditions (1 M Cu) to observe the relative differences of NO release from the test samples in a shorter time period. Release profiles, measured by chemiluminescence for 6 h, indicate four distinct phases of NO-release (Figure 4B). A recovery of 31% with SNAP-PCU was obtained.22 However, the DD–SNAP demonstrated a recovery of 49% and OD–SNAP demonstrated a recovery of 54%, which is attributed to its significantly high hydrophobicity and therefore greater retention within the polymer. DD–SNAP-P and OD–SNAP-P showed an initial burst of NO release recorded within the first 3 min after initiation. In contrast, the SNAP–PCU control showed a rapid release for the first 200 min. The second phase of NO release from DD–SNAP-P and OD–SNAP-P is unique and distinct from the control, with a characteristic significantly slower rate of continuous NO release. Phase III occurs after 2.2 h, where the rate of NO release gradually declines for all three groups. However, the SNAP–PCU control enters this reduced release rate at ~115 min: an earlier point in time than DD–SNAP-P and OD–SNAP-P. The final phase, Phase IV, occurs after 4.5 h and is a stable release phase for both control and modified SNAP groups.
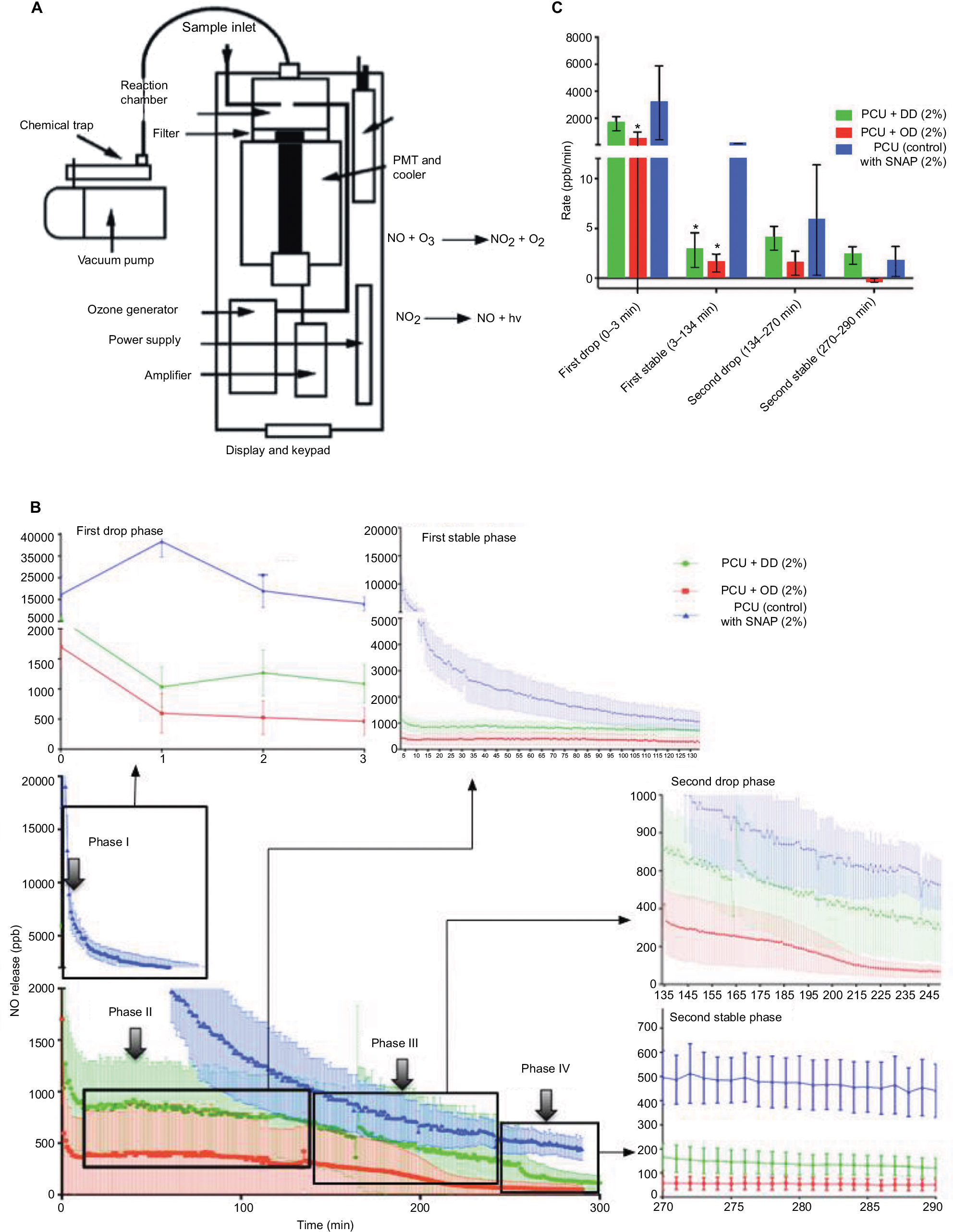 | Figure 4 Details of chemiluminescence studies. Notes: (A) Schematic representation of chemiluminescence set-up for NO release measurement. (B) Four distinct phases of average NO release catalyzed with 1 M copper sulfate over a 5 h period for DD–SNAP, OD–SNAP, and SNAP–PCU (control). Values are shown as mean ± SD. (C) Rates of NO release in parts per billion per minute illustrating relative NO release during the four distinct phases. Values are shown as mean ± SD. Statistical significance is indicated with an *. Abbreviations: DD, dodecylamine; NO, nitric oxide; NO2, nitrogen dioxide; OD, octadecylamine; O3, ozone; PCU, poly(carbonate-urea); PMT, photomultiplier tube; SNAP, S-nitro-N-acetylpenicillamine. |
The average rate of NO in ppb/min per phase was calculated for each group (Figure 4C). During Phase I, we demonstrated a relatively high rate of NO release from all samples within the initial 3 minutes, but OD–SNAP-P was nevertheless significantly lower from average rate of release from the control during the same phase. However, during Phase II for the next 134 minutes, the rates of both OD–SNAP-P and DD–SNAP-P are almost completely stable with a relatively low release rates 1.53 ppb/min and 2.85 ppb/min compared to 91.33 ppb/min for free SNAP–PCU. There is therefore a statistically significant difference between the NO-release of DD–SNAP-P and OD–SNAP-P with the control but not between each of these LCAH-modified SNAP groups. Despite no statistically significant difference between the LCAH-modified SNAP groups and control during Phase III, OD–SNAP-P and DD–SNAP-P continue to release NO at a reduced rate in comparison to SNAP–PCU. During Phase IV, OD–SNAP-P, DD–SNAP-P and SNAP–PCU show a low release rate. An interesting observation was the burst release occurring during Phase I for both DD–SNAP-P and OD–SNAP-P, and a subsequently similar release rate between Phase II and Phase IV. In comparison, SNAP–PCU initiates with a burst release, but the rate does not stabilize until Phase IV, which is ~4 hours after the initiation of release. The relatively slower release of NO achieved by both OD–SNAP-P and DD–SNAP-P in comparison to SNAP–PCU during Phase II confirms that the conjugation of LCAHs to SNAP has conferred better retention of the RSNOS within the polymer. All samples tested after 7 days indicated no significant difference in total volume of NO.
Moreover, in addition to variations in the duration and the rate of release of NO, the successful outcomes of NO bio-functionalization rely on the concentration of release. This is due to the fact that the optimum concentration of NO delivery varies according to the application for which it is warranted. For example, the exploitation of the antithrombogenic properties of NO requires a concentration of NO release between 0.3 and 0.6 fmol/s/cm2.23 This is in contrast with the application of NO in xerogels for antibacterial effect, which requires a concentration of NO release between 1 and 20 pmol/s/cm2.24 Previous research has made the use of a similar concept to the current study, whereby hydrophobic particles are implemented for a prolonged and stable delivery of NO. An example of such a study involves the use of a micellar system based on an amphiphilic copolymer, which has been shown to deliver NO via diazeniumdiolation enabling a stable 16-day release without the use of a catalyst.25 Another example of the modification of NONOates is their conjugation to NO prodrugs rendering greater hydrophobicity. This modified NONOate was incorporated into the core of polymeric nanoparticles protecting the core from the aqueous environment.26 S-nitrosoglutathione has also been conjugated to hydrophobic polymeric nanoparticles, thus showing a release profile for 10 days. However, the release profile from this delivery system is not stable and the quantity of NO drops by half within the first 5 h after the initiation of release.27 In comparison to the present study, these studies have used higher quantities of donors compared with different or no catalysts. An additional advantage of our novel delivery system in comparison to previous micellar and nanoparticle-based delivery systems is its potential for use in implantable devices with the implementation of NO delivery system in the biofunctionalization of biomaterials. Interestingly, the unique “reverse micelle” forming OD/DD–SNAP syntheses may show more promise in structural stability and biocompatibility.
Furthermore, the polymer’s ability to coagulate into sheets and to shape can accommodate for the desired function, such as incorporation into grafts and stents for cardiovascular applications. This can also reduce the rate of re-stenosis in vascular grafts due to its antithrombotic properties, hence negating the need for patients to take life-long antiplatelet therapy.
Various groups have documented the efficacy of NO release from polymeric systems for antimicrobial properties in vitro and in vivo to prevent potential nosocomial infections by pathogens such as Staphylococcus aureus.10 The formation of biofilm in implanted grafts has been an issue in biomaterials, where patients have to be treated in a sterile environment, ie, theater. Hence, the synthesis of a hydrophobic material coupled with NO release would therefore have a range of applications including antibacterial properties for wound healing and for use in the respiratory and cardiovascular system from air-borne and blood-borne infections. The novel NO delivery system defined in the current study, comprising amine LCAH chains conjugated to an NO-releasing donor, could be potentially modified to deliver a range of bioactive functional groups.
Conclusion
We have successfully functionalized RSNOS with LCAH in order to increase its hydrophobicity. We have confirmed the functionalization and passive integration of this RSNOS–LCAH conjugate within a biocompatible polyurethane polymer. We have investigated the physicochemical properties of this biofunctionalized polymer and determined NO release profile in vitro, verifying that DD–SNAP and OD–SNAP can elute NO at a slower rate in comparison to alternative biomaterial delivery systems. OD–SNAP is more favorable compared with DD–SNAP as OD–SNAP can be relatively easily separated out of the reaction mixture in addition to the abovementioned slowest rate of NO release. We believe that this novel NO delivery system shows great promise for use in surgical implants and for other regenerative medicine applications.
Acknowledgments
The authors gratefully acknowledge BioHORIEN Biochemical Technology Co-Ltd for their sponsorship to support KJT. AdM is a co-investigator of Engineering and Physical Sciences Research Council (EPSRC) project EP/L020904/1. The authors would like to thank Arnold Darbyshire for synthesizing and providing PCU, which was used as a base material for this study, Dr Bala Ramesh for expert advice on conjugation chemistry, Dr Odlyha for electrical conductivity evaluation of materials, Dr Adam Strange of Dr Bozec’s group for AFM facilities, evaluation, and AFM analysis of the material. Mr Jafree was involved in proof reading of the article and assisting with formatting of an earlier version of the article.
Author contributions
All authors contributed toward data analysis, drafting and revising the paper and agree to be accountable for all aspects of the work.
Disclosure
The authors report no conflicts of interest in this work.
References
Tsai KJ, Dixon S, Hale LR, Darbyshire A, Martin D, de Mel A. Biomimetic heterogenous elastic tissue development. NPJ Regen Med. 2017;2:16. | ||
Wang T, van der Vlies AJ, Uyama H, Hasegawa U. Nitric oxide-releasing polymeric furoxan conjugates. Polym Chem. 2015;6:7737–7748. | ||
Garthwaite J. New insight into the functioning of nitric oxide-receptive guanylyl cyclase: physiological and pharmacological implications. Mol Cell Biochem. 2010;334(1–2):221–232. | ||
Moore C, Tymvios C, Emerson M. Functional regulation of vascular and platelet activity during thrombosis by nitric oxide and endothelial nitric oxide synthase. Thromb Haemost. 2010;104(2):342–349. | ||
Ozuyaman B, Ebner P, Niesler U, et al. Nitric oxide differentially regulates proliferation and mobilization of endothelial progenitor cells but not of hematopoietic stem cells. Thromb Haemost. 2005;94(4):770–772. | ||
Parent M, Boudier A, Fries I, et al. Nitric oxide-eluting scaffolds and their interaction with smooth muscle cells in vitro. J Biomed Mater Res A. 2015;103(10):3303–3311. | ||
de Mel A, Murad F, Seifalian AM. Nitric oxide: a guardian for vascular grafts? Chem Rev. 2011;111(9):5742–5767. | ||
Calabrese V, Mancuso C, Calvani M, Rizzarelli E, Butterfield DA, Stella AM. Nitric oxide in the central nervous system: neuroprotection versus neurotoxicity. Nat Rev Neurosci. 2007;8(10):766–775. | ||
Brisbois EJ, Davis RP, Jones AM, et al. Reduction in thrombosis and bacterial adhesion with 7 day implantation of S-nitroso-N-acetylpenicillamine (SNAP)-doped elast-eon E2As catheters in sheep. J Mater Chem B. 2015;3(8):1639–1645. | ||
Engelsman AF, Krom BP, Busscher HJ, van Dam GM, Ploeg RJ, van der Mei HC. Antimicrobial effects of an NO-releasing poly(ethylene vinylacetate) coating on soft-tissue implants in vitro and in a murine model. Acta Biomater. 2009;5(6):1905–1910. | ||
Choudhari SK, Chaudhary M, Bagde S, Gadbail AR, Joshi V. Nitric oxide and cancer: a review. World J Surg Oncol. 2013;11:118. | ||
Broniowska KA, Diers AR, Hogg N. S-nitrosoglutathione. Biochim Biophys Acta. 2013;1830(5):3173–3181. | ||
Joslin JM, Lantvit SM, Reynolds MM. Nitric oxide releasing Tygon materials: studies in donor leaching and localized nitric oxide release at a polymer-buffer interface. ACS Appl Mater Interfaces. 2013;5(19):9285–9294. | ||
Van Wagner M, Rhadigan J, Lancina M, et al. S-nitroso-N-acetylpenicillamine (SNAP) derivatization of peptide primary amines to create inducible nitric oxide donor biomaterials. ACS Appl Mater Interfaces. 2013;5(7):8430–8439. | ||
Koh A, Carpenter AW, Slomberg DL, Schoenfisch MH. Nitric oxide-releasing silica nanoparticle-doped polyurethane electrospun fibers. ACS Appl Mater Interfaces. 2013;5(16):7956–7964. | ||
Hwa Yu S, Hu J, Ercole F, et al. Transformation of RAFT polymer end groups into nitric oxide donor moieties: en route to biochemically active nanostructures. ACS Macro Lett. 2015;4(11):1278–1282. | ||
Suchyta DJ, Schoenfisch MH. Encapsulation of N-diazeniumdiolates within liposomes for enhanced nitric oxide donor stability and delivery. Mol Pharm. 2015;12(10):3569–3574. | ||
de Mel A, Naghavi N, Cousins BG, et al. Nitric oxide-eluting nanocomposite for cardiovascular implants. J Mater Sci Mater Med. 2014;25(3):917–929. | ||
Everett W, Scurr DJ, Rammou A, Darbyshire A, Hamilton G, de Mel A. A material conferring hemocompatibility. Sci Rep. 2016;6:26848. | ||
Stasko NA, Fischer TH, Schoenfisch MH. S-nitrosothiol-modified dendrimers as nitric oxide delivery vehicles. Biomacromolecules. 2008;9:834–841. | ||
Liu H, Ding Y, Ao Z, Zhou Y, Wang S, Jiang L. Fabricating surfaces with tunable wettability and adhesion by ionic liquids in a wide range. Small. 2015;11(15):1782–1786. | ||
Naghavi N, Seifalian AM, Hamilton G, de Mel A. Evaluation of experimental methods for nitric oxide release from cardiovascular implants; bypass grafts as an exemplar. Ther Adv Cardiovasc Dis. 2015;9(6):375–388. | ||
Kobsar A, Simonis S, Klinker E, et al. Specific inhibitory effects of the NO donor MAHMA/NONOate on human platelets. Eur J Pharmacol. 2014;735:169–176. | ||
Riccio DA, Dobmeier KP, Hetrick EM, Privett BJ, Paul HS, Schoenfisch HM. Nitric oxide-releasing S-nitrosothiol-modified xerogels. Biomaterials. 2009;30(27):4494–4502. | ||
Jo YS, Van Der Vlies A, Gantz J, et al. Micelles for delivery of nitric oxide. J Am Chem Soc. 2009;131(40):14413–14418. | ||
Kumar V, Hong SY, Maciag AE, et al. Stabilization of the nitric oxide (NO) prodrugs and anticancer leads, PABA/NO and double JS-K, through incorporation into PEG-protected nanoparticles. Mol Pharm. 2010;7(1):291–298. | ||
Duong HTT, Kamarudin ZM, Erlich RB, et al. Intracellular nitric oxide delivery from stable NO-polymeric nanoparticle carriers. Chem Commun (Camb). 2013;49(39):4190–4192. | ||
Carpenter AW, Slomberg DL, Rao KS, Schoenfisch MH. Influence of scaffold size on bactericidal activity of nitric oxide releasing silica nanoparticles. ACS Nano. 2011;5(9):7235–7244. | ||
Chaves C, Gao C, Hunckler J, et al. Dual-acting biofunctionalised scaffolds for applications in regenerative medicine. J Mater Sci Mater Med. 2017;28(2):32. |
Supplementary materials
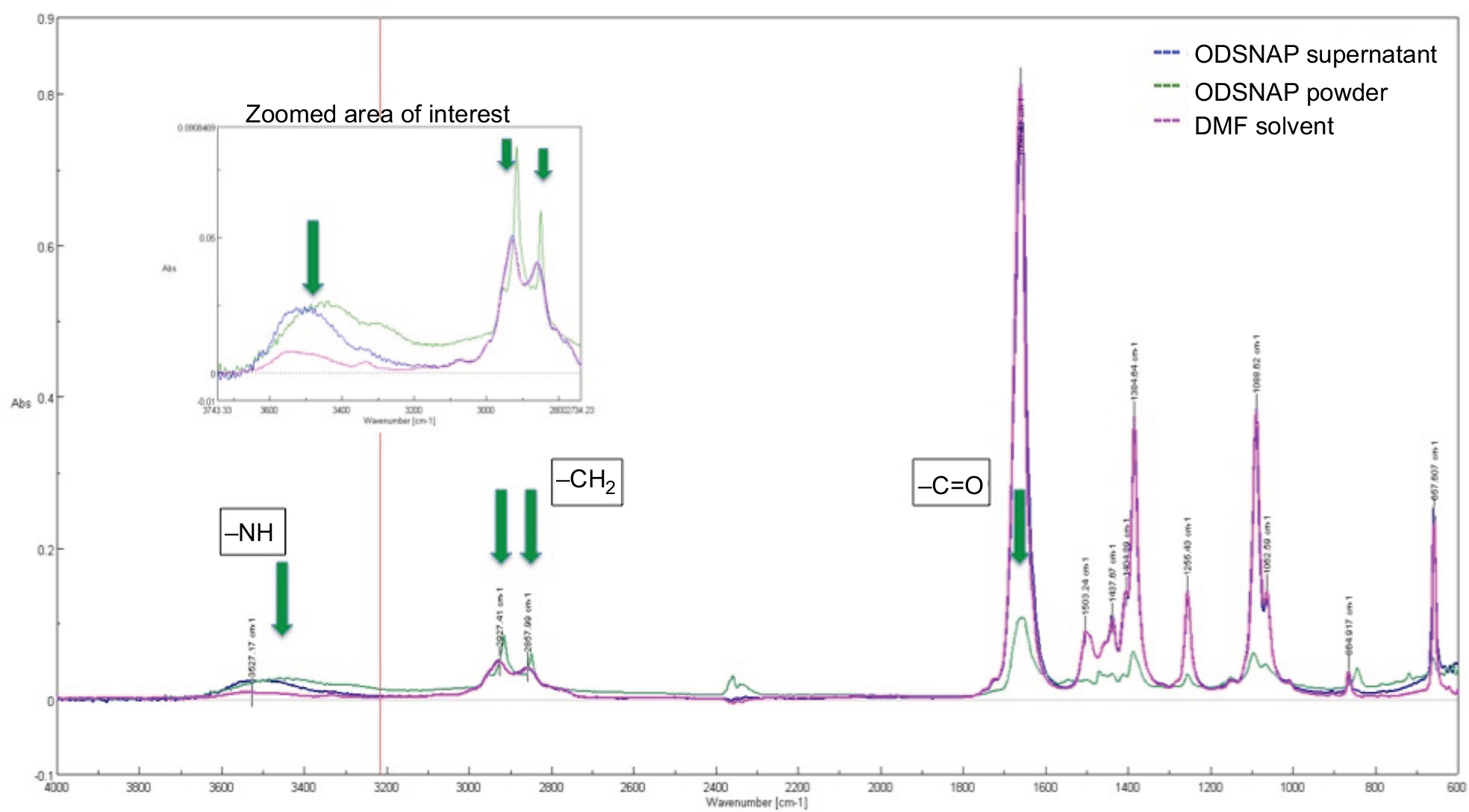 | Figure S1 FTIR spectra of DMF, OD–SNAP, and OD–SNAP. Notes: Arrows indicate the peaks of interest. FTIR was used to characterize the amide bond formation in both the DD and OD mixtures. In the DD–SNAP sample, DMF that has amide bonds has masked the amide bond formation of the DD–SNAP despite vacuum drying the traces of DMF. In the case of OD–SNAP, the same masking effect is can be seen. As OD is insoluble, it was easier to separate out. Peaks at 2850 and 2927 that correspond to the alkane groups (-CH2) of the long hydrocarbon chain. A peak at 1690 corresponds to the C=O of the amide bond. There is a relatively small but very broad peak ~3450, which could be attributed to the N–H of the amide bond. Abbreviations: DD, dodecylamine; DMF, dimethylformamide; FTIR, Fourier transform infrared; OD, octadecylamine; SNAP, S-nitro-N-acetylpenicillamine. |
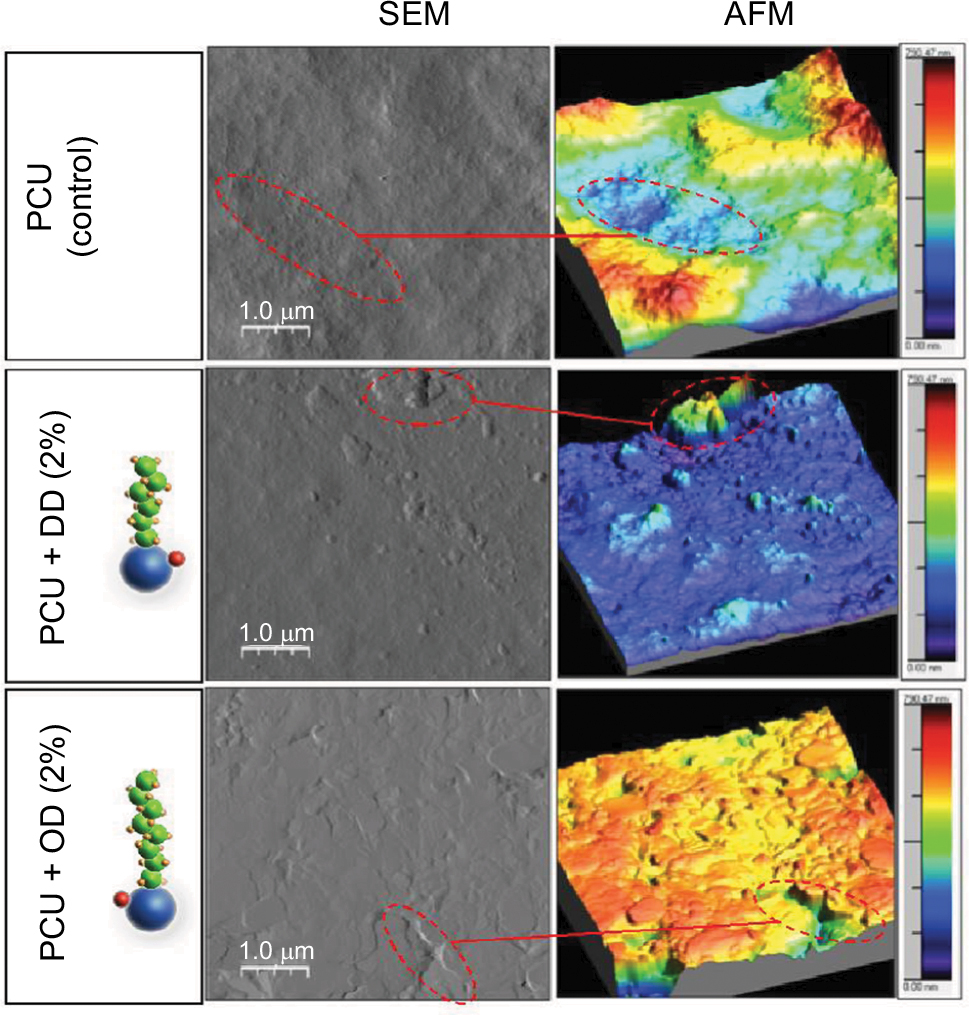 | Figure S2 Characterization of LCAH–SNAP–PCU: AFM images showing changes in topography after the addition of OD/DD–SNAP. Abbreviations: AFM, atomic force microscopy; DD, dodecylamine; LCAH, long-chain aliphatic hydrocarbon; OD, octadecylamine; PCU, poly(carbonate-urea); SEM, scanning electron microscopy; SNAP, S-nitro-N-acetylpenicillamine. |
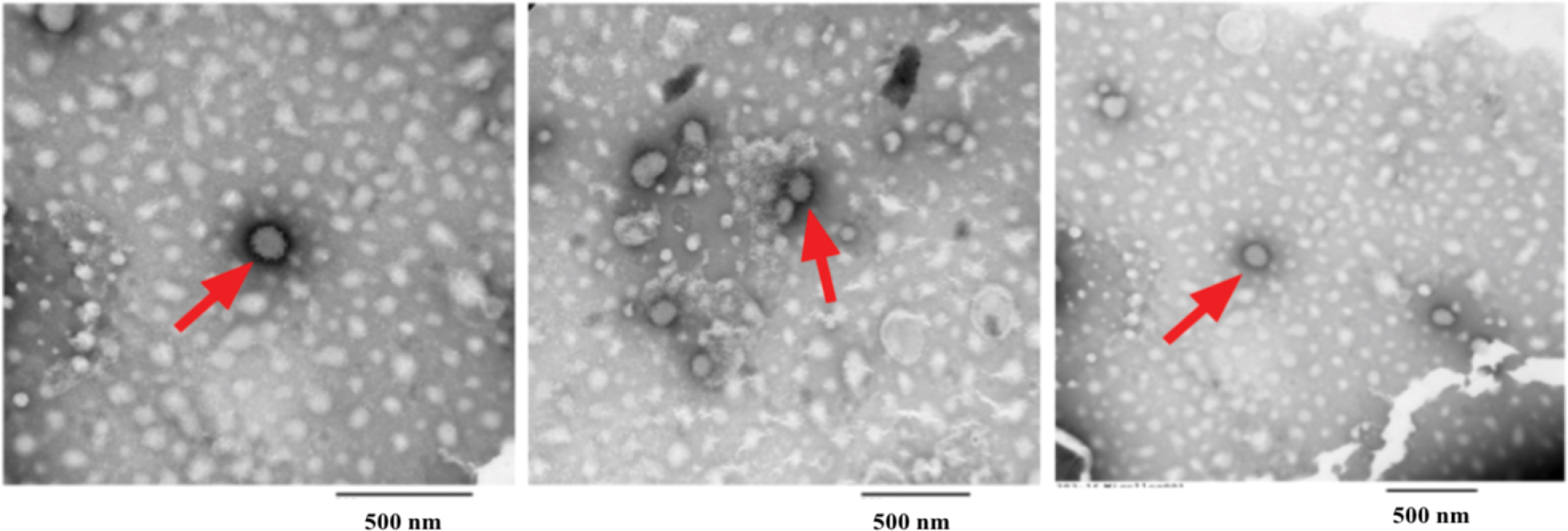 | Figure S3 Negatively stained TEM imaging of aliphatic hydrocarbon globules in solvent (1% uranyl acetate). Red arrows indicate the presence of hydrocarbon globules. Abbrevaition: TEM, transmission electron microscope. |
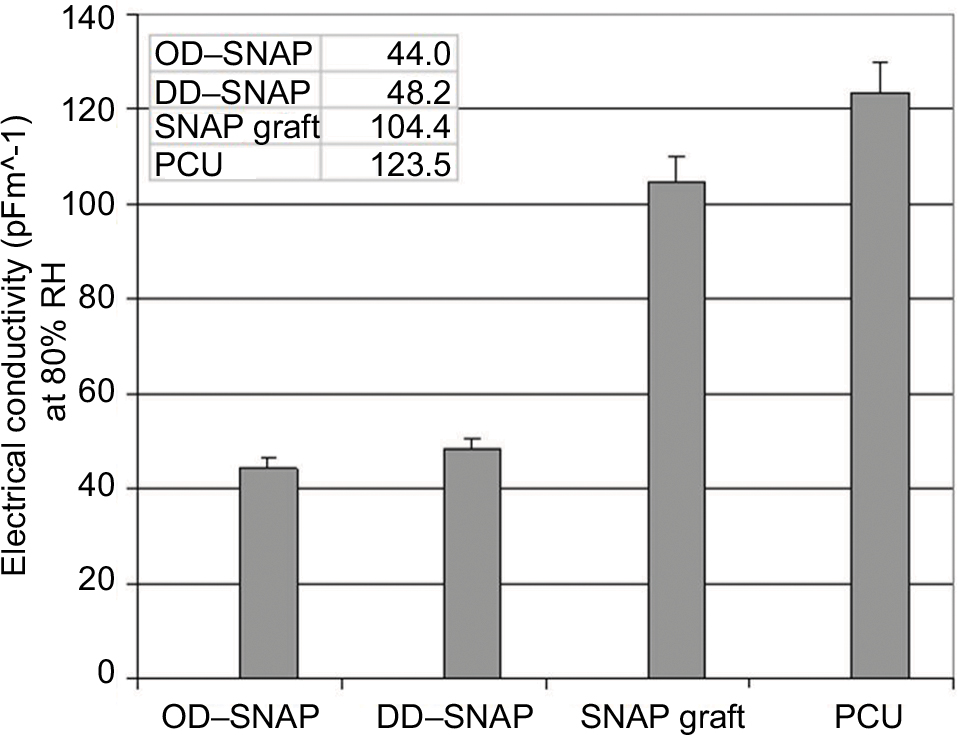 | Figure S4 Values for electrical conductivity (pFm^-1) at 80% RH measured after subjecting polymers to increase in RH at 1%/min from 20 to 80% RH and then leaving polymer at 80% RH for 30 min. Notes: Image presents a comparison of dielectric values for polyurethanes with modifications with DD/OD–SNAP and native SNAP compared to polyurethane graft in its natural state. Lowest value for OD–SNAP–PCU indicates most hydrophobic material. Abbreviations: DD, dodecylamine; OD, octadecylamine; PCU, poly(carbonate-urea); SNAP, S-nitro-N-acetylpenicillamine. |
 © 2018 The Author(s). This work is published by Dove Medical Press Limited, and licensed under a
Creative Commons Attribution License.
The full terms of the License are available at http://creativecommons.org/licenses/by/4.0/.
The license permits unrestricted use, distribution, and reproduction in any medium, provided the
original author and source are credited.
© 2018 The Author(s). This work is published by Dove Medical Press Limited, and licensed under a
Creative Commons Attribution License.
The full terms of the License are available at http://creativecommons.org/licenses/by/4.0/.
The license permits unrestricted use, distribution, and reproduction in any medium, provided the
original author and source are credited.