Back to Journals » Drug Design, Development and Therapy » Volume 17
Bioequivalence Study of Vortioxetine Hydrobromide Tablets in Healthy Chinese Subjects Under Fasting and Fed Conditions
Authors Bai W ![]() , Song H, Hu Y, Zhang X, Wang X, Guo C, Qiu B, Dong Z
, Song H, Hu Y, Zhang X, Wang X, Guo C, Qiu B, Dong Z
Received 4 July 2023
Accepted for publication 23 September 2023
Published 29 September 2023 Volume 2023:17 Pages 3035—3046
DOI https://doi.org/10.2147/DDDT.S428771
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Tuo Deng
Wanjun Bai,1 Haojing Song,1 Yiting Hu,1 Xueyuan Zhang,2 Xiaoru Wang,3 Caihui Guo,1 Bo Qiu,1 Zhanjun Dong1
1Department of Pharmacy, Hebei General Hospital, Shijiazhuang, Hebei, People’s Republic of China; 2Shanghai Innovstone Therapeutics Limited, Shanghai, People’s Republic of China; 3CSPC Zhongqi Pharmaceutical Technology (Shijiazhuang) Co., Ltd, Shijiazhuang, People’s Republic of China
Correspondence: Zhanjun Dong, Department of Pharmacy, Hebei General Hospital, No. 348 Heping West Road, Xinhua District, Shijiazhuang City, Hebei Province, 050051, People’s Republic of China, Tel +86 311 85988604, Email [email protected]
Objective: This study compared the pharmacokinetic and safety profiles of generic and original vortioxetine hydrobromide tablets under fasting and fed conditions, and evaluated the bioequivalence of two vortioxetine formulations to obtain sufficient evidence for abbreviated new drug application.
Methods: A randomized, open-label, two-formulation, single-dose, two-period crossover bioequivalence study was conducted under fasting and fed conditions (n = 32 per study). Eligible healthy Chinese subjects received a single 10-mg dose of the test or reference vortioxetine hydrobromide tablet, followed by a 28-day washout interval between periods. Serial blood samples were collected up to 72 h after administration in each period, and the plasma concentrations of vortioxetine were detected using a validated method. The primary pharmacokinetic (PK) parameters were calculated using the non-compartmental method. The geometric mean ratios for the PK parameters of the test drug to the reference drug and the corresponding 90% confidence intervals were acquired for bioequivalence analysis. A safety evaluation was performed throughout the study.
Results: Under fasting and fed conditions, the PK parameters of the test drug were similar to those of the reference drug. The 90% confidence intervals (CIs) of the geometric mean ratios of the test to reference formulations were 96.44– 105.81% for peak concentration (Cmax), 97.94– 105.05% for the area under the curve truncated at 72 hours (AUC0-72 h) under fasting conditions, 93.92– 104.15% for Cmax, and 96.67– 102.55% for AUC0-72 h under fed conditions, all of which were within the accepted bioequivalence range of 80.00– 125.00%. Both the test and reference formulations were well-tolerated, and no serious adverse events related to the study drug were reported during the study.
Conclusion: The PK bioequivalence of the test and reference vortioxetine hydrobromide tablets in healthy Chinese subjects was established under fasting and fed conditions, which met the predetermined regulatory criteria. Both formulations were safe and well tolerated.
Keywords: vortioxetine, bioequivalence, pharmacokinetics, safety
Introduction
Major depressive disorder (MDD) is considered one of the most frequent psychiatric disorders and is characterized by depressed mood, decreased interest, cognitive dysfunction, and vegetative symptoms.1,2 MDD is a highly prevalent disease that affects approximately 6% of the adult population worldwide each year,1 and women have a two-fold increased risk of developing MDD compared with men.3 Approximately 20% of all individuals are estimated to fulfill the criteria for MDD at some point during their lifetime.1 MDD negatively impacts the quality of life and is considered the highest contributor to disease burden and years lived with disability.4 In addition to suicide, MDD has been linked to an increased risk of cardiovascular disease, diabetes, stroke, Alzheimer’s disease, and cancer.1,5,6 Currently, available antidepressants are widely used as effective treatments for MDD today. Classical tricyclic antidepressants (TCA) are regarded as reference compounds for the treatment.7 However, TCAs’ high adverse effect burden limits their widespread use.1,2 Serotonin-selective reuptake inhibitors (SSRIs) are generally better tolerated than TCAs and are generally comparable in antidepressant efficacy.2
Vortioxetine, an SSRI with several other potentially relevant effects on serotoninergic receptors, has been approved as a novel antidepressant for MDD.8,9 Many previous studies have shown that vortioxetine exerts multimodal antidepressant effects by inhibiting the 5-HT transporter and 5-HT1D, 5-HT3, and 5-HT7 receptors as well as agonists of 5-HT1A receptors and partial agonists of 5-HT1B receptors.8,10 Preclinical and clinical data indicated that vortioxetine showed convincing clinical efficacy, enhanced cognitive function,11 decreased the risk of recurrence, and good safety and tolerability in patients with MDD,12 which differentiated vortioxetine from currently used SSRI and serotonin noradrenaline reuptake inhibitor (SNRI) antidepressants.10 Therefore, Vortioxetine received its first approval for MDD by the US Food and Drug Administration (FDA) in September 2013 and regulatory approval for its use in this indication by the European Medicines Agency (EMA).8
The PK profile of vortioxetine hydrobromide tablet (Brintellix®), originally developed by H. Lundbeck A/S, exhibits an absolute bioavailability of 75%, time to reach Cmax (Tmax) of 7 ~ 11 h, terminal elimination half-life (T1/2) of 57 ~ 66 h, and no food effect.9,13 Vortioxetine is extensively metabolized to inactive metabolites Lu AA34443,14 primarily through oxidation by cytochrome P450 2D6 (CYP2D6) in the liver.13 However, considering the variations in vortioxetine across different races and ethnicities, it is important to evaluate the clinical PKs and pharmacodynamics of vortioxetine in diverse populations. The clinical PKs, safety, and tolerability of vortioxetine have been investigated in the United States, Europe, and Japanese populations.12,14,15 However, data from PK studies of the Chinese population are limited. Recently, a generic vortioxetine hydrobromide tablet was developed by the CSPC Ouyi Pharmaceutical Co., Ltd. (Hebei, China). Consistent with the National Medical Products Administration (NMPA) guidelines, this study compared the PK parameters of the new test, vortioxetine hydrobromide, with those of the reference product (Brintellix®). To support the marketing approval of the newly developed generic formulation in China, a bioequivalence study was performed in healthy Chinese subjects under fasting and fed conditions.
Materials and Methods
Ethics Statement of Human Rights
The study was registered at the Drug Clinical Trial Registration and Information Publicity Platform [http://www.chinadrugtrials.org.cn/index.html] (number: CTR20201062, date: June 9, 2020) and was retrospectively registered in the Chinese Clinical Trial Registry [https://www.chictr.org.cn/] (number: ChiCTR2300073164, date: July 3, 2023). The study protocol and informed consent forms (ICFs) were reviewed and approved by the independent ethics committee of Hebei General Hospital [Ethics Number: (2020) (05-01)]. This study was conducted in compliance with the Guidelines for Good Clinical Practice recommended by NMPA, the International Conference on Harmonization Good Clinical Practice Guidelines, and the ethical principles of the Declaration of Helsinki. The participants were free to withdraw from the study at any time.
Study Drugs
The test formulation was a vortioxetine hydrobromide tablet provided by CSPC Ouyi Pharmaceutical Co., Ltd., Hebei, China (10 mg/tablet, batch number: Q61200301; expiry date: March 2, 2024). The reference formulation was Brintellix® from Lundbeck A/S, Valby, Denmark (10 mg/tablet, batch number 2611271; expiry date: December 2022). Each subject took a single oral dose of a 10 mg vortioxetine hydrobromide tablet as a test or reference formulation during each treatment period.
Subjects
All participants signed ICFs after a full understanding of the study’s objectives, content, procedures, and possible risks. Male and female subjects aged over 18 years old, with weights higher than 50 and 45 kg, respectively, and with body mass index (BMI) between 19.0 and 28.0 kg/m2, were enrolled in the screening period. All subjects were judged to be healthy by physicians based on their medical history, vital signs, clinical examinations, clinical laboratory tests (including routine analyses of the blood, coagulation function, blood glucose, liver function, renal function, routine urinalysis, immunological examination for hepatitis B surface antigen, hepatitis C antibody, syphilis antibody, and human immunodeficiency virus antibody), 12-lead electrocardiography, and chest radiography. Subjects were excluded if they had any history or evidence of the following: clinically relevant acute or chronic diseases; drug, nicotine, or alcohol abuse; allergic constitution, especially allergy to any ingredient in vortioxetine hydrobromide tablet; history of donation or acute loss of blood (more than 400 mL) for 3 months; or those taking any medications or supplements within 30 days before the first dosing. Female subjects who were pregnant or lactating during the study period, or planned pregnancy one month before dosing to six months after the end of the study were excluded.
Study Design
This study comprised two independent clinical trials (fasting and fed study), each of which was a randomized, open-label, two-formulation, single-dose, two-sequence, two-period crossover bioequivalence study performed at the Phase Ι Clinical Research Center of Hebei General Hospital, Shijiazhuang, Hebei, China. All subjects in each trial were randomly assigned to either the T-R or R-T group (T was the test product and R was the reference product) at a 1:1 ratio according to a random number table generated by SAS statistical software (v9. 4) Group T-R subjects who received the test product in the first treatment period received the reference product in the second treatment period, whereas Group R-T had the opposite administration sequence. A 28-day washout period was used between the two treatment periods. Under fasting conditions, each subject received a single dose of the test or reference tablet, administered orally with 240 mL warm water after at least a 10-hour overnight fast, whereas the subjects under fed conditions received a standard high-fat (800–1000 kcal) breakfast (approximately 150 kcal of protein, 250 kcal of carbohydrate, and 500–600 kcal of fat) 30 min before each drug administration and followed the same scheme. Water intake was prohibited for 1 h before and after administration. Standardized lunches and dinners were provided at 4 and 10 hours after drug administration. Safety assessments were conducted during the study period.
According to the clinical pharmacology and biopharmaceutical reviews of vortioxetine by FDA,16 the individual variation in vortioxetine was < 30%. Considering racial differences, the coefficient of intra-individual variation of Cmax and AUC0-72 h for vortioxetine in the fasting and fed groups was set at 22.5%. Assuming a one-sided test with α = 0.05, and power of 0.8 (β = 0.2), the geometric mean ratios of the test and reference formulations were expected to be 0.95~1.05, and 90% CI of 80.00%~125.00% for bioequivalence, 24 samples were required for the fasting and fed study. After considering the dropout rate, 32 participants were enrolled for each study.
Blood Sampling and Determination of Concentration
Based on the low intra-subject variability and long half-life,8 a 72-h truncation of the AUC for vortioxetine was used for bioequivalence assessment according to EMA guidelines.17 Previous clinical trials have indicated that vortioxetine has a Tmax of 7–11 hours and an average terminal half-life of approximately 66 hours.8 Slightly modified with reference to the blood collection point design of previous studies,14 serial blood samples were collected within 1 hour before drug administration (baseline) and at 1.0, 2.0, 3.0, 4.0, 5.0, 6.0, 7.0, 8.0, 9.0, 10.0, 11.0, 12.0, 14.0, 16.0, 20.0, 24.0, 36.0, 48.0, 60.0, and 72.0 h after dosing in the study under both fasting and fed conditions. The Blood samples were centrifuged at 1700 × g for 10 min at 4 °C, and plasma was obtained from the supernatants and stored at −60 °C until use. Plasma samples were analyzed in a specialized analytical laboratory (Suzhou Haike Pharmaceutical Technology Co. Ltd, Suzhou, China), where the plasma concentrations of vortioxetine were detected using a validated high-performance liquid chromatography-tandem mass spectrometry method according to the Chinese NMPA and FDA guidelines.
Pharmacokinetic and Statistical Analysis
Based on the plasma concentration data, the PK parameters of vortioxetine were calculated using a non-compartmental model using Phoenix WinNonlin software (Pharsight Corporation, Mountain View, CA, USA; version 8.2). Cmax and Tmax were determined directly from the observed plasma concentration-time profiles. AUC0-72 h was calculated using the trapezoidal method. The elimination rate constant (λz) was determined by log-linear regression of the plasma concentration over time in the terminal phase. T1/2 was calculated to be 0.693/λz.
After the transformation of Cmax and AUC0-72 h to their natural logarithmic values, analysis of variance (ANOVA) was performed using a linear mixed model to evaluate the effects of formulation, trial period, dosing sequence, and subjects. To assess the bioequivalence between the test and reference formulations, 90% CIs of the geometric least-squares mean ratios of the test and reference formulations were calculated. The acceptance criteria for bioequivalence were that 90% of CIs were completely within the range of 80.00–125.00% for Cmax and AUC0-72 h. The Wilcoxon signed-rank test was used to analyze the Tmax between the two formulations.
Safety Assessment
Safety assessments included adverse events (AEs), severe adverse events (SAEs), combined medications, laboratory tests, vital sign measurements, physical examinations, and 12-lead electrocardiography (ECG). Vital signs (blood pressure, heart rate, and body temperature) were monitored pre-dose (within 1 h), as well as at 2.0, 12.0, 24.0, 48.0, and 72.0 h after each drug administration in each treatment period. Laboratory tests, physical examinations, and 12-lead ECG were performed for each participant at screening and during the follow-up period. All AEs were monitored throughout the trial by the research doctors and spontaneously reported by the subjects. The severity of AEs was graded according to the Common Terminology Criteria for Adverse Events (CTCAE) v5.0 from the National Cancer Institute.
Results
Study Population
This study was conducted between June 18, 2020, and October 16, 2020. As shown in Figure 1, a total of 271 potential Chinese adults were screened. Of these, 64 healthy subjects met the eligibility criteria for the protocol and were selected for the fasting and fed studies (n = 32 per study). Table 1 summarizes the demographic characteristics of all subjects. The age, sex, weight, height, BMI, and race of the subjects were similar between the two parts of the study. Under fasting conditions, one subject discontinued the study because of elevated HCG levels before admission to the ward during the second period. One participant withdrew for personal reasons before period 2. The remaining 30 subjects completed both periods. In the fed condition, two subjects withdrew for personal reasons before period 2, one subject discontinued because of a fracture at work outside during the washout period, and one subject dropped out because of vomiting within 2 h after dosing in the second period. Ultimately, 28 subjects completed both periods.
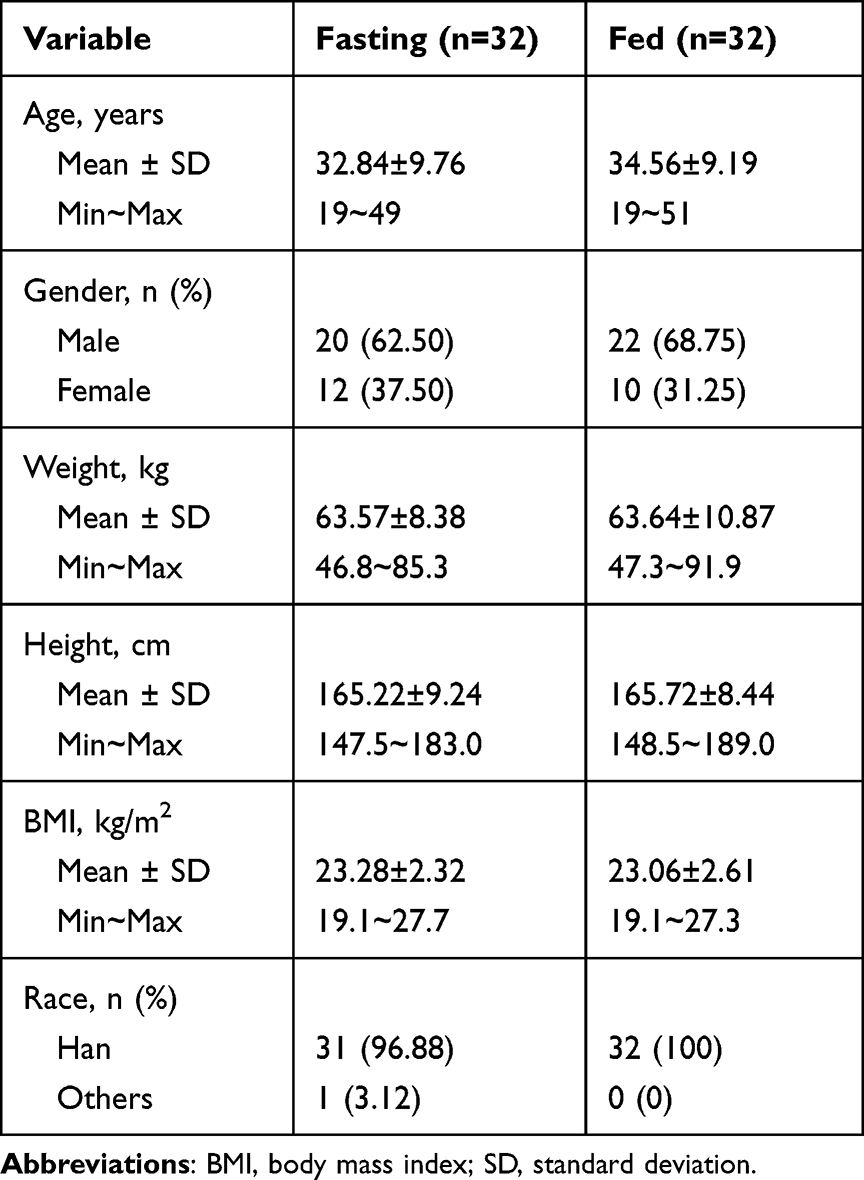 |
Table 1 Demographic Characteristics of the Healthy Subjects Under Fasting and Fed Condition |
 |
Figure 1 Study design and disposition of subjects. |
Pharmacokinetics and Bioequivalence
All PK parameters were analyzed based on the pharmacokinetics concentration set (PKCS) and pharmacokinetics parameter set (PKPS). Under fasting conditions, two subjects in the R-T group withdrew in period 2, which were excluded from the PKCS and PKPS. Four subjects in the T-R group in period 2 were also excluded from the PKPS because the drug plasma concentration before dosing was not BQL and greater than Cmax×5%. Therefore, 30 and 28 subjects were included in the PK analyses of the test and reference products, respectively. For the fed condition, 2 subjects in the T-R group and 2 subjects in the R-T group in period 2 were excluded from the PKPS. Likewise, the pre-dose plasma concentrations of the two subjects in the R-T group in period 2 were greater than Cmax×5% and were excluded from the PKPS. Therefore, 28 subjects for the test product and 30 subjects for the reference product were included in the PK analyses. The major PK parameters of vortioxetine under fasting and fed conditions were calculated using a non-compartmental model, and are summarized in Table 2. The mean Tmax of vortioxetine was about 6.99 to 7.49 h, which indicated that vortioxetine was slowly absorbed. The mean T1/2 was approximately 79.28 to 96.84 h, which showed that vortioxetine was eliminated slowly, in accordance with the characteristics of drugs with a long half-life. As shown in Figures 2 and 3, the mean plasma concentration-time curves of the test and reference products under fasting and fed conditions were consistent. These results suggest that the in vivo disposal process of the test product was similar to that of the reference product.
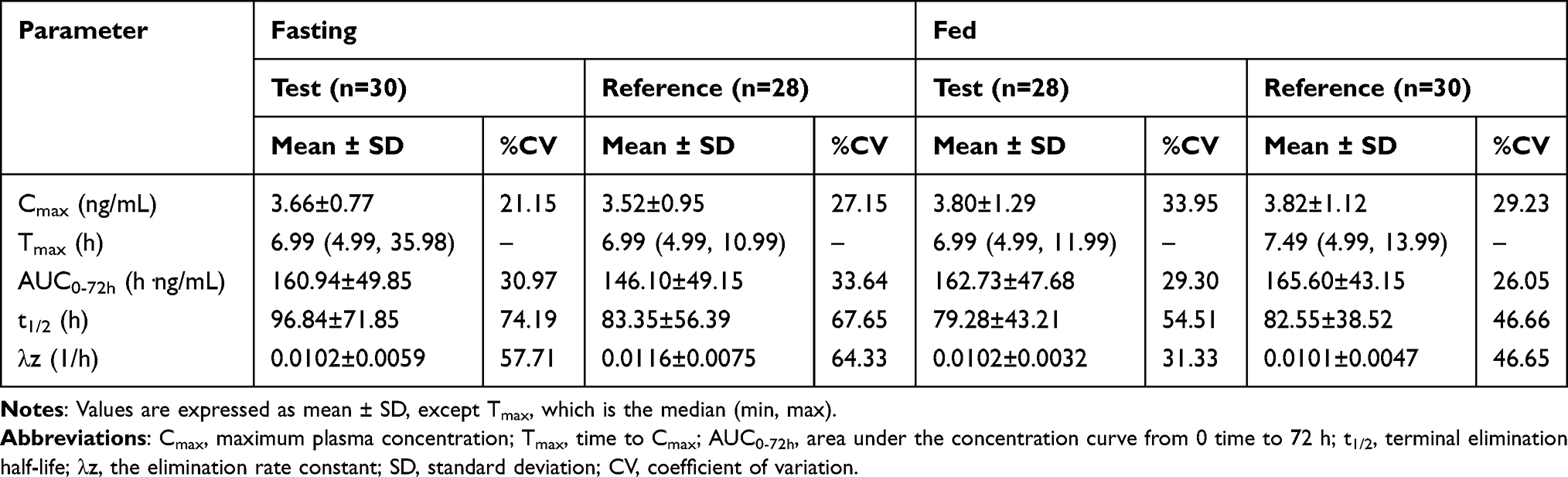 |
Table 2 Pharmacokinetic Parameters of Vortioxetine After Single Oral Administration of 10 Mg of the Test and Reference Vortioxetine Hydrobromide Tablets in Healthy Subjects Under Fasting and Fed Condition |
 |
Figure 2 Mean plasma concentration-time profiles of vortioxetine after oral administration of 10 mg of the test (n=30) and reference (n=28) vortioxetine hydrobromide tablets in healthy subjects under fasting conditions. Data represent the mean value, and error bars represent the SD. |
 |
Figure 3 Mean plasma concentration-time profiles of vortioxetine after oral administration of 10 mg of the test (n=28) and reference (n=30) vortioxetine hydrobromide tablets in healthy subjects under fed condition. Data represent the mean value, and error bars represent the SD. |
For PK bioequivalence evaluation, the subjects were enrolled in the bioequivalence analysis set (BES) as with PKPS under fasting and fed conditions. The results of the fasting test are shown in Table 3. No formulation, period, or sequence effects were found for Cmax and AUC0-72 h by ANOVA. A comparison of the primary PK parameters of vortioxetine showed that the geometric least-squares mean (GLSM) ratios of the test to reference products for Cmax and AUC0-72 h were 101.02% and 101.43%, respectively (Table 3). The corresponding 90% CIs were 96.44% to 105.81% for Cmax and 97.94% to 105.05% for AUC0-72 h (Table 3), which were within the accepted bioequivalence range of 80.00% to 125.00%, indicating that the test and reference products of vortioxetine were considered bioequivalent under fast conditions. There was a significant difference in Tmax between the two products according to the Wilcoxon signed-rank test (p = 0.0368), which did not affect the bioequivalence decision. Under fed conditions, the comparison of primary PK parameters between the test and reference products showed that the GLSM ratios for Cmax and AUC0-72 h were 98.90% and 99.56% (Table 3), and the corresponding 90% CIs were 93.92–104.15% and 96.67–102.55% (Table 3), respectively. Both 90% CIs were within the predefined equivalence margin of 80.00% to 125.00%, which showed that the test and reference products of vortioxetine were also considered bioequivalent under the fed conditions.
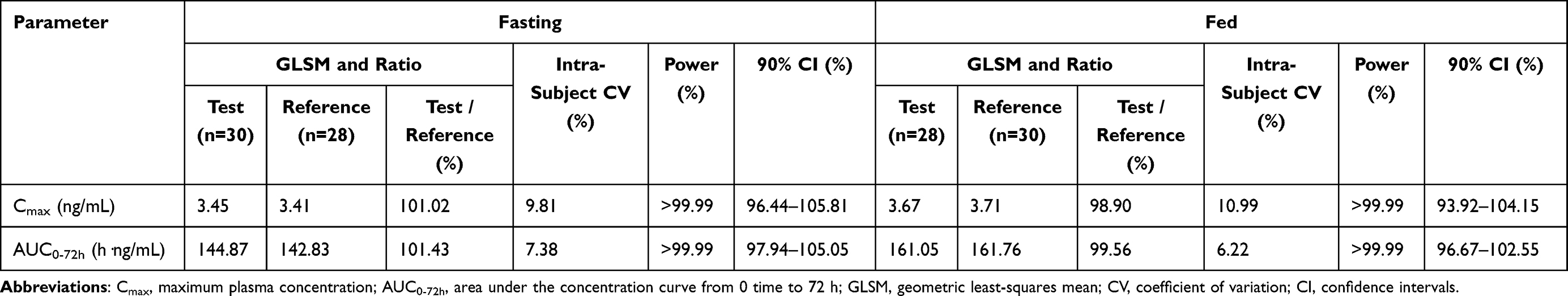 |
Table 3 Bioequivalence Assessment of the Primary Pharmacokinetic Parameters of Vortioxetine After Single Oral Administration of 10 Mg of the Test and Reference Vortioxetine Hydrobromide Tablets in Healthy Subjects Under Fasting and Fed Condition |
Safety Assessment
The safety and tolerability of vortioxetine were assessed based on the safety analysis set (SS), in which all subjects received at least one dose of the study drug. 32 subjects were included in SS in the fasting study, 15 AEs for 7 subjects were reported, and the incidence of AEs was 21.88% (7/32) (Table 4). Of these, seven AEs were reported for two subjects in the test product; the incidence of AEs was 6.67% (2/30) (Table 4), whereas eight AEs were reported for five subjects and an incidence of 15.63% (5/32) in the reference product (Table 4). All AEs were reported as grade 1 and spontaneously resolved without any specific treatment; one subject underwent an induced abortion due to pregnant endometrial cavity fluid, ovarian cyst, and cervical canal cyst and withdrew before admission to the ward in period 2 of the R-T group. No adverse drug reactions (ADR) or SAEs occurred during the fasting period. Similarly, 32 subjects were included in the SS group in the fed study, 32 AEs were reported in 15 subjects, and the incidence of AEs was 46.88% (15/32) (Table 4). The incidence rates of AEs in the test and reference products were 32.26% (10/31) and 20.00% (6/30), respectively (Table 4). All AEs were mild and reported as grade 1 or 2, except for 4 cases of AE reported as grade 3, which was judged to be unrelated to the study drug because one subject working outside underwent the lumbar vertebral fracture, surface injury, and pelvic fracture during the washout period. Considering that the subject with fractures was hospitalized, although certainly unrelated to the study drug, 4 SAEs and an incidence of 3.23% (1/31) were reported in the test product. Six AEs in four subjects were defined as ADRs, and the incidence of ADRs was 12.50% (4/32) under fed conditions. All ADRs, such as nausea, vomiting, and abdominal discomfort, were mild, judged as grade 1, and spontaneously resolved without any specific treatment. The results indicated that both the test and reference products of vortioxetine were safe and well-tolerated in healthy subjects under fasting and fed conditions.
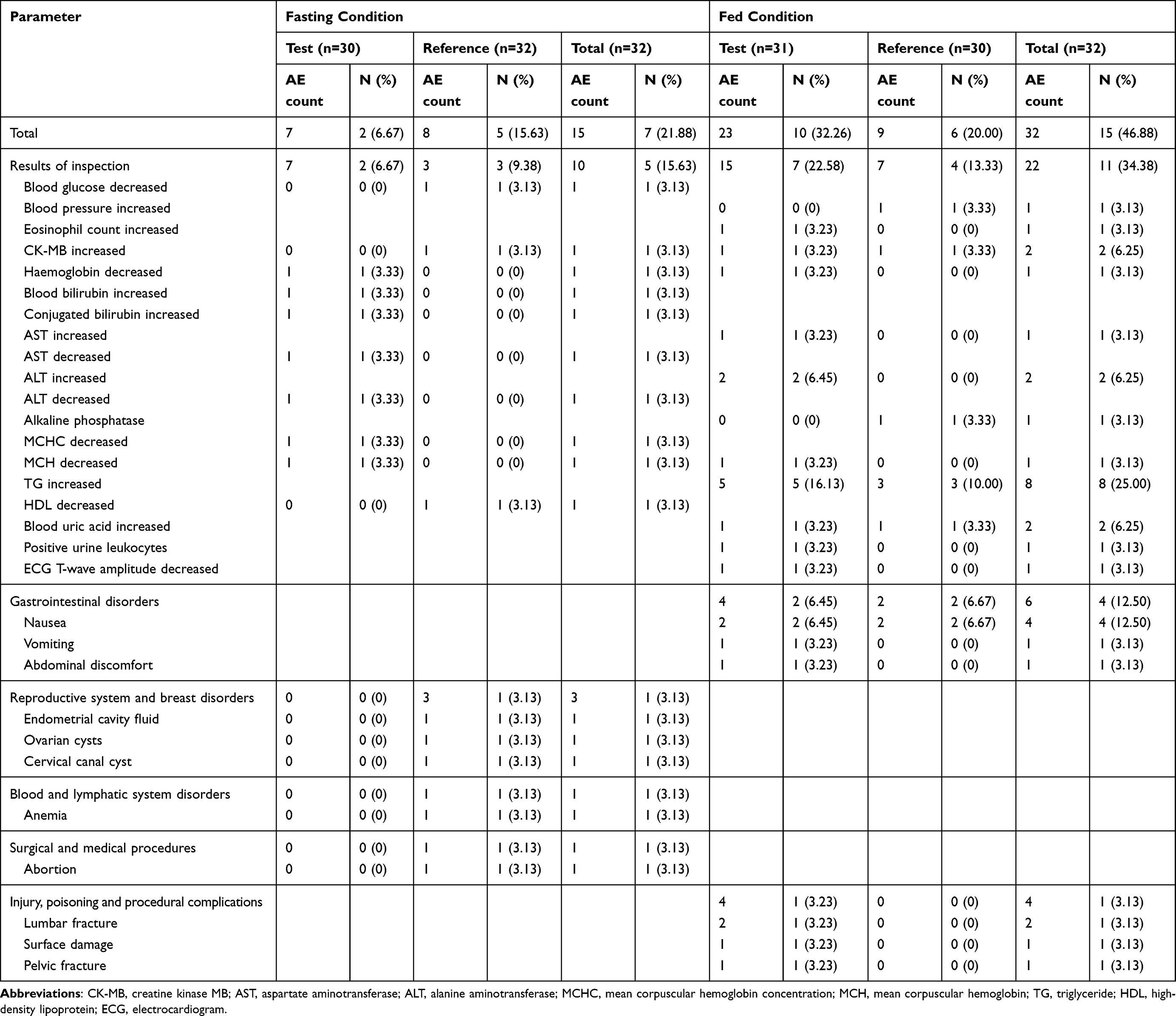 |
Table 4 Summary of AEs in Healthy Subjects Under Fasting and Fed Condition |
Discussion
In the present study, we provided evidence for the bioequivalence of test and reference vortioxetine hydrobromide tablets in healthy Chinese subjects under fasting and fed conditions that met the acceptance criteria and were considered bioequivalent. Both formulations were well tolerated and no SAEs related to the study drug were reported.
According to the bioequivalence guidelines of FDA18 and EMA,19 the truncated AUC0-72 h can be used instead of AUC0-t or AUC0-inf in the bioequivalence evaluation of drugs with a long elimination half-life (> 24 h) and exhibiting low intra-subject variability. Vortioxetine has a longer half-life, with a T1/2 of 66 hours.9 The intra-subject variability, which reflects the residual variability apart from sequence, period, formulation, and inter-subject variations,20 was 14.28% for Cmax and 10.81% for AUC0-inf in the FDA clinical pharmacology review of vortioxetine.16 Consistent with FDA data, the intra-subject coefficients of variation for Cmax and AUC0-72 h under the fasting condition were 9.81% and 7.38% (Table 3), respectively, and 10.99% and 6.22% (Table 3) under the fed condition in our study. These results demonstrated the low intra-subject variability of vortioxetine, Cmax, and AUC0-72 h were selected as the primary parameters for assessing bioequivalence, and the present results showed the similarity of the PK properties between a generic formulation and the original drug in fasting and fed conditions. Furthermore, our results also suggest that it would be reasonable and advantageous to limit the PK sample collection period to 72 h in bioequivalence and bioavailability studies for vortioxetine, consistent with EMA recommendations.17
Comparing the PK parameters of vortioxetine with those of previous studies,9,14,15 the observed Cmax and Tmax values for vortioxetine in the Chinese population were similar to those previously reported in Caucasian or Japanese population,14,15 which validated our results. However, T1/2 was somewhat longer in our study than in the Caucasian or Japanese population reported,14,15 suggesting that there are differences in the metabolic processes of vortioxetine in different populations. Vortioxetine is primarily metabolized through CYP2D621 and poor metabolizers have a longer T1/2 than extensive metabolizers because of genetic polymorphisms in CYP2D6. Therefore, the racial differences in CYP2D6 among Caucasian, Japanese, and Chinese populations may be a possible reason. In addition, Tmax under fasting conditions differed between the generic and original products. The main reason for this was that the Tmax of one subject in the test group was 35.98 hours, which was much longer than that of the other. Such outliers might be due to individual differences in the extended absorption phase14 of vortioxetine.
Regarding safety, one pregnant subject in the fasting study reported a pregnant endometrial cavity fluid, ovarian cyst, cervical canal cyst, and subsequent induced abortion, but these were all reported as AEs due to mild intensity and were not considered to be associated with vortioxetine. Under the fed condition, although lumbar fractures, surface damage, and pelvic fractures were all reported as SAEs, they were judged to be unrelated to vortioxetine because of occupational injuries. Therefore, none of the SAEs related to vortioxetine occurred in either fasting or feeding studies. Consistent with previous tolerability results,9,15 gastrointestinal disorders such as nausea, vomiting, and abdominal discomfort have been reported in feeding studies, these common acute treatment adverse effects were considered to be associated with the mechanism of serotonin reuptake inhibition of vortioxetine.2
Conclusion
Based on the results of this study, generic vortioxetine hydrobromide tablets in China are bioequivalent to the reference formulation (Brintellix®) in terms of the rate and extent of absorption under fasting and fed conditions. Both formulations were generally well tolerated in the healthy Chinese population and can be used interchangeably in the clinic to relieve the economic burden on MDD patients in China.
Data Sharing Statement
Individual-identified subject data is not going to be shared because of confidentiality.
Acknowledgments
The authors thank the participants and the staff who participated in this study.
Author Contributions
All authors made a significant contribution to the work reported, whether that is in the conception, study design, execution, acquisition of data, analysis and interpretation, or in all these areas; took part in drafting, revising or critically reviewing the article; gave final approval of the version to be published; have agreed on the journal to which the article has been submitted; and agree to be accountable for all aspects of the work.
Funding
This study was sponsored and funded by CSPC Ouyi Pharmaceutical Co., Ltd. (Hebei, China).
Disclosure
The authors declare that there are no conflicts of interest in this study.
References
1. Otte C, Gold SM, Penninx BW., et al. Major depressive disorder. Nat Rev Dis Primers. 2016;2(1):16065. doi:10.1038/nrdp.2016.65
2. Hong J, Vernon D, Kunovac J, Stahl S. Emerging drugs for the treatment of major depressive disorder. Expert Opin Emerg Drugs. 2022;27(3):263–275. doi:10.1080/14728214.2022.2117297
3. Seedat S, Scott KM, Angermeyer MC, et al. Cross-national associations between gender and mental disorders in the World Health Organization World Mental Health Surveys. Arch Gen Psychiatry. 2009;66(7):785–795. doi:10.1001/archgenpsychiatry.2009.36
4. Disease GBD, Injury I, Prevalence C. Global, regional, and national incidence, prevalence, and years lived with disability for 328 diseases and injuries for 195 countries, 1990–2016: a systematic analysis for the Global Burden of Disease Study 2016. Lancet. 2017;390(10100):1211–1259. doi:10.1016/S0140-6736(17)32154-2
5. Whooley MA, Wong JM. Depression and cardiovascular disorders. Annu Rev Clin Psychol. 2013;9(1):327–354. doi:10.1146/annurev-clinpsy-050212-185526
6. Penninx BW, Guralnik JM, Pahor M, et al. Chronically depressed mood and cancer risk in older persons. J Natl Cancer Inst. 1998;90(24):1888–1893. doi:10.1093/jnci/90.24.1888
7. Guaiana G, Barbui C, Hotopf M. Amitriptyline for depression. Cochrane Database Syst Rev. 2007;3:CD004186. doi:10.1002/14651858.CD004186.pub2
8. Gibb A, Deeks ED. Vortioxetine: first global approval. Drugs. 2014;74(1):135–145. doi:10.1007/s40265-013-0161-9
9. Zhang J, Mathis MV, Sellers JW, et al. The US Food and Drug Administration’s perspective on the new antidepressant vortioxetine. J Clin Psychiatry. 2015;76(1):8–14. doi:10.4088/JCP.14r09164
10. Sanchez C, Asin KE, Artigas F. Vortioxetine, a novel antidepressant with multimodal activity: review of preclinical and clinical data. Pharmacol Ther. 2015;145:43–57. doi:10.1016/j.pharmthera.2014.07.001
11. Mork A, Montezinho LP, Miller S, et al. Vortioxetine (Lu AA21004), a novel multimodal antidepressant, enhances memory in rats. Pharmacol Biochem Behav. 2013;105:41–50. doi:10.1016/j.pbb.2013.01.019
12. Boulenger JP, Loft H, Florea I. A randomized clinical study of Lu AA21004 in the prevention of relapse in patients with major depressive disorder. J Psychopharmacol. 2012;26(11):1408–1416. doi:10.1177/0269881112441866
13. Dubovsky SL. Pharmacokinetic evaluation of vortioxetine for the treatment of major depressive disorder. Expert Opin Drug Metab Toxicol. 2014;10(5):759–766. doi:10.1517/17425255.2014.904286
14. Areberg J, Sogaard B, Hojer AM. The clinical pharmacokinetics of Lu AA21004 and its major metabolite in healthy young volunteers. Basic Clin Pharmacol Toxicol. 2012;111(3):198–205. doi:10.1111/j.1742-7843.2012.00886.x
15. Matsuno K, Nakamura K, Aritomi Y, Nishimura A. Pharmacokinetics, safety, and tolerability of vortioxetine following single- and multiple-dose administration in healthy Japanese adults. Clin Pharmacol Drug Dev. 2018;7(3):319–331. doi:10.1002/cpdd.381
16. US Food and Drug Administration. Clinical pharmacology and biopharmaceutics review(s). Available from: https://www.accessdata.fda.gov/drugsatfda_docs/nda/2013/204447Orig1s000ClinPharmR.pdf.
17. European Medicines Agency. Vortioxetine hydrobromide immediate release tablets 5 mg, 10 mg, 15 mg, and 20 mg; vortioxetine lactate oral drops solution 20 mg/mL product-specific bioequivalence guidance. Available from: https://www.ema.europa.eu/en/documents/scientific-guideline/vortioxetine-hydrobromide-immediate-release-tablets-5-mg-10-mg-15-mg-20-mg-vortioxetine-lactate-oral/mL-product-specific-bioequivalence-guidance_en.pdf.
18. US Food and Drug Administration. Bioavailability and bioequivalence studies submitted in NDAs or INDs—general considerations; 2014. Available from: https://www.fda.gov/media/88254/download.
19. European Medicines Agency. Guideline on the investigation of bioequivalence; 2014. Available from: https://www.gmp-compliance.org/files/guidemgr/2016_EMEA_Bioequivalence.pdf.
20. Gadiko C, Tippabhotla SK, Thota S, Battula R, Khan SM, Vobalaboina V. A randomized, crossover, single-dose bioequivalence study of two extended-release tablets of donepezil 23 mg in healthy human volunteers under fasting and fed states. Sci Pharm. 2013;81(3):777–791. doi:10.3797/scipharm.1302-13
21. Hvenegaard MG, Bang-Andersen B, Pedersen H, Jorgensen M, Puschl A, Dalgaard L. Identification of the cytochrome P450 and other enzymes involved in the in vitro oxidative metabolism of a novel antidepressant, Lu AA21004. Drug Metab Dispos. 2012;40(7):1357–1365. doi:10.1124/dmd.112.044610
 © 2023 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2023 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.