")
Back to Journals » Drug Design, Development and Therapy » Volume 11
Bioequivalence study of a new sildenafil 100 mg orodispersible film compared to the conventional film-coated 100 mg tablet administered to healthy male volunteers
Authors Radicioni M, Castiglioni C , Giori A , Cupone I , Frangione V , Rovati S
Received 6 October 2016
Accepted for publication 14 December 2016
Published 11 April 2017 Volume 2017:11 Pages 1183—1192
DOI https://doi.org/10.2147/DDDT.S124034
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Sukesh Voruganti
Milko Radicioni,1 Chiara Castiglioni,1 Andrea Giori,2 Irma Cupone,3 Valeria Frangione,4 Stefano Rovati4
1CROSS Research S.A., Phase I Unit, Arzo, Switzerland; 2IBSA Farmaceutici Italia, Lodi, Italy; 3Bouty S.p.A., Strada Padana Superiore, Cassina De’ Pecchi, Italy; 4IBSA Institut Biochimique S.A., Pambio-Noranco, Switzerland
Abstract: A new orodispersible film formulation of the phosphodiesterase type 5 inhibitor, sildenafil, has been developed to examine the advantages of an orally disintegrating film formulation and provide an alternative to the current marketed products for the treatment of erectile dysfunction. The pharmacokinetics of the sildenafil 100 mg orodispersible film (IBSA) was compared to that of the conventional marketed 100 mg film-coated tablet (Viagra®) after single-dose administration to 53 healthy male volunteers (aged 18–51 years) in a randomized, open, two-way crossover bioequivalence study. Each subject received a single oral dose of 100 mg of sildenafil as test or reference formulation administered under fasting conditions at each of the two study periods according to a randomized crossover design. There was a washout interval of ≥7 days between the two administrations of the investigational medicinal products. Blood samples for pharmacokinetic analysis were collected up to 24 h post-dosing. The primary objective was to compare the rate (peak plasma concentration; Cmax) and extent (area under the curve [AUC] from administration to last observed concentration time; AUC0–t) of sildenafil absorption after single-dose administration of test and reference. Secondary endpoints were observed to describe the plasma pharmacokinetic profiles of sildenafil and its metabolite N-desmethyl-sildenafil relative bioavailability and safety profile after single-dose administration. The mean sildenafil and N-desmethyl-sildenafil plasma concentration–time profiles up to 24 h after single-dose administration of sildenafil 100 mg orodispersible film and film-coated tablet were nearly superimposable. The bioequivalence test was fully satisfied for sildenafil and N-desmethyl-sildenafil in terms of rate and extent of bioavailability. Adverse events occurred at similar rates for the two formulations and were of mild-to-moderate severity. The results suggest that the new orodispersible film formulation can be used interchangeably with the conventional film-coated formulation.
Keywords: sildenafil, pharmacokinetics, bioequivalence, orodispersible film, PDE5 inhibitor, N-desmethyl-sildenafil
Introduction
The first-in-class selective inhibitor of cyclic guanosine monophosphate-specific phosphodiesterase type 5 (PDE5), sildenafil, has become well established as a safe and effective treatment for male erectile dysfunction since the approval of Viagra® (Pfizer Ltd, Tadworth, UK) by the US Food and Drug Administration (FDA) in 1998. Guidelines on male sexual dysfunction established by the European Association of Urology recommend oral pharmacotherapy with PDE5 inhibitors as first-line therapy for erectile dysfunction.1
The PDE5 inhibitors sildenafil, tadalafil, vardenafil, and avanafil are currently approved for use for erectile dysfunction. Each PDE5 inhibitor has an individual pharmacokinetic and side effects profile, although no significant differences in efficacy among therapies are evident.1–5 The onset of action after oral administration of the available PDE5 inhibitors may be within 30 min; however, most men require at least 60 min delay after taking the medication (up to 2 h with tadalafil, while avanafil and vardenafil have the shortest onsets of action). Avanafil reaches peak plasma concentration (Cmax) 30–45 min after oral dosing and has a t1/2 of 1.1–1.23 h. Tadalafil has the longest t1/2 (~17.5 h), and the t1/2 for sildenafil and vardenafil is ~4 h.
More than 25 years of clinical experience has confirmed the risk/benefit profile of sildenafil and established it as an effective treatment option for erectile dysfunction. Sildenafil could also be taken by men with comorbidities, such as hypertension, stable coronary artery disease, congestive heart failure, diabetes mellitus, traumatic spinal cord injury, obstructive sleep apnea, multiple sclerosis, renal dysfunction, prostate cancer, Parkinson’s disease, depression, and traumatic stress disorders.6 It is usually administered in doses of 25, 50, and 100 mg, with the recommended starting dose of 50 mg to be adapted according to response and side effects.1 Commonly reported adverse events (AEs) with sildenafil, as with the other available PDE5 inhibitors, include headache, flushing, dyspepsia, dizziness, and vision disturbances and are generally mild in nature and self-limiting.1
Since the expiry of Pfizer’s patent on sildenafil citrate for the treatment of erectile dysfunction in a number of European countries in 2013, several different formulations of sildenafil have become available. Orally disintegrating film and tablet formulations of PDE5 inhibitors that disintegrate in the patient’s mouth without the need for swallowing with water have been developed.7–12 Orodispersible films are defined as single or multilayer sheets of suitable materials, to be placed in the mouth where they disperse rapidly,13 requiring only a small amount of saliva on the tongue to dissolve within a few minutes of administration. In addition to not requiring water for administration, orodispersible films share the advantages of tablet formulations (accurate dosage and ease of administration) with those of liquid dosing forms (ease of swallowing and rapid bioavailability due to bypassing the hepatic first-pass effect when the absorption of active substance occurs mainly through the oral mucosae).
Although orodispersible formulations have particular relevance by improving compliance in special patient populations, such as children, geriatric patients, and dysphasic patients who have difficulty in swallowing tablets or capsules, their convenience, superior dosing accuracy, and rapid onset of action contribute to strong patient preference over conventional solid dosage forms across a wide range of patient groups.14–16 Rapid dissolution, absorption, and onset of drug action are useful in motion sickness and sudden episodes of allergic attack of coughing, bronchitis, and asthma. Some drugs are absorbed from the mouth, pharynx, and esophagus: pre-gastric absorption can improve bioavailability with consequent reduction of dosage and unwanted effects.
Orodispersible films have advantages over orally disintegrating tablet formulations, which may require more complicated and expensive manufacturing processes, have issues of hardness and friability during manufacturing, storage, handling and administration, and may present a risk of choking.16 Orodispersible films are thin and flexible, can be manufactured in a range of sizes and shapes, and are easily transported and stored. Research has shown that four out of five patients prefer orally disintegrating dosage forms over conventional solid oral dosage forms.15 As orodispersible film formulations do not require administration with water, they may provide additional benefit where comorbid conditions such as renal impairment, congestive heart failure, or other disorders impose a restriction on fluid intake, or where dysphagia creates difficulty with swallowing conventional film tablet dosage forms. Drugs that have potency at low doses are most suitable for orodispersible film administration, as technical considerations limit the incorporation of the drug; generally, between 1% and 30% w/w of the active pharmaceutical ingredient can be introduced in film formulation.17
Taste is an important factor in the development of oral pharmaceutical products to ensure patient acceptability and compliance and is one of the prime factors determining market penetration and commercial success of oral formulations. Palatability and pleasant taste are necessary for fast dissolving films, and various techniques are available to mask drug taste. Flavors, sweeteners, and amino acids can be added to the formulation, generally in association with other taste-masking components. The classical sources of sweeteners are sucrose, dextrose, fructose, glucose, liquid glucose, and maltose.
A pharmacokinetic comparison of an orally disintegrating film formulation of sildenafil marketed in South Korea suggested that the pharmacokinetics of the orodispersible formulation was similar to that of the conventional film-coated tablet and met the criterion of assumed bioequivalence according to Korean FDA regulatory criteria.18 In the study, the 90% confidence interval (CI) of the geometric mean ratio of test/reference for the area under the curve (AUC0–last) for the sildenafil 100 mg dose was 101.68%–114.78% and the 90% CI was 93.76%–109.76% for Cmax.18 Both the formulations were well tolerated, and no serious AEs were observed. Roh et al noted that the new formulation could be used interchangeably with the tablet formulation and may be preferred because of enhanced dosing convenience.18
An orodispersible film formulation of sildenafil 100 mg has been developed by Institut Biochimique SA (IBSA, Pambio-Noranco, Switzerland). The aim of this study was to assess the bioequivalence between the new sildenafil 100 mg orodispersible film and the conventional marketed 100 mg film-coated tablet after single-dose administration to healthy male volunteers.
Materials and methods
Materials
According to the Biopharmaceutics Classification System, sildenafil is classified as a class II drug substance (high permeability and low solubility). The new sildenafil orodispersible film developed by IBSA is approved in Europe in doses of 25, 50, 75, and 100 mg. Each orodispersible film contained 140.4 mg of sildenafil citrate, equivalent to 100 mg of sildenafil in the form of a rectangular, flexible, opaque, light blue film 40×45 mm. Details of the pharmaceutical excipients and their functions in the formulation are presented in Table 1. The product is an innovative, patented formulation developed in accordance with patents EP 1689374 (self-supporting films for pharmaceutical and food use) and WO 2014/049548 (orodispersible films having quick dissolution times for therapeutic and food use).
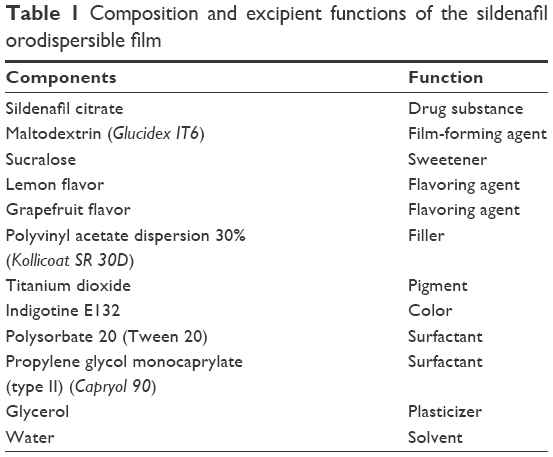 | Table 1 Composition and excipient functions of the sildenafil orodispersible film |
The preparation process according to the patent WO 2014/049548 comprises, briefly, the following steps:
- Maltodextrin, plasticizer, active ingredient, and the other excipients are solubilized/dispersed in water.
- The mixture is coated onto a release liner and dried in the oven controlling for temperature, air circulation, and coating speed.
- The dried mass is cut into reels and the films are then punched, pouched, and sealed in suitable single-dose sachets.
Study design
This bioequivalence study was a single-center, single-dose, randomized, open, two-way crossover study performed between the sildenafil 100 mg orodispersible film (IBSA, test) and the marketed Viagra 100 mg film-coated tablet (Pfizer, reference). The study, performed between February 2014 and March 2014, consisted of a screening phase, an interventional (treatment) phase of two periods with a washout interval of at least 7 days between the two administrations of the investigational medicinal products.
The sildenafil IBSA 100 mg orodispersible film test treatment (batch number L3002V21, expiry date April 2014) was administered to a subject without water, placed directly on the tongue by the investigator after the subject had swallowed 20 mL of water to pre-wet his/her mouth.
The sildenafil 100 mg reference formulation (Viagra film-coated tablet; batch number A325702G, expiry date February 2018) was administered to a subject under fasting conditions together with 240 mL of water. The tablet was not chewed.
Eligible subjects received a single oral dose of 100 mg of sildenafil as test or reference formulation administered to the subjects under fasting conditions in each of the two study periods according to a randomized crossover design. The sildenafil dose, 100 mg, was chosen on the basis of clinical practice and the recommended dose range of 25–100 mg.
Administration under fasting conditions was undertaken in compliance with the European regulatory guidelines for determining bioequivalence, as this is considered to be the most sensitive condition to detect a potential difference between formulations.
Subjects
Subjects were eligible for inclusion in the study if they met the following inclusion criteria: male, aged 18–55 years (inclusive); informed consent, including signed written informed consent before inclusion in the study; body mass index of 18.5–30 kg/m2 (inclusive); vital signs (systolic blood pressure 100–139 mmHg, diastolic blood pressure 50–89 mmHg, heart rate 50–90 bpm, measured after 5 min at rest in the sitting position); the ability to fully comprehend the nature and aims of the study, including possible risks and side effects, and to cooperate with the investigator and comply with the study requirements.
Subjects with the following characteristics were excluded: clinically significant cardiac abnormalities assessed by 12-lead electrocardiography; clinically significant abnormal physical findings; clinically significant abnormal laboratory values indicative of physical illness, in particular sickle cell anemia, or history of other diseases that could interfere with the aim of the study; ascertained or presumptive hypersensitivity to the active principle and/or ingredients of the formulations or a history of anaphylaxis to drugs or allergic reactions in general; participation in the evaluation of any investigational product for 3 months before this study.
The study was conducted in accordance with the principles of the Declaration of Helsinki and the International Conference on Harmonization Harmonized Tripartite Guidelines for Good Clinical Practice. The study protocol, investigator’s brochure, and all other relevant documentation were reviewed and approved by an independent ethics committee and the Swiss Federal Health Authorities approved the study (reference number 2014DR1005).
Randomization, treatment assignment, and assessment
Subjects who met the eligibility criteria at the screening visit returned on study day 1 for confinement at the clinical center where they were assigned a consecutive randomization number recorded on a computer-generated randomization list prepared by the Contract Research Organization Biometry Unit using the PLAN procedure of the validated SAS® for Windows version 9.1.3 Service Pack 4 (10) and supplied to the study site prior to study start. As this was an open study, no masking procedure was applied.
Two single doses of 100 mg sildenafil (one with the test and one with the reference investigational medicinal product) were administered under fasting conditions to each subject at 8:00±1 h on day 1 of each study period, according to the randomization list. Palatability was assessed immediately after test product administration, according to a 5-point scale where 0= very unpleasant and 4= very good. There was a washout interval of at least 7 days between the two administrations. Blood samples were collected up to 24 h post-dosing. Vital signs (blood pressure and heart rate) were measured on days 1–2 of each study period at pre-dose, 1.5, and 24 h post-dose. The 24-h post-dose vital signs check during period 2 corresponded to the final assessment. A 12-lead resting electrocardiogram was recorded at screening, on day 1 of each study period, and at the final visit. Blood and urine samples were collected for routine hematology, blood chemistry, virology, and urinalysis at screening and final visit.
Safety/AEs were assessed throughout the study. A full physical examination was performed by the investigator at screening and at the final visit.
Sample collection and drug concentration analysis
Venous blood samples (10 mL) for the pharmacokinetic analysis of sildenafil and N-desmethyl-sildenafil were collected in heparinized tubes via an indwelling catheter inserted into a forearm vein using a standardized procedure to avoid any contamination of the sample with heparin. The blood samples were collected pre-dose (0), and at 6, 15, 30, and 45 min and at 1, 1.25, 1.5, 2, 2.5, 3, 4, 6, 8, 12, 16, and 24 h post-dose on days 1–2 of each study period, for a total of 17 samples in each study period. The blood sampling time points were selected on the basis of the known sildenafil pharmacokinetic profile. The blood samples were stored in ice for a maximum of 20 min and then centrifuged at 4°C for 10 min at 2,500 × g to obtain plasma, which was stored frozen at ≤−20°C in pre-labeled polypropylene tubes until analyzed.
The concentration of sildenafil (free base) and its metabolite N-desmethyl-sildenafil in plasma aliquots was determined at an analytical laboratory in the Netherlands (Analytisch Biochemisch Laboratorium BV [ABL]) using a fully validated liquid chromatography tandem mass spectrometry method with a lower quantification limit of 0.5 ng/mL. The method was validated according to the most recent European regulatory guidelines on bioanalytical method validations for determining bioequivalence.
The analyses were performed in compliance with the general principles of Good Laboratory Practice regulations.
The complete details of the analytical procedures used to determine drug concentrations are presented in the Supplementary materials.
Data quality was assured by Clinical Medical Services Sagl, Switzerland, who conducted regular onsite monitoring visits and regular inspections of the case report forms. All study documentation and results were reviewed according to the quality assurance standard operating procedures of CROSS Research, Switzerland.
Outcomes
The primary objective of this study was to compare the rate (Cmax) and extent (AUC0–t) of sildenafil absorption after single-dose administration of test and reference. Secondary endpoints were to describe the plasma pharmacokinetic profiles of sildenafil and its metabolite N-desmethyl-sildenafil after single-dose administration of test and reference, including Cmax, AUC from administration to the last observed concentration time (AUC0–t), AUC extrapolated to infinity (AUC0–8), t½, time to Cmax (tmax), terminal elimination constant rate (λz), and relative bioavailability (Frel), and to evaluate the sildenafil safety profile after single-dose administration.
Safety evaluations consisted of treatment-emergent AEs (TEAEs), vital signs (blood pressure and heart rate), physical examination and body weight, laboratory parameters, and electrocardiogram.
Statistical analysis
The sample size for the study was determined with reference to the relevant, recent literature available on the pharmacokinetics of sildenafil, in particular the results of a study conducted after administration of two 25 mg capsules of Viagra film-coated tablets in a population of 12 male subjects.19 The highest coefficient of variance for the pharmacokinetic parameters Cmax and AUC was estimated to be 0.383. The sample size was calculated using the SAS 9.1.3 software (SAS Institute, Cary, NC, USA) Power Procedure with paired means option. Fixing the significance level α at 5% and the hypothesized test/reference mean ratio to 1, 50 subjects were considered sufficient to attain a power of 80% to correctly conclude the bioequivalence between the two formulations within the range 80.00%–125.00% for all parameters (Cmax and AUC).
Data were described using classic descriptive statistics for quantitative variables and frequencies for qualitative variables. The pharmacokinetic analysis and the statistical analysis of pharmacokinetic parameters were performed using Phoenix WinNonlin® version 6.3 (Pharsight Corporation, Mountain View, CA, USA) and SAS version 9.3 (TS1M1). Sildenafil and N-desmethyl-sildenafil rate (Cmax) and extent (AUC) of absorption were compared between test and reference using analysis of variance for a crossover design on log-transformed data. Period, treatment, sequence, and subject within sequence were taken into account as sources of variation.
The acceptance criterion for bioequivalence was to have all 90% CIs of the test/reference ratios of sildenafil AUC and Cmax geometric means within the range of 80%–125%. tmax was analyzed using the nonparametric Friedman test.
Results
Subjects
A total of 54 subjects met the eligibility criteria and were included in the study and randomized, and 53 of them received both test and reference treatments and completed the study per protocol. The flow of subjects throughout the study is shown in Figure 1. One subject prematurely discontinued the study on day 1 of period 1 before administration of the study medication due to pretreatment AEs. All the 53 subjects were included in the safety and pharmacokinetic analysis sets. Minor protocol deviations (vital signs measured earlier than the specified 10 min before scheduled time point) were observed for 15/54 subjects and were not considered relevant. Although the exact film disintegration time was not recorded, complete dissolution in 1 min after administration was confirmed in all the volunteers (the study protocol required the investigator to check the oral cavity exactly 1 min after drug intake to confirm oral film dissolution).
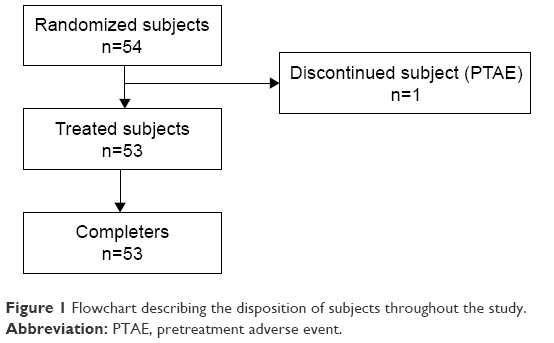 | Figure 1 Flowchart describing the disposition of subjects throughout the study. |
Demographics of the subjects are summarized in Table 2. The mean age of the subjects was 36.3 years (range 18–51 years) and the majority were Caucasian (98.1%). All enrolled subjects were in good physical and mental health, as determined on the basis of medical and surgical history and physical examination. Mean body weight was 75.91 kg (range 56.2–99.6 kg).
 | Table 2 Demographic characteristics of the safety and pharmacokinetic sets (n=53) at baseline |
Pharmacokinetics
The mean ± standard deviation (SD) of plasma sildenafil pharmacokinetic parameters and the results of their statistical comparisons for the pharmacokinetic set (n=53) are summarized in Table 3. Figure 2 shows the sildenafil pharmacokinetic profiles of the two investigational medicinal products up to 24 h after sildenafil 100 mg test and reference treatments. The mean plasma concentration–time profiles up to 24 h of sildenafil 100 mg orodispersible film and the film-coated tablet were nearly superimposable.
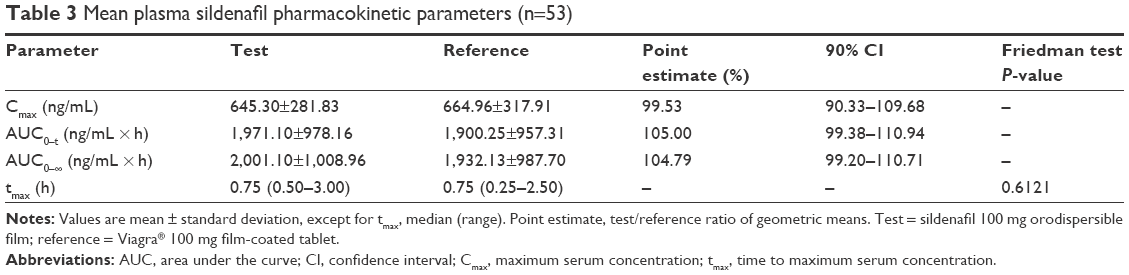 | Table 3 Mean plasma sildenafil pharmacokinetic parameters (n=53) |
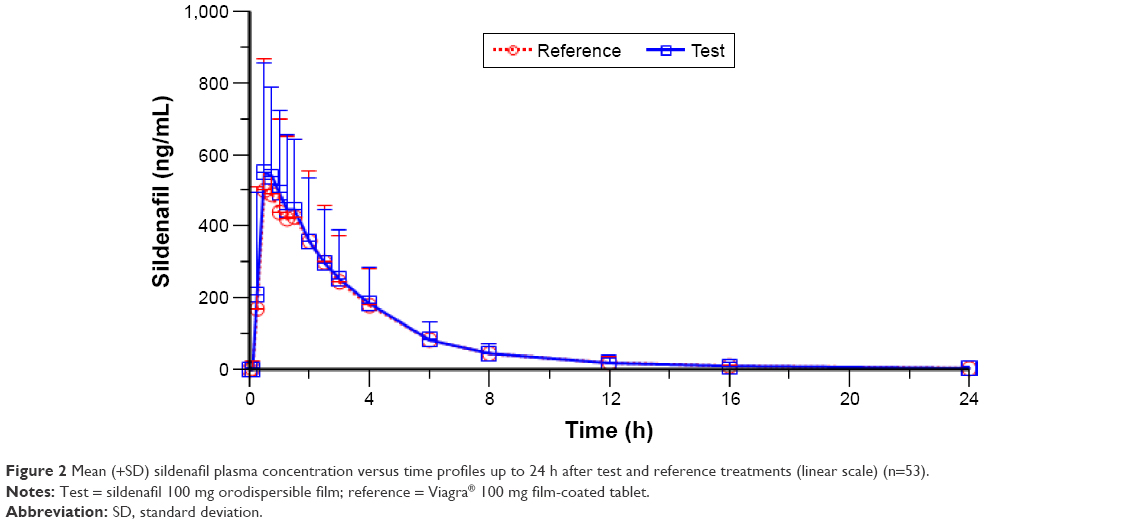 | Figure 2 Mean (+SD) sildenafil plasma concentration versus time profiles up to 24 h after test and reference treatments (linear scale) (n=53). |
The mean ± SD of plasma N-desmethyl-sildenafil pharmacokinetic parameters and the results of their statistical comparisons for the pharmacokinetic set (n=53) are summarized in Table 4. The mean plasma concentration–time profiles of N-desmethyl-sildenafil were nearly superimposable (Figure 3).
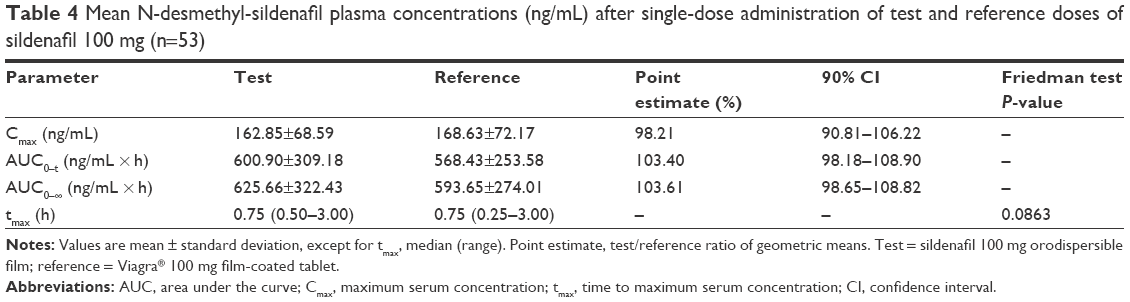 | Table 4 Mean N-desmethyl-sildenafil plasma concentrations (ng/mL) after single-dose administration of test and reference doses of sildenafil 100 mg (n=53) |
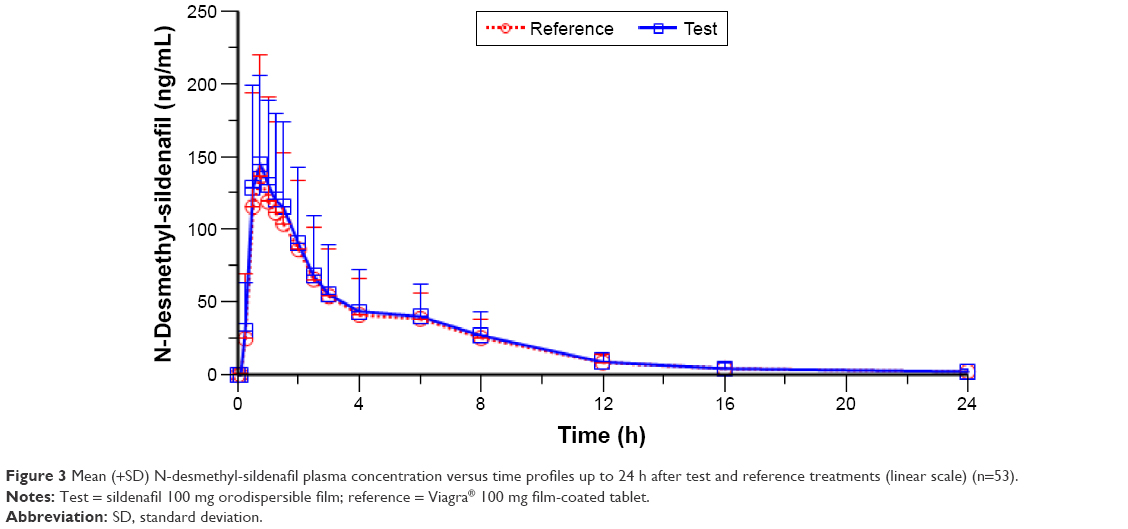 | Figure 3 Mean (+SD) N-desmethyl-sildenafil plasma concentration versus time profiles up to 24 h after test and reference treatments (linear scale) (n=53). |
The bioequivalence test was fully satisfied for sildenafil and N-desmethyl-sildenafil in terms of rate and extent of bioavailability (Cmax, AUC0–t, and AUC0–f). Specifically, 90% CIs of the test/reference ratio of geometric means were fully comprised within the bioequivalence acceptance range 80%–125% (Table 3, sildenafil; Table 4, N-desmethyl-sildenafil).
Palatability
The majority of subjects (28/53; 52.8%) judged the palatability of the sildenafil 100 mg orodispersible film to be good/acceptable.
Safety
Sildenafil administered as a single dose of test or reference product was well tolerated. With the exception of a transient aspartate aminotransferase increase, possibly related to the reference product, in one subject, no unexpected TEAEs were observed. Overall, 37 TEAEs were reported by 22 (41.5%) subjects: 20 TEAEs were experienced by 14 (26.4%) subjects with the test product and 17 TEAEs were experienced by 13 (24.5%) subjects with the reference product (Table 5).
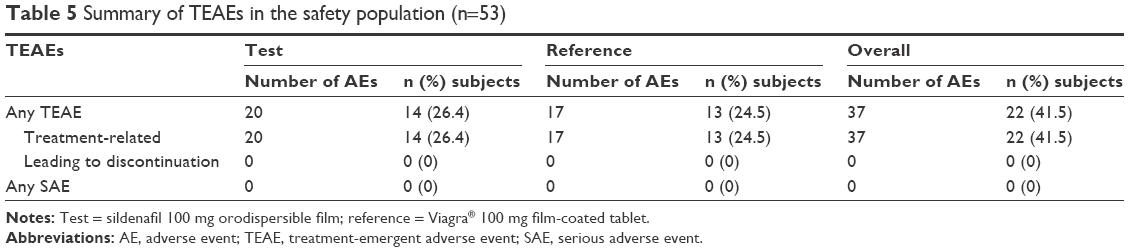 | Table 5 Summary of TEAEs in the safety population (n=53) |
All reported TEAEs were deemed related to the treatment by the investigator. The most common TEAE was headache, which was observed in 7 (13.2%) subjects with test and 9 (17.0%) subjects with reference. No subjects withdrew from the study as a consequence of a TEAE. One subject discontinued the study due to pretreatment AEs. No serious AEs or other significant AEs occurred in the study.
Apart from the single transient increase in aspartate aminotransferase levels, no significant effects of treatment on blood pressure, heart rate, body weight, electrocardiogram, or other laboratory parameters were observed.
Discussion
Over recent years, various innovative drug delivery systems, including films, gels, chewing gums, and drug-loaded micro- and nanoparticles, have been developed as alternatives to conventional dosage forms with the aim of improving patient convenience and acceptability and enhancing compliance. Orodispersible films (variously termed as quick dissolving film, thin strips, oral films, orally dissolving film, etc) disintegrate in patients’ mouth without the need for water and consist of a thin oral strip comprising a hydrophilic, film-forming polymer, and the active pharmaceutical ingredient. The film is hydrated by saliva when placed on the tongue or oral cavity, and disintegrates rapidly to release the medication for local (oromucosal) and systemic (gastrointestinal) absorption.
Ideally, film-forming hydrophilic polymers for use in orodispersible films should be nonirritant, nontoxic, and tasteless; quickly disintegrate, possess sufficient peel, shear, and tensile strength; and inexpensive and stable.14 The hydrophilic polymers are combined with other excipients to improve the physical properties (eg, plasticizers), to incorporate active pharmaceutical substances (eg, surfactants) and to improve palatability. The films are formulated using various methods, including solvent casting, hot-melt extrusion, semisolid casting, solid dispersion extrusion, and rolling; solvent casting and spraying methods are most commonly used.14,16
The pharmaceutical development of the IBSA 100 mg orodispersible film was based on the use of maltodextrins as the film-forming agent, glycerin as a plasticizer, and water as a solvent. Polyvinyl acetate was introduced to improve the mechanical properties and tensile strength of the films. The sildenafil IBSA 100 mg orodispersible film was prepared by the solvent casting method, consisting of spreading and drying of a viscous mixture composed of active principle and excipients in a suitable solvent. The mixture, homogeneous and without bubble and clumps, is spread on a liner and dried, allowing the production of a film with a homogeneous and smooth surface and a thickness of ~220 μm. The resulting film is then cut into pieces of required size. The novelty of the IBSA orodispersible film is described in the patent WO 2014/049548.
Unlike many of the films that have been considered to be state-of-the-art, the sildenafil IBSA orodispersible film does not use pullulan, an ingredient that is expensive and difficult to source, as the film-forming component. Formulations based on other film-forming polymers are available, but with some limitations in terms of physical properties or dissolution behavior. Maltodextrins, defined as non-sweet, nutritive saccharide mixtures with a dextrose equivalent (DE) of <20,20 are obtained from the partial hydrolysis of starch with suitable acids and/or enzymes and are a suitable ingredient for manufacturing orodispersible films. They can differ in average molecular size, with the DE value (ie, the proportion of reducing sugars relative to dextrose expressed as a percentage on a dry basis) commonly used in classification. However, maltodextrins usually require the addition of a hydrocolloid at concentrations >10% to reduce dissolution time. However, hydrocolloids tend to become a gel when they are in contact with saliva; thus, they give the sensation of poor dissolution in the mouth. On the contrary, oral films based on maltodextrins without hydrocolloids tend to have poor physical properties, with hardening during storage. The current IBSA formulation avoids hardening of the films based on maltodextrin and plasticizer by incorporating a homopolymer of vinyl acetate into the composition and is thus able to replace pullulan while maintaining its benefits, conferring advantages in terms of solubility and film formation to the sildenafil orodispersible film product.
Manufacturing an orodispersible film with the tensile properties required for packaging and handling procedures is a critical issue in the development of orodispersible films. The flexibility and strength of the film could affect cutting, film formation, and packaging during the manufacturing process. To facilitate handling, the oral film should be flexible while exhibiting a tensile strength that guarantees a suitable stiffness. Elongation at break should be low to avoid deformation of films during the manufacturing process. During the development of the IBSA orodispersible film, tensile properties were evaluated, and tensile strength, elongation at break, and tensile energy to break were calculated to determine the appropriate tensile properties.
The study described in this paper was conducted to compare the pharmacokinetic and safety profiles of the new sildenafil 100 mg orodispersible film formulation in healthy male volunteers. In the study, the 90% CIs for sildenafil Cmax, AUC0–t, and AUC0–f were within the range of 80%–125% used for the assessment of bioequivalence according to the current European guidance on the investigation of bioequivalence (CPMP/EWP/QWP/1401/98 Rev. 1/Corr**, January 20, 2010). Therefore, the sildenafil 100 mg orodispersible film (test) and the 100 mg film-coated tablet (reference) were found to be bioequivalent with respect to sildenafil rate and extent of absorption. Bioequivalence was also shown in terms of rate and extent of bioavailability of the sildenafil metabolite, N-desmethyl-sildenafil. The time to reach peak drug concentration of sildenafil and N-desmethyl-sildenafil did not differ significantly between the two investigational products.
Safety data confirmed a favorable safety profile of the test and reference investigational products, administered as a single oral dose of 100 mg sildenafil. AEs occurred at similar rates between the two formulations and all AEs were of mild or moderate severity. There were no serious AEs and no subjects withdrew because of treatment-related AEs.
Patient satisfaction with erectile dysfunction treatments is a complex and personal issue, and there is no clearly established and robust evidence base to demonstrate a preference between different PDE5 inhibitors.3,21–24 The needs and expectations of patients vary widely, and involving the patient and, if possible, his sexual partner in the decision-making process by providing relevant information about the therapy options, including expected benefits and possible adverse effects or complications, is important in deciding on the most appropriate approach for treating erectile dysfunction.1 Patient satisfaction is determined not only in terms of erectile response and side effects, but also in how well the treatment meets patient needs and expectations and the dynamics of the relationship. Therefore, follow-up evaluation to review the success of the intervention, identify any side effects, discuss patient satisfaction with treatment, and revisit their expectations are essential to ensure optimal long-term treatment strategy.
The availability of alternative oral dosage forms that offer advantages over conventional tablet formulations broadens the prescribing options of physicians. The IBSA sildenafil orodispersible film was developed to provide a safe, efficacious, and stable orodispersible film formulation that was pharmaceutically equivalent to the conventional marketed branded film-coated tablets. As noted, orodispersible films have the advantages of conventional tablets (precise dosage and ease of administration) and those of liquid dosage forms (ease of swallowing and rapid bioavailability). The IBSA formulation offers a convenient, discrete method of intake with a rapid onset of action and the added patient convenience and acceptability of a dosage form that does not require administration with water, of particular benefit for men who have difficulty with swallowing conventional film tablet dosage forms or in whom daily fluid intake is restricted, as the orodispersible film does not require administration with water.
The orodispersible film formulation of sildenafil 100 mg was developed by IBSA to recognize the advantages of an orodispersible formulation that dissolved rapidly in the oral cavity without drinking or chewing and to provide an alternative to the current marketed products for the treatment of erectile dysfunction. This study has demonstrated that the sildenafil IBSA 100 mg orodispersible film can be used interchangeably with the conventional film-coated tablet formulation. However, as is the nature of most bioequivalence and pharmacokinetic studies, this study was conducted in a limited number of healthy, relatively young, subjects; therefore, the findings cannot be directly extended to patients with comorbidities or the elderly.
Conclusion
The pharmacokinetics of the sildenafil 100 mg orodispersible film formulation and the 100 mg film-coated tablet were shown to be without statistically significant difference. The results suggest that the new orodispersible film formulation could represent a valid alternative to the current marketed products for the treatment of erectile dysfunction.
Acknowledgments
The authors thank Ray Hill, an independent medical writer, who provided medical writing support funded by IBSA Institut Biochimique SA, Pambio-Noranco, Switzerland. This study was also funded by IBSA Institut Biochimique SA. All the authors contributed equally to this work.
Disclosure
AG, IC, VF, and SR are IBSA employees. The other authors report no conflicts of interest in this work.
References
Hatzimouratidis K, Eardley I, Giuliano F, et al. Guidelines on male sexual dysfunction: erectile dysfunction and premature ejaculation. 2014. Available from: https://uroweb.org/guideline/male-sexual-dysfunction/. Accessed June 20, 2016. | ||
Hakky TS, Jain L. Current use of phosphodiesterase inhibitors in urology. Turk J Urol. 2015;41(2):88. | ||
Smith WB 2nd, McCaslin IR, Gokce A, Mandava SH, Trost L, Hellstrom WJ. PDE5 inhibitors: considerations for preference and long-term adherence. Int J Clin Pract. 2013;67(8):768–780. | ||
Alwaal A, Al-Mannie R, Carrier S. Future prospects in the treatment of erectile dysfunction: focus on avanafil. Drug Des Devel Ther. 2011;5:435–443. | ||
Limin M, Johnsen N, Hellstrom WJ. Avanafil, a new rapid-onset phosphodiesterase 5 inhibitor for the treatment of erectile dysfunction. Expert Opin Investig Drugs. 2010;19(11):1427–1437. | ||
Leoni LA, Leite GS, Wichi RB, Rodrigues B. Sildenafil: two decades of benefits or risks? Aging Male. 2013;16(3):85–91. | ||
Goel H, Rai P, Rana V, Tiwary AK. Orally disintegrating systems: innovations in formulation and technology. Recent Pat Drug Deliv Formul. 2008;2(3):258–274. | ||
Heinig R, Weimann B, Dietrich H, Bottcher MF. Pharmacokinetics of a new orodispersible tablet formulation of vardenafil: results of three clinical trials. Clin Drug Investig. 2011;31(1):27–41. | ||
Debruyne FM, Gittelman M, Sperling H, Borner M, Beneke M. Time to onset of action of vardenafil: a retrospective analysis of the pivotal trials for the orodispersible and film-coated tablet formulations. J Sex Med. 2011;8(10):2912–2923. | ||
Sperling H, Debruyne F, Boermans A, Beneke M, Ulbrich E, Ewald S. The POTENT I randomized trial: efficacy and safety of an orodispersible vardenafil formulation for the treatment of erectile dysfunction. J Sex Med. 2010;7(4 Pt 1):1497–1507. | ||
Gittelman M, McMahon CG, Rodriguez-Rivera JA, Beneke M, Ulbrich E, Ewald S. The POTENT II randomised trial: efficacy and safety of an orodispersible vardenafil formulation for the treatment of erectile dysfunction. Int J Clin Pract. 2010;64(5):594–603. | ||
Sperling H, Gittelman M, Norenberg C, Ulbrich E, Ewald S. Efficacy and safety of an orodispersible vardenafil formulation for the treatment of erectile dysfunction in elderly men and those with underlying conditions: an integrated analysis of two pivotal trials. J Sex Med. 2011;8(1):261–271. | ||
Thakur N, Bansal M, Sharma N, Yadav G, Khare P. Overview “a novel approach of fast dissolving films and their patents”. Adv Biol Res (Rennes). 2013;7(2):50–58. | ||
Irfan M, Rabel S, Bukhtar Q, Qadir MI, Jabeen F, Khan A. Orally disintegrating films: a modern expansion in drug delivery system. Saudi Pharm J. 2015;24(5):537–546. | ||
Kathpalia H, Gupte A. An introduction to fast dissolving oral thin film drug delivery systems: a review. Curr Drug Deliv. 2013;10(6):667–684. | ||
Hoffmann EM, Breitenbach A, Breitkreutz J. Advances in orodispersible films for drug delivery. Expert Opin Drug Deliv. 2011;8(3):299–316. | ||
European Pharmacopoeia. 2014, 8th Edition, Orodispersible Films. Available from: http://online6.edqm.eu/ep802/. Accessed July 8, 2016. | ||
Roh H, Son H, Lee D, et al. Pharmacokinetic comparison of an orally disintegrating film formulation with a film-coated tablet formulation of sildenafil in healthy Korean subjects: a randomized, open-label, single-dose, 2-period crossover study. Clin Ther. 2013;35(3):205–214. | ||
Muirhead GJ, Wilner K, Colburn W, Haug-Pihale G, Rouviex B. The effects of age and renal and hepatic impairment on the pharmacokinetics of sildenafil. Br J Clin Pharmacol. 2002;53(Suppl 1):21S–30S. | ||
Garnero C, Chattah AK, Longhi M. Supramolecular complexes of maltodextrin and furosemide polymorphs: a new approach for delivery systems. Carbohydr Polym. 2013;94(1):292–300. | ||
Doggrell S. Do vardenafil and tadalafil have advantages over sildenafil in the treatment of erectile dysfunction? Int J Impot Res. 2007;19(3):281–295. | ||
Hackett GI. Patient preferences in treatment of erectile dysfunction: the continuing importance of patient education. Clin Cornerstone. 2005;7(1):57–65. | ||
Mirone V, Fusco F, Rossi A, Sicuteri R, Montorsi F. Tadalafil and vardenafil vs sildenafil: a review of patient-preference studies. BJU Int. 2009;103(9):1212–1217. | ||
Raheem AA, Kell P. Patient preference and satisfaction in erectile dysfunction therapy: a comparison of the three phosphodiesterase-5 inhibitors sildenafil, vardenafil and tadalafil. Patient Prefer Adherence. 2009;3:99–104. |
 © 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms.php
and incorporate the Creative Commons Attribution
- Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms.php
and incorporate the Creative Commons Attribution
- Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.