")
Back to Journals » Drug Design, Development and Therapy » Volume 15
Bioequivalence and Pharmacokinetic Evaluation of Two Oral Formulations of Regorafenib: An Open-Label, Randomised, Single-Dose, Two-Period, Two-Way Crossover Clinical Trial in Healthy Chinese Volunteers Under Fasting and Fed Conditions
Authors Zhang Q, Wang Z , Wu J, Zhou Z, Zhou R, Hu W
Received 9 June 2021
Accepted for publication 22 July 2021
Published 29 July 2021 Volume 2021:15 Pages 3277—3288
DOI https://doi.org/10.2147/DDDT.S323169
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Jianbo Sun
Qian Zhang,1,2 Zhiqiang Wang,1– 3 Jingying Wu,1,2 Zhen Zhou,3 Renpeng Zhou,1,2 Wei Hu1,2
1Department of Clinical Pharmacology, The Second Hospital of Anhui Medical University, Hefei, People’s Republic of China; 2Anhui Provincial Institute of Translational Medicine, Hefei, People’s Republic of China; 3Menzies Institute for Medical Research, University of Tasmania, Hobart, Tasmania, Australia
Correspondence: Renpeng Zhou; Wei Hu Email [email protected]; [email protected]
Background: Regorafenib is an oral multi-kinase inhibitor approved for the treatment of solid tumours, but the pharmacokinetic profile of regorafenib in the Chinese population is unclear.
Objective: The aim of this study was to examine the pharmacokinetics, bioequivalence, and safety of two formulations of regorafenib 40 mg in healthy Chinese volunteers under fed and fasting conditions.
Methods: A single-centre, randomised, open-label, two-period, two-way crossover phase 1 trial was conducted by randomising a single oral dose of test (T) or reference (R, Stivarga®) regorafenib (40 mg) to healthy Chinese volunteers under both fasting and fed conditions (high-fat and high-calorie diet). Pharmacokinetic parameters were calculated using non-compartmental methods. Adverse events were recorded to assess drug safety.
Results: Sixty-six participants were enrolled for both fasting and fed treatments. The 90% CIs geometric least-square means of ratioT/R for regorafenib were completely contained within the equivalence margin of 80– 125% under both fasting and fed conditions. Both formulations displayed similar and generally good safety profiles.
Conclusion: Single oral dose of the T (40 mg) and R (40 mg) regorafenib was bioequivalent under fasting and fed conditions and had similar favourable safety profiles among healthy Chinese volunteers.
Keywords: regorafenib, phase 1, pharmacokinetic, bioequivalence, Chinese healthy volunteers
Introductions
Regorafenib is an oral multi-kinase inhibitor approved for the treatment of a range of advanced solid tumours, including refractory metastatic colorectal cancer (mCRC),1 advanced gastrointestinal stromal tumours (GISTs),2 and unresectable hepatocellular carcinoma (HCC).3 These tumours are the leading causes of death globally,4 and their prevalence continues to increase in China.5 Regorafenib inhibits tumour formation, angiogenesis, tumour microenvironment formation and metastasis through inhibiting a broad range of kinases and modulating immune systems.6
Regorafenib is recommended to be taken with a low-fat meal or a meal that contains less than 600 calories and 30% fat.7 Compared with high-fat breakfast or fasting conditions, regorafenib and its major pharmacologically active metabolites (M-2, nitrogen oxides; and M-5, nitrogen oxides and N-demethylation) have the highest concentration after a low-fat breakfast.8,9 After a single oral dose of 160 mg (4 × 40 mg tablets), regorafenib can reach an average peak plasma concentration of about 2.5 mg/L in 3 to 4 hours.8,9 Regorafenib is mainly metabolised in the liver by CYP3A4-mediated oxidative metabolism and UGT1A9-mediated glucuronidation.10,11 M-2 and M-5 reach similar concentrations to regorafenib at a steady state in plasma and exhibit similar kinase inhibition profiles and comparable potency to regorafenib.10,12 Enterohepatic circulation (EHC) is a major disposition pathway for regorafenib, where the unbound regorafenib or its metabolites are hydrolysed by the microbial in the gastrointestinal tract and then reabsorbed.13
A previous multicentre, single-arm, phase 1 trial suggested that 160 mg regorafenib taken orally once daily for the first 21 days of each 28-day cycle showed acceptable tolerability and antitumor activity in fifteen Japanese patients who had solid tumours without undue toxicity.14 However, the pharmacokinetics and bioequivalence of oral generic regorafenib (produced by Yangzijiang and Bayer) in healthy Chinese subjects and the effect of food on their pharmacokinetics have not yet been investigated. Evaluation of the effect of food on the PK of oral oncology drugs, such as regorafenib, is essential to inform dosing in future pivotal trials.15 Usually, a single-dose, two-period, two-treatment, two-sequence crossover study is recommended for food-effect bioequivalence studies.16 Thus, the aim of this study was to examine the pharmacokinetics of regorafenib (40 mg) tablets taken orally under fasting and fed conditions (high-fat and high-calorie diet) in healthy Chinese volunteers and to evaluate the bioequivalence and safety of the two formulations, test (T) and reference (R), of regorafenib produced by different pharmaceuticals.
Methods
Study Design
This was a single-centre, single-dose, randomized, open, two-period, two-way cross-over phase 1 trial. This trial was registered in the Centre for Drug Evaluation, National Medical Products Administration (NMPA) (http://www.chinadrugtrials.org.cn/; CTR20201844). Ethical approval for the trial conduct was obtained from the Ethics Committee of the Second Affiliated Hospital of Anhui Medical University (Hefei, China). The study was conducted in the Phase 1 Clinical Trial Unit, Second Affiliated Hospital of Anhui Medical University (Hefei, China) between 10 August 2020 and 14 December 2020. The study was conducted in full compliance with the International Conference on Harmonisation Guidelines for Good Clinical Practice and the ethical standards of the Declaration of Helsinki. Written informed consent was obtained from all subjects before enrolment.
Inclusion and Exclusion Criteria
The inclusion criteria were as follows: (1) male or female volunteers 18 years of age and over; (2) body mass index (BMI) between 19.0 and 26.0 kg/m2; (3) having no fertility plan and take effective contraceptive measures voluntarily from 2 weeks before screening to 6 months after the last administration of the study drug and no sperm or egg donation plan; and (4) willing and able to provide informed consent and willing to accept the trial requirement. Exclusion criteria were based on medical history, physical examination, electrocardiograms, and routine laboratory tests (detailed in Supplementary S1).
Randomisation and Blinding
Random table was generated by the statistical unit on the computer using SAS (version 9.4) in 1:1 block. The random table was reproducible, and the set of initial seed parameters of the random number was saved. Subjects were given a unique number in the order of signing the informed consent form. The subjects were randomly divided into two dosing sequence groups (TR or RT group) according to the pre-made random table and received the corresponding study drugs according to the corresponding dosing sequence. The study was conducted in two periods, the wash-out period of the two periods was 14 days (more than 7 terminal half-lives period). Personnel who were responsible for sample testing were blinded to the subject’s drug administration. Clinical investigators, project managers, project supervisors, data management and statistical analysis personnel were not blinded.
Drug Product and Administration
The T preparation was regorafenib tablets (40 mg, batch number: 20032521, content: 99.0%, expiration date: 2022.02; produced by Yangzijiang Pharmaceutical Group Co., Ltd). The R preparation was regorafenib tablets (Stivarga®) (40 mg, batch number: BXJ8S52, content: 100.4%, expiration date: 2022.02.24) produced by Bayer AG. All drugs were provided by Yangzijiang Pharmaceutical Group Co., Ltd., and sealed and stored below 25°C.
The recommended clinical dosage of this product in the reference preparation instructions is 160 mg (4 tablets, each containing 40 mg regorafenib).17 Considering the safety of the drug in healthy subjects and the declared specification of the tested preparations (40 mg), the dosage in this trial was set at a single oral dose of 40 mg (1 tablet) per period.
This study had two parts: fasting bioequivalence trial and fed (high-fat and high-calorie diet) bioequivalence trial. In the fasting trial, after fasting overnight for at least 10 hours, the subject took one tablet of the T preparation or the R preparation under the condition of an empty stomach, and the drug was taken with 240mL of water without chewing. After taking the medication, the researcher checked the subject’s mouth, hands, and medicine container to ensure that the medicine had been taken correctly. Lunch was given 4 hours after the medication taking. In the fed trial, after fasted overnight for at least 10 hours, the subjects consumed a high-fat and high-calorie meal 30 minutes before the administration. At 30min (±1min) from the start of mealtime, the subjects took one tablet of T preparation or R preparation with 240mL of water. The subjects were forbidden to drink water from 1h before and after taking the medicine. Lunch was given 4 hours after taking the medication.
Outcomes
The primary outcomes were the pharmacokinetic parameters of regorafenib (Cmax, AUC0-144h, AUC0~∞). The secondary outcomes were the pharmacokinetic parameters of regorafenib (Tmax, t1/2z, AUC_%Extrap, λz) and metabolite M-2 (Cmax, AUC0-144h, AUC0~∞, Tmax, t1/2z, AUC_%Extrap, λz). The reason we have chosen to evaluate M-2 rather than M-5 was that previous study has shown higher inter-individual CV (>100%) for M-5.18 In addition, the 2014 Food and Drug Administration (FDA) draft guidance on regorafenib also recommended evaluation of both regorafenib and M-2.19
Blood Collection
The Tmax of the prototype drug of 160 mg regorafenib in healthy male volunteers was 4.0 h in the fasting state and was 6.0 h in the high-fat fed state.20 The mean elimination half-life (t1/2) of regorafenib and M-2 were 28 h and 25 h, respectively.20 According to the requirements of NMPA guidelines, dense sampling was conducted around Tmax, and the last sampling time was 144 h (larger than 3 elimination half-life). In the fasting condition, venous blood was collected at 26 time points: 0h before each cycle (within 1.0h before administration) and 30min, 1.0h, 1.5h, 2.0h, 2.5h, 3.0h, 3h 20min, 3h 40min, 4.0h, 4h 20min, 4h 40min, 5.0 h, 6.0h, 8.0h, 10.0h, 12.0h, 24.0h, 27.0h, 30.0h, 36.0h, 48.0h, 72.0h, 96.0h, 120.0h, and 144.0h after administration.
In the fed condition, venous blood was collected at 27 time points: 0h before each cycle (within 1.0h before administration) and 30min, 1.0h, 1.5h, 2.0h, 2.5h, 3.0h, 3.5h, 4.0h, 4.5h, 5.0 h, 5.5h, 6.0h, 7.0h, 8.0h, 9.0h, 10.0h, 12.0h, 24.0h, 27.0h, 30.0h, 36.0h, 48.0h, 72.0h, 96.0h, 120.0h, and 144.0h after administration. Blood sample (4 mL) was collected at each collection time point to EDTA-K2 anticoagulation vacuum blood collection tube.
Analytical Method
As per the FDA’s guidelines on bioequivalence studies of regorafenib, this study used a validated liquid chromatography-tandem mass spectrometry (LC-MS/MS) to determine the concentration of regorafenib and its main metabolite M-2 (regorafenib N oxide, C21H15ClF4N4O4) in plasma. LC–MS/MS analysis was carried out using an LC 30AD system (Shimadzu, Japan) coupled with a QTRAP 5500 triple quadrupole MS (AB Sciex, Canada) equipped with an ESI ion spray source. Detailed analytical methods and method validation were provided in Supplementary S2. The detection range of regorafenib was 2.00~1300 ng/mL, and the detection range of metabolite M-2 was 1.00~650 ng/mL. All plasma samples were detected with quality control samples evenly distributed among the tested samples. Internal standard (regorafenib-13C-d3 and M-2-13C-d3) matrix effects, lipemic effects, and haemolytic matrix effects did not interfere with the accurate quantification of regorafenib and its metabolite M-2 (regorafenib N-oxide) in plasma, which met the NMPA requirements of biological sample analysis. The incurred sample reanalysis indicated that the LC-MS/MS method was reproducible.
Adverse Events
The adverse events after taking regorafenib tablets were recorded according to the changes in clinical laboratory results (blood routine, blood biochemistry, urine routine, coagulation function, and thyroid function), vital signs, physical examination results, 12-lead electrocardiogram, and other indicators. Treatment emergent adverse event (TEAE) was coded by the MedDRA version 23.0,21 and the severity of adverse events was determined according to the NCI-CTCAE version 5.0.22 The strength of the relation between adverse events and the tested drug was divided into five levels including: definite, probable, possible, unlikely, and not unrelated. Events that were classified as being definite, probable, and possible related to the tested drugs were recorded as adverse drug reactions (ADRs).
Sample Size Calculations
The intra-individual variation of regorafenib in healthy people was 28.3%.20 Assuming unilateral α = 0.05, β = 0.1, and intra-CV = 30%, the geometric mean ratio of the T preparation and the R preparation was 0.95~1.05, the bioequivalence interval was 80.00%~125.00%, and the minimum sample size of the two-period crossover test was 52 cases. Considering a 20% dropout rate, 66 subjects were included for both fasting and fed conditions, and a total of 132 volunteers were included in the entire trial. PASS (version 11.0. 7) software was used for a sample size calculation.
Pharmacokinetic and Statistical Analyses
Pharmacokinetic parameters were analysed using data from subjects who received at least one study drug. The pharmacokinetic parameters of regorafenib and metabolite M-2 were calculated by Phoenix WinNonlin (version 8.1, Certara, Princeton, NJ, USA) using a non-compartmental analysis (NCA) model. All pharmacokinetic parameters are presented as means ± standard deviation (SD), CV and geometric mean ratio (GMR), except for Tmax values, which are reported as the median, maximum, and minimum values.
Statistical analysis was conducted by SAS (version 9.4) software. Differences in natural logarithmic-transformed pharmacokinetic parameters (Cmax, AUC0-144h, AUC0-∞) between the two formulations were evaluated by linear mixed effects models, in which the sequence, drugs, and period were modelled as fixed effects, and participant ID was modelled as a random effect. The 90% confidence intervals (CIs) of the geometric mean for the T/R ratios (Cmax, AUC0-144h, AUC0-∞) were obtained to determine the bioequivalence, considering the acceptance range of 80~125%. Randomised participants whose data of serum concentration of regorafenib and M-2 were collected for at least one time were included in the PK analysis.
Sensitivity analyses of AUC0-∞ for regorafenib and M-2 bioequivalence between the T and R preparations were conducted, respectively. Participants with the AUC_%Extrap larger than 20% were excluded in the sensitivity analysis of PK parameters of the current period. The main parameters, such as Cmax, AUC0-144h, AUC0-∞, were regressed on age, sex, and BMI univariately to explore possible associations. All participants who were randomised and received a study drug were included in the safety analysis. A P value <0.05 was considered statistically significant.
Results
Subject Screening, Recruitment, and Compliance
A total of 358 healthy subjects were initially screened, with 66 participants (11 females 16.7%) were enrolled for the fasting period and 66 participants (13 females 19.7%) for the fed period, respectively. The mean (SD) age of participants was 29 (5.9) years old, and most of the participants were Han Chinese. Baseline demographics were similar between groups under the fasting and fed conditions (Table 1). All participants completed the fasting part, and only one participant withdrew from the second period of the fed condition due to an intestinal disorder (Figure 1).
 |
Table 1 Baseline Demographics of Full Analysis Set (FAS) |
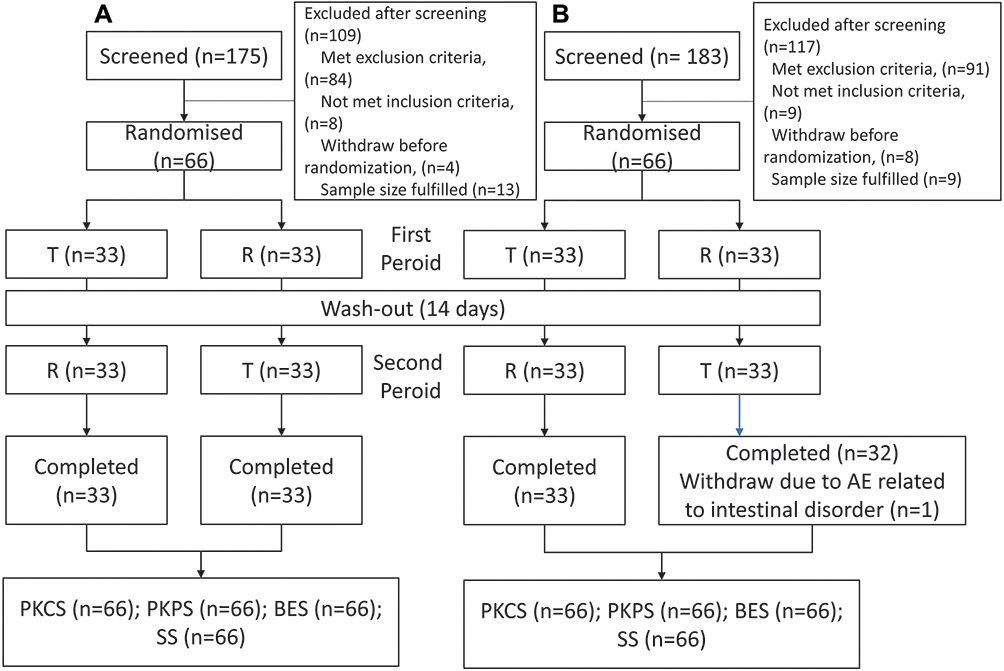 |
Figure 1 Flow charts of trials. (A) Fasting trial. (B) Fed trial. (One participant in fed trial withdrew due to intestinal disorder in second period (T drug), and the blood was taken only till 30h after drug administration. The participant reached Cmax on 4.5h, and the median Tmax was 5h. This participant was included in PKPS for Cmax and Tmax parameters and was only include in BES for Cmax parameter.). Abbreviations: T, test drug; R, reference drug; PKCS, pharmacokinetic concentration analysis set; PKPS, pharmacokinetic parameter analysis set; BES, bioequivalence analysis set; SS, safety analysis set. |
Plasma Concentration–Time Profiles
In the first period of the fasting condition, one participant vomited after taken the R treatment (within the two times median Tmax) and thus was not included in the pharmacokinetic parameter and bioequivalence analysis set. The linear plasma concentration and semi-logarithmic concentration plots were shown for the fasting (Figure 2) and fed conditions (Figure 3), respectively. The mean plasma concentration–time profiles of regorafenib and metabolite M-2 were superimposable between the two treatments under both fasting and fed conditions, which were characterised by a rapid increase in plasma concentration followed by a multiphasic decline.
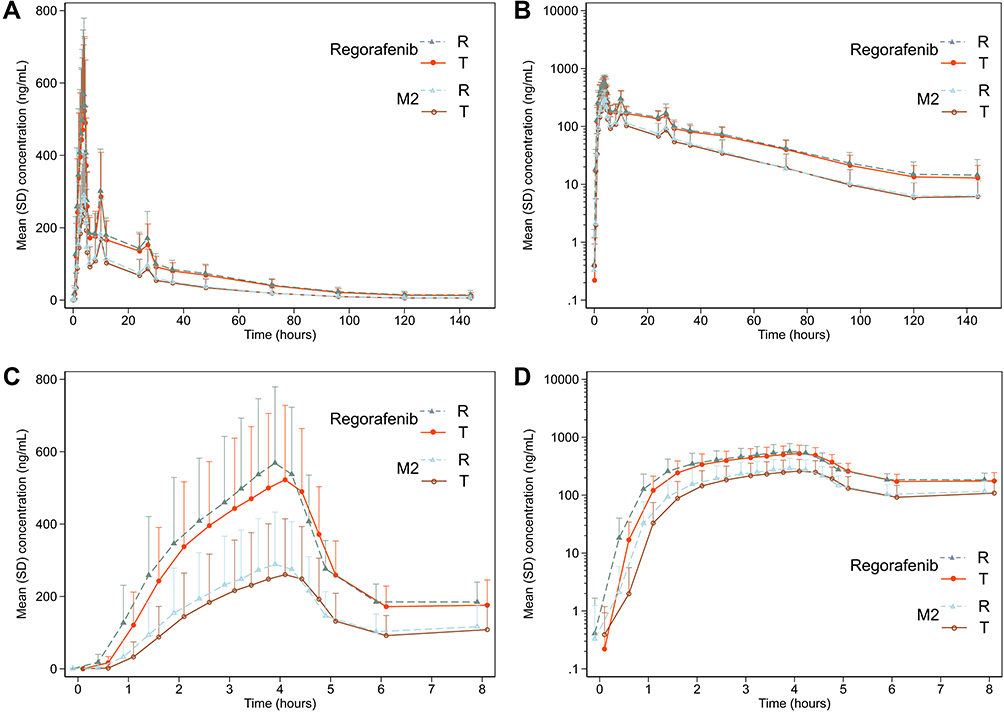 |
Figure 2 Plasma concentration–time plots of regorafenib and metabolite M2 of single oral regorafenib under the fasting condition on linear and semi-logarithmic scale (the test and reference treatments were jittered on time scale for better comparison). (A) Mean plasma concentration–time plot on linear scale; (B) mean plasma concentration–time on semilogarithmic scale; (C) mean plasma concentration–time plot on linear scale of first 8 hours; (D) mean plasma concentration–time on semilogarithmic scale of first 8 hours. Abbreviations: T, test drug; R, reference drug. |
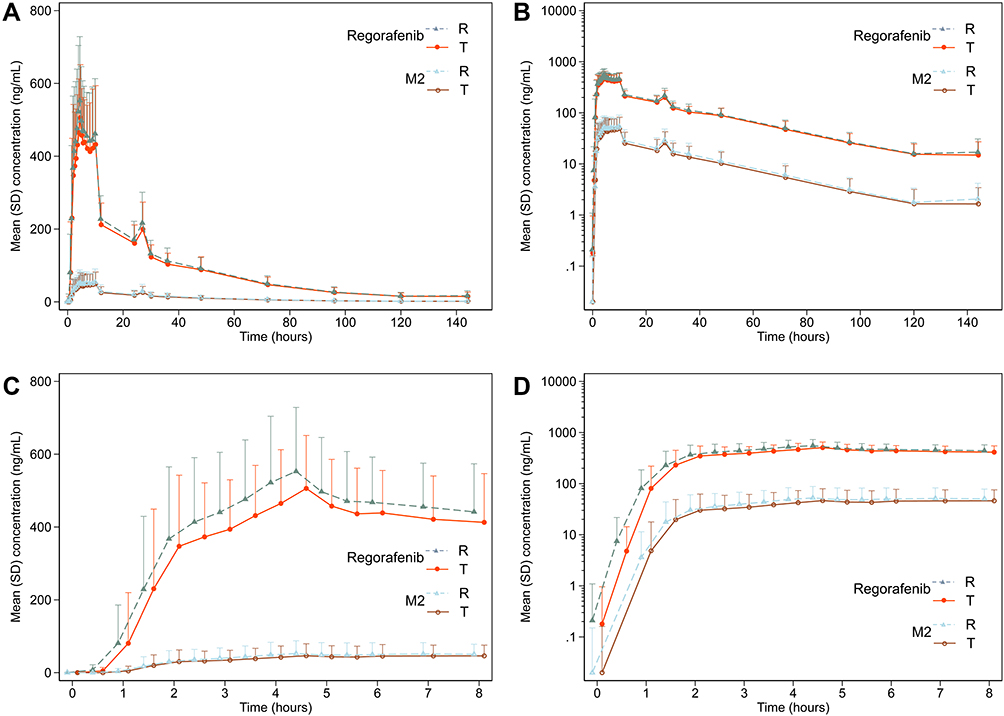 |
Figure 3 Plasma concentration–time plots of regorafenib and metabolite M2 of single oral regorafenib under the fed condition on linear and semi-logarithmic scale (the test and reference treatments were jittered on time scale for better comparison). (A) Mean plasma concentration–time plot on linear scale; (B) mean plasma concentration–time on semilogarithmic scale; (C) mean plasma concentration–time plot on linear scale of first 8 hours; (D) mean plasma concentration–time on semilogarithmic scale of first 8 hours. Abbreviations: T, test drug; R, reference drug. |
The PK parameters of regorafenib and M-2 for T and R treatments are listed in Table 2. Under both fasting and fed conditions, the mean values of the regorafenib PK parameters, including Cmax, AUC0-144h, AUC0-∞, t1/2z, and λz, were similar between the two treatments. The median Tmax of plasma regorafenib concentrations was 3.99 h under fasting condition and was 4.99 h under fed condition.
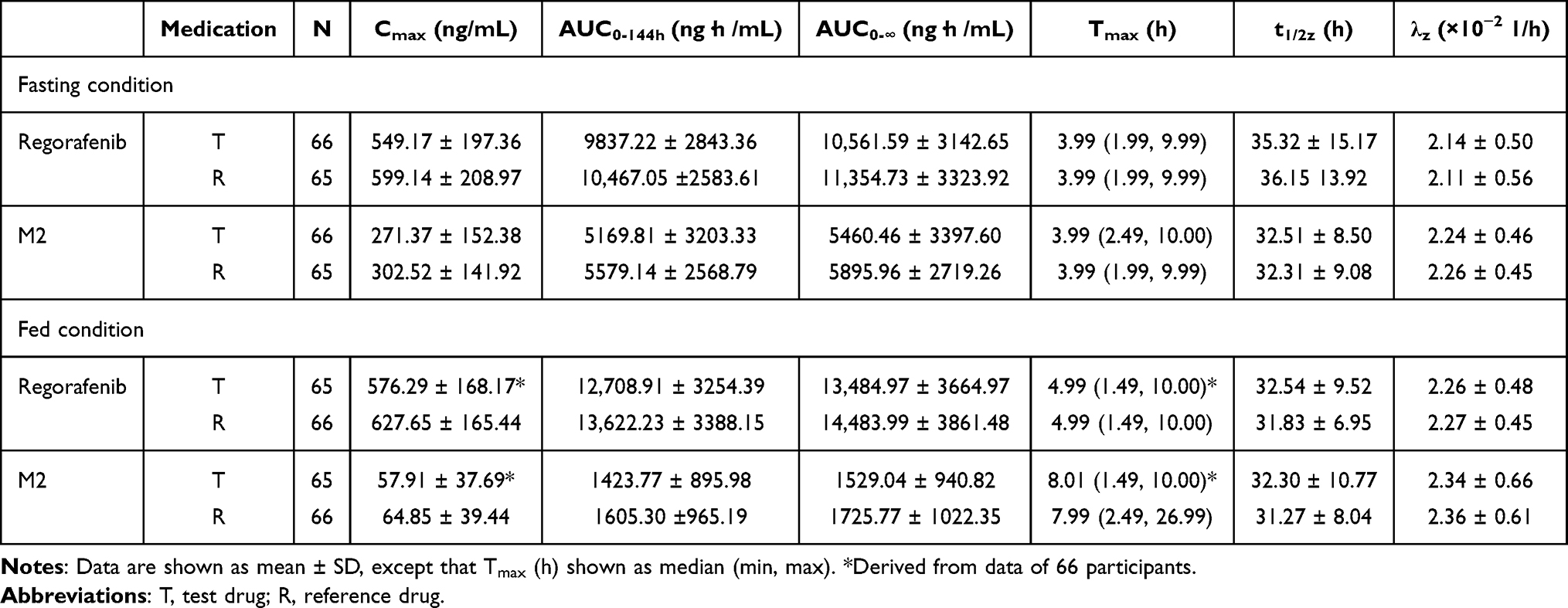 |
Table 2 The Pharmacokinetic Parameters of Regorafenib and Metabolite M2 |
Bioavailability and Bioequivalence Analysis
The geometric least square mean ratios (90% CI) for the logarithmically transformed pharmacokinetic parameters of regorafenib and metabolite M-2 were shown for the fasting period (Table 3) and fed period (Table 4), respectively. The corresponding 90% CIs for regorafenib were completely contained within the equivalence margin of 80–125% under both fasting and fed conditions. However, most of the corresponding 90% CIs for metabolite M-2 transcended the lower equivalence margin of 80%. Sensitivity analyses of AUC0-∞ showed similar results for regorafenib and were similar for metabolite M-2 except that AUC0-∞ of metabolite M-2 became bioequivalent for T and R treatments under the fed condition.
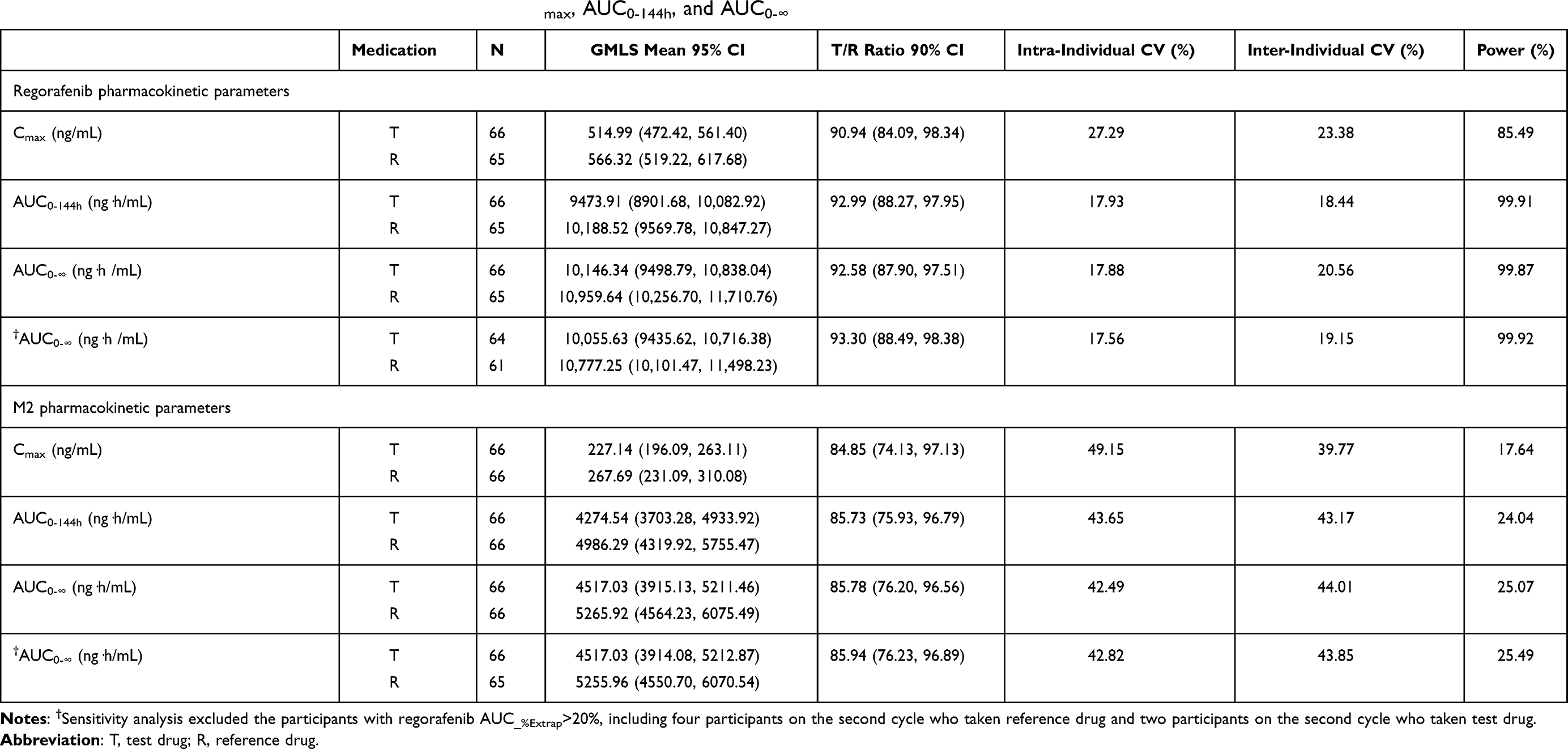 |
Table 3 Bioequivalence Analysis of Fasting Trial in Terms of Cmax, AUC0-144h, and AUC0-∞ |
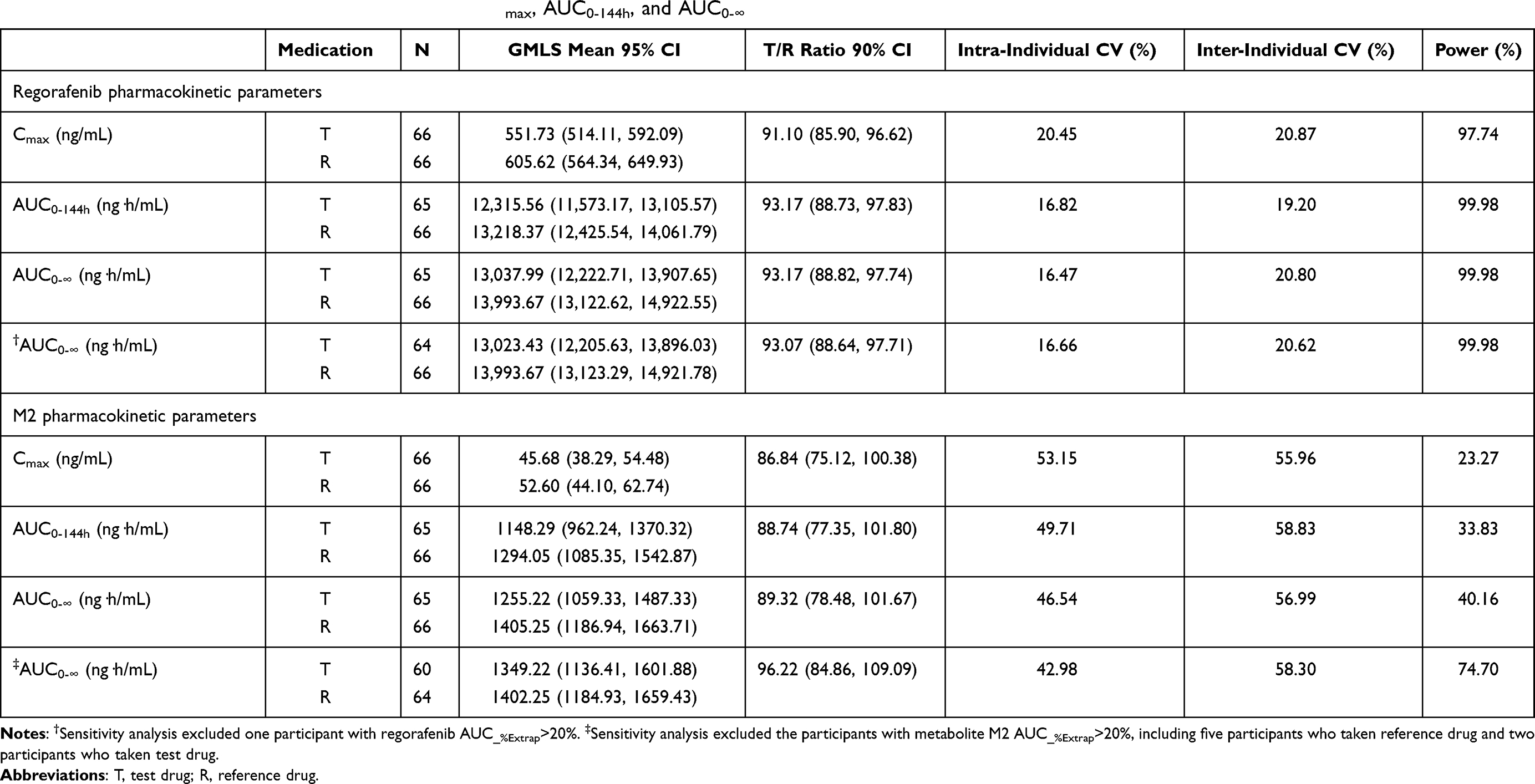 |
Table 4 Bioequivalence Analysis of Fed Trial in Terms of Cmax, AUC0-144h, and AUC0-∞ |
Exploratory Analysis
Compared to the fasting condition, a high-fat and high-calorie meal increased the exposure of plasma concentration of regorafenib (AUC0-144h 2924.45 ng·h/mL, 95% CI [2043.32, 3841.58]) but decreased the plasma concentration of M-2 (AUC0-144h −3836.82 ng·h/mL, 95% CI [−4477.33, −3196.31]). Compared to male, female participants had a shorter Tmax but a longer t1/2z for serum regorafenib. Higher BMI was associated with the shorter Tmax and slower λz for plasma regorafenib levels (Supplementary Table S1).
Tolerability and Safety Analysis
All participants (n = 132) who received assigned medications were included in the safety analysis (Table 5). When taking medications under the fasting condition, 18 participants reported TEAE, of which 10 (15.2%) reported the events in test treatment cycle and 13 (19.7%) in reference treatment cycle. When taking medications under the fed condition, 20 participants reported TEAE, of which 12 (18.2%) participants reported in the test treatment cycle and 11 (16.7%) reported in the reference treatment cycle. In the fasting trial, all AEs were CTCAE grade 1 except two AEs were grade 2, which were “elevated thyroid stimulating hormone” and “reduced haemoglobin”. In the fed trial, all AEs were grade 1 except two AEs were grade 2, which were “lumbar sprain” and “intestinal disorder”. All AEs were followed up until recovery or improvement. No severe adverse events were recorded under both conditions, and the safety profile of both treatments was generally good under both fasting and fed conditions.
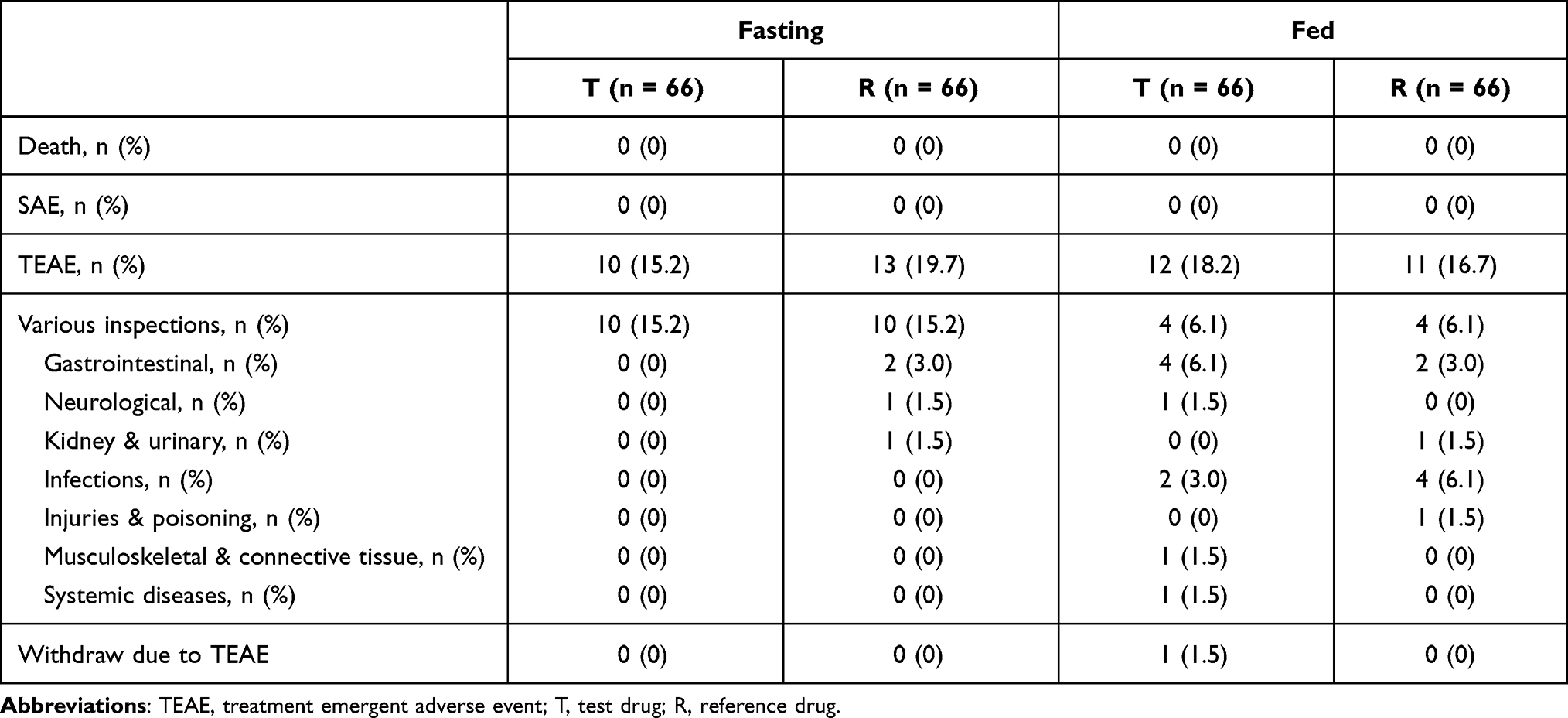 |
Table 5 Adverse Events |
Discussion
To our best knowledge, this study was the first bioequivalence trial conducted for two oral formulations of regorafenib among healthy Chinese volunteers under both fasting and fed conditions. The Cmax, AUC0-144h, and AUC0-∞ for regorafenib plasma concentration were bioequivalent between the T and R treatments under both fasting and fed conditions. This study provides novel evidence for the pharmacokinetic parameters of regorafenib among a healthy Chinese population.
According to the FDA reviews of data for 24 healthy American subjects,20 the Cmax and AUC0-144h for a single oral intake of 160 mg regorafenib (4 × 40 mg) were 1250 ng/mL and 45,390 ng·h/mL under fasted state and were 2160 ng/mL and 67,270 ng·h/mL under high-fat state. These data were higher than the parameters in the current study (40 mg) due probably to different doses administered. Compared to the fasted state, previous data showed a 48% of regorafenib exposure if taken under high-fat state.20 However, in the current study, fed regorafenib exposure increased 30.1% (R) and 29.2% (T) compared to fasting condition. Previous data suggested metabolite M-2 exposure decreased 20% on high-fat state compared to fasted state,20 and the current study reported more decrease (71.2% for R and 72.5% for T). These differences may possibly be explained by a different subject group (western versus Chinese population), the proportion of fat in the meal (the current study used high-fat and high-calorie meal), and different dose used.
The results of the secondary endpoints PK parameters were in agreement with previous data.10 We found that the median Tmax were 3.99 hours for regorafenib and metabolite M-2 under fasting condition. The mean t1/2z was around 36 hours for regorafenib and around 32 for metabolite M-2 under a fasting condition and was around 32 hours for both regorafenib and metabolite M-2 under a fed condition. The 90% CI of geometric mean for the T/R ratios of M-2 exceeded the range of 80%–120%, which may be caused by high intra- and inter-individual variability of M-2, and plasma levels of M-2 were significantly lower than regorafenib. The high intra- and inter-individual variability also caused the discrepancy in the power analysis (Tables 3 and 4). Bioequivalence should be based on the parent compound of regorafenib because sample size calculations were based on regorafenib rather than M-2, and the primary outcomes were PK parameters of regorafenib. Moreover, the 2021 updated draft guidance on regorafenib recommends detection of only regorafenib for the bioequivalence study.17
In the phase 1 trial of regorafenib in Japanese patients with solid tumours, after a single dose (160 mg), the Tmax for both regorafenib and metabolite M-2 was around 4 hours.14 One phase 1 dose–escalation study suggested that plasma concentration profile of regorafenib and its metabolite M-2 at steady state were multipeak shaped with an initial maximum Tmax of 1 to 6 hours.23 Our exploratory analysis suggests that gender and BMI may associate with the pharmacokinetic parameters of regorafenib, which was also reported in a previous study conducted on solid tumour patients.13
Previous trials have shown that regorafenib exhibits survival benefit compared to placebo in Asian patients with treatment-refractory mCRC,24 in whom dose-escalation strategy of regorafenib (80+, regorafenib at 80 mg per day, weekly dose escalation if no significant drug-related toxicities, up to 160 mg per day) may be a preferred option.25 A recent cost-effectiveness study suggested that regorafenib provided 0.79 quality adjusted life years (QALYs) at a cost of CNY 226657 (USD231697), which is less cost-effective than fruquintinib as a third-line treatment for patients with mCRC in China.26 However, the domestically produced test regorafenib, if approved, may have great implications for healthcare costs at both societal and individual levels.
The safety profiles of the test and reference treatments were similar and generally good. A single oral 40 mg regorafenib has an acceptable safety profile as the majority of AEs were CTCAE grade 1 in severity and resolved quickly. In the fasting trial, the most prevalent AEs were hypertension (T 3.0%; R 6.1%) and increased heart rate (T 3.0%; R 3.0%). While in the fed trial, the most prevalent AEs were urinary tract infection (T 3.0%; R 6.1%), abdominal pain (T 3.0%; R 3.0%), and diarrhea (T 3.0%; R 3.0%). The physical examination, 12-lead electrocardiogram, and other inspections of the subjects were normal or abnormal but with no clinical significance.
Key strengths of this study were its rigorous design, large sample size, and being the first bioequivalence trial of regorafenib conducted in a Chinese population. This study also has several limitations. First, the genotype of cytochrome P450 enzyme was not included in the trial, which makes comparing pharmacokinetic parameters between different CYP isoforms impossible. Second, regorafenib was recommended to be administered with low-fat diet,27 while we did not include an additional low-fat diet group. Third, we employed NCA models that did not take into account an EHC pathway. Since EHC is a significant pathway in which the regorafenib is metabolised and disposed,13 future investigations using pharmacokinetic models that consider EHC are needed.
Conclusion
Single oral intake of test (40 mg) and the reference (40 mg) regorafenib treatments were bioequivalent under fasting and fed conditions and had similar favourable safety profiles among healthy Chinese subjects.
Data Sharing Statement
The data that support the findings of this study are available from the corresponding authors upon reasonable request for legitimate research purpose.
Acknowledgments
We thank the participants for involving in this trial. This study was funded by Yangtze River Pharmaceutical Group Co., Ltd.; National Major Scientific and Technological Special Project (2020ZX09201014).
Disclosure
The authors declare no conflicts of interest for this work.
References
1. Dhillon S. Regorafenib: a review in metastatic colorectal cancer. Drugs. 2018;78(11):1133–1144. doi:10.1007/s40265-018-0938-y
2. Shirley M, Keating GM. Regorafenib: a review of its use in patients with advanced gastrointestinal stromal tumours. Drugs. 2015;75(9):1009–1017. doi:10.1007/s40265-015-0406-x
3. Heo YA, Syed YY. Regorafenib: a review in hepatocellular carcinoma. Drugs. 2018;78(9):951–958. doi:10.1007/s40265-018-0932-4
4. Bray F, Ferlay J, Soerjomataram I, Siegel RL, Torre LA, Jemal A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin. 2018;68(6):394–424. doi:10.3322/caac.21492
5. Chen W, Zheng R, Baade PD, et al. Cancer statistics in China, 2015. CA Cancer J Clin. 2016;66(2):115–132. doi:10.3322/caac.21338
6. Grothey A, Blay JY, Pavlakis N, Yoshino T, Bruix J. Evolving role of regorafenib for the treatment of advanced cancers. Cancer Treat Rev. 2020;86:101993. doi:10.1016/j.ctrv.2020.101993
7. Parsad S, Ratain MJ. Food effect studies for oncology drug products. Clin Pharmacol Ther. 2017;101(5):606–612. doi:10.1002/cpt.610
8. Stivarga® (regorafenib) [prescribing information]. FDA; 2015. Available from: https://www.accessdata.fda.gov/drugsatfda_docs/label/2012/203085lbl.pdf. Accessed July 01, 2021.
9. Stivarga: summary of product characteristics. Stivarga, Bayer Healthcare; 2015. Available from: https://www.ema.europa.eu/en/documents/product-information/stivarga-epar-product-information_en.pdf. Accessed July 01, 2021.
10. Gerisch M, Hafner FT, Lang D, et al. Mass balance, metabolic disposition, and pharmacokinetics of a single oral dose of regorafenib in healthy human subjects. Cancer Chemother Pharmacol. 2018;81(1):195–206. doi:10.1007/s00280-017-3480-9
11. Tlemsani C, Huillard O, Arrondeau J, et al. Effect of glucuronidation on transport and tissue accumulation of tyrosine kinase inhibitors: consequences for the clinical management of sorafenib and regorafenib. Expert Opin Drug Metab Toxicol. 2015;11(5):785–794. doi:10.1517/17425255.2015.1030392
12. Zopf D, Fichtner I, Bhargava A, et al. Pharmacologic activity and pharmacokinetics of metabolites of regorafenib in preclinical models. Cancer Med. 2016;5(11):3176–3185. doi:10.1002/cam4.883
13. Keunecke A, Hoefman S, Drenth HJ, Zisowsky J, Cleton A, Ploeger BA. Population pharmacokinetics of regorafenib in solid tumours: exposure in clinical practice considering enterohepatic circulation and food intake. Br J Clin Pharmacol. 2020;86(12):2362–2376. doi:10.1111/bcp.14334
14. Sunakawa Y, Furuse J, Okusaka T, et al. Regorafenib in Japanese patients with solid tumors: phase I study of safety, efficacy, and pharmacokinetics. Invest New Drugs. 2014;32(1):104–112. doi:10.1007/s10637-013-9953-8
15. Farha M, Masson E, Tomkinson H, Mugundu G. Food effect study design with oral drugs: lessons learned from recently approved drugs in oncology. J Clin Pharmacol. 2019;59(4):463–471.
16. FDA. Guidance for industry bioavailability and bioequivalence studies for orally administered drug products – general considerations; 2002. Available from: https://www.fda.gov/files/drugs/published/Guidance-for-Industry-Bioavailability-and-Bioequivalence-Studies-for-Orally-Administered-Drug-Products—General-Considerations.PDF.
17. FDA. Draft guidance on regorafenib; 2021. Available from: https://www.accessdata.fda.gov/drugsatfda_docs/psg/PSG_203085.pdf.
18. Strumberg D, Scheulen ME, Schultheis B, et al. Regorafenib (BAY 73-4506) in advanced colorectal cancer: a phase I study. Br J Cancer. 2012;106(11):1722–1727. doi:10.1038/bjc.2012.153
19. FDA. Draft guidance on regorafenib; 2014. Available from: https://www.accessdata.fda.gov/drugsatfda_docs/psg/Regorafenib_tab_203085_RC12-14.pdf.
20. FDA. Clinical pharmacology and biopharmaceutics review(S) [Stivarga]; 2012. Available from: https://www.accessdata.fda.gov/drugsatfda_docs/nda/2012/203085Orig1s000ClinPharmR.pdf.
21. MedDRA MS. Introductory guide for standardised MedDRA queries (SMQs) Version 23.1. Available from: https://admin.new.meddra.org/sites/default/files/guidance/file/SMQ_intguide_23_1_English.pdf.
22. Dueck AC, Mendoza TR, Mitchell SA, et al. Validity and reliability of the US National Cancer Institute’s Patient-Reported Outcomes Version of the Common Terminology Criteria for Adverse Events (PRO-CTCAE). JAMA Oncol. 2015;1(8):1051–1059. doi:10.1001/jamaoncol.2015.2639
23. Mross K, Frost A, Steinbild S, et al. A phase I dose-escalation study of regorafenib (BAY 73-4506), an inhibitor of oncogenic, angiogenic, and stromal kinases, in patients with advanced solid tumors. Clin Cancer Res. 2012;18(9):2658–2667. doi:10.1158/1078-0432.CCR-11-1900
24. Li J, Qin S, Xu R, et al. Regorafenib plus best supportive care versus placebo plus best supportive care in Asian patients with previously treated metastatic colorectal cancer (CONCUR): a randomised, double-blind, placebo-controlled, Phase 3 trial. Lancet Oncol. 2015;16(6):619–629. doi:10.1016/S1470-2045(15)70156-7
25. Sonbol MB, Benkhadra R, Wang Z, et al. A systematic review and network meta-analysis of regorafenib and TAS-102 in refractory metastatic colorectal cancer. Oncologist. 2019;24(9):1174–1179. doi:10.1634/theoncologist.2019-0189
26. Guan X, Li H, Xiong X, et al. Cost-effectiveness analysis of fruquintinib versus regorafenib as the third-line therapy for metastatic colorectal cancer in China. J Med Econ. 2021;24(1):339–344. doi:10.1080/13696998.2021.1888743
27. STIVARGA® product monograph. Bayer, Inc; 2013. Available from: https://omr.bayer.ca/omr/online/stivarga-pm-en.pdf.
 © 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.