Back to Journals » International Journal of Nanomedicine » Volume 12
Biodegradable polymeric micelles coencapsulating paclitaxel and honokiol: a strategy for breast cancer therapy in vitro and in vivo
Authors Wang N, Wang Z, Nie S, Song L, He T, Yang S, Yang X, Yi C, Wu Q, Gong C ![]()
Received 17 October 2016
Accepted for publication 16 January 2017
Published 23 February 2017 Volume 2017:12 Pages 1499—1514
DOI https://doi.org/10.2147/IJN.S124843
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Professor Lei Yang
Ning Wang, Zhihan Wang,* Shihong Nie,* Linjiang Song, Tao He, Suleixin Yang, Xi Yang, Cheng Yi, Qinjie Wu, Changyang Gong
State Key Laboratory of Biotherapy and Cancer Center, Collaborative Innovation Center for Biotherapy, West China Hospital, Sichuan University, Chengdu, People’s Republic of China
*These authors contributed equally to this work
Abstract: The combination of chemotherapy drugs attracts more attention in clinical cancer trials. However, the poor water solubility of chemotherapeutic drugs restricts their anticancer application. In order to improve antitumor efficiency and reduce side effects of free drugs, we prepared paclitaxel (PTX) and honokiol (HK) combination methoxy poly(ethylene glycol)–poly(caprolactone) micelles (P–H/M) by solid dispersion method against breast cancer. The particle size of P–H/M was 28.7±2.5 nm, and transmission electron microscope image confirmed that P–H/M were spherical in shape with small particle size. After being encapsulated in micelles, the release of PTX or HK showed a sustained behavior in vitro. In addition, both the cytotoxicity and the cellular uptake of P–H/M were increased in 4T1 cells, and P–H/M induced more apoptosis than PTX-loaded micelles or HK-loaded micelles, as analyzed by flow cytometry assay and Western blot. Furthermore, the antitumor effect of P–H/M was significantly improved compared with PTX-loaded micelles or HK-loaded micelles in vivo. P–H/M were more effective in inhibiting tumor proliferation, inducing tumor apoptosis, and decreasing the density of microvasculature. Moreover, bioimaging analysis showed that drug-loaded polymeric micelles could accumulate more in tumor tissues compared with the free drug. Our results suggested that P–H/M may have potential applications in breast cancer therapy.
Keywords: paclitaxel, honokiol, micelles, codelivery, breast cancer
Introduction
As one of the most common malignant tumors, breast cancer has shown an increasing incidence in recent years, and it is the most common cancer among women with an estimated 231,840 new cancer cases diagnosed in the USA up till 2015.1 Currently, chemotherapy remains one of the major treatments for patients with breast cancer in different stages. However, a large percentage of patients suffer undesirable severe side effects, low bioavailability, or drug resistance, resulting in therapeutic failure in clinical practice.2 Therefore, discovering more efficient and less toxic therapeutic strategies for breast cancer is getting popular attention.
Paclitaxel (PTX), a microtubule-stabilizing agent, is one of the most effective anticancer drugs in many tumors, including ovarian cancer, breast cancer, non-small-cell lung cancer, and head and neck carcinomas.3–5 However, the poor aqueous solubility of PTX has limited its clinical application. Cremophor EL and dehydrated alcohol (1:1, v/v) were used in the current clinical formulation of PTX to improve its solubility before diluting 5- to 20-fold in normal saline (NS) or dextrose solution. Unfortunately, Cremophor EL releases histamine after degradation in the body, causing serious side effects, such as hypersensitivity.6 It is imperative to develop novel strategies to enhance aqueous solubility of PTX, enhance antitumor effect, and avoid adverse effects, including those combining natural products or compounds with PTX or other standard chemotherapy reagents.
Honokiol (HK) is an active component isolated and purified from the roots, stem bark, or seed cone of the Chinese traditional herb magnolia. HK has long been known to have antithrombosis, antibacterial, and anxiolytic effects.7–9 In recent years, HK has been demonstrated to inhibit growth and induce apoptosis of different cancer cell lines, and was used as a potential antitumor agent.10–12 Furthermore, HK exhibits potent anticancer activities and further enhances conventional chemotherapies in a variety of preclinical models of human cancer, including chronic lymphocytic leukemia, prostate cancer, and multiple myeloma.13 HK used in this study had been extracted in our laboratory by high-capacity high-speed countercurrent chromatography (high-capacity HSCCC) with purities of 98.6%.14
Over the past few decades, polymeric micelles have attracted increasing attention and played an important role in drug delivery systems.15 The micelles formed a core-shell structure with hydrophobic drugs combining in the hydrophobic core and hydrophilic shell stabilizing the appearance in aqueous solution.16 This feature exhibits a crucial advantage to develop aqueous formulations for hydrophobic drugs.17 Therefore, polymeric micelles could encapsulate hydrophobic drugs, including even the combination of different chemotherapy drugs, to form stable and homogeneous aqueous solutions for intravenous applications.18–20 Besides, polymeric micelles exhibited good biodegradability and biocompatibility, which would avoid side effects on normal cells and also prolong the half-life of drugs in vivo.21
In this study, PTX and HK combination methoxy poly(ethylene glycol)–poly(caprolactone) (MPEG–PCL) micelles (P–H/M) were prepared using a nontoxic and biodegradable amphiphilic MPEG–PCL diblock copolymer. The experiments for detecting its antitumor activity were conducted in vitro and in vivo. Cytotoxicity, cellular uptake, apoptosis, and expression of caspase family in vitro were studied in detail. Subsequently, subcutaneous 4T1 tumor models were used to evaluate the antitumor activity of P–H/M. Microvasculature density of tumor, tumor proliferation, and tumor apoptosis assay were investigated in vivo. Furthermore, the target ability of the polymeric micelles was compared in vivo. Our findings indicated that P–H/M showed great antitumor activity and might have potential applications in breast cancer therapy.
Materials and methods
Materials, cell lines, and animals
PTX (Shanxi SENFU Biotechnology, Shanxi, People’s Republic of China), methyl thiazolyl tetrazolium (MTT; Sigma-Aldrich, St Louis, MO, USA), MPEG (molecular weight; Mn =2,000; Fluka, St Louis, MO, USA), ε-caprolactone (ε-CL; Alfa Aesar, Ward Hill, MA, USA), stannous octoate (Sn(Oct)2; Sigma-Aldrich), acetone (KeLong Chemicals, ChengDu, People’s Republic of China), dimethyl sulfoxide (DMSO; KeLong Chemicals), Annexin V-fluorescein isothiocyanate (FITC) Apoptosis Detection Kit (KeyGEN Biotech, Nanjing, People’s Republic of China), polyoxyethylene sorbitan monooleate (Tween 80; KeLong Chemicals), and antigens for Western blot (Abcam, San Francisco, CA, USA) were used as received. HK was purified in our laboratory.20 All the materials used in this article were analytic reagent (AR) grade and used as received.
Murine breast carcinoma cell line (4T1) and human embryonic kidney 293 (HEK 293) cell line were obtained from the American Type Culture Collection (ATCC; Rockville, MD, USA). The cells were grown in Roswell Park Memorial Institute-1640 (Gibco, Waltham, MA, USA), containing 10% heat-inactivated fetal bovine serum (FBS; Caoyuan Lvye, Huhht, People’s Republic of China) and 100 U/mL of penicillin and streptomycin.
Female Balb/c mice and female Balb/c nude mice were purchased from the Laboratory Animal Center of Sichuan University (Chengdu, People’s Republic of China). The animals were housed at a controlled temperature of 20°C–22°C and a relative humidity of 50%–60% with 12 h light–dark cycles. All the animals were in quarantine for a week before treatment. All animal procedures were performed following the protocol and guidelines of the Animal Welfare Act. All animal experiments were approved by the Institutional Animal Care and Treatment Committee of Sichuan University (Chengdu, People’s Republic of China).
Synthesis of the MPEG–PCL copolymer and preparation of the P–H/M
The MPEG–PCL diblock copolymer (Mn =4,000) was synthesized by ring-open polymerization of ε-CL on MPEG–PCL using Sn(Oct)2 as catalyst according to our previous reports.22,23 In brief, the MPEG and ε-CL were placed simultaneously into a dry glass ampule under nitrogen atmosphere, and a calculated amount of Sn(Oct)2 was added under mild magnetic stirring. The reaction system was kept at 130°C for 6 h. The final products were dissolved in dichloromethane and reprecipitated from filtrate using diethyl ether. Then, the MPEG–PCL copolymer was dialyzed molecular weight cutoff (MWCO): 3,000 Da) against double distilled water for 3 days, lyophilized, and stored in a glass desiccator before further use. The molecular weight of the obtained MPEG–PCL copolymer was 3,950 Da (2,000–1,950 Da), calculated by 1H nuclear magnetic resonance (400 spectrometer; Varian Medical Systems, Inc., Palo Alto, CA, USA).
P–H/M were prepared by solid dispersion method. Briefly, HK, PTX, and MPEG–PCL copolymers were dissolved in acetone and evaporated in a rotary evaporator at 60°C. Subsequently, the coencapsulation of drug and micelles was dissolved in water or saline at 60°C to self-assemble for P–H/M. The PTX-loaded micelles (PTX/M) and HK-loaded micelles (HK/M) were prepared in the same way. All the micelles were filtered through a 0.22 μm syringe filter (Millex-LG; Millipore Co., Billerica, MA, USA) and were lyophilized and stored at 4°C before use.
Characterization of the micelles
The particle size and zeta potential of the P–H/M were determined using the Malvern Nano ZS90 laser particle size analyzer. The samples were diluted with pure water for detection. The temperature was kept at 25°C during the measurement process. All results were the mean of three test runs, and all data were expressed as the mean ± standard deviation (SD).
The morphology of P–H/M was observed under a transmission electron microscope (TEM) (H-6009IV; Hitachi, Tokyo, Japan). The micelles were diluted with distilled water and placed on a copper grid covered with nitrocellulose. The samples were negatively stained with phosphotungstic acid and dried at room temperature.
Crystallographic study was performed on empty or drug-loaded micelles powder using X-ray diffractometer (XRD) (X’Pert Pro MPD; Philips, Amsterdam, the Netherlands). The samples were scanned by 2θ of angle measurement range 1°–160°.
The concentrations of PTX or HK were determined by a high-performance liquid chromatography (HPLC; LC-20AD; Shimadzu, Kyoto, Japan) instrument. The samples were diluted before measurement. Each sample was assayed three times. The drug loading (DL) efficiency and encapsulation efficiency (EE) of the P–H/M were determined according to the following equations. Briefly, 20 mg of lyophilized drug-loaded micelles was dissolved in 0.2 mL of acetonitrile. The DL and EE of P–H/M were calculated according to the following equations:
|
|
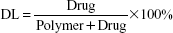
|
|
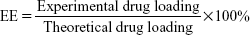
In vitro drug release behavior
To determine the release kinetics of HK or PTX from the micelles, we used a modified dialysis method as in previous work.24 Free PTX or free HK was dissolved in DMSO–water solution, and the concentration of PTX or HK was 1 mg/mL, which was the same dose as HK/M, PTX/M, and P–H/M. The solution was placed in a dialysis bag (MWCO is 3.5 kDa), which was incubated in phosphate-buffered solution (pH 7.4) containing Tween 80 (0.5%, w/w) with gentle shaking (100 rpm) in a 15 mL BD tube (Becton, Dickinson and Comany, Franklin Lakes, NY, USA) at 37°C. At predetermined time points, 1 mL of incubation medium was collected and the remaining medium was replaced with fresh medium. After centrifugation for 10 min, the supernatants of the removed release media were collected and stored at −20°C. The release behavior of PTX or HK was quantified using HPLC. All data are expressed as the mean ± SD.
Cytotoxicity assay
MTT method was used to study the cytotoxicity of PTX/M, HK/M, and P–H/M against 4T1 cells. Briefly, the 4T1 cells were plated in 96-well plates. After 12 h, the cells were exposed to PTX/M, HK/M, or P–H/M at various concentrations (0–40 μg/mL) for 48 h. The cell viability assay was performed using the MTT method. The experiment was performed in triplicate. The synergic index was calculated using the formula methods. In addition, cytotoxicity evaluation of the blank micelles (0–2,000 μg/mL) was conducted on 293T cells and 4T1 cells as control. All data are expressed as the mean ± SD.
Cellular uptake of PTX and HK
Quantification of cellular uptake of PTX and HK, PTX/M, HK/M, or P–H/M was evaluated on 4T1 cells at different time intervals (2 and 4 h). The drug concentration of PTX or HK was based on the half maximal inhibitory concentration (IC50) value calculated from the cell cytotoxicity test. The cells were cultured in a six-well plate for 24 h, and then the growth medium was replaced with serum-free medium containing a certain amount of PTX/M, HK/M, and P–H/M (25, 25, and 25+25 μg/mL). After incubation for 2 and 4 h, the cells were collected and lysed by radio-immunoprecipitation assay (RIPA) buffer (Sigma-Aldrich) for 30 min on ice, and PTX or HK was extracted by the mixture of ethyl acetate and n-hexane (1:3). The samples were measured by HPLC (LC-20AD).
Cell apoptosis assay
Apoptosis assay was performed by flow cytometry (FCM) (BD, FACSCalibur™; BD Biosciences, Lake Franklin, NJ, USA) using FITC-conjugated Annexin V/propidium iodide (PI) (KeyGEN Biotech) staining as per the manufacturer’s instructions. Briefly, 4T1 cells were incubated in a six-well plate for 12 h. Then, the cells were exposed to serum-free medium containing a certain amount of PTX/M, HK/M, or P–H/M (25, 25, and 25+25 μg/mL) for 24 h. The extent of apoptosis was evaluated by FCM. Both early apoptotic (Annexin V+/PI−) and late apoptotic (Annexin V+/PI+) cells were included in cell apoptosis determinations.
Western blot
To determine the inhibitory effect of P–H/M on the apoptosis signal pathway, 4T1 cells were cultured in dishes for 12 h. Then, the cells were treated with serum-free medium containing a certain amount of PTX/M, HK/M, and P–H/M (25, 25, and 25+25 μg/mL). After incubating for 24 h, the 4T1 cells were lysed by RIPA buffer for 30 min on ice. After centrifugation for 15 min at 4°C, the supernatant was collected and boiled with Sodium dodecyl sulfate (SDS) buffer for 5 min. Then, the samples were stored at −20°C for later use. These different protein samples were detected by SDS-polyacrylamide gel electrophoresis and incubated with antibodies (Abcam), including caspase 3, cleaved caspase 3, caspase 8, cleaved caspase 8, and caspase 9, cleaved caspase 9. Western blots were developed with horseradish peroxidase (HRP)-conjugated secondary antibodies and chemiluminescent substrate on Kodak X-ray films.
In vivo tumor model
Female BALB/c mice (aged 6–8 weeks), weighing 18±2 g, were used for in vivo antitumor tests. The mice were injected with 4T1 cell suspension (5×105 cells) into the right buttock. When the mean tumor volume reached 100 mm3, the tumor-bearing mice were randomly divided into five groups (six mice per group). The mice were injected intravenously every 3 days with 100 μL of PTX/M (the concentration of PTX was 10 mg/kg body weight), HK/M (the concentration of HK was 10 mg/kg body weight), or P–H/M (the concentration of HK or PTX was 10 mg/kg body weight). The NS and blank micelles groups were used as control. The tumor length and width were measured every 3 days, and the tumor volume was calculated using the equation Vol =0.5× length × width2. When the control mice became weak, the mice were sacrificed and their tumors were removed for immunohistochemical analysis and terminal deoxynucleotidyl transferase dUTP nick end labeling (TUNEL) assay.
Assays of apoptosis, proliferation, and microvessels in tumor tissues
The 4T1 tumors were fixed in 4% paraformaldehyde solution and then embedded in paraffin. A commercially available TUNEL kit (Promega, Madison, WI, USA) was used to identify apoptotic cells in the tumors. This analysis was performed following the manufacturer’s protocol, and the samples were then examined using a fluorescence microscope (×400).
The proliferation of tumor cells was detected by Ki-67 staining method. The primary antibody was rat antimouse monoclonal antibody Ki-67 (GeneTex Inc., Irvine, CA), and the secondary antibody was biotinylated goat antimice immunoglobulin (BD Biosciences). The Ki-67 labeling index (LI) was calculated as the number of Ki-67-positive cells/total number of cells counted under magnification (×400). The data were counted in five randomly selected areas in each tumor sample by two independent investigators.
For blood vessel staining, the tumor tissues were immunostained with epithelial cell marker goat antimouse CD31 antibody (dilution 1:100; Santa Cruz Biotechnology, Santa Cruz, CA, USA) and rabbit antigoat FITC (dilution 1:100; Santa Cruz Biotechnology) secondary antibody. Microvessel density was determined as the average number of small CD31-positive vessels in a high-power (×400) field.
Bioimaging
The cell uptake and in vivo behavior of drug-loaded micelles were investigated using fluorescent dye, 1,1′-dioctadecyl-3,3,3′,3′- tetramethylindodicarbocyanine, 4-chlorobenzenesulfonate salt (DiD), as the cell membrane fluorescent probe. The particle size of DiD/M was determined by Malvern Nano ZS90 laser particle size analyzer. The DiD/M were diluted with pure water for detection. All results were the mean of three test runs, and all data are expressed as the mean ± SD. For the in vivo animal tests, 5-week-old female nude BALB/c mice were bearing breast cancer cells (1.0×107) in the right buttock. When the tumor volume reached ~300–400 mm3, DiD-loaded MPEG–PCL micelles (DiD/M) or free DiD was intravenously injected through the tail vein. After injection, optical fluorescence imaging was performed by positioning each mouse on an animal plate in the IVI Spectrum system (Caliper Life Sciences, Hopkington, MA, USA). The images were obtained 1, 4, 8, and 12 h after injection of the test drugs.
Statistical analysis
All data are expressed as mean ± SD. Statistical analysis was performed by one-way analysis of variance. Significant differences between groups are indicated by *P<0.05, **P<0.01, and ***P<0.001.
Results
Preparation and characterization of P–H/M
Self-assembly behavior of P–H/M is presented in Figure 1A. PTX and HK were distributed into the PCL hydrophobic core, while PEG was the hydrophilic segment outside the micelles. After purification, MPEG–PCL copolymer was characterized. Then, we prepared different kinds of drug-loaded micelles samples and evaluated different mass ratios, especially the mass ratio of P–H/M, and the results are shown in Table 1. Considering this, the S2 sample had the most appropriate ratio for the P–H/M combination system. The EE of S2 sample was 98.2%±0.2%, and the DL was 9.4%±0.3%. The particle size of S2 was 28.7±2.5 nm with the polydispersity index (PDI) of 0.06±0.01, and the zeta potential was −1.42±0.21 mV (Figure 1B and C). In Figure 2A, the TEM image demonstrated that P–H/M were a homogeneous spherical shape with small particle sizes ~20 nm, which revealed that P–H/M could be homodispersed and stable in water solution. As shown in Figure 2B, PTX and HK could be coencapsulated into polymeric micelles and they formed a stable micelles system.
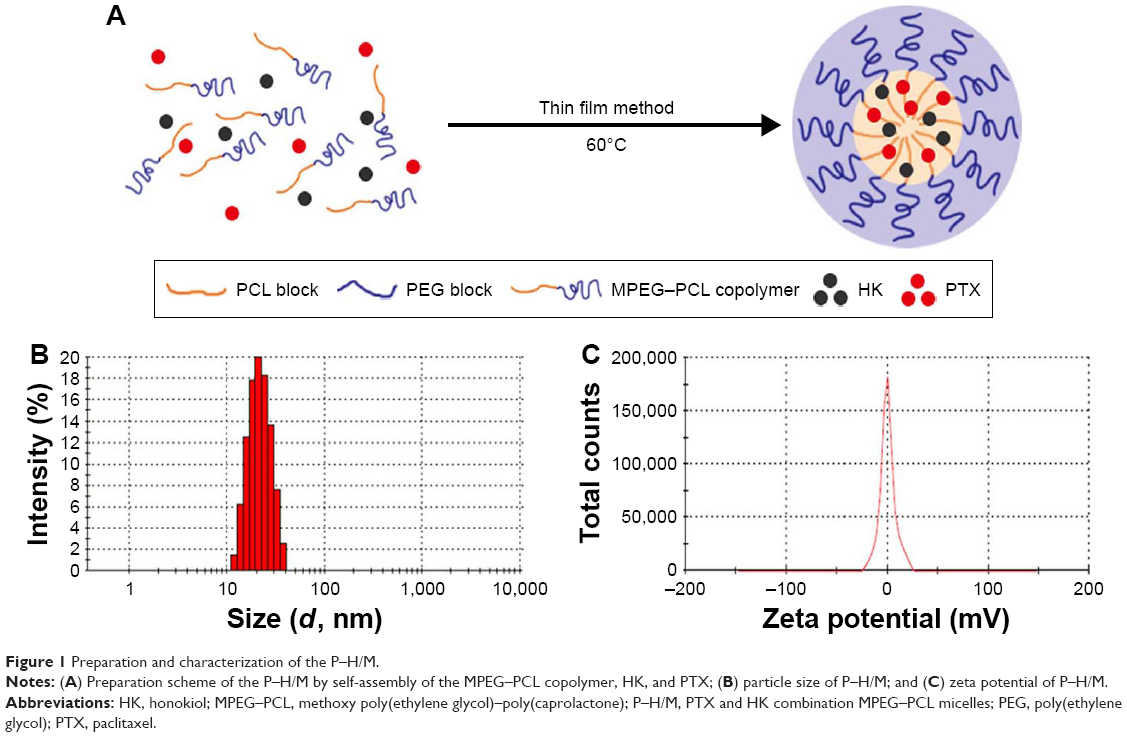 | Figure 1 Preparation and characterization of the P–H/M. |
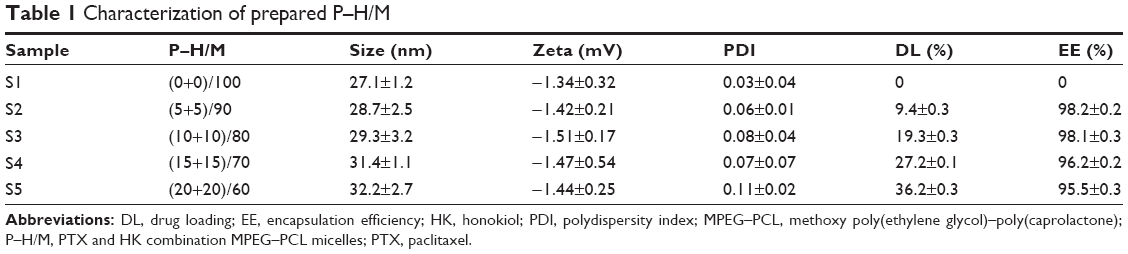 | Table 1 Characterization of prepared P–H/M |
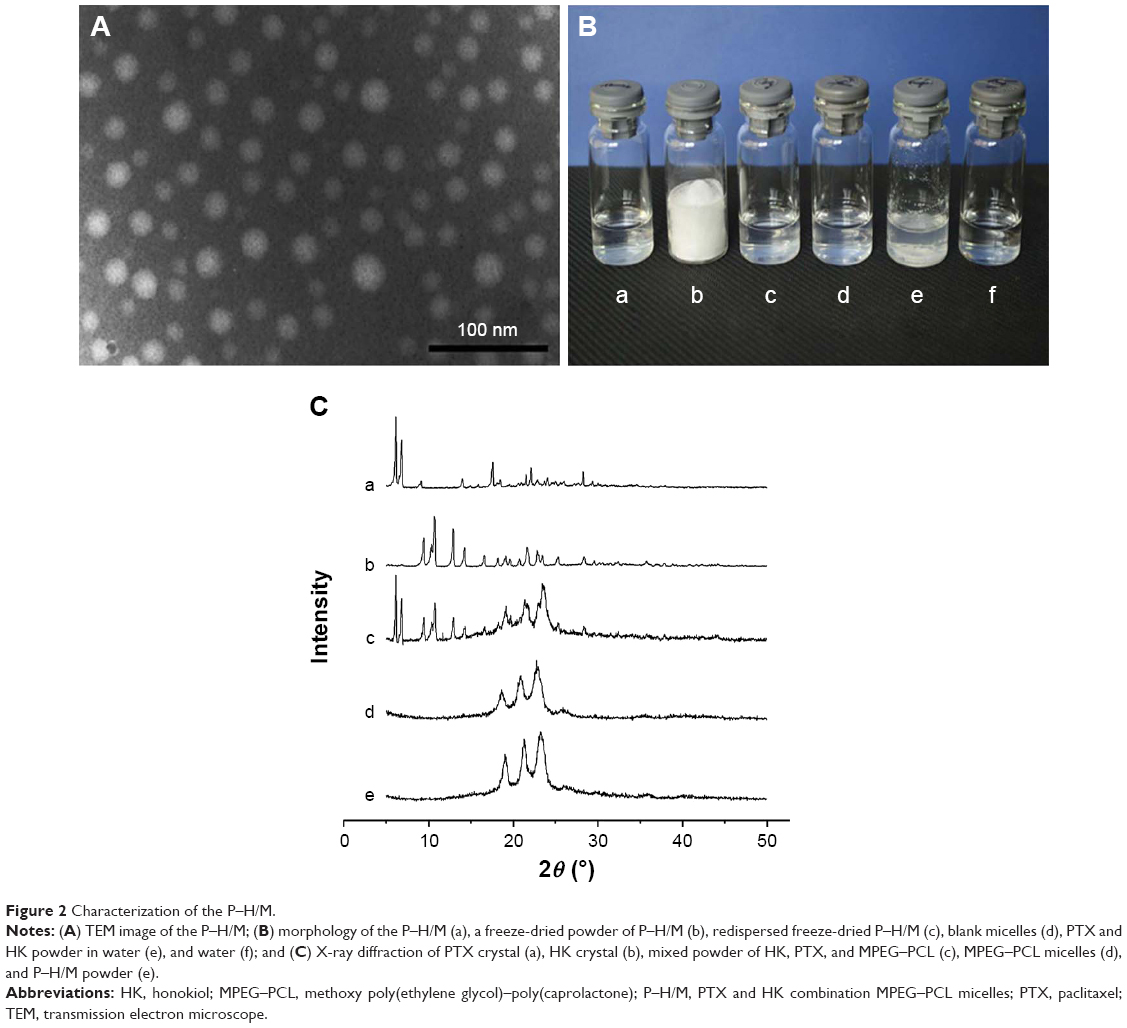 | Figure 2 Characterization of the P–H/M. |
Crystallographic assay was performed by XRD, and the spectra are shown in Figure 2C. It was found that the characteristic diffraction peaks of PTX and HK in P–H/M disappeared in the XRD spectrum and there were only peaks of MPEG–PCL copolymer, which indicated that PTX and HK were incorporated into the MPEG–PCL micelles.
Results in Figures 1 and 2 suggested that MPEG–PCL copolymer and drug-loaded micelles were prepared successfully. P–H/M were prepared without using any surfactants or toxic organic solvents and were stable in aqueous solution with small particle size, were nearly neutral and had high drug loading, which indicated that P–H/M could be applied to in vitro experiments and administrated intravenously in vivo.
In vitro drug release behavior
The release profiles of the drug-loaded micelles were detected by HPLC. As shown in Figure 3A, ~87.3%±2.1% of the PTX was released to media from the free PTX group in 48 h, whereas only ~32.0%±1.6% or 25.0%±2.3% was released from the PTX/M or P–H/M group. As shown in Figure 3B, free HK was rapidly released, 88% in 48 h, whereas HK was released only ~45.3%±1.2% or 44.2%±3.1% from the HK/M or P–H/M group. After 2 weeks, 62.5%±1.3% of the PTX was released from the PTX/M group and 58.4%±2.1% of the PTX was released from the P–H/M group, and 69%±2.42% of the HK was released from the HK/M group and 71.2%±3.1% of the HK was released from the P–H/M group. The results indicated that cumulative release rate in drug-loaded micelles was much lower than that in the free drug group. The drug-loaded micelles showed a sustained-release behavior compared to free drug, and the release behavior of PTX and HK did not impact on each other in P–H/M. The release behavior suggested that a much slower release behavior of drug-loaded micelles could be attributed to the core-shell structure of polymeric micelles.
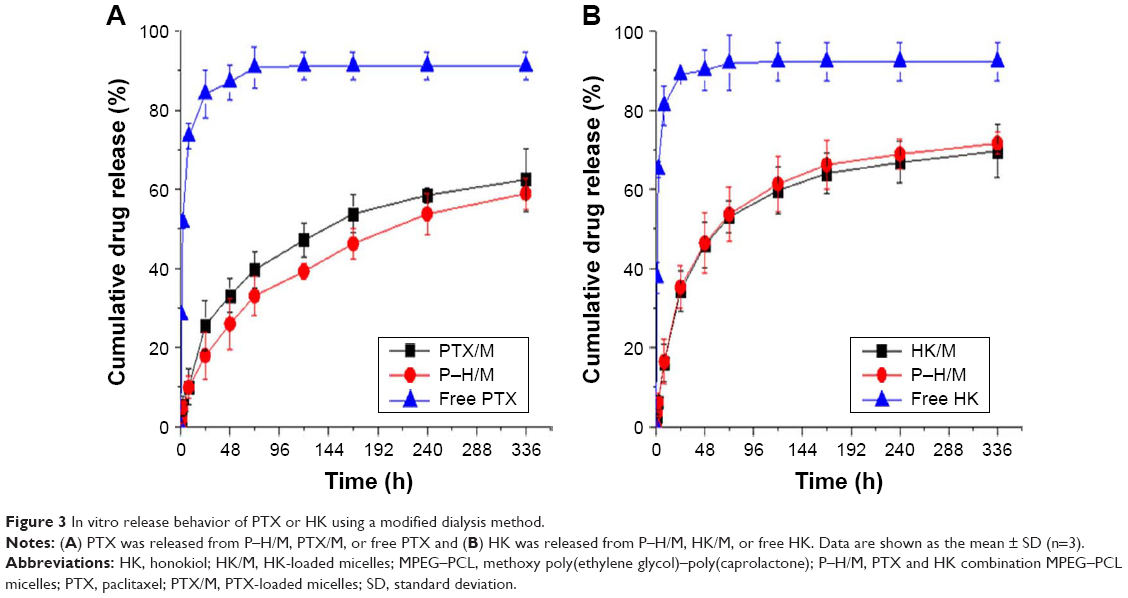 | Figure 3 In vitro release behavior of PTX or HK using a modified dialysis method. |
In vitro cytotoxicity study
Cytotoxicity assay of blank micelles, PTX/M, HK/M, and P–H/M against normal cells and cancer cells was performed using the MTT method. As displayed in Figure 4A and B, the results suggested that blank micelles had low cytotoxicity on 4T1 and HEK 293 cell lines and the cell viability was ~>90% after 24 or 48 h. To verify the cytotoxic activity of P–H/M, we conducted MTT assay on the 4T1 cells. As shown in Figure 4C, cell viability decreased in each group with a dose-dependent relationship with an increase in the concentration of drug-loaded micelles. We found that the IC50 value of P–H/M was much lower than that of PTX/M or HK/M (25.0 versus 42.4 and 65.1 μg/mL), which indicated that P–H/M improved cytotoxicity against 4T1 cells.
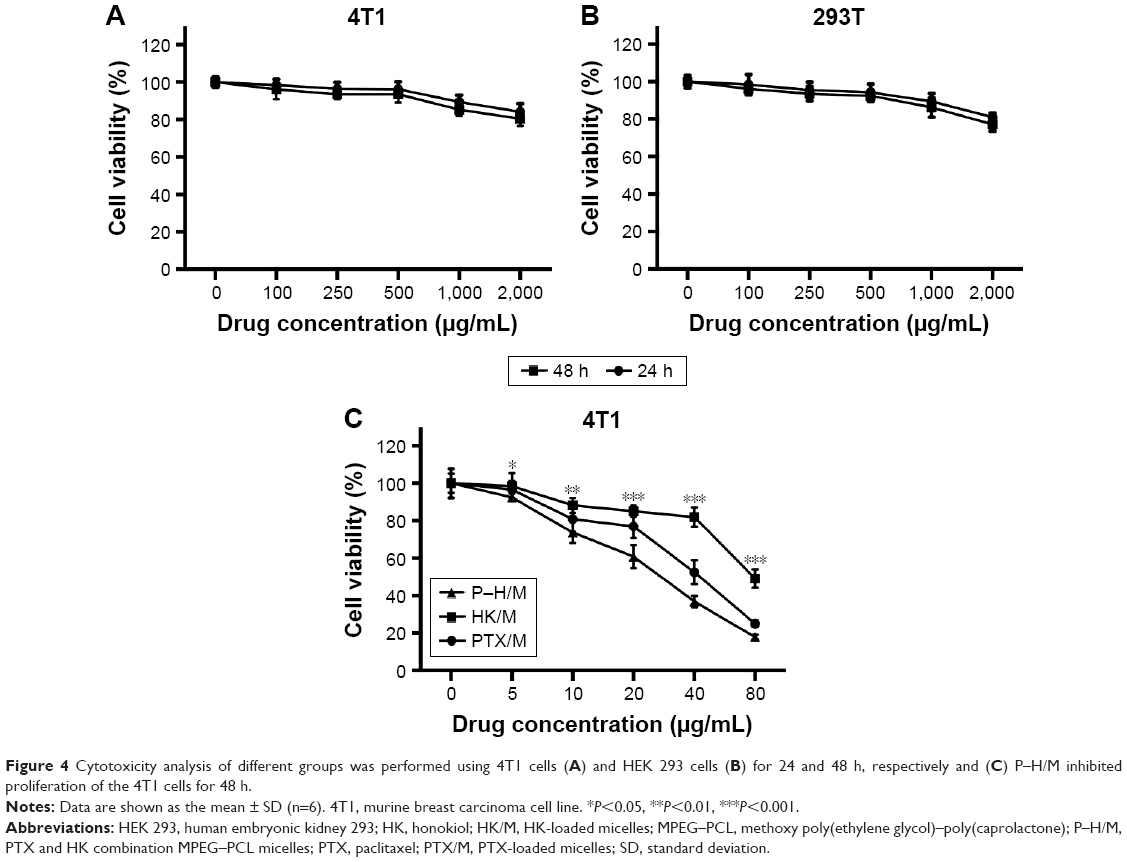 | Figure 4 Cytotoxicity analysis of different groups was performed using 4T1 cells (A) and HEK 293 cells (B) for 24 and 48 h, respectively and (C) P–H/M inhibited proliferation of the 4T1 cells for 48 h. |
In cytotoxicity studies, we found that the blank micelles had low cytotoxicity and good biocompatibility. The P–H/M displayed an enhanced toxicity compared to PTX/M and HK/M against 4T1 cell lines. All the results revealed that coencapsulated PTX and HK could improve cytotoxicity of drugs against breast cancer cells.
In vitro cellular uptake analysis
In order to study the progress of the improved cytotoxicity effect of P–H/M on 4T1 cells, we used a quantitative method to investigate the increasing amount of cellular uptake of PTX and HK. As shown in Figure 5A, PTX accumulated similarly in the PTX/M or P–H/M group in 4T1 cells after incubation for 2 or 4 h. Similarly, HK was taken up in an analogous way in both the P–H/M and HK/M groups after 2 or 4 h incubation in 4T1 cells (Figure 5B). The results indicated that both HK and PTX could accumulate in cancer cells, and each one did not affect the uptake of the other, increasing the cytotoxicity of the P–H/M dual–drug delivery system.
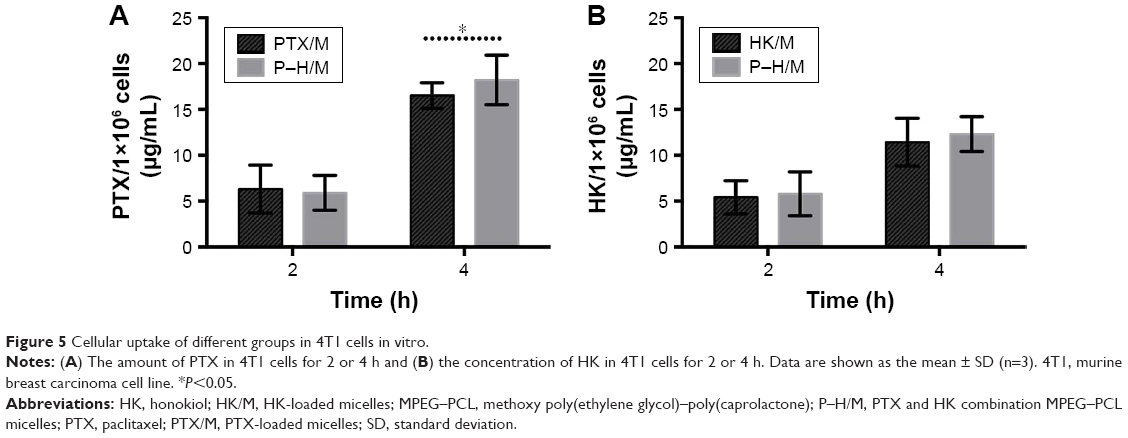 | Figure 5 Cellular uptake of different groups in 4T1 cells in vitro. |
Apoptosis assay of 4T1 cells
The apoptosis induction of P–H/M against 4T1 cells was analyzed by FCM assay. As shown in Figure 6, the percentage of apoptotic cells was 34.02%±0.05% in the P–H/M group, which was higher than that in the PTX/M (26.37%±0.08%) group and the HK/M (20.15%±0.11%) group. This indicated that P–H/M increased the apoptosis of 4T1 cancer cells more effectively than other groups in vitro (P<0.05). The results indicated that P–H/M could induce more apoptosis than control groups.
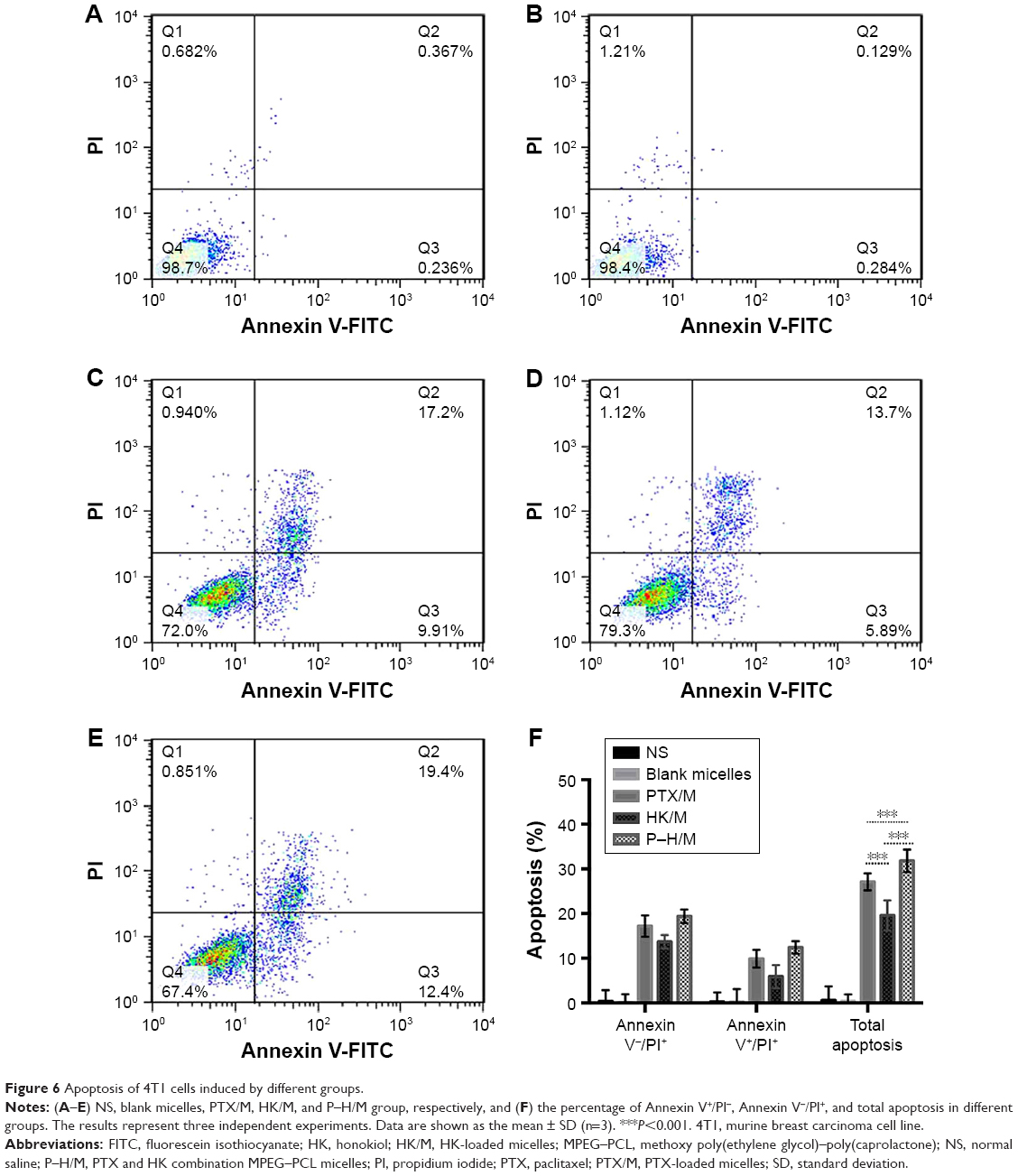 | Figure 6 Apoptosis of 4T1 cells induced by different groups. |
Western blot analysis
We used Western blot to detect the expression of apoptosis-related proteins in 4T1 cell lines. According to Figure 7, caspase family proteins (cleaved caspase 3, cleaved caspase 8, and cleaved caspase 9) were upregulated more in the micelles groups than in the NS group and the blank micelles group. In addition, the activation of cleaved caspase apoptosis protein was much more in the P–H/M group than in the PTX/M and HK/M groups. The NS group and the blank micelles group were used as controls. Western blot indicated that P–H/M increased more activation of caspase 3 (35 kDa), caspase 8 (57 kDa), and caspase 9 (45 kDa) compared to cleaved caspase 3 (17 and 19 kDa), cleaved caspase 8 (10 kDa), and cleaved caspase 9 (35 kDa) than others.
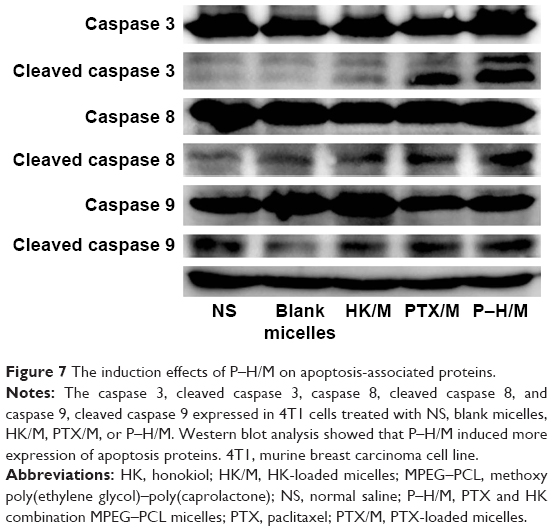 | Figure 7 The induction effects of P–H/M on apoptosis-associated proteins. |
Anticancer activity of P–H/M in vivo
We used Balb/c mice bearing subcutaneous 4T1 breast carcinoma to compare the antitumor effect of P–H/M in vivo with PTX/M, HK/M, blank micelles, and NS groups. As shown in Figure 8, we found that P–H/M (557.64±243.81 mm3) was more efficient in inhibiting’ tumor growth compared to PTX/M (932.70±256.43 mm3) and HK/M (1,463.77±148.74 mm3). The blank micelles (2,178.35±247.41 mm3) had no antitumor effect compared to the NS group (2,207.62±214.17 mm3). Meanwhile, the tumor weight of P–H/M was lower than that of PTX/M and HK/M groups, whereas blank micelles were similar to the NS group. The body weight measurements (Figure 8) showed no significant differences between the groups throughout the study. All the results in Figure 8 suggested that P–H/M were more efficient in suppressing tumor growth than PTX/M and HK/M.
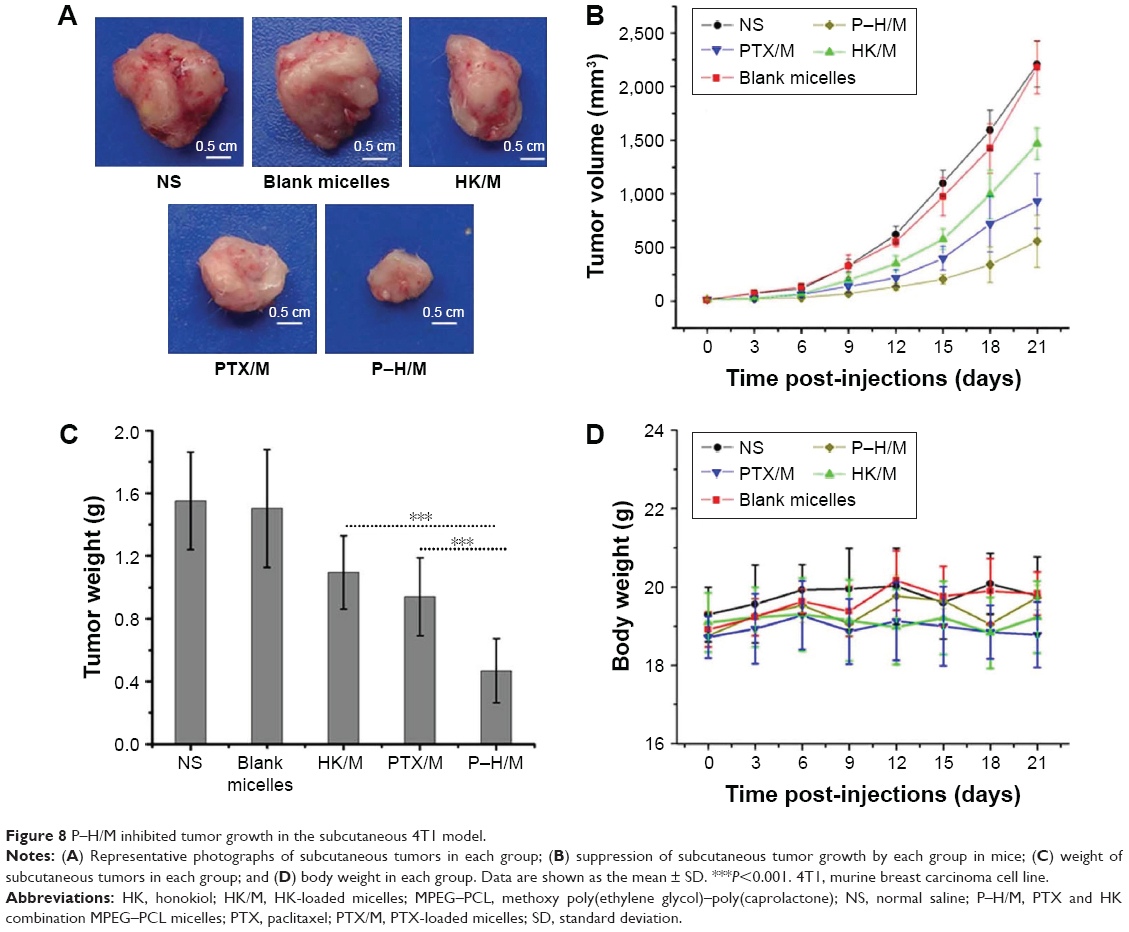 | Figure 8 P–H/M inhibited tumor growth in the subcutaneous 4T1 model. |
Hematoxylin-eosin (H&E) staining was performed for histological analysis on tumor tissues (Figure 9). Compared to the PTX/M and HK/M groups, the P–H/M group exhibited more necrosis areas. These results indicated that the P–H/M suppressed tumor cell proliferation and enhanced higher anticancer efficacy. In addition, the results showed no pathologic changes in the lung, heart, kidney, spleen, or liver, which proved that MPEG–PCL polymer had low side effects and good biocompatibility on normal tissues.
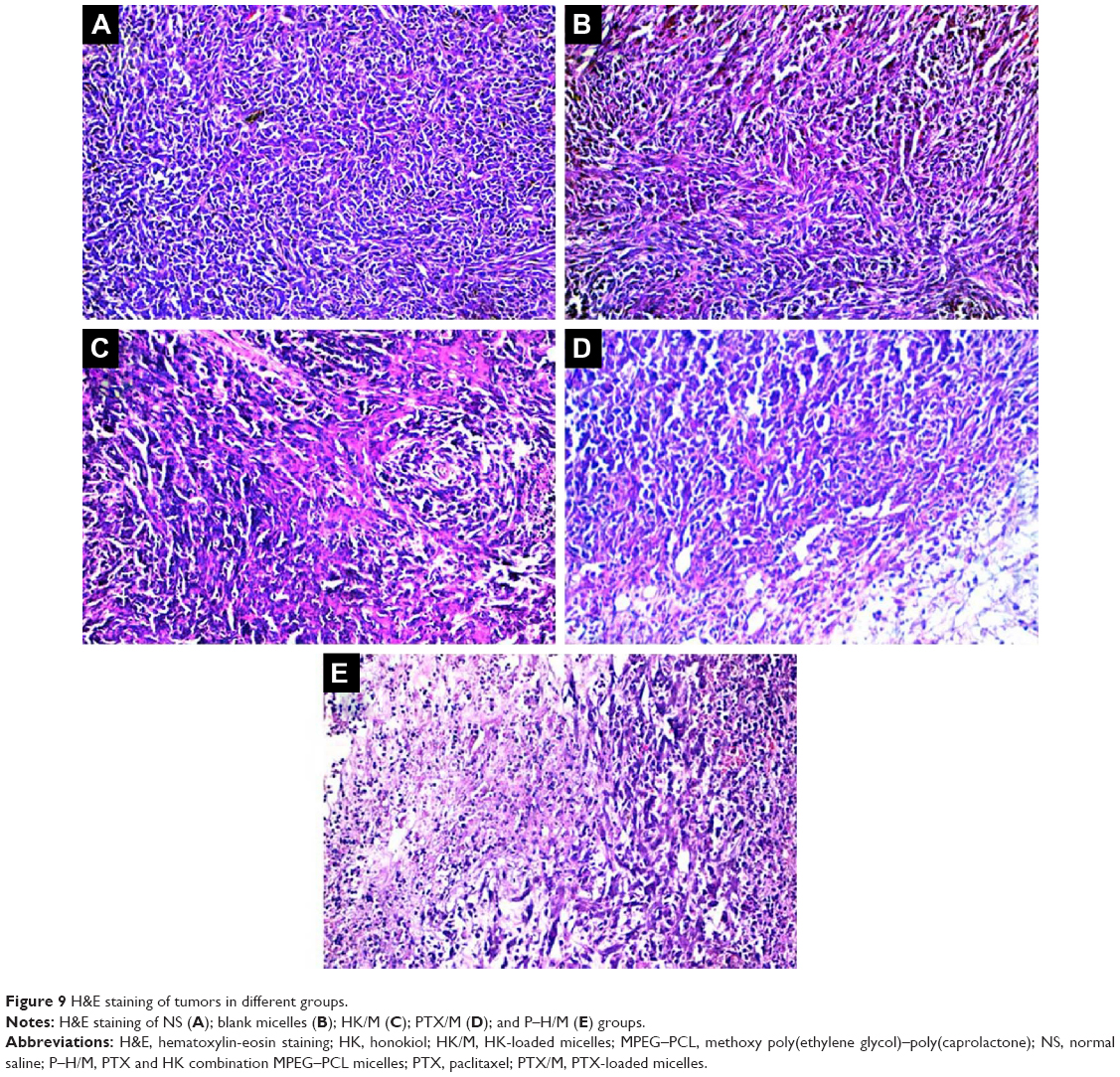 | Figure 9 H&E staining of tumors in different groups. |
Antiangiogenic effect in vivo
The vessel density of tumor tissues was tested by immunohistochemical CD31 staining on different groups in 4T1 breast cancer model. As displayed in Figure 10, P–H/M (18.54%±3.24%) appeared to significantly decrease the density of vessels compared to the PTX/M (43.72%±4.53%, P<0.05) and HK/M (47.14%±6.25%, P<0.05) groups. The NS group (85.37%±9.14%) and the blank micelles group (79.15%±7.82%) were used as controls. The results suggested that P–H/M dramatically decreased tumor vessel density compared to the other groups.
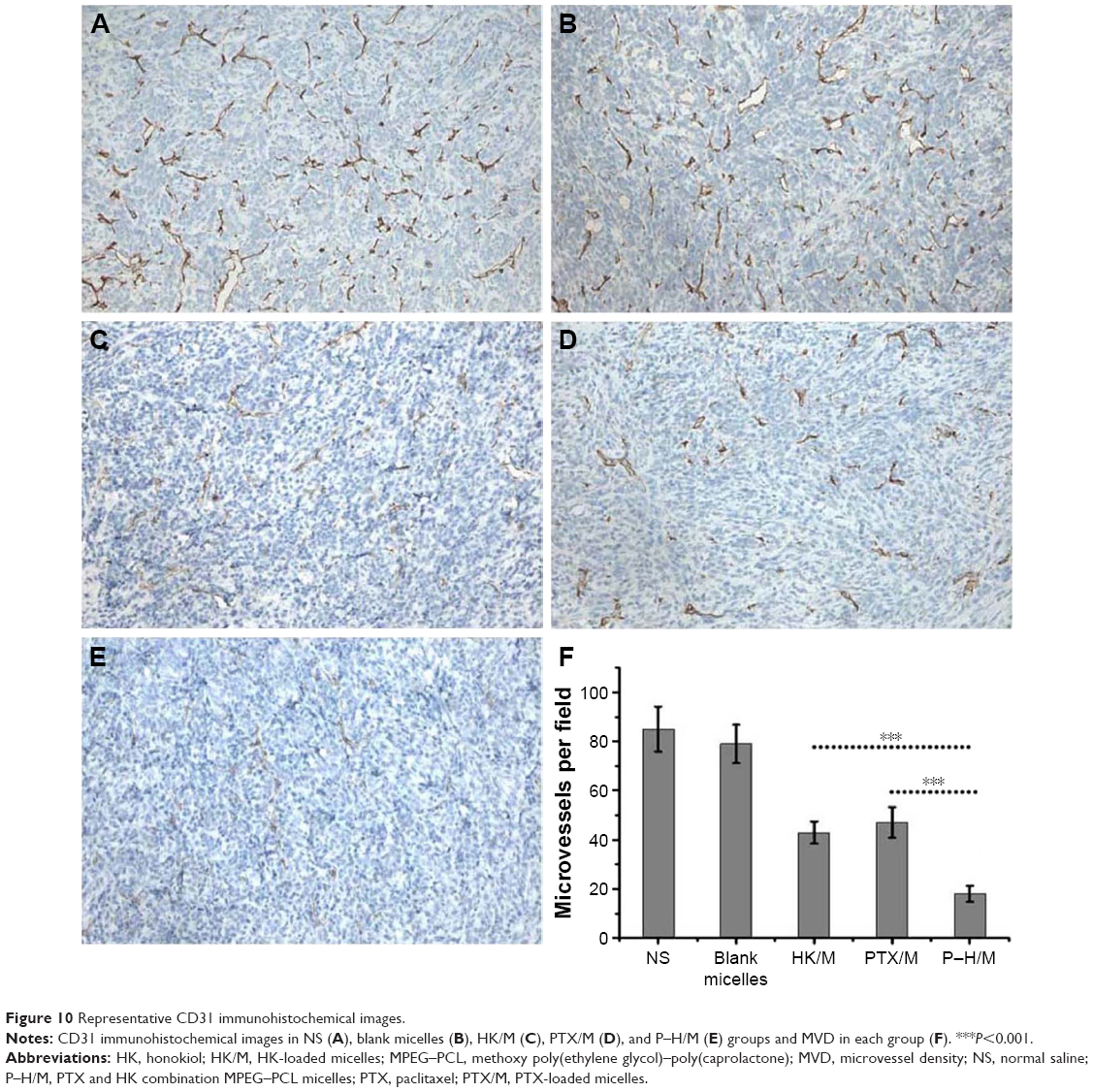 | Figure 10 Representative CD31 immunohistochemical images. |
Proliferation of tumor cells in vivo
Ki-67 immunohistochemical staining was used to examine the tumor proliferation activity of P–H/M in vivo. As shown in Figure 11, more Ki-67-positive cells could be observed in tumor tissue treated with the blank micelles group (78.60%±4.46%) and the NS group (74.31%±4.74%), whereas Ki-67-positive cells reduced significantly in the P–H/M group (16.52%±3.15%) compared to in the PTX/M (43.26%±5.32%, P<0.05) and HK/M (38.74%±4.27%, P<0.05) groups. The results implied that P–H/M could significantly inhibit tumor cell proliferation in vivo.
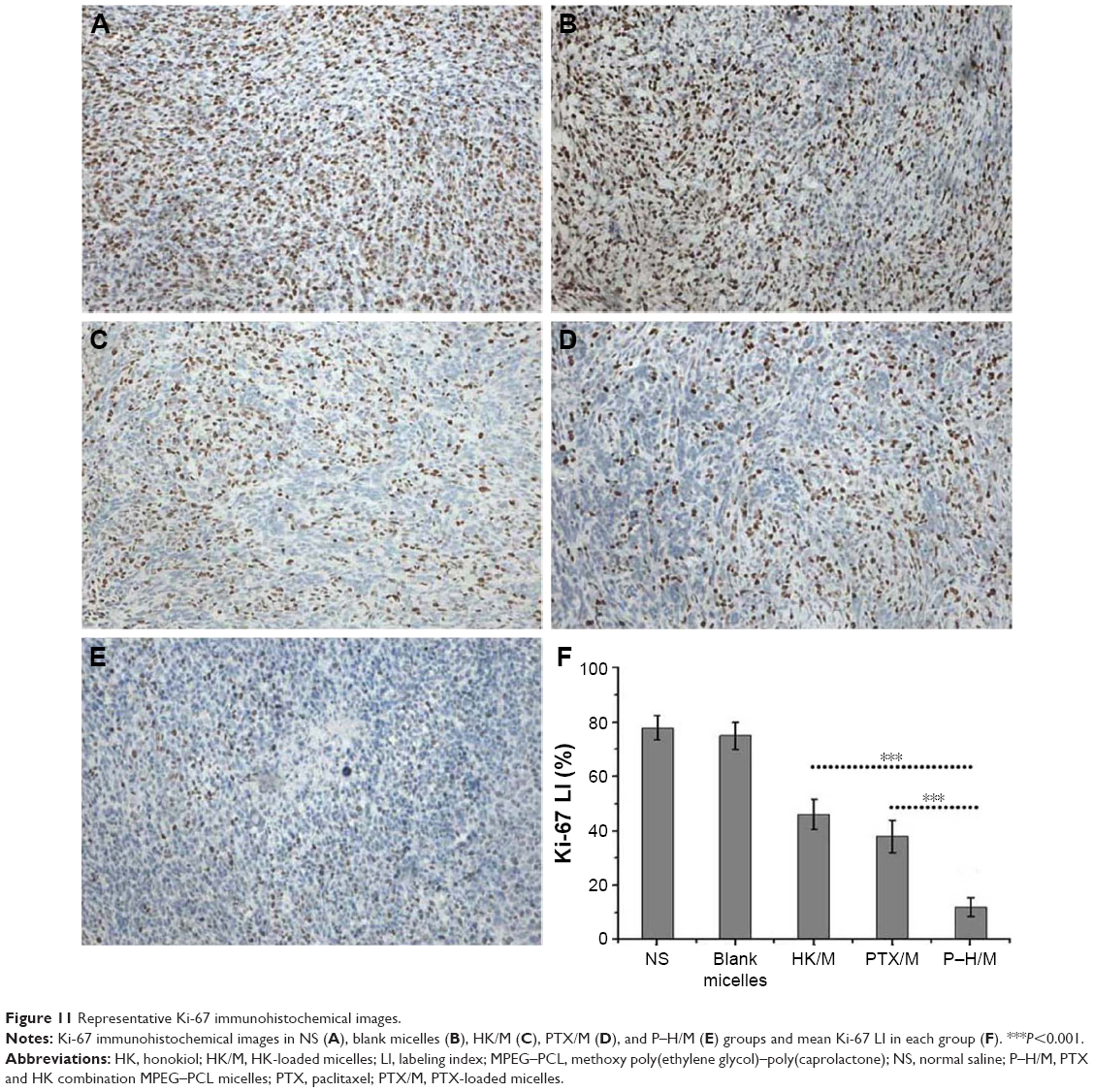 | Figure 11 Representative Ki-67 immunohistochemical images. |
Induction of apoptosis in vivo
We analyzed the apoptosis effect of P–H/M in 4T1 tumors by the assay of TUNEL staining. TUNEL-positive cells were counted only in regions of intact tumor cells, in which the central necrosis did not interfere with the quantification of apoptosis index. As shown in Figure 12, more apoptotic cells in tumor tissues were observed in the P–H/M group (55.82%±6.53%) than in the PTX/M (27.20%±3.11%, P<0.05) and HK/M (16.80%±3.03%, P<0.05) groups. The results revealed that the combination of PTX and HK may attribute to increasing the apoptosis in vivo.
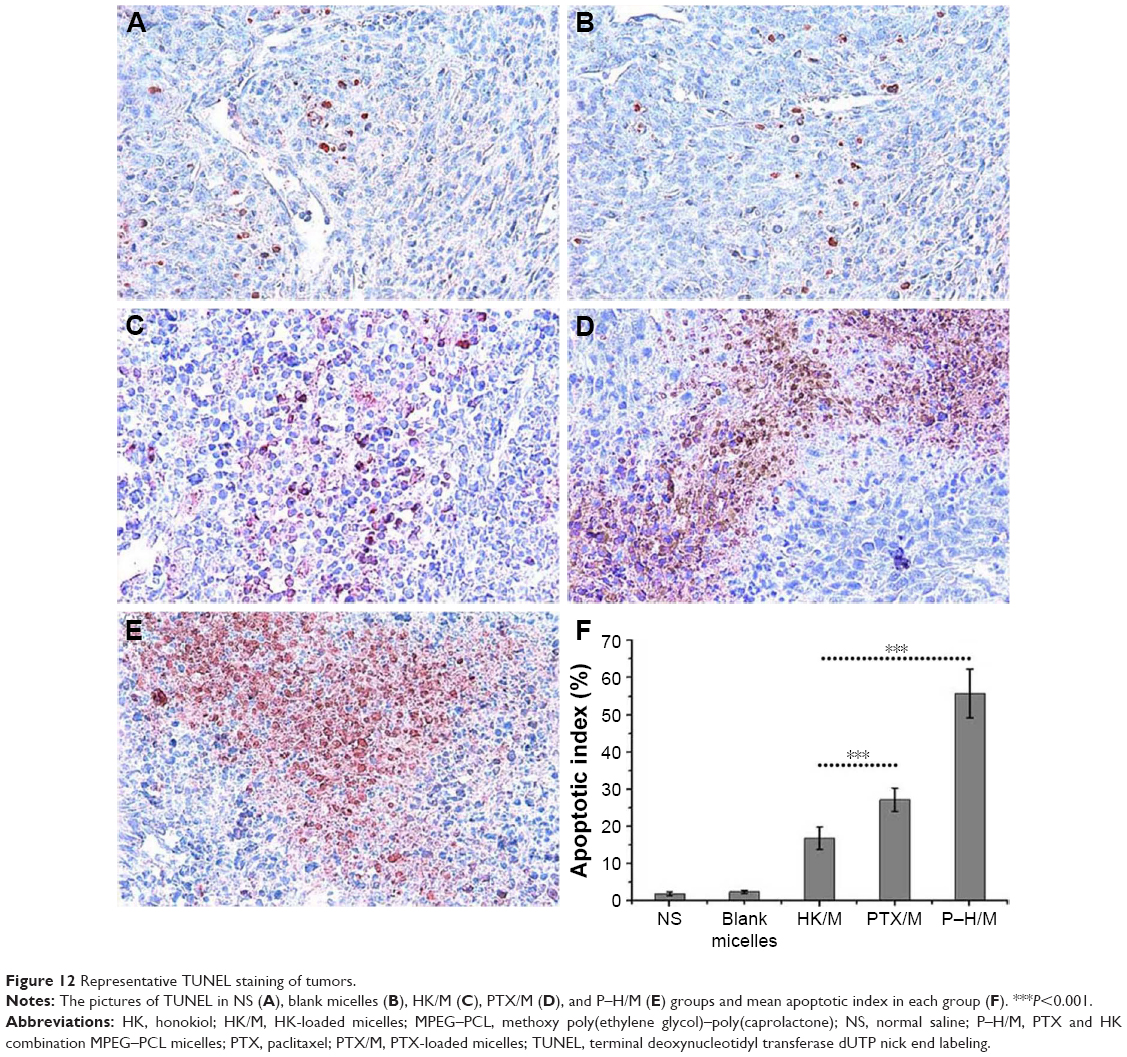 | Figure 12 Representative TUNEL staining of tumors. |
Bioimaging analysis
The particle size of DiD/M was 31.2±3.4 nm with the PDI of 0.03±0.01. The Balb/c nude mice bearing breast tumors were injected with free DiD or DiD/M and imaged at predetermined time points using a bioimaging system (Figure 13). Nontreated mice were used as control. The fluorescence signal in the tumors of DiD/M group was stronger than that in the tumors of free DiD group, suggesting that DiD/M enhanced more accumulation of DiD in the tumor.
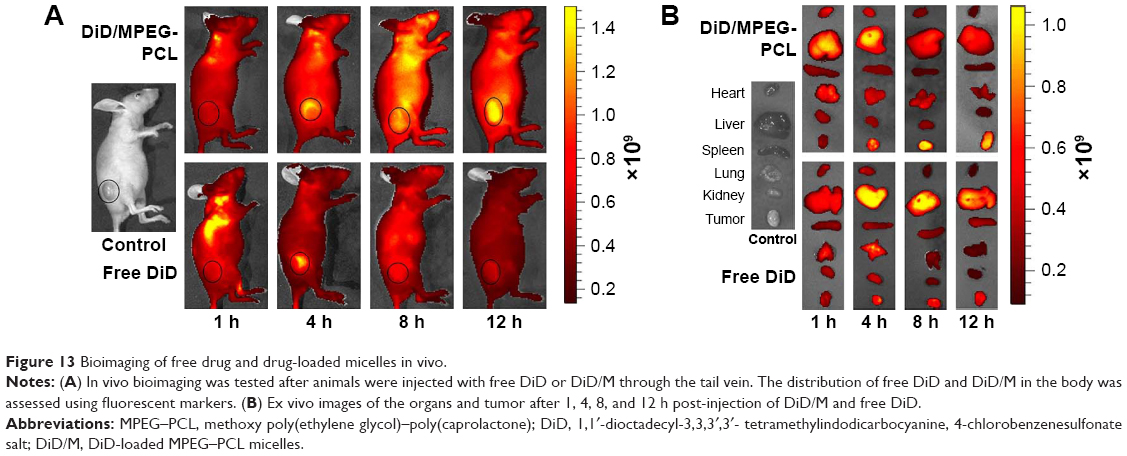 | Figure 13 Bioimaging of free drug and drug-loaded micelles in vivo. |
Unlike being tightly connected in normal vessels, vessels in tumor tissues were connected weakly with large gaps (~200–1,200 nm). Micelles with a small particle size (<200 nm) passively accumulated in tumors because of the enhanced permeability and retention (EPR) effect. Free drug also could permeate into both tumor and normal tissues and permeated out of tumors very easily because of its extremely small size, which reduced drug concentration in the tumor, and had more side effects against normal tissues. In this study, we found that DiD/M produced more DiD accumulation in the tumor tissues than free DiD and decreased drug extravasation into the normal tissues. Moreover, free drug accumulated mainly in liver, which would cause liver toxicity. The results indicated that the MPEG–PCL micelles had a slow-release feature, enhanced drug concentration in tumor, and could reduce the distribution in normal tissues.
Discussion
PTX and HK were used as chemotherapeutic agents in the treatment of pulmonary cancer, breast cancer, gastric cancer, ovarian cancer, head and neck squamous cell cancer, and a variety of other solid tumors.13,23–28 Intravenous administration of chemotherapeutic agents has been accepted as a conventional approach for antitumor chemotherapy. However, the hydrophobicity of PTX and HK had limited its use in clinical practice.29 Fortunately, MPEG–PCL micelles were able to solve the problem and also proved the combination drug delivery system with PTX and HK effective.
In our previous studies, different approaches were used to prepare the P–H/M, and the solid dispersion method was finally chosen and used.30,31 After evaluating the properties of different mass ratios of P–H/M from 100 (0+0) to 60 (20+20), we found 90 (5+5) as the most appropriate mass ratio. The concentration of PTX and HK in aqueous solvent could become much higher. It is notable that MPEG–PCL encapsulating PTX and HK showed small particle size (28.7±2.5 nm), good homogeneity (a mean PDI of 0.06±0.01), high EE (98.2%±0.2%), and high DL (9.4%±0.3%), as well as slower in vitro release behavior (Figure 3). What is more, the surface charge of P–H/M was nearly neutral (zeta potential is −1.42±0.2 mV) (Table 1). Thus, a core-shell structure should contribute to the stability of P–H/M.
In this study, MPEG–PCL was an amphiphilic diblock copolymer with the “core-shell” nanostructure in water, which allowed the slow-release behaviors of drug-loaded micelles in vitro and in vivo, indicating that drug-loaded micelles were stable and well dispersed in aqueous solution (Figures 3 and 13). Thus, the drug concentration in plasma could be kept at a maintained level to achieve enough concentration of drug in tumors. The superiority of P–H/M was confirmed accurately both in vitro and in vivo. A cell cytotoxicity study suggested that P–H/M had an enhanced cytotoxicity compared to PTX/M and HK/M (the IC50 value was 25 versus 41.4 and 64.1 μg/mL). Hence, we chose 25 μg/mL as the concentration of PTX or HK for in vitro experiments. Furthermore, 4T1 cells treated with P–H/M exhibited more apoptosis (Figures 6 and 7). The phenomenon may be dependent on the enhanced amount of PTX and HK taken up by cancer cells (Figure 5). The in vivo antitumor studies further proved that P–H/M possessed a better tumor inhibition and prolonged animals’ survival. Tumor sections were analyzed by H&E staining, immunohistochemical detection of cell proliferation and tumor vessel density, and the TUNEL assay, which illustrated that in comparison with PTX/M and HK/M, P–H/M had enhanced antiproliferation, angiogenesis effects, and apoptosis induction in the animal tumor model.
In addition, different from normal tissues, these newly formed tumor vessels are usually abnormal in form and architecture. The gaps between adjacent endothelial cells are large enough for polymer micelles to enter and accumulate in tumor tissues.32 This phenomenon is referred to as the “EPR effect” in solid tumors. The EPR effect is the property by which molecules of certain sizes (typically liposomes, nanoparticles, and macromolecular drugs) tend to accumulate much more in tumor tissues than in normal tissues.33–35 In this study, we examined in vivo tumor targeting of nanoparticles with DiD as a fluorescent probe and we found that DiD/M accumulated at a higher concentration in the tumor than free DiD, while at a lower concentration in normal tissues. Because of the extremely small size of free DiD, it could not only permeate into both tumor tissue and normal tissue but also permeate out of tumors very easily. For this reason, the in vivo antitumor study further demonstrated the superior antitumor activity of P–H/M, because of the long circulation time in blood, high concentration accumulation in tumor tissue, and low concentration in normal tissues, indicating the benefits of nanoparticles. In addition, our results confirmed that passive targeting of polymeric micelles to solid tumors could be achieved through the EPR effect.
The antitumor therapeutic effectiveness of P–H/M has been successfully proved in our study, which makes P–H/M a promising formulation for breast cancer therapy with superior antitumor efficiency and low systemic toxicity. Although further investigations are still needed, the excellent properties of P–H/M suggested that P–H/M could serve as a pontential candidate for future antitumor chemotherapeutic application.
Conclusion
In this study, we designed P–H/M combination drug delivery system for breast cancer therapy in vitro and in vivo. The encapsulation of PTX and HK simultaneously in polymeric micelles could enhance cytotoxicity, cellular uptake, and apoptosis of 4T1 cells in vitro. P–H/M could block tumor growth and reduced tumor volume in vivo. In addition, P–H/M prepared in our study inhibited angiogenesis and induced more apoptosis in vivo. Furthermore, bioimaging analysis showed that drug-loaded polymeric micelles could accumulate more in tumor tissues compared with free drug, suggesting that the MPEG–PCL polymeric micelles had good target ability because of the EPR effect in the tumor. In conclusion, we have shown that P–H/M may have excellent potential applications clinically for breast cancer.
Acknowledgments
This study was financially supported by the National Natural Science Foundation of China (NSFC31400814), the Distinguished Young Scholars of Sichuan University (2015SCU04A42), and the National Young Top-notch Talent Program. We thank Mr Chao Liu for his kind help in revising the article.
Author contributions
All authors contributed toward data analysis, drafting and critically revising the paper and agree to be accountable for all aspects of the work.
Disclosure
The authors report no conflicts of interest in this work.
References
Siegel RL, Miller KD, Jemal A. Cancer statistics, 2015. CA Cancer J Clin. 2015;65(1):5–29. | ||
Wang H, Zhao Y, Wu Y, et al. Enhanced anti-tumor efficacy by co-delivery of doxorubicin and paclitaxel with amphiphilic methoxy PEG-PLGA copolymer nanoparticles. Biomaterials. 2011;32(32):8281–8290. | ||
Singla AK, Garg A, Aggarwal D. Paclitaxel and its formulations. Int J Pharm. 2002;235(1–2):179–192. | ||
Spencer CM, Faulds D. Paclitaxel. A review of its pharmacodynamic and pharmacokinetic properties and therapeutic potential in the treatment of cancer. Drugs. 1994;48(5):794–847. | ||
Weiss RB, Donehower RC, Wiernik PH, et al. Hypersensitivity reactions from taxol. J Clin Oncol. 1990;8(7):1263–1268. | ||
Liou KT, Shen YC, Chen CF, Tsao CM, Tsai SK. The anti-inflammatory effect of honokiol on neutrophils: mechanisms in the inhibition of reactive oxygen species production. Eur J Pharmacol. 2003;475(1–3):19–27. | ||
Zhang X, Chen S, Wang Y. Honokiol up-regulates prostacyclin synthease protein expression and inhibits endothelial cell apoptosis. Eur J Pharmacol. 2007;554(1):1–7. | ||
Lo YC, Teng CM, Chen CF, et al. Magnolol and honokiol isolated from Magnolia officinalis protect rat heart mitochondria against lipid peroxidation. Biochem Pharmacol. 1994;47(3):549–553. | ||
Dikalov S, Losik T, Arbiser JL. Honokiol is a potent scavenger of superoxide and peroxyl radicals. Biochem Pharmacol. 2008;76(5):589–596. | ||
Li Z, Liu Y, Zhao X, et al. Honokiol, a natural therapeutic candidate, induces apoptosis and inhibits angiogenesis of ovarian tumor cells. Eur J Obstet Gynecol Reprod Biol. 2008;140(1):95–102. | ||
Liu H, Zang C, Emde A, et al. Anti-tumor effect of honokiol alone and in combination with other anti-cancer agents in breast cancer. Eur J Pharmacol. 2008;591(1–3):43–51. | ||
Ishitsuka K, Hideshima T, Hamasaki M, et al. Honokiol overcomes conventional drug resistance in human multiple myeloma by induction of caspase-dependent and -independent apoptosis. Blood. 2005;106(5):1794–1800. | ||
Gong C, Xie Y, Wu Q, et al. Improving anti-tumor activity with polymeric micelles entrapping paclitaxel in pulmonary carcinoma. Nanoscale. 2012;4(19):6004–6017. | ||
Chen L, Zhang Q, Yang G, et al. Rapid purification and scale-up of honokiol and magnolol using high-capacity high-speed counter-current chromatography. J Chromatogr A. 2007;1142(2):115–122. | ||
Gao X, Wang B, Wu Q, et al. Combined delivery and anti-cancer activity of paclitaxel and curcumin using polymeric micelles. J Biomed Nanotechnol. 2015;11(4):578–589. | ||
Li X, Zhang Z, Li J, Sun S, Weng Y, Chen H. Diclofenac/biodegradable polymer micelles for ocular applications. Nanoscale. 2012;4(15):4667–4673. | ||
Park EK, Kim SY, Lee SB, et al. Folate-conjugated methoxypoly(ethylene glycol)/poly(epsilon-caprolactone) amphiphilic block copolymeric micelles for tumor-targeted drug delivery. J Control Release. 2005;109(1–3):158–168. | ||
Li L, Braiteh FS, Kurzrock R. Liposome-encapsulated curcumin: in vitro and in vivo effects on proliferation, apoptosis, signaling, and angiogenesis. Cancer. 2005;104(6):1322–1331. | ||
Gou M, Men K, Shi H, et al. Curcumin-loaded biodegradable polymeric micelles for colon cancer therapy in vitro and in vivo. Nanoscale. 2011;3(4):1558–1567. | ||
Thipparaboina R, Chavan RB, Kumar D, Modugula S, Shastri NR. Micellar carriers for the delivery of multiple therapeutic agents. Colloids Surf B Biointerfaces. 2015;135:291–308. | ||
Gong C, Wei X, Wang X, et al. Biodegradable self-assembled PEG-PCL-PEG micelles for hydrophobic honokiol delivery: I. Preparation and characterization. Nanotechnology. 2010;21(21):215103. | ||
Fang J, Nakamura H, Maeda H. The EPR effect: unique features of tumor blood vessels for drug delivery, factors involved, and limitations and augmentation of the effect. Adv Drug Deliv Rev. 2011;63(3):136–151. | ||
Wang X, Yang L, Chen ZG, et al. Application of nanotechnology in cancer therapy and imaging. CA Cancer J Clin. 2008;58(2):97–110. | ||
Gong C, Deng S, Wu Q, et al. Improving antiangiogenesis and anti-tumor activity of curcumin by biodegradable polymeric micelles. Biomaterials. 2013;34(4):1413–1432. | ||
Zhang Y, Zhang H, Wang X, Wang J, Zhang X, Zhang Q. The eradication of breast cancer and cancer stem cells using octreotide modified paclitaxel active targeting micelles and salinomycin passive targeting micelles. Biomaterials. 2012;33(2):679–691. | ||
Li X, Lu X, Xu H, et al. Paclitaxel/tetrandrinecoloaded nanoparticles effectively promote the apoptosis of gastric cancer cells based on “oxidation therapy”. Mol Pharm. 2012;9(2):222–229. | ||
Wang N, He T, Shen Y, et al. Paclitaxel and tacrolimus coencapsulated polymeric micelles that enhance the therapeutic effect of drug-resistant ovarian cancer. ACS Appl Mater Interfaces. 2016;8(7):4368–4377. | ||
Xie L, Jiang F, Zhang X, et al. Honokiol sensitizes breast cancer cells to TNF-alpha induction of apoptosis by inhibiting Nur77 expression. Br J Pharmacol. 2016;173(2):344–356. | ||
Yan B, Peng ZY. Honokiol induces cell cycle arrest and apoptosis in human gastric carcinoma MGC-803 cell line. Int J Clin Exp Med. 2015;8(4):5454–5461. | ||
Singh T, Gupta NA, Xu S, et al. Honokiol inhibits the growth of head and neck squamous cell carcinoma by targeting epidermal growth factor receptor. Oncotarget. 2015;6(25):21268–21282. | ||
Gong C, Shi S, Wang X, et al. Novel composite drug delivery system for honokiol delivery: self-assembled poly(ethylene glycol)-poly(epsilon-caprolactone)-poly(ethylene glycol) micelles in thermosensitive poly(ethylene glycol)-poly(epsilon-caprolactone)-poly(ethylene glycol) hydrogel. J Phys Chem B. 2009;113(30):10183–10188. | ||
Vuorelaa P, Leinonenb M, Saikkuc P, et al. Natural products in the process of finding new drug candidates. Curr Med Chem. 2004;11(11):1375–1389. | ||
Bindseil KU, Jakupovic J, Wolf D, Lavayre J, Leboul J, van der Pyl D. Pure compound libraries; a new perspective for natural product based drug discovery. Drug Discov Today. 2001;6(16):840–847. | ||
Maeda H, Wu J, Sawa T, Matsumura Y, Hori K. Tumor vascular permeability and the EPR effect in macromolecular therapeutics: a review. J Control Release. 2000;65(1–2):271–284. | ||
Danafar H, Davaran S, Rostamizadeh K, Valizadeh H, Hamidi M. Biodegradable m-PEG/PCL core-shell micelles: preparation and characterization as a sustained release formulation for curcumin. Adv Pharm Bull. 2014;4(suppl 2):501–510. |
 © 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.