Back to Journals » Diabetes, Metabolic Syndrome and Obesity » Volume 12
Beta-cell function in type 2 diabetic patients who failed to maintain good glycemic status with a combination of maximum dosages of metformin and sulfonylurea
Authors Kunavisarut T ![]() , Sriussadaporn S, Lertwattanarak R
, Sriussadaporn S, Lertwattanarak R ![]()
Received 12 February 2019
Accepted for publication 18 April 2019
Published 21 May 2019 Volume 2019:12 Pages 761—770
DOI https://doi.org/10.2147/DMSO.S204439
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Konstantinos Tziomalos
Tada Kunavisarut, Sutin Sriussadaporn, Raweewan Lertwattanarak
Division of Endocrinology and Metabolism, Siriraj Hospital, Mahidol University, Bangkok, Thailand
Background: The aim of this study was to investigate beta-cell function and examine whether sulfonylureas (SUs) are still useful in patients with type 2 diabetes (T2DM) who failed to maintain optimal glycemic control with a combination of maximum dosages of metformin and SU.
Method: T2DM who had HbA1c >8% during treatment with a combination of maximum dosages of metformin and SU were studied. After enrollment, the patients were assigned to continue maximum dosages of SU and metformin for 2 weeks and then underwent the first oral glucose tolerance test (OGTT), the Max-SU OGTT. After the Max-SU OGTT, SUs were discontinued for 4 weeks and the second OGTT, the Discont-SU OGTT, was performed. After the Discont-SU OGTT, the same SU was restarted at 25% of the maximum dosage (25%Max-SU). After taking 25%Max-SU for 4 weeks, the third OGTT, the 25%Max-SU OGTT, was performed. Metformin at the same dosage was continued throughout the study. Normal OGTT (NGT) subjects, matched for age and body mass index (BMI), were also studied.
Results: There were 25 T2DM and 28 NGT subjects. There was no difference in age and BMI between the two groups. The beta-cell function during Max-SU was 0.1, which was higher than 0.06 during Discont-SU (p<0.001) and also higher than 0.09 during 25%Max-SU (p=0.269). The beta-cell function during 25%Max-SU was higher than during Discont-SU (p<0.001). The beta-cell function of the NGT group was 0.34 and higher than during Max-SU (p<0.001). Fasting capillary blood glucose (FCBG) levels during Discont-SU (14.2±3.7 mmol/L) were higher than during 25%Max-SU (12.3±3.4 mmol/L) and during Max-SU (10.3±2.4 mmol/L) (p<0.05). In addition, the FCBG during Discont-SU was higher than that during 25%Max-SU (p<0.05).
Conclusion: In T2DM patients who failed to achieve glycemic control with a combination of maximum dosages of metformin and SU, the beta-cell function declined compared to NGT subjects. However, the beta-cells were still responsive to SUs, which play a significant role in glycemic control.
Keywords: beta-cell function, sulfonylureas, sulfonylurea failure, type 2 diabetes mellitus
Introduction
Sulfonylureas (SUs) are a class of oral hypoglycemic agent that exert their glucose-lowering action by stimulating insulin secretion via reducing the activity of ATP-sensitive potassium (KATP) channels at the plasma membrane of pancreatic islet beta-cells.1 In pharmacological management of type 2 diabetes mellitus (T2DM), SUs are commonly used conventionally as the first-line monotherapy,2,3 and currently as a second-line drug in combination with metformin.4–6 A problem with SU monotherapy commonly observed in clinical practice is that SUs can initially control blood glucose levels very effectively, but subsequently become less or no longer effective and must be replaced or added to with insulin.2,7 The reduction in or loss of the glucose-lowering effect with duration of SU monotherapy has been defined as secondary sulfonylurea failure (SUF).7 Previous clinical, in vivo and in vitro studies on beta-cell function in patients on SU monotherapy who had SUF showed that the failure of beta-cells to respond to SU monotherapy may be due to a decrease in either number or function of beta-cells, or both, resulting in insulin deficiency.2,8,9 Showing the same problem as observed in SU monotherapy, many T2DM patients cannot achieve or maintain optimal glycemic control despite receiving dual therapy with the maximum dosage of an SU in combination with metformin, and subsequently need triple therapy by adding other agents.4 It is not known whether beta-cell function in T2DM patients who failed to maintain optimal glycemic status during treatment with a combination of metformin and SU is similar to or different from beta-cell function in patients who developed SUF during SU monotherapy. To the best of our knowledge, and following a review of the literature, there have been few studies on beta-cell function in T2DM patients who failed to maintain optimal glycemic control with combined SU and metformin therapy. The objectives of this study were to investigate beta-cell function and to examine whether SUs are still useful in glycemic control in T2DM patients who could not maintain optimal glycemic status with a combination of the maximum dosages of SU and metformin (MFM-SUF).
Method
Participants
T2DM patients, older than 18 years, who failed to maintain optimal glycemic status during treatment with a combination of maximum dosages of metformin and SU (MFM-SUF group), were studied. MFM-SUF was defined by the presence of past medical records showing good glycemic control with HbA1c levels of <53 mmol/mol (<7%) for years, and subsequently the glycemic status worsening, with HbA1c levels of >64 mmol/mol (>8%) for >6 months during treatment with a combination of the recommended maximum daily dosages of SU and metformin. Patients who had one or more of the following conditions at the time of recruitment were excluded: acute illnesses and infections, serum creatinine levels of >114.4 μmol/L, and serum aspartate transferase (AST) and/or alanine transferase (ALT) levels of >3 times higher than the upper limit of normal ranges, and history of diabetic ketoacidosis or hyperosmolar hyperglycemic state, glucocorticoid therapy, and currently being treated with insulin or other classes of oral hypoglycemic agent. Age- and body mass index (BMI)-matched healthy non-diabetic subjects with normal oral glucose tolerance test (OGTT) results (NGT group) were also studied for comparison with the MFM-SUF group.
Study protocol
At the time of recruitment, each patient in MFM-SUF group was assessed for body weight, height, BMI, biochemical profile, including fasting plasma glucose, HbA1c, serum creatinine, AST and ALT levels, and anti-glutamic acid decarboxylase (anti-GAD) autoantibody, and was assigned to perform standard lifestyle management and fasting capillary blood glucose (FCBG) measurement at home every other day throughout the study. After the initial assessment, the patients were assigned to continue maximum daily dosage of the same SU and the same dosages of metformin for 2 weeks, and then underwent the first 75 g OGTT, the Max-SU OGTT. After finishing the Max-SU OGTT, the patients were assigned to discontinue their SU but continue taking the same dosages of metformin. After discontinuation of SU for 4 weeks, the second OGTT, the Discont-SU OGTT, was performed and then the same SU was restarted at the dosage of 25% of the recommended maximum dose. After taking 25%Max-SU for 4 weeks, the third OGTT, the 25%Max-SU OGTT, was performed. The study protocol is summarized in Figure 1. The healthy non-diabetic subjects underwent an OGTT once.
 | Figure 1 Study protocol in type 2 diabetic patients who failed to maintain optimal glycemic control with a combination of maximum dosages of metformin and sulfonylurea (MFM-SUF group). Notes: After enrollment, the patients were standardized for lifestyle management and were assigned to continue the maximum daily dosage of the same sulfonylurea and the same dosages of metformin for 2 weeks, and then underwent the first OGTT, the Max-SU OGTT. After the Max-SU OGTT, the patients were assigned to discontinue their SU but continue taking the same dosages of metformin. After discontinuation of SU for 4 weeks, the second OGTT, the Discont-SU OGTT, was performed and then the same SU was restarted at 25% of the recommended maximum dose. After taking 25%Max-SU for 4 weeks, the third OGTT, the 25%Max-SU OGTT, was performed. Abbreviations: CBG, capillary blood glucose; OGTT, oral glucose tolerance test; SU, sulfonylurea. |
All participants provided written informed consent. This study was conducted in accordance with the Declaration of Helsinki and was approved by the Institutional Review Board of the Faculty of Medicine Siriraj Hospital, Mahidol University.
OGTT
The 75 g OGTT was performed according to the method described by the WHO in 1985.10 Venous blood samples were collected before (at 0 minutes) and at 30, 60, 90, and 120 minutes after drinking the glucose solution. The blood samples were immediately processed for measurement of plasma glucose levels and the plasma samples were stored at −20°C for subsequent plasma insulin assay.
Laboratory analyses
Plasma glucose levels were measured by the hexokinase method using an automated analyzer Modular P800 (Roche/Hitachi, Indianapolis, IN, USA). Plasma insulin levels were measured by radioimmunoassay (insulin-CT; MP Biomedicals, Santa Ana, CA, USA). FCBG levels were measured using glucometers (Accu-Chek Performa; Roche, Basel, Switzerland).
Calculation formulae
The plasma glucose and insulin levels obtained at each time-point during the OGTT were used for assessment of beta-cell function, insulin resistance determined by the Homeostasis Model Assessment of Insulin Resistance (HOMA-IR), and insulin sensitivity determined by the Matsuda index.11 Beta-cell function was assessed by the area under the curve (AUC) of plasma insulin levels at 0–120 minutes divided by the AUC of plasma glucose levels at 0–120 minutes (beta-cell function = AUC insulin0–120/AUC glucose0–120). AUC was calculated by the trapezoidal rule. Insulin resistance was assessed by HOMA-IR (HOMA-IR = Fasting plasma glucose (mmol/L) × Fasting plasma insulin (pmol/L)/22.5). Insulin sensitivity was assessed by the Matsuda index (Matsuda index = 10,000/√G0 × I0 × Mean G × Mean I; where G and I are plasma glucose and insulin values, respectively, at the time indicated).12
Statistical analysis
Descriptive statistics including frequency and percentage were used for categorical variables. Normality of distribution of variables was examined by the Kolmogorov–Smirnov test. Continuous variables were expressed as mean ± SD for normally distributed variables such as age, HbA1c, and FCBG; and as median (IQR) (25th percentile, 75th percentile) for non-normally distributed variables, such as duration of diabetes, AUC insulin0–120/glucose0–120, HOMA-IR, and Matsuda index. Comparisons of categorical variables between groups were performed using the chi-squared test or Fisher’s exact test. Continuous variables were compared using Student’s t-test or Mann–Whitney U-test. For all tests performed, a two-tailed p-value of <0.05 was considered statistically significant. All statistical analyses were performed using Predictive Analytics SoftWare (PASW) version 18.0 (SPSS, Chicago, IL, USA).
Results
There were 25 T2DM patients in the MFM-SUF group, comprising 11 males and 14 females, recruited in this study. Baseline clinical and biochemical characteristics of T2DM patients in the MFM-SUF group and healthy non-diabetic subjects in the NGT group are shown in Table 1. In the MFM-SUF group, the age was 56.8±9.8 years, median duration of diabetes was 12 years (IQR 7.5, 14), and BMI was 25.8±4.7 kg/m2. All patients had a negative anti-GAD 65 autoantibody. Fasting plasma glucose and HbA1c levels were 11.0±2.2 mmol/L and 80±7.3 mmol/mol or 9.5±1.4%, respectively. The daily dose of metformin was 2.2±0.8 g/day. There were 28 healthy non-diabetic subjects in the NGT group (two males and 26 females), aged 53.8±6.7 years, with a BMI of 26.4±3.6 kg/m2. There were no statistically significant differences in age (p=0.29) or BMI (p=0.77) between the MFM-SUF and NGT groups.
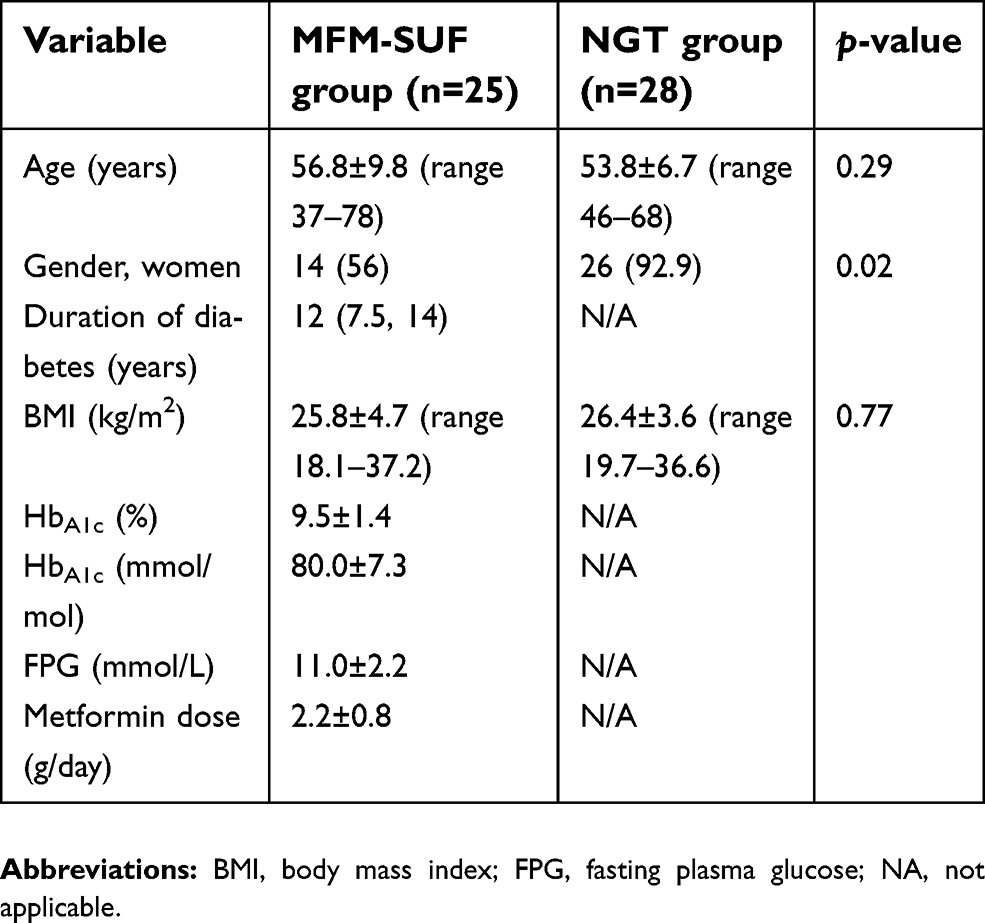 | Table 1 Baseline characteristics of type 2 diabetic patients who failed to maintain optimal glycemic control with a combination of maximum dosage of metformin and sulfonylurea (MFM-SUF group), and the normal glucose tolerance (NGT) group |
All patients had worsening glycemic status after discontinuation and decreasing dosage of the SU. Home FCBG levels during Discont-SU (14.2±3.7 mmol/L) were significantly higher than those during 25%Max-SU (12.3±3.4 mmol/L) and during Max-SU (10.3±2.4 mmol/L) (both p<0.05). In addition, home FCBG levels during Discont-SU (14.2±3.7 mmol/L) were significantly higher than during 25%Max-SU (12.3±3.4 mmol/L) (p<0.05).
Plasma glucose and insulin levels in response to OGTT are shown in Figure 2. Median beta-cell function during treatment with Max-SU was 0.1 (IQR 0.08, 0.17), which was significantly higher than during Discont-SU (0.06; IQR 0.04, 0.09) (p<0.001) and also higher, but not significantly, than during 25%Max-SU (0.09; IQR 0.01, 0.32) (p=0.269). Median beta-cell function during 25%Max-SU was 0.09 (IQR 0.01, 0.32), which was significantly higher than during Discont-SU (0.06; IQR 0.04, 0.09) (p<0.001). Median beta-cell function during Max-SU was 0.1 (IQR 0.08, 0.17), which was significantly lower than in the NGT group (0.34; IQR 0.24, 0.44) (p<0.001) (Figure 3 and Table 2).
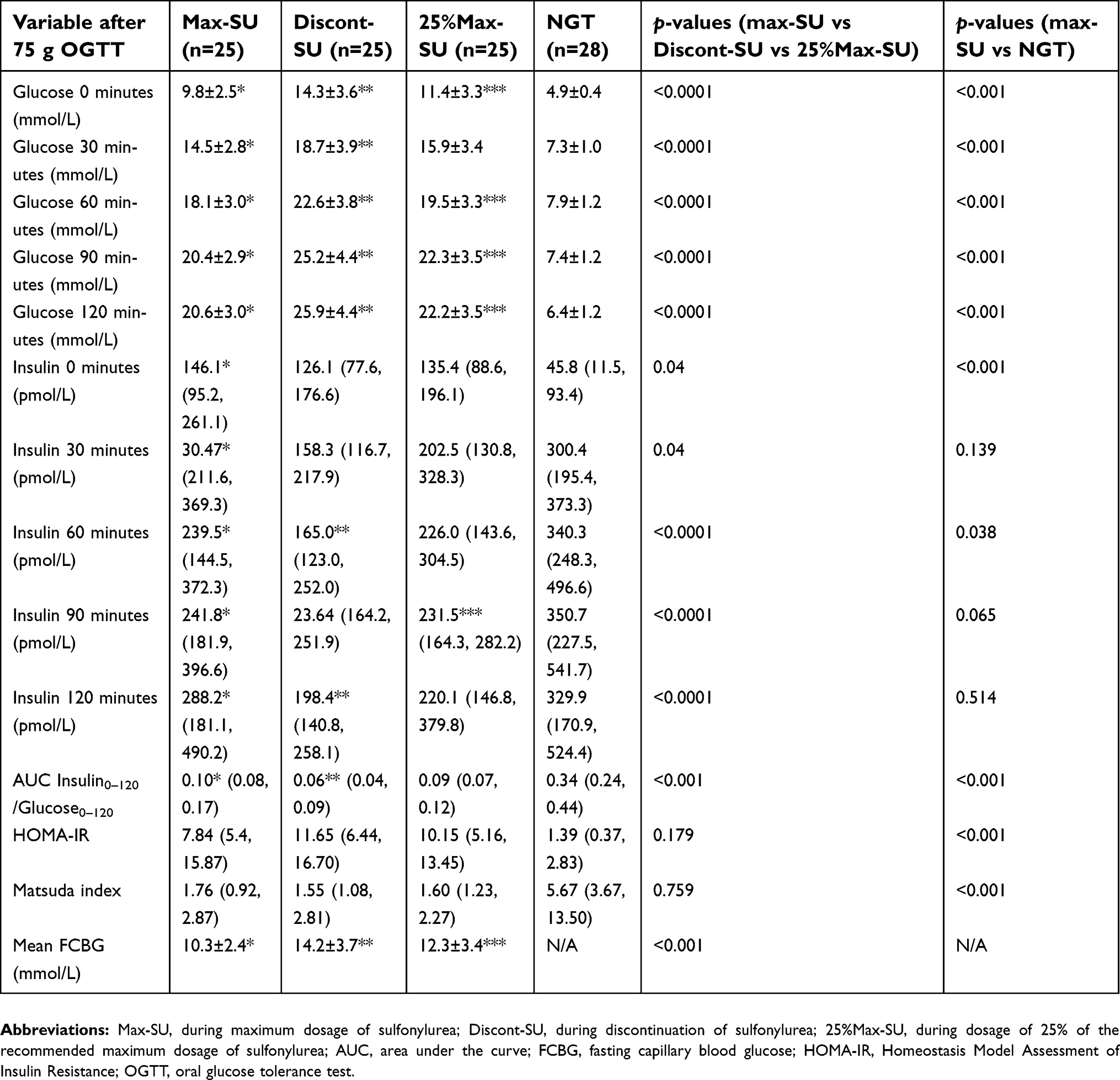 | Table 2 Plasma glucose, insulin, Beta-cell function, insulin resistance indexes and mean fasting capillary blood glucose in the two groups during the OGTT |
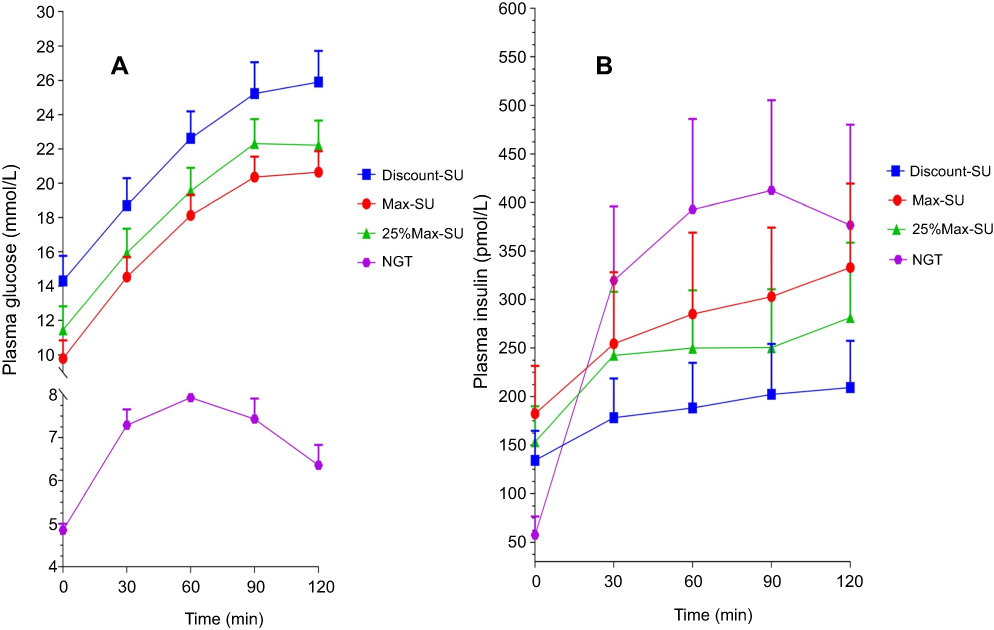 | Figure 2 (A) Plasma glucose and (B) plasma insulin levels during OGTTs in patients in the MFM-SUF and NGT groups. Notes: For the MFM-SUF group, the OGTT was performed three times, while taking the maximum dosage of sulfonylurea (Max-SU), discontinuing sulfonylurea (Discont-SU), and taking 25% of the maximum dosage of sulfonylurea (25%Max-SU). During the treatment with Max-SU, all MFM-SUF patients had significantly higher fasting plasma insulin levels than did the subjects with NGT, but the plasma insulin response to OGTT in Max-SU was much lower than in the NGT group. Abbreviations: MSM-SUF, maximum dosages of metformin and sulfonylurea; NGT, normal glucose tolerance; OGTT, oral glucose tolerance test; SU, sulfonylurea. |
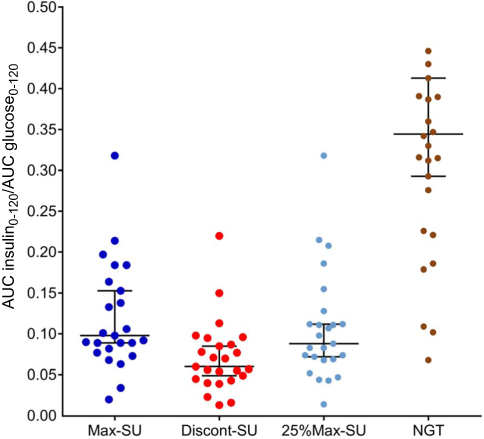 | Figure 3 Beta-cell function during OGTTs in the MFM-SUF and NGT groups. Notes: For the MFM-SUF group, the OGTT was performed three times, while taking the maximum dosage of sulfonylurea (Max-SU), discontinuing sulfonylurea (Discont-SU), and taking 25% of the maximum dosage of sulfonylurea (25%Max-SU). Median (IQR) beta-cell function during treatment with Max-SU was 0.1 (0.08, 0.17), which was significantly higher than during Discont-SU (0.06; 0.04, 0.09) (p<0.001), and also higher, but not significantly so, than during 25%Max-SU (p=0.269). Median beta-cell function during 25%Max-SU was 0.09 (0.01, 0.32), which was significantly higher than during Discont-SU (0.06; 0.04, 0.09) (p<0.001). Median beta-cell function during Max-SU was 0.1 (0.08, 0.17), which was significantly lower than in the NGT group (0.34; 0.24, 0.44) (p<0.001). Abbreviations: AUC, area under the curve; MSM-SUF, maximum dosages of metformin and sulfonylurea; NGT, normal glucose tolerance; OGTT, oral glucose tolerance test; SU, sulfonylurea. |
There were no significant differences in Matsuda index or HOMA-IR during Max-SU, Discont-SU, and 25%Max-SU (7.84 vs 11.65 vs 10.15, respectively; p=0.179; and 1.76 vs 1.55 vs 1.60, respectively; p=0.759) (Table 2). However, Max-SU had a lower Matsuda index (1.76 vs 5.67; p<0.001) and a higher HOMA-IR (7.84 vs 1.39; p<0.001) than the NGT group.
Discussion
Current guidelines suggest MFM as the first-line medical therapy for T2DM, and if the HbA1c target of <53 mmol/mol (<7%) is not achieved with MFM monotherapy, the addition of a second-line glucose-lowering agent, such as SUs, dipeptidyl peptidase-4 (DPP-4) inhibitors, thiazolidinediones, sodium-glucose cotransporter-2 (SGLT2) inhibitors, or insulin, to MFM is recommended.6,13 A survey of the characteristics and treatment patterns of patients with newly diagnosed T2DM in a large US integrated health system showed that SUs were most commonly selected as a second-line glucose-lowering agent as they are not only effective in improving glycemic control but also low cost.3 The WHO 2018 guideline recommends SUs as second-line treatment owing to their similar efficacy in lowering HbA1c to other classes of oral glucose-lowering agents with a not very high risk of hypoglycemia and low price.5 When an optimal HbA1c target is not achieved or maintained with combined MFM and SU therapy, adding the third glucose-lowering agent, either a DPP-4 inhibitor, thiazolidinedione, SGLT2 inhibitor, or an insulin, is recommended; the selection of one of these agents is based on plasma glucose and HbA1c levels, glucose-lowering efficacy, risk of developing hypoglycemia, effect on body weight, cost of medication, and simplicity or complexity of drug administration, but is not based on the patient's islet beta-cell function.13 Knowing the beta-cell function in T2DM patients who have MFM-SUF may be helpful in the selection of the third glucose-lowering agent and deciding whether SU should be continued or discontinued, especially when insulin therapy is considered.
The present study was conducted to examine beta-cell function in T2DM patients who failed to maintain an optimal glycemic control during treatment with a combination of maximum dosages of metformin and SU. The absence of anti-GAD autoantibody and the presence of high fasting plasma insulin levels that further increased after the oral glucose load in all patients (as shown in Table 2) indicated that the MFM-SUF in our patients was not due to type 1 diabetes mellitus or latent autoimmune diabetes of adults.
The mechanisms of failure to achieve or maintain an optimal glycemic status with SU monotherapy that have been previously proposed included worsening of insulin resistance,7 decrease in beta-cell function,2,7,14 and/or decrease in beta-cell mass or number, either along the natural course of T2DM15 or induced by prolonged exposure to SU,16 and decreased pancreatic beta-cell response to insulin-stimulating activity of SU after prolonged exposure to SU, known as SU desensitization.16–19 However, to the best of our knowledge, these mechanisms have not been described in patients who failed to maintain optimal glycemic control during long-term treatment with a combination of maximum dosages of metformin and an SU.
SUs have been shown to possess extrapancreatic insulin-mimetic activity in vitro,20 and to increase insulin sensitivity and insulin-dependent glucose disposal in type 1 diabetic patients,21 whereas some studies have shown that SUs have neutral or no effect on insulin sensitivity.2,22 The absence of significant change, either increase or decrease in insulin resistance status, determined by the HOMA-IR and Matsuda index during Max-SU, 25%Max-SU, and Discont-SU in the MFM-SUF group suggested that long-term SU therapy per se had a neutral or no negative effect on insulin resistance status. However, the lack of change in insulin resistance index after decreasing the dosage and discontinuation of SU observed in our study may be due to the more potent insulin-sensitizing effect of metformin, which may obscure the less potent insulin-sensitizing effect of SU.
Decreases in islet beta-cell function7,14 and/or islet beta-cell mass or number,15 related to long-term exposure to SU, have been demonstrated in vitro,23 but not in in vivo studies.24 Our study has shown that T2DM patients who failed to achieve an optimal glycemic status during long-term treatment with the maximum dosage of SU and MFM had decreased beta-cell function, determined by lower AUC insulin/AUC glucose ratio in response to oral glucose load during Discont-SU than in NGT subjects, as shown in Table 2. Our findings are consistent with a study by Pontiroli et al in T2DM patients receiving SU monotherapy, showing that secondary SUF was related to decreased islet beta-cell function as determined by reduced insulin release on fasting, 6 minutes after intravenous glucagon, and post-prandially.7,14 The decreased insulin secretion in response to oral glucose load in our patients who were receiving long-term SU and MFM therapy may be due to the decrease in either beta-cell function or beta-cell number, or both, that related to the deterioration of beta-cells along the natural course of T2DM or the deleterious effect of SU per se on beta-cells. The AUC insulin/AUC glucose ratio in response to OGTT, which decreased during Discont-SU compared to during Max-SU, and increased after resumption of SU at 25% of the maximum dosage observed in our study, suggested that the impaired beta-cell function was not directly induced by long-term SU therapy and was still able to be stimulated by SU.
An in vitro study in clonal pancreatic BRIN-BD11 beta-cells by Brennan et al showed that SU actively stimulated insulin secretion from the BRIN-BD11 beta-cells during initial exposure to the SU, but the insulin-stimulating activity subsequently decreased during prolonged exposure.17 This observation was defined as SU desensitization.17 The results of our study suggest that SU desensitization may not exist in patients with MFM-SUF since the insulin secretion apparently decreased during both fasting and oral glucose loading states, as shown in Table 2, instead of being unchanged, as would be the case if desensitization did exist, and the blood glucose levels increased in all patients after discontinuation of SU, suggesting that long-term treatment with SU was still able to stimulate insulin secretion, which has a certain role in glycemic control. Moreover, when SU were restarted with 25% of the maximum dosage after discontinuation of SU, the insulin secretion and glucose control were worse than during Max-SU administration. Our findings correspond to those of Grunberger, who showed that continuous administration of glibenclamide for 16 weeks provided better glycemic control than did intermittent administration with 2 weeks on treatment and 2 weeks off treatment.25 The UK Prospective Diabetes Study Group (UKPDS) showed that beta-cell function progressively deteriorated in all T2DM patients irrespective of the therapy used, either diet control alone or monotherapy with MFM or an SU after 6 years of diagnosis of T2DM.2 Conforming to the UKPDS findings, our study showed that the median beta-cell function of 0.06 in our patients with MFM-SUF, whose duration of diabetes ranged from 4 to 38 years with a median of 12 years, during discontinuation of SU was 17.6% of that of age- and BMI-matched NGT subjects (0.34), suggesting that the beta-cell function should gradually decline with the duration of diabetes. In addition, the significant decrease in median beta–cell function during Discont-SU compared to during Max-SU (0.06 vs 0.1, p<0.001) and the significant increase in median beta-cell function during resumption of 25%Max-SU compared to during Discont-SU (0.09 vs 0.06, p<0.001) suggested that the decrease in beta-cell function in our T2DM patients with MFM-SUF was not caused by SU therapy per se. In addition, the beta-cells were still able to respond to the insulin-stimulatory effect of SU. The increase in home FCBG levels and the decrease in beta-cell function observed after discontinuation and decreasing the dosage of SU from Max-SU to 25%Max-SU indicated that SUs were still able to stimulate insulin secretion in MFM-SUF patients in a dose-dependent manner and still possessed a significant role in glycemic control in T2DM patients with MFM-SUF. The inability to maintain optimal glycemic control can be explained by the decline in beta-cell function along the natural course of T2DM.2 Our study also showed that during the treatment with Max-SU, all MFM-SUF patients had significantly higher fasting plasma insulin levels than did the subjects with NGT, but the plasma insulin response to OGTT in Max-SU was much lower than in the NGT group, suggesting that SUs were able to stimulate insulin secretion from beta-cells, which resulted in a significant impact on glycemic control but not enough to maintain optimal glycemic targets.
The use of SU in the later stage of diabetes is controversial, especially when insulin therapy is considered. In T2DM patients who have SUF, there is no clear consensus among current guidelines as to whether SU should be continued when insulin therapy is initiated. The American Association of Clinical Endocrinologists guideline 2017 suggests discontinuing or reducing the dosage of SU when basal insulin is started, to reduce the risk of hypoglycemia.26 The American Diabetes Association guideline 2018 recommends that SU should be stopped once more complex insulin regimens beyond the basal insulin regimen are used.13 Nevertheless, these recommendations were based principally on expert opinions about the risk of hypoglycemia, which may outweigh the doubtful beneficial effect in glycemic control if an SU is continued. A 2016 Cochrane meta-analysis showed that combination treatment with insulin and SU resulted in significantly lower HbA1c levels compared to insulin monotherapy, with more body weight gain and a higher number of mild hypoglycemic episodes.27 According to the results of our study, showing that SUs are still able to stimulate the beta-cells despite being administered for several years and that glycemic control was worse after discontinuing them, SU should be therefore reasonably continued, with precautions against hypoglycemia, when the third oral glucose-lowering agent or insulin therapy is considered in T2DM patients with MFM-SUF. In addition, our study supports the results of previous studies in patients treated with combined insulin and SU demonstrating that discontinuation of SU resulted in worsening glycemic control,28–30 and the addition of SU in patients treated with all insulin regimens resulted in significant reductions in HbA1c levels and decrease in insulin dosage by 20%.31
The strength of this study is that, to our best knowledge, this is the first study to demonstrate beta-cell function in T2DM patients who failed to maintain optimal glycemic status with a combination of maximum dosages of metformin and SU. However, our study has some limitations. First, this study had quite a small sample size, which may lead to the argument that the results may not be generalizable. However, as the glycemic status got worse after discontinuation of SU and improved after resumption of SU in all of our T2DM patients with MFM-SUF, the results should be applicable to most, but not all, other T2DM patients with MFM-SUF who are commonly found in general practice. Second, all of our patients were Asian, so the findings may not be generalizable to other ethnic populations, since Asian T2DM patients have been shown to have more impaired beta-cell function than Caucasian patients in the pathogenesis of T2DM.32 Asian populations may respond to SU differently from Caucasians. Third, we did not study T2DM patients who had good glycemic control during treatment with a combination of metformin and an SU. Data on beta-cell function in this group of patients would be valuable in examining the effect of SU on beta-cell function compared with patients who had MFM-SUF. Fourth, there were more females in the NGT group, which therefore may not be the ideal group for comparing to our T2DM patients. The last limitation was the use of the OGTT; this may not be the perfect method for assessment of beta-cell function in T2DM patients with MFM-SUF, as the impaired beta-cell function may, to some degree, be due to glucotoxicity, especially during discontinuation of SU. A better model for evaluation of beta-cell function to correct for the confounding effects of glucotoxicity, such as inducing normoglycemia by adding insulin and subsequently reassessing beta-cell function, could be performed. However, a randomized controlled study conducted by our group (Srivanichakorn et al), in T2DM patients who achieved optimal glycemic control during long-term treatment with combined SU and insulin, showed that withdrawal of SU resulted in worsening of glycemic status.29 The findings from Srivanichakorn et al and the present study confirm that SU still plays a significant role in glycemic control in both hyperglycemic and optimal glycemic states.
Conclusion
In conclusion, in T2DM patients who fail to maintain optimal glycemic status with a combination of maximum dosages of metformin and an SU, the beta-cell function is reduced compared to non-diabetic subjects. The declined beta-cell function in this group of patients is not directly caused by SU. In addition, the beta-cells are still responsive to the insulin-stimulatory action of SU, which has a significant role in glycemic control. SUs should therefore be continued, with precautions against hypoglycemia, when other oral glucose-lowering agents or insulin is considered.
Abbreviations list
ALT, alanine transferase; AST, aspartate transferase; AUC, area under the curve; BMI, body mass index; DPP-4, dipeptidyl peptidase-4; FCBG, fasting capillary blood glucose; GAD, glutamic acid decarboxylase; HOMA-IR, Homeostasis Model Assessment of Insulin Resistance; NGT, normal glucose tolerance; SU, sulfonylurea; Max-SU, maximum dosage of sulfonylurea; Discont-SU, discontinuation of sulfonylurea; 25%Max-SU, dosage of 25% of the recommended maximum dose of sulfonylurea; SUF, sulfonylurea failure; T2DM, type 2 diabetes mellitus; UKPDS, Prospective Diabetes Study Group.
Data availability
The data sets generated and analyzed during the current study are available from the corresponding author on reasonable request.
Acknowledgments
We would like to thank Professor Apichati Vichayanrat and Associate Professor Apiradee Sriwijitkamol, Division of Endocrinology and Metabolism, Department of Medicine, Faculty of Medicine, Siriraj Hospital, for their valuable comments. We also thank Ms Khemjira Karaketklang of the Division of Clinical Research and Academic, Department of Medicine, Faculty of Medicine, Siriraj Hospital, for her assistance with statistical analysis. This study was supported by Siriraj Research Development Fund, managed by the Routine to Research (R2R) Project, Faculty of Medicine, Siriraj Hospital, Mahidol University.
Author contributions
All authors contributed toward data analysis, drafting and revising the paper, gave final approval of the version to be published and agree to be accountable for all aspects of the work.
Disclosure
The authors report no conflicts of interest in this work.
References
1. Aquilante CL. Sulfonylurea pharmacogenomics in type 2 diabetes: the influence of drug target and diabetes risk polymorphisms. Expert Rev Cardiovasc Ther. 2010;8(3):359–372. doi:10.1586/erc.09.154
2.
3. Pantalone KM, Hobbs TM, Wells BJ, et al. Changes in characteristics and treatment patterns of patients with newly diagnosed type 2 diabetes in a large united states integrated health system between 2008 and 2013. Clin Med Insights Endocrinol Diabetes. 2016;9:23–30. doi:10.4137/CMED.S39761
4.
5.
6. Davies MJ, D‘Alessio DA, Fradkin J, et al. Management of hyperglycaemia in type 2 diabetes, 2018. A consensus report by the American Diabetes Association (ADA) and the European Association for the Study of Diabetes (EASD). Diabetologia. 2018;61(12):2461–2498. doi:10.1007/s00125-018-4729-5
7. Pontiroli AE, Calderara A, Pozza G. Secondary failure of oral hypoglycaemic agents: frequency, possible causes, and management. Diabetes Metab Rev. 1994;10(1):31–43.
8. Matthews DR, Cull CA, Stratton IM, Holman RR, Turner RC. UKPDS 26: sulphonylurea failure in non-insulin-dependent diabetic patients over six years. UK Prospective Diabetes Study (UKPDS) Group. Diabetic Med. 1998;15(4):297–303. doi:10.1002/(SICI)1096-9136(199804)15:4<297::AID-DIA572>3.0.CO;2-W
9. Rosengren A, Jing X, Eliasson L, Renstrom E. Why treatment fails in type 2 diabetes. PLoS Med. 2008;5(10):e215. doi:10.1371/journal.pmed.0050215
10. Alberti KG, Zimmet PZ. Definition, diagnosis and classification of diabetes mellitus and its complications. Part 1: diagnosis and classification of diabetes mellitus provisional report of a WHO consultation. Diabetic Med. 1998;15(7):539–553. doi:10.1002/(SICI)1096-9136(199807)15:7<539::AID-DIA668>3.0.CO;2-S
11. Pacini G, Mari A. Methods for clinical assessment of insulin sensitivity and beta-cell function. Best Pract Res Clin Endocrinol Metab. 2003;17(3):305–322. doi:10.1016/S1521-690X(03)00042-3
12. Matsuda M, DeFronzo RA. Insulin sensitivity indices obtained from oral glucose tolerance testing: comparison with the euglycemic insulin clamp. Diabetes Care. 1999;22(9):1462–1470.
13.
14. Pontiroli AE, Calderara A, Maffi P, et al. Secondary failure to oral hypoglycaemic agents in non-obese patients with non-insulin-dependent diabetes is related to reduced insulin release. Diabete Metab. 1989;15(2):79–84.
15. Butler AE, Janson J, Bonner-Weir S, Ritzel R, Rizza RA, Butler PC. Beta-cell deficit and increased beta-cell apoptosis in humans with type 2 diabetes. Diabetes. 2003;52(1):102–110.
16. Takahashi A, Nagashima K, Hamasaki A, et al. Sulfonylurea and glinide reduce insulin content, functional expression of K(ATP) channels, and accelerate apoptotic beta-cell death in the chronic phase. Diabetes Res Clin Pract. 2007;77(3):343–350. doi:10.1016/j.diabres.2006.12.021
17. Brennan L, Hewage C, Malthouse JP, McClenaghan NH, Flatt PR, Newsholme P. Investigation of the effects of sulfonylurea exposure on pancreatic beta cell metabolism. FEBS J. 2006;273(22):5160–5168. doi:10.1111/j.1742-4658.2006.05513.x
18. Remedi MS, Nichols CG, Groop L. Chronic antidiabetic sulfonylureas in vivo: reversible effects on mouse pancreatic beta-cells. PLoS Med. 2008;5(10):e206. doi:10.1371/journal.pmed.0050206
19. Karam JH, Sanz N, Salamon E, Nolte MS. Selective unresponsiveness of pancreatic beta-cells to acute sulfonylurea stimulation during sulfonylurea therapy in NIDDM. Diabetes. 1986;35(12):1314–1320.
20. Muller G. The molecular mechanism of the insulin-mimetic/sensitizing activity of the antidiabetic sulfonylurea drug Amaryl. Mol med. 2000;6(11):907–933.
21. Pernet A, Trimble ER, Kuntschen F, Assal JP, Hahn C, Renold AE. Sulfonylureas in insulin-dependent (type I) diabetes: evidence for an extrapancreatic effect in vivo. J Clin Endocrinol Metab. 1985;61(2):247–251. doi:10.1210/jcem-61-2-247
22. Keller U, Muller R, Berger W. Sulfonylurea therapy fails to diminish insulin resistance in type I-diabetic subjects. Horm Metab Res. 1986;18(9):599–603. doi:10.1055/s-2007-1012384
23. Maedler K, Carr RD, Bosco D, Zuellig RA, Berney T, Donath MY. Sulfonylurea induced beta-cell apoptosis in cultured human islets. J Clin Endocrinol Metab. 2005;90(1):501–506. doi:10.1210/jc.2004-0699
24. Donath MY, Ehses JA, Maedler K, et al. Mechanisms of β-cell death in type 2 diabetes. Diabetes. 2005;54(suppl2):S108–S113.
25. Grunberger G. Maintenance of sulfonylurea responsiveness in NIDDM. Randomized double-blind study of intermittent glyburide therapy. Diabetes Care. 1992;15(5):696–699.
26. Garber AJ, Abrahamson MJ, Barzilay JI, et al. Consensus statement by the American Association of Clinical Endocrinologists and American College of Endocrinology on the comprehensive type 2 diabetes management algorithm - 2017 executive summary. Endocr Pract. 2017;23(2):207–238. doi:10.4158/EP161682.CS
27. Vos RC, van Avendonk MJ, Jansen H, et al. Insulin monotherapy compared with the addition of oral glucose-lowering agents to insulin for people with type 2 diabetes already on insulin therapy and inadequate glycaemic control. Cochrane Database Syst Rev. 2016;9:CD006992.
28. Nyback-Nakell A, Adamson U, Lins PE, Landstedt-Hallin L. Glycaemic responsiveness to long-term insulin plus sulphonylurea therapy as assessed by sulphonylurea withdrawal. Diabetic Med. 2007;24(12):1424–1429. doi:10.1111/j.1464-5491.2007.02286.x
29. Srivanichakorn W, Sriwijitkamol A, Kongchoo A, et al. Withdrawal of sulfonylureas from patients with type 2 diabetes receiving long-term sulfonylurea and insulin combination therapy results in deterioration of glycemic control: a randomized controlled trial. Diabetes Metab Syndr Obes. 2015;8:137–145. doi:10.2147/DMSO.S78008
30. Yokoyama H, Sone H, Yamada D, Honjo J, Haneda M. Contribution of glimepiride to basal-prandial insulin therapy in patients with type 2 diabetes. Diabetes Res Clin Pract. 2011;91(2):148–153. doi:10.1016/j.diabres.2010.10.007
31. Nyback-Nakell A, Adamson U, Lins PE, Landstedt-Hallin L. Adding glimepiride to insulin+metformin in type 2 diabetes of more than 10 years‘ duration–a randomised, double-blind, placebo-controlled, cross-over study. Diabetes Res Clin Pract. 2014;103(2):286–291. doi:10.1016/j.diabres.2013.12.062
32. Yoon KH, Lee JH, Kim JW, et al. Epidemic obesity and type 2 diabetes in Asia. Lancet. 2006;368(9548):1681–1688. doi:10.1016/S0140-6736(06)69703-1
 © 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.