")
Back to Journals » International Journal of Nanomedicine » Volume 10 » Issue 1
Berberine-loaded solid lipid nanoparticles are concentrated in the liver and ameliorate hepatosteatosis in db/db mice
Authors Xue M, Zhang L, Yang M, Zhang W, Li X, Ou Z, Li Z, Liu S, Li X, Yang S
Received 13 March 2015
Accepted for publication 11 May 2015
Published 6 August 2015 Volume 2015:10(1) Pages 5049—5057
DOI https://doi.org/10.2147/IJN.S84565
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 5
Editor who approved publication: Professor Lei Yang
Mei Xue,1,2,* Liang Zhang,3,* Ming-xing Yang,1 Wei Zhang,1 Xiu-min Li,1,2 Zhi-min Ou,1 Zhi-peng Li,1,2 Su-huan Liu,1 Xue-jun Li,1 Shu-yu Yang1
1Xiamen Diabetes Institute, the First Affiliated Hospital of Xiamen University, Xiamen, 2Department of Pharmacology, Beijing University of Chinese Medicine, Chao Yang District, Beijing, 3Department of Pharmacology, Nanjing University of Chinese Medicine, Nanjing, People’s Republic of China
*These authors contributed equally to this work
Abstract: Berberine (BBR) shows very low plasma levels after oral administration due to its poor absorption by the gastrointestinal tract. We have previously demonstrated that BBR showed increased gastrointestinal absorption and enhanced antidiabetic effects in db/db mice after being entrapped into solid lipid nanoparticles (SLNs). However, whether BBR-loaded SLNs (BBR-SLNs) also have beneficial effects on hepatosteatosis is not clear. We investigated the effects of BBR-SLNs on lipid metabolism in the liver using histological staining and reverse transcription polymerase chain reaction analysis. The results showed that oral administration of BBR-SLNs inhibited the increase of body weight and decreased liver weight in parallel with the reduction of serum alanine transaminase and liver triglyceride levels in db/db mice. The maximum drug concentration in the liver was 20-fold higher than that in the blood. BBR-SLNs reduced fat accumulation and lipid droplet sizes significantly in the liver, as indicated by hematoxylin and eosin and Oil Red O staining. The expression of lipogenic genes, including fatty acid synthase (FAS), stearoyl-CoA desaturase (SCD1), and sterol regulatory element-binding protein 1c (SREBP1c) were downregulated, while lipolytic gene carnitine palmitoyltransferase-1 (CPT1) was upregulated in BBR-SLN-treated livers. In summary, we have uncovered an unexpected effect of BBR-SLNs on hepatosteatosis treatment through the inhibition of lipogenesis and the induction of lipolysis in the liver of db/db mice.
Keywords: berberine, solid lipid nanoparticles, fatty liver, hepatosteatosis
Introduction
Berberine (BBR), a major pharmacological component of the Chinese herb Coptis chinensis originally used to treat bacterial diarrhea, has been recently demonstrated to have great effects on type 2 diabetes.1,2 BBR improved glucose and lipid metabolism in patients and animal models, possibly through the activation of adenosine monophosphate-activated protein kinase (AMPK) and improvement in insulin sensitivity.3,4 However, the clinical application of BBR is greatly limited due to its poor absorption in the gastrointestinal tract and its resulting low plasma levels and poor bioavailability (<5%) after oral administration. Besides, high doses (0.9–1.5 g/day in clinic) of BBR usually causes gastrointestinal side effects due to its poor absorption and long-term administration.5–7
Recently, the application of solid lipid nanoparticles (SLNs) as carriers in facilitating the absorption of active ingredients in traditional Chinese medicine has been widely studied, such as silymarin, triptolide, paclitaxel, etc.8 We have previously reported the preparation method, characterization, and pharmacokinetic features of BBR-loaded SLNs (BBR-SLNs); the results showed that after being entrapped into SLNs, BBR’s bioavailability was greatly increased.9 Furthermore, the oral administration of BBR-SLNs significantly suppressed body weight increases, fasting blood glucose levels, the homeostasis assessment of insulin resistance, and it ameliorated impaired glucose tolerance and insulin tolerance in db/db mice.9
Nonalcoholic fatty liver disease (NAFLD) has been recognized as a major health issue.10 It is caused by excess liver lipid accumulation, hepatic inflammation, hepatic insulin resistance, and nonalcoholic steatohepatitis that usually culminates in hepatic fibrosis or cirrhosis.11–13 Diabetic fatty liver is one of the common complications of diabetes, with an incidence of around 50% among diabetic patients. Particularly for patients with obesity, high plasma insulin levels increase the accumulation of nonesterified fatty acids (NEFA) in the liver and, in turn, fatty liver aggravates insulin resistance, glucose metabolic disorders, and other pathological conditions.14,15 Since BBR-SLNs are effective in hyperglycemia, we aimed to investigate the function of BBR-SLNs on hepatosteatosis in db/db mice in the present study.
Materials and methods
Drug and chemicals
Reference-grade BBR (purity quotient 99.8%) was purchased from Sigma-Aldrich Co. (St Louis, MO, USA). BBR-SLNs were prepared using our patented method16 and details were listed in our previous study.9 The composition of the blank nanoparticles is a mixture of glycerol tripalmitate and soybean phospholipid (1:1). All other reagents were high-performance liquid chromatography grade, or they were of the highest purity commercially available.
Experimental animals
Animal experiments in this study were conducted in strict accordance with the standard ethical guidelines and were approved by Xiamen University Animal Care and Use Committee. Forty male db/db mice, weighing 30–40 g, were purchased from the Experimental Animal Center of PLA Military Academy of Medical Sciences (Beijing, People’s Republic of China). After 1 week of acclimatization, the mice were randomly divided into one of four groups (ten mice per group): control group; BBR group (100 mg/kg); high-dose BBR-SLN group (100 mg/kg); and low-dose BBR-SLN group (50 mg/kg). Animals in the control group were treated with vehicle (saline), and the other groups were treated with their respective drugs (BBR or BBR-SLNs) via intragastric gavage for 4 weeks.
Measurement of tissue distribution of BBR-SLNs
At the end of the experiment, the mice were anesthetized; blood samples were drawn from the inferior vena cava and were then centrifuged at 3,000 rpm for 10 minutes within 1 hour after blood collection. Liver, kidney, subcutaneous adipose, epididymal adipose, pancreas, heart, brain, muscle, and jejunum tissues were harvested, weighed, and homogenized in triple-distilled water (0.4 mL/200 mg tissue). Tetrahydropalmatine (40 μL of a 1 μg/mL solution) was added as an internal standard and extracted with 2 mL of ethyl acetate by vortexing for 3 minutes, centrifuged at 3,500 rpm for 10 minutes, and 1.8 mL of the upper organic phase was carefully transferred to a 2 mL tube and evaporated by nitrogen. After drying, the residue was reconstituted in 50 μL of 0.1% formic acid: acetonitrile (50:50, v/v), vortexed for 3 minutes, and centrifuged at 15,000 rpm for 10 minutes at 4°C. The BBR concentration of the samples was analyzed by liquid chromatography–tandem mass spectrometry. The chromatographic system and instrumentation for detection were prepared as previously described.9
Measurement of serum ALT, AST, NEFA levels, and liver TG content
The levels of serum alanine transaminase (ALT) and aspartate transaminase (AST) were measured using an automatic biochemistry analyzer (ARCHITECT c8000) following the manufacturer’s instructions. The levels of serum NEFA were measured by the Non-Esterified Free Fatty Acids Assay Kit (Jiancheng, People’s Republic of China). For hepatic triglyceride (TG) content measurement, 100 mg of liver was homogenized to release the lipid, which was further extracted by the Triglyceride Quantification Kit (BioVision, Inc., Milpitas, CA, USA). Total lipid was suspended in 5% NP-40 in water, and TG concentrations were then measured according to the manufacturer’s procedures.9
Histological staining
The liver morphology was visualized by hemotoxylin and eosin (H&E) staining. Liver tissue samples were fixed in 4% polyformaldehyde, dehydrated, embedded in paraffin, and stained with H&E. For Oil Red O staining, a stock solution of Oil Red O (0.5 g/100 mL) in isopropanol was prepared, stored, and protected from light. Liver tissues were embedded in Optimal Cutting Temperature gel. Air-dried tissue sections of 7 μm were dipped in formalin, washed with Oil Red O without counterstaining with hematoxylin.9
RNA extraction and reverse transcription polymerase chain reaction
Total RNA was isolated from hepatic tissues using the standard TRIzol method according to the manufacturer’s instructions (Thermo Fisher Scientific, Waltham, MA, USA). Messenger (m)RNA was reverse-transcribed into complementary DNA using a reverse transcription kit (Promega Corporation, Fitchburg, WI, USA) following the manufacturer’s instructions. Quantitative real-time polymerase chain reaction was performed using SYBR Green Premix Ex Taq (Takara Bio Inc., Shiga, Japan) on LightCycler® 480 (Hoffman-La Roche Ltd., Basel, Switzerland). The primer sequences used are available upon request (details in Table 1). The relative mRNA levels of sterol regulatory element-binding protein 1c (SREBP1c), fatty acid synthase (FAS), stearoyl-CoA desaturase 1 (SCD1), carnitine palmitoyltransferase-1 (CPT1), PPAR, acyl-CoA oxidase 1 (ACOX1), and MCAD were calculated according to the 2 −ΔΔCp (Cp = crossing point) method.
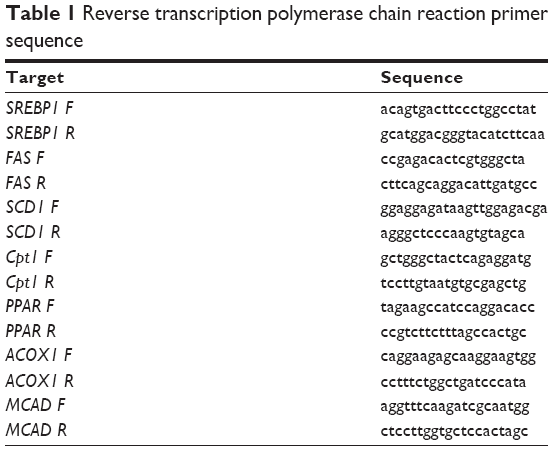 | Table 1 Reverse transcription polymerase chain reaction primer sequence |
Statistical analysis
All data were presented as the mean ± standard error of the mean. Statistical significance was determined by one-way analysis of variance. The differences were considered significant when P<0.05.
Results
BBR-SLNs were predominantly concentrated in the liver
The concentrations of BBR from the BBR-SLN (100 mg/kg) group had increased significantly in the liver compared to the control group (P<0.05) (Figure 1). The maximum drug concentration in the liver was 20 times more than that in the blood in the BBR-SLN group (Figure 1B), implying a predominant accumulation of BBR-SLNs in the liver. There was no difference observed in the other tissues (Figure 1A). It is worth mentioning that the BBR concentration was increased in the brain, suggesting that BBR-SLNs may promote the drug’s ability to penetrate the blood–brain barrier. Changes and characteristics of tissue distribution are substantial for the pharmacodynamic effects of BBR-SLNs. The accumulation of BBRs in the jejunum showed that the drug’s intestinal first-pass effect was obvious (Figure 1C).
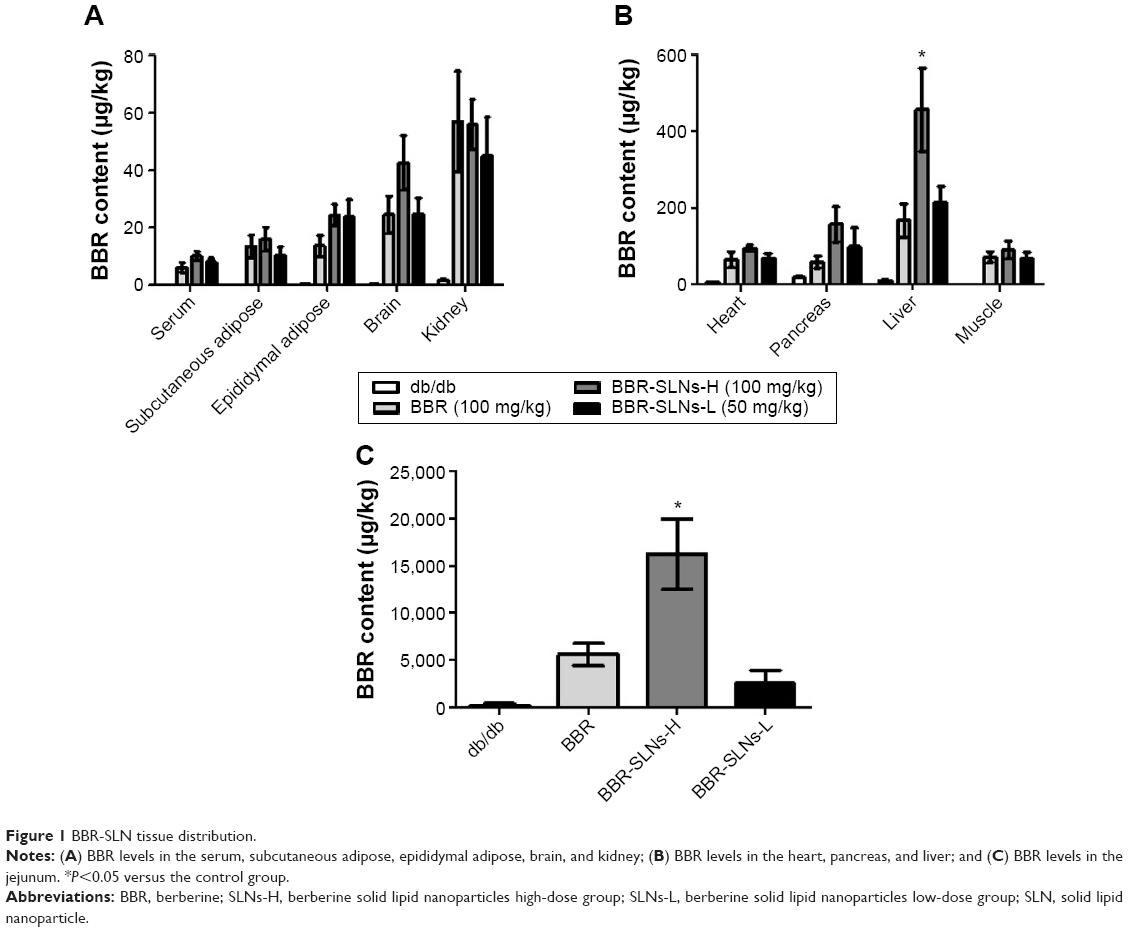 | Figure 1 BBR-SLN tissue distribution. |
Effects of BBR-SLNs on body weight and liver weight
Compared with the control group, BBR-SLNs at the dose of 100 mg/kg suppressed the gain of body weight, which was not seen in the mice treated with BBR 100 mg/kg or BBR-SLNs 50 mg/kg. The liver weight of mice in the BBR-SLN 100 mg/kg group was significantly lower (P<0.05) than that of the BBR group, shown as the ratio of liver weight to body weight (Figure 2).
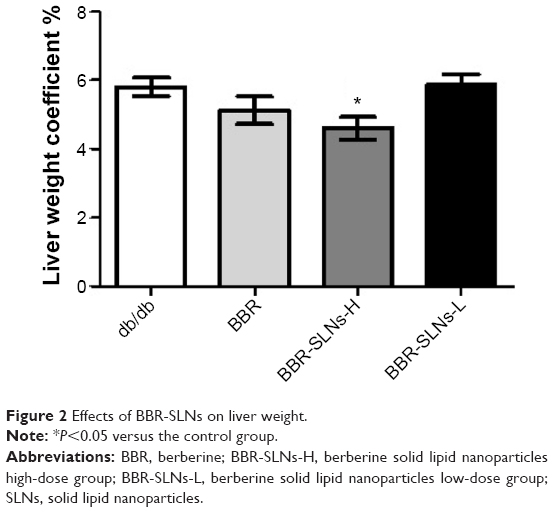 | Figure 2 Effects of BBR-SLNs on liver weight. |
Effects of BBR-SLNs on serum ALT, AST, and NEFA levels, and liver TG content
BBR-SLNs tended to lower the ALT level and hepatic TG content in db/db mice (P<0.05) (Figure 3), implying a liver protective effect. AST and NEFA levels were not significantly different among the vehicle and the BBR or BBR-SLN-treated group.
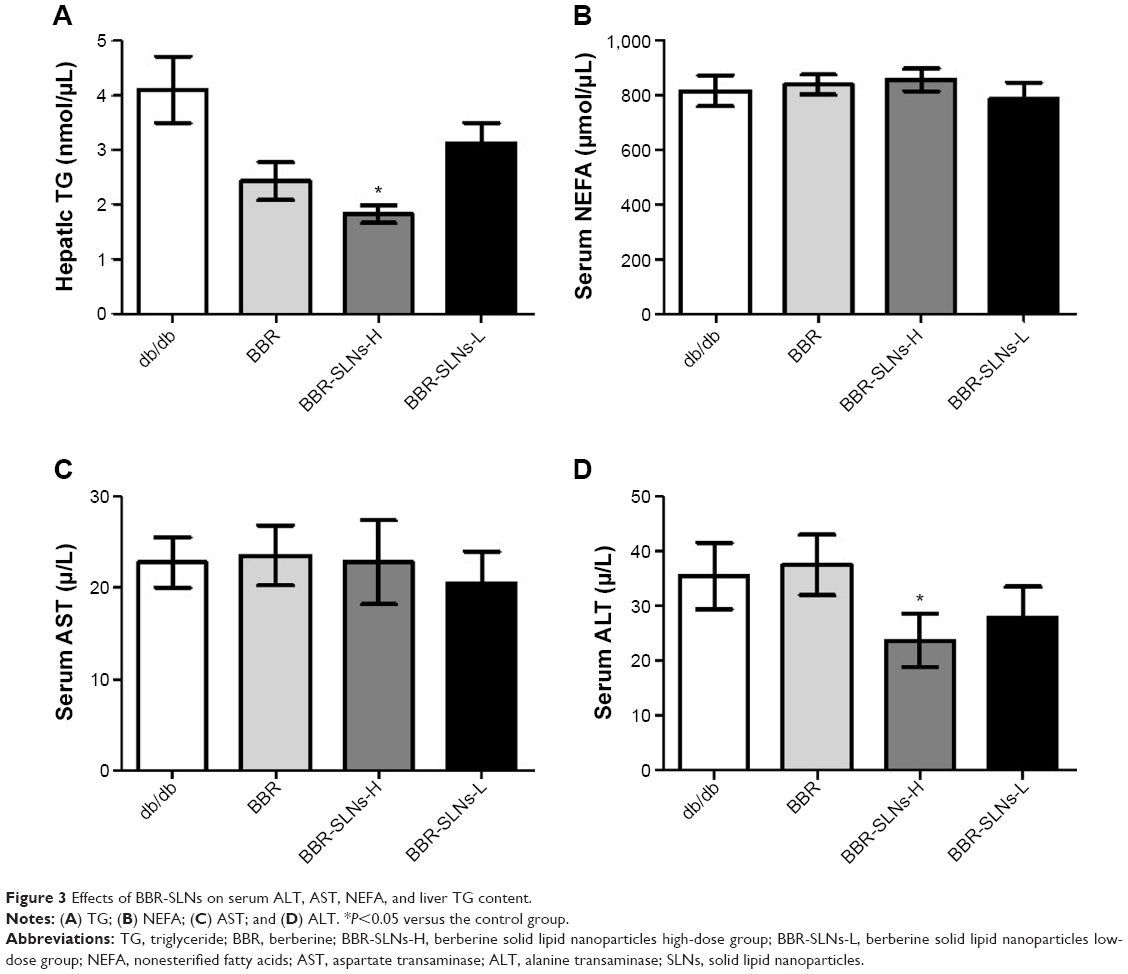 | Figure 3 Effects of BBR-SLNs on serum ALT, AST, NEFA, and liver TG content. |
BBR-SLNs inhibited lipid accumulation in the liver
Morphological changes of the liver were shown in Figure 4, where lipid accumulation in the liver resulted in pale discoloration (Figure 4A, B, D). As shown in Figure 4C, liver lipid content and lipid droplet size were significantly attenuated by BBR-SLN treatment. This finding was further confirmed by the assessment of lipid accumulation using Oil Red O staining (Figure 5). BBR-SLNs (100 mg/kg) exerted more potent effects than the BBR group at an equivalent dose (P<0.01).
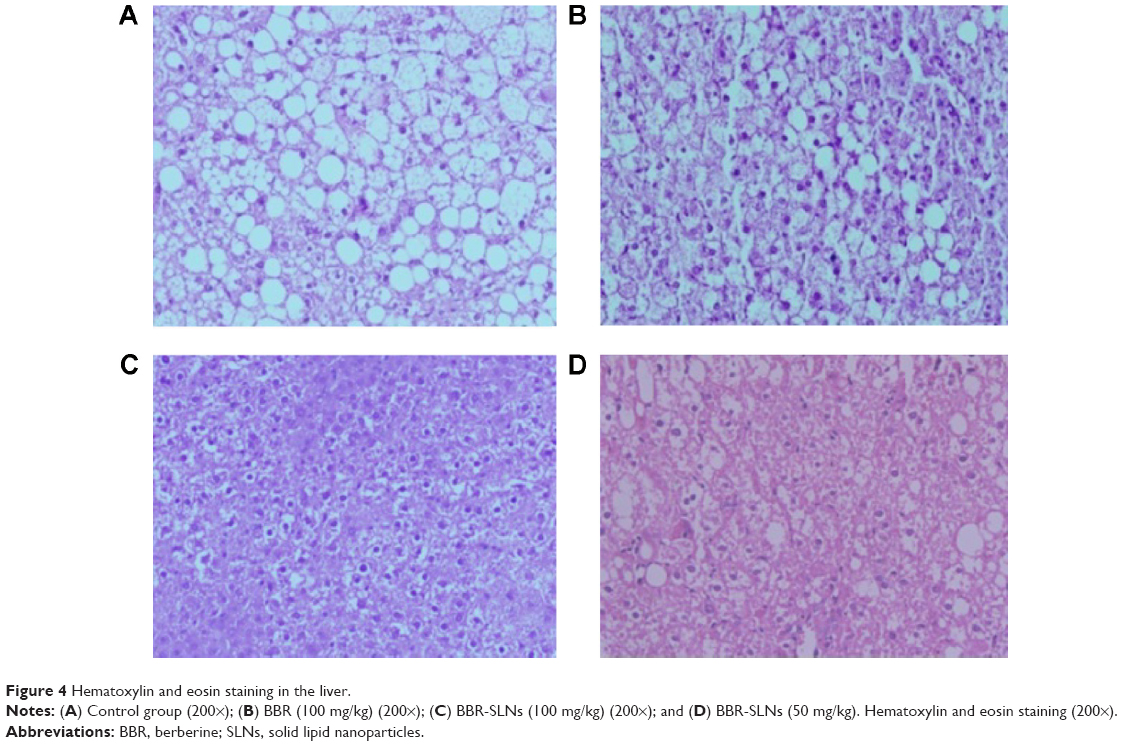 | Figure 4 Hematoxylin and eosin staining in the liver. |
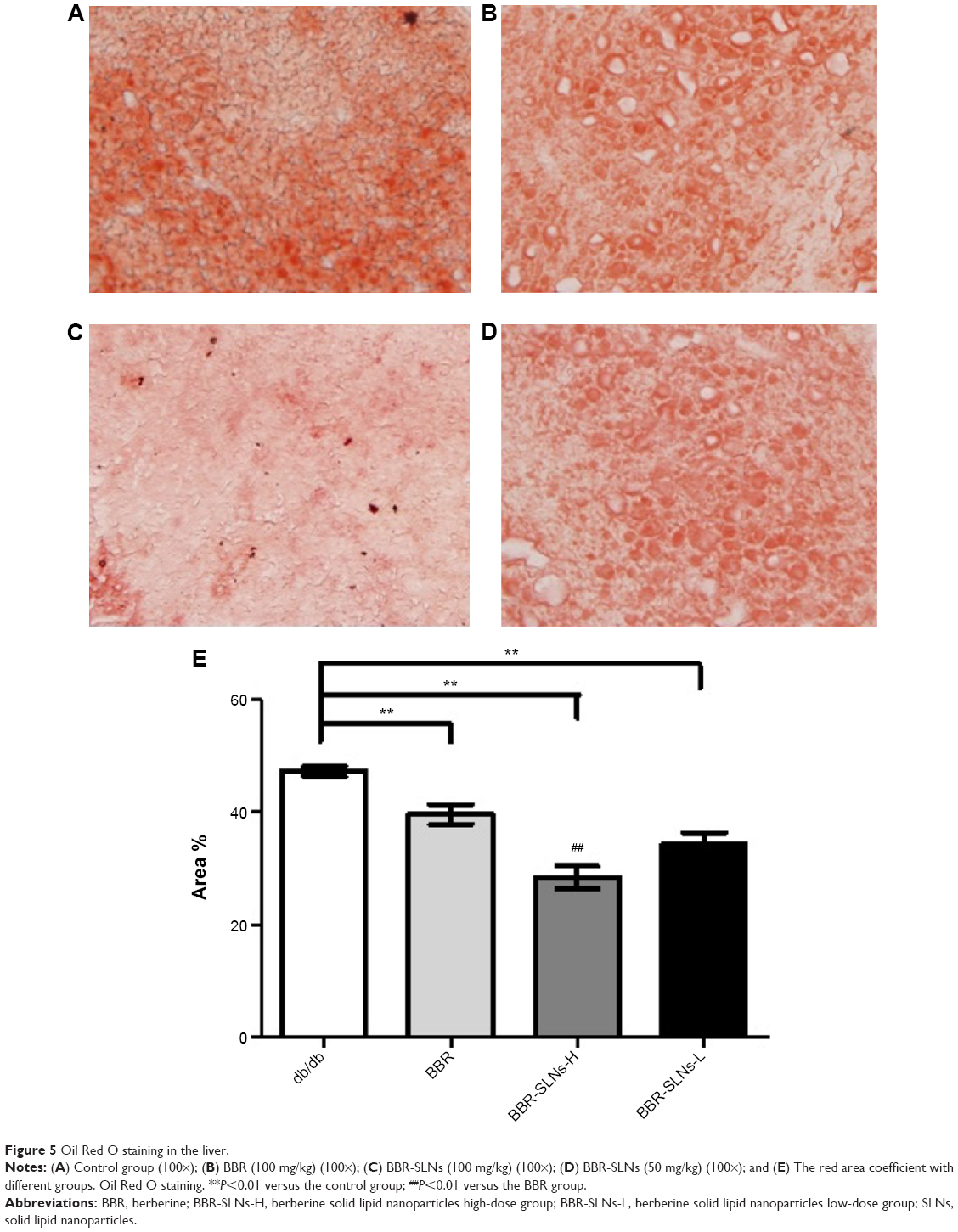 | Figure 5 Oil Red O staining in the liver. |
BBR-SLNs improved lipid metabolism
As shown in Figure 6, the mRNA expression of SCD1, FAS, and SREBP1c in the liver were significantly downregulated, while the CPT1 mRNA level was increased compared to the control mice (P<0.05). The mRNA levels of PPAR, ACOX1, and MCAD of the BBR-SLN groups tended to be lower than that of the control group, although not significantly (P>0.05).
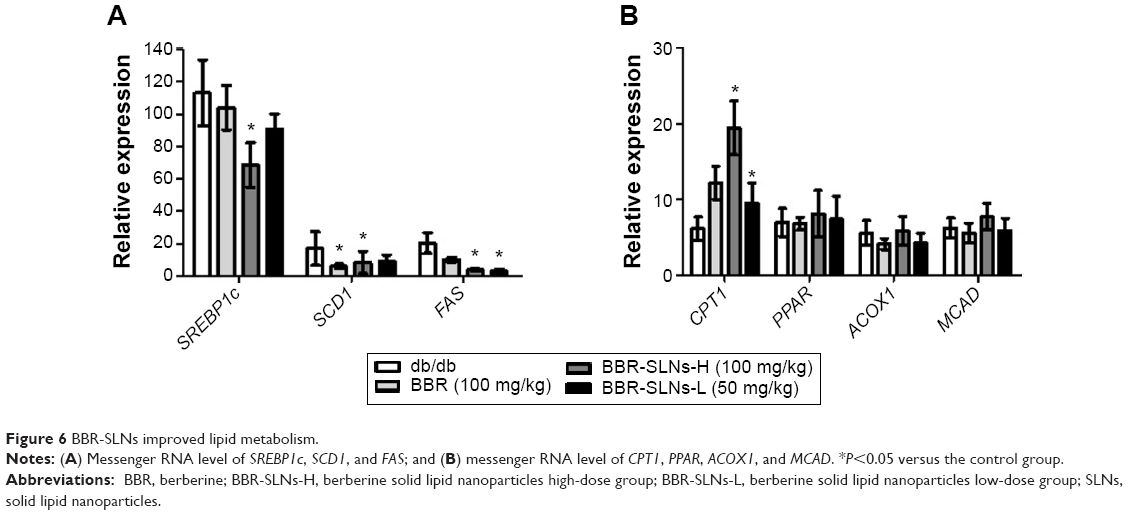 | Figure 6 BBR-SLNs improved lipid metabolism. |
Discussion
Previously, we have found that BBR-SLNs increased plasma BBR concentration and bioavailability, which further enhanced the antidiabetic effects of BBR-SLNs in vivo.9 Changes in the pharmacokinetic and tissue distribution characteristics in vivo are believed to be responsible for the enhanced efficacy and reduced toxicity of nanoformulations. Experimental evaluation of tissue distribution may reveal a basis for target organ efficacy. In certain target organs and active sites in the body, the distribution of BBR can also be used as evidence to explain why very low exposure of an oral drug is associated with the achievement of a certain efficacy of BBR. This is because the pharmacological effects are exerted by the unbound fraction of the drug, which is absorbed into the blood and then distributed into the target tissues to exert its effects.7,17 In the present study, a relatively dominant tissue distribution of BBR was observed in the liver (Figure 1B). The drug concentration of liver in the BBR-SLN group was nearly twofold higher than that of the BBR group. After oral administration, nanoparticles can undergo direct gastrointestinal transit or adhere to the mucosa before elimination in feces.9 The drug’s intestinal first-pass effect is obvious. Excretion of BBR to the intestinal lumen, bile, and urine may be a second pharmacokinetics, which leads to the very low absorption and exposure of BBR after oral intake.7 It has been reported that intravenous coadministration with cyclosporine, an inhibitor of P-gp, breast cancer resistance protein, organic anion-transporting polypeptide, and CYP3A, led to a dramatic decrease in the unbound concentrations of BBR in the liver and bile, but not in blood.18–20 Therefore, the drug transporters may play an important role in the entire first-pass elimination of BBR that would be more complicated than we assumed. In short, we have shown that extensive elimination in the small intestine and high hepatic distribution are the major causes for the low plasma concentrations of BBR, which further attenuates a hepatic steatosis effect in mice.7
The lower liver weight coefficient of mice in the BBR-SLN 100 mg/kg group was first observed. TG is thought to be a surrogate marker of disrupted insulin signal. We demonstrated that, after being entrapped into SLNs, BBR-SLNs were predominantly accumulated in the liver and significantly lowered live TG content, indicating that BBR-SLNs had beneficial effects in improving hepatosteatosis in db/db mice. In addition, BBR-SLNs tended to decrease the serum ALT levels, implying a potential liver protective effect without hepatotoxicity. With higher drug concentrations in the liver tissue, the attenuation of hepatic steatosis should be observed more significantly in the BBR-SLN-treated mice. Morphologically, the livers of the BBR-SLN-treated mice had fewer lipid droplets, suggesting a beneficial effect of BBR-SLNs on preventing lipid accumulation and reversing the disrupted structure of the liver. The effects of BBR-SLNs in ameliorating hepatosteatosis were at least partly due to the inhibition of lipogenesis while inducting lipolysis in the liver.
Previous findings mentioned that BBR may be a potential drug for NAFLD, and that the beneficial role of BBR on NAFLD might be achieved through multiple mechanisms, including mediating insulin resistance, regulating the AMPK pathway, modifying the gut microenvironment, and so on.21 Increasing empirical evidence implied that AMPK is a crucial player in BBR’s ability to dissipate stored fat and elevated glucose levels. More importantly, AMPK specifically binds to and directly phosphorylates SREBP-1c and SREBP-2. The Ser372 phosphorylation of SREBP-1c by AMPK could inhibit the proteolytic cleavage and nuclear translocation of SREBP-1c in hepatocytes, thereby preventing its autoregulation and transcription of target lipogenic genes (acetyl-CoA carboxylase 1 [ACC1], FAS, and SCD1).22 To elucidate the mechanisms of BBR-SLNs in decreasing liver lipid accumulation, we examined the mRNA levels of genes related to fatty acid transport and lipid metabolism. SREBP-1c is a crucial transcription factor that controls the expression of the genes involved in the biosynthesis of TG.9,23 FAS, ACC, and SCD1 are key enzymes controlling the lipogenic pathway. PPARα-targeted genes involved in β-oxidation are CPT1, acyl-CoA oxidase (ACO), ACOX1, etc.24 ACOX1 is the first and a rate-limiting enzyme of the PPARα-regulated and peroxisome proliferator-inducible fatty acid β-oxidation system. Our results showed that BBR-SLNs effectively inhibited SCD1, FAS, and SREBP1c expression (P<0.05) following the downregulation of SCD1 and FAS, which suggested that the increased hepatic expression of SREBP-1c might lead to fatty liver and increase rate of hepatic fatty acid synthesis with an increase in lipogenic genes like FAS, ACC, and SCD. CPT1 mRNA levels were increased in the BBR-SLN group mice compared to the control db/db mice. These genes were increased in the BBR-SLN group mice compared to the control db/db mice (P<0.05), indicating increased lipid catabolism. PPARs and COX1 are proposed to play a central role in the signaling system that controls lipid homeostasis.25–27 However, the levels of mPPAR, mAcox1, and mMCAD expression in the BBR-SLN group tended to be lower than that of the db/db group, although it did not reach statistical significance (P>0.05).
Although the beneficial action of BBR-SLNs is obvious, as a new dosage form, preclinical studies of BBR-SLNs are still incomplete and more chemical and physical properties of BBR-SLNs should be elucidated. New drug safety evaluation is necessary; in particular, the acute toxicity of the oral drug and the long-term toxicity of BBR-SLNs should also be rigorously investigated. Further studies should focus on the toxicology, other possible mechanisms, and the reality of dosage in human beings with NAFLD. The clinical effects in humans, as associated with the use of BBR, was already hypothesized, including liver steatosis.28 However, there is still a long way to go before BBR-SLNs become produced for the market.
Conclusion
In summary, we demonstrated the important role of BBR-SLNs (especially in 100 mg/kg dose group) in inhibiting hepatic steatosis in db/db mice via the induction of CPT1 expression and the inhibition of SREBP1c, SCD1, and FAS expression. The predominant accumulation of BBR-SLNs in the liver is an important material foundation for the enhanced pharmacodynamic effects of BBR-SLNs in ameliorating hepatosteatosis in db/db mouse models.
Acknowledgments
This work was supported by grants from the National Natural Science Foundation to Shu-yu Yang (30973912), Su-huan Liu (81270901), and Xue-jun Li (81073113); the Key project of Fujian Provincial Science and Technology Planning programs (2012D60) and the Xiamen Innovation Program for Outstanding Youth Scientist (2011S0446) to Su-huan Liu; as well as the Xiamen Science and Technology Bureau (Xiamen Research Platform for Systems Biology of Metabolic Disease; 3502Z20100001).
Disclosure
The authors report no conflicts of interest in this work.
References
Yang J, Ljn J. [Advance on study in anti-tumor mechamism of bererine (Ber)]. Zhongguo Zhong Yao Za Zhi. 2007;32(10):881–883, 934. Chinese. | ||
Tang J, Feng Y, Tsao S, Wang N, Curtain R, Wang Y. Berberine and Coptidis rhizoma as novel antineoplastic agents: a review of traditional use and biomedical investigations. J Ethnopharmacol. 2009;126(1):5–17. | ||
Yin J, Gao Z, Liu D, Liu Z, Ye J. Berberine improves glucose metabolism through induction of glycolysis. Am J Physiol Endocrinol Metab. 2008;294(1):E148–E156. | ||
Li X, Li Z, Xue M, et al. Fructus Xanthii attenuates hepatic steatosis in rats fed on high-fat diet. PLoS One. 2013;8(4):e61499. | ||
Hua W, Ding L, Chen Y, Gong B, He J, Xu G. Determination of berberine in human plasma by liquid chromatography-electrospray ionization-mass spectrometry. J Pharm Biomed Anal. 2007;44(4):931–937. | ||
Liu Y, Hao H, Xie H, Lv H, Liu C, Wang G. Oxidative demethylenation and subsequent glucuronidation are the major metabolic pathways of berberine in rats. J Pharm Sci. 2009;98(11):4391–4401. | ||
Liu YT, Hao HP, Xie HG, et al. Extensive intestinal first-pass elimination and predominant hepatic distribution of berberine explain its low plasma levels in rats. Drug Metab Dispos. 2010;38(10):1779–1784. | ||
Ajazuddin, Saraf S. Applications of novel drug delivery system for herbal formulations. Fitoterapia. 2010;81(7):680–689. | ||
Xue M, Yang MX, Zhang W, et al. Characterization, pharmacokinetics, and hypoglycemic effect of berberine loaded solid lipid nanoparticles. Int J Nanomedicine. 2013;8:4677–4687. | ||
Lazo M, Clark JM. The epidemiology of nonalcoholic fatty liver disease: a global perspective. Semin Liver Dis. 2008;28(4):339–350. | ||
Browning JD, Horton JD. Molecular mediators of hepatic steatosis and liver injury. J Clin Invest. 2004;114(2):147–152. | ||
Jaskiewicz K, Rzepko R, Sledzinski Z. Fibrogenesis in fatty liver associated with obesity and diabetes mellitus type 2. Dig Dis Sci. 2008;53(3):785–788. | ||
Fon Tacer K, Rozman D. Nonalcoholic Fatty liver disease: focus on lipoprotein and lipid deregulation. J Lipids. 2011;2011:783976. | ||
Iizuka K, Horikawa Y. ChREBP: a glucose-activated transcription factor involved in the development of metabolic syndrome. Endocr J. 2008;55(4):617–624. | ||
Nagle CA, Klett EL, Coleman RA. Hepatic triacylglycerol accumulation and insulin resistance. J Lipid Res. 2009;50 Suppl:S74–S79. | ||
Xue-jun Li, Mei Xue, Shu-yu Yang, inventor. The First Affiliated Hospital of Xiamen University, assignee. A kind of berberine hydrochloride solid lipid nanoparticle pharmaceutics and its preparation method. China Patent CN201210495674.4. 2013 Mar 6. | ||
Cummins CL, Jacobsen W, Christians U, Benet LZ. CYP3A4-transfected Caco-2 cells as a tool for understanding biochemical absorption barriers: studies with sirolimus and midazolam. J Pharmacol Exp Ther. 2004;308(1):143–155. | ||
Tsai PL, Tsai TH. Hepatobiliary excretion of berberine. Drug Metab Dispos. 2004;32(4):405–412. | ||
Xia CQ, Liu N, Miwa GT, Gan LS. Interactions of cyclosporin a with breast cancer resistance protein. Drug Metab Dispos. 2007;35(4):576–582. | ||
Shitara Y, Nagamatsu Y, Wada S, Sugiyama Y, Horie T. Long-lasting inhibition of the transporter-mediated hepatic uptake of sulfobromophthalein by cyclosporin a in rats. Drug Metab Dispos. 2009;37(6):1172–1178. | ||
Liu Y, Zhang L, Song H, Ji G. Update on berberine in nonalcoholic fatty liver disease. Evid Based Complement Alternat Med. 2013;2013:308134. | ||
Li Y, Xu S, Mihaylova MM, et al. AMPK phosphorylates and inhibits SREBP activity to attenuate hepatic steatosis and atherosclerosis in diet-induced insulin-resistant mice. Cell Metab. 2011;13(4):376–388. | ||
Sun W, Bi Y, Liang H, et al. Inhibition of obesity-induced hepatic ER stress by early insulin therapy in obese diabetic rats. Endocrine. 2011;39(3):235–241. | ||
Osborne TF. Sterol regulatory element-binding proteins (SREBPs): key regulators of nutritional homeostasis and insulin action. J Biol Chem. 2000;275(42):32379–32382. | ||
Shimano H, Horton JD, Shimomura I, Hammer RE, Brown MS, Goldstein JL. Isoform 1c of sterol regulatory element binding protein is less active than isoform 1a in livers of transgenic mice and in cultured cells. J Clin Invest. 1997;99(5):846–854. | ||
Jeong HW, Lee JW, Kim WS, et al. A newly identified CG301269 improves lipid and glucose metabolism without body weight gain through activation of peroxisome proliferator-activated receptor alpha and gamma. Diabetes. 2011;60(2):496–506. | ||
Xue M, Zhao Y, Li XJ, et al. Comparison of toxicokinetic and tissue distribution of triptolide-loaded solid lipid nanoparticles vs free triptolide in rats. Eur J Pharm Sci. 2012;47(4):713–717. | ||
Caliceti C, Rizzo P, Cicero AF. Potential benefits of berberine in the management of perimenopausal syndrome. Oxid Med Cell Longev. 2015;2015:723093. |
 © 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms.php
and incorporate the Creative Commons Attribution
- Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms.php
and incorporate the Creative Commons Attribution
- Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.