Back to Journals » Drug Design, Development and Therapy » Volume 14
Berberine Induces Autophagic Cell Death in Acute Lymphoblastic Leukemia by Inactivating AKT/mTORC1 Signaling
Authors Liu J ![]() , Liu P, Xu T, Chen Z, Kong H, Chu W, Wang Y, Liu Y
, Liu P, Xu T, Chen Z, Kong H, Chu W, Wang Y, Liu Y
Received 19 November 2019
Accepted for publication 21 April 2020
Published 12 May 2020 Volume 2020:14 Pages 1813—1823
DOI https://doi.org/10.2147/DDDT.S239247
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 4
Editor who approved publication: Dr Sukesh Voruganti
Jian Liu,1 Peng Liu,2 Tiantian Xu,1 Zhiwei Chen,1 Huimin Kong,1 Weihong Chu,1 Yingchao Wang,1 Yufeng Liu1
1Department of Pediatrics, The First Affiliated Hospital of Zhengzhou University, Zhengzhou 450052, People’s Republic of China; 2Department of Pediatric Intensive Care Unit, The First Affiliated Hospital of Zhengzhou University, Zhengzhou 450052, People’s Republic of China
Correspondence: Jian Liu
The First Affiliated Hospital of Zhengzhou University, 1 East Jianshe Road, Zhengzhou 450052, People’s Republic of China
Email [email protected]
Introduction: Berberine has been reported to inhibit cancer cell growth by apoptosis induction and exhibits a protective role against cancer progression. The current study aims to investigate the effects of berberine on acute lymphoblastic leukemia (ALL) and the mechanism beyond apoptosis.
Methods: Cell viability was determined in ALL cell lines EU-6 and SKW-3 using trypan blue staining. Cell autophagy was determined by immunofluorescence and Western blot. ALL xenograft mice were established to investigate the anti-tumor effects of BBR. The molecular mechanism was explored in ALL cell lines using siRNA and signaling inhibitors.
Results: Herein, we show that berberine treatment significantly inhibits ALL cell viability and promotes cell death by inducing autophagy in a dose-dependent manner. Moreover, berberine significantly alleviates the aggressive pathological condition in ALL xenograft mice. Mechanistic studies exhibit that berberine induces autophagic death in ALL cells by inactivating AKT/mTORC1 signaling. Chemically targeting AKT/mTORC1 signaling controls berberine-induced cell autophagy in vitro, and blockade of autophagic process blunts berberine-alleviated pathological condition in vivo.
Discussion: In conclusion, our study reveals that berberine could induce ALL cell autophagic death by inactivating AKT/mTORC1 signaling that could be used to develop small molecule drug for ALL treatment.
Keywords: acute lymphoblastic leukemia, berberine, AKT/mTORC1, autophagy
Introduction
Acute lymphoblastic leukemia (ALL) is an aggressive hematological malignancy caused by both B-cell and T-cell lymphoid lineage disorders. Even though most ALL patients show better prognosis in children, long-term survival remains poor in adult patients.1,2 In adults, about 75% of patients are developed from B-cell lymphoid lineage disorders, while the others are generated from T-cell lymphoid lineage disorders.3 There are several symptoms of ALL: frequent or severe nose bleeds, bleeding from the gums, bone pain, lumps caused by swollen lymph nodes in and around the neck, underarm, abdomen or groin as well as fever and shortness of breath.4 Furthermore, the infiltration of lymph nodes, liver, brain and spleen commonly occurs at the stage of diagnosis resulting in great challenges in the following treatment.5 In recent years, the 5-year survival rate for ALL patients has been improved owing to the enhanced supportive care and novel therapies, however, continuous therapy could also lead to adverse effects.6 As a consequence, it is urgent to uncover novel pathogenic mechanisms and develop related drugs for ALL treatment.
Berberine (BBR), a natural alkaloid compound that existed in traditional Chinese medicine Coptis chinensis, shows remarkable pharmacological properties in the treatment of various diseases.7 For instance, BBR has been used as a hypolipidemic drug on diabetic mellitus for years.8 In addition, BBR performs anti-inflammatory and anti-thrombotic activities through inhibiting lipoxygenase and antioxidant properties.9 It has also been reported that BBR has the ability to suppress cell proliferation by inhibiting DNA and protein synthesis in vascular smooth muscle cells.10 Furthermore, BBR-induced cell cycle arrest at G1 phase and decreased the percentage of G2/M phase in lymphocytic Jurkat cells.11
Autophagy is a multistep process that characterized by bulk autophagosomes in the cytoplasm.12 Autophagy is identified to participate in the cellular homeostasis maintenance in normal cellular processes.13 Recently, signaling pathways that involve in the autophagy have been implicated. For instance, activation of ROS/JNK prominently induced autophagy in glioma cells.14 Protein disulfide isomerase family 6 (PDIA6) inhibits autophagy of non-small cell lung cancer cells through activating MAP4K1/JNK signaling.15 In addition, inactivation of PI3K/AKT/mTOR is proved to contribute to autophagy process in the mouse cerebral cortex and in human ALL.16,17 The role of BBR on autophagy has been widely studied on various disorders, including mitochondria dysfunction,18 neurodegenerative disease,19 heart disease,20 as well as cancers.21 The autophagy-related pathway AMPK/mTOR plays an important role on BBR ameliorating inflammation and cell apoptosis.22,23 However, it is unclear whether AKT/mTOR signaling mediates BBR-mediated autophagy on ALL.
Protein kinase B (PKB, also known as AKT) hyperactivation exists in the primary bone marrow samples from patients with ALL.24 The serine kinase mTOR, a downstream effector of AKT, controls cell proliferation in various cell processes. Various studies have identified that the inhibitors of mTORC1, such as rapamycin or RAD001 show anti-ALL activities.25 PI3K/AKT/mTOR had frequently been served as a target for ALL therapy26,27 and mediates autophagy process in various cell types.28,29
In this study, our aims are to investigate the effects of BBR on ALL. We find BBR caused ALL cell death by inducing autophagy. We also investigate the underlying mechanism responsible for BBR-induced autophagy. The findings will provide crucial insight into the application of BBR on ALL treatment.
Patients and Methods
Patients
A total of 26 patients aged between 4 and 71 years, already diagnosed with ALL at the First Affiliated Hospital of Zhengzhou University, were enrolled in this study. All the patients were diagnosed according to the cytomorphology, cytochemistry, molecular genetics, multipara meter flow cytometry and immunology.30 The details of the patients’ information are presented in Supplemental Table 1. This study was approved by the Ethical Committee of the First Affiliated Hospital of Zhengzhou University (No: 20170853), and all experiments were conducted according to the Declaration of Helsinki principles. All participants and their legal guardians signed written informed consent before the experiments.
Cell Culture and Transfection
ALL cell line EU-6 was purchased from American Type Culture Collection (ATCC). SKW-3 and Jurkat cells were purchased from the Cell Bank of the Chinese Academy of Science in Shanghai, and the phenotypes of the three cell lines were described previously.31 All cells were cultured in RPMI-1640 medium supplemented with 10% fetal bovine serum at 37°C and 5% CO2. For transfection, the siRNA targeting ATG5 (5ʹ-ACCGGAAACUCAUGGAAUA-3ʹ) and the negative control were constructed and transfected into Jurkat, EU-6 and SKW-3 cells using Lipofectamine 3000 following the manufacture’s protocol. Briefly, cells were cultured in Opti-MEM® I Reduced Serum Medium (Gibco, Thermo Fisher Scientific) containing 4 μg Lipofectamine®RNAiMAX (Invitrogen, ThermoFisher Scientific) with 100 nM of ATG5 siRNA or control siRNA for 24 h. Then, the medium was removed and replaced by RPMI containing 10% FBS for other 48 h culture.
Peripheral Blood Mononuclear Cells (PBMCs) Separation
PBMCs were isolated according to the previous study.32 Briefly, Ficoll-Hypaque density gradient separation (Sigma-Aldrich) was used to isolate PBMCs, which were then cultured in plastic dishes to remove adherent cells. After that cells were cultured in RPMI-1640 medium supplemented with 10% fetal bovine serum at 37°C and 5% CO2.
Cell Viability
Cells were placed in a 96-well plate with a density of 2000 cells per well and cultured with different concentrations (0, 12.5, 25, 50 and 100 μM) of berberine chloride (St. Louis, MO, USA) according to previous studies.33 For mechanistic experiments, cells were treated with 100 μM BBR in the presence or absence of autophagy inhibitors 3-MA (1 mM) or Bafilomycin A1 (Baf A1, 0.5 μM), mTORC1 signaling inhibitor rapamycin (0.5 μM) or MHY1485 (2 μM) to validate the contribution of autophagy and AKT/mTORC1 signaling to the effects of BBR on cell autophagy. After 24 h culture, 10 μL Cell Counting Kit-8 (CCK-8) was added in each well and incubated for 1 h. Cell viability was detected using a microplate reader at the absorbance of 450 nm. The cells collected from different groups were stained by trypan blue (Sigma, USA) and evaluated under a microscope according to the manufacture’s instruction. The dead cells were stained in blue. The percentage of death cells was determined by counting total and blue cells in the five random microscopic fields.
Western Blot
After treatment, cells were harvested and lysed by RIPA buffer (Invitrogen). Total protein was measured by BCA protein assay kit (Beyotime Biotechnology, Shanghai, China), separated on 10% SDS-PAGE, and then transferred onto polyvinylidene fluoride (PVDF) and blocked with 5% skimmed milk for 1 h at room temperature. After three times washing with 1×TBST buffer, the PVDF membrane was incubated with the primary antibodies (anti-S6, 1:1000, Abcam; anti-p-S6, 1:1000, Abcam; anti-AKT, 1:500, Abcam; anti-p-AKT (T308), 1:500, Abcam; anti-p-AKT (S473), 1:1000, Abcam; LC3-I/II, 1:500, Abcam; GAPDH, 1:1000, Abcam; Beclin-1, 1:2000, Abcam; anti-ATG5, 1:1000, Abcam) at 4°C for 24 h. Next, the membrane was washed using 1×TBST and followed by incubation with horseradish peroxidase (HRP)-conjugated secondary antibody at room temperate for 1 h. The protein bands were visualized using enhanced chemiluminescence. GAPDH was used as an internal control.
Immunofluorescence Assay
For immunofluorescence, cells were fixed with PHEMO buffer for 10 min and permeabilized with 0.5% Triton X-100 at room temperature for 15 min. After washing with PBS three times, cells were incubated with 3% BSA for 30 min. The anti-LC3 antibody (Santa Cruz) at an appropriate concentration was incubated with the cells overnight at 4°C. Afterward, cells were incubated with Alexa Flour-conjugated goat anti-rabbit IgG at room temperature for 1 h. Images were visualized using confocal microscopy (Leica STED, Germany).
Leukemia Engraftment
NOD-SCID mice (6–8 weeks) were housed in the ventilated cage with a temperature of 22± 2°C and a 12:12 h light/dark cycle. All mice were irradiated with a sublethal dose of 250 cGy γ-radiation using a radiation source before cell transplant as described previously.34 For WT ALL mice, NOD-SCID mice were intravenously (i.v.) injected with 1×106 WT ALL cells. To generate ATG5−/- ALL mice, NOD-SCID mice were i.v. injected with 1×106 siATG5 ALL cells. The different group mice were then orally administrated with BBR at a dose of 10 mg/kg/d for 2 weeks according to the previous study.35,36 The control mice were received the same volume of DMSO. The mice were assessed by flow cytometric analysis for human CD45+ cells in the bone marrow and spleen at 1 week after completion of treatment. The blood was sampled from the orbital venous, and the survival condition was recorded every day for 8 weeks. This study was approved by the Ethic Committee of The First Affiliated Hospital of Zhengzhou University (No: 20170881), and all experiments were handled in accordance with the animal care and use guidelines.
Hematologic Parameter Test
The blood parameters, such as red blood cells (RBC), white blood cells (WBC) and hemoglobin, were detected using an automatic blood cell analyzer according to the manufacture’s instruction.
Statistical Analysis
All experiments were repeated at least three independent times. Results were presented as mean±SD. Student’s t-test was used for comparisons between groups, while Statistical significance among multiple groups was analyzed using a one-way analysis of variance. p<0.05 was considered as statistically significant.
Results
BBR Promotes ALL Cell Death
To determine the potential effects of BBR on ALL, ALL cell lines Jurkat, EU-6 and SKW-3 were treated by BBR with different concentrations of 0, 12.5, 25, 50 and 100 μM. Cell viability and death were determined using CCK-8 and trypan blue staining, respectively. It showed that BBR treatment induced a significantly reduced viability of Jurkat, EU-6 and SKW-3 in a dose-dependent manner (Figure 1A). To confirm the effects of BBR on ALL, the peripheral blood mononuclear cells (PBMCs) were separated from peripheral blood of ALL patients and subjected to BBR for in vitro treatment. The results revealed that BBR treatment incurred a significantly reduced viability of PBMCs in a dose-dependent manner (Figure 1A). Consistently, BBR treatment led to an increased death of Jurkat, EU-6, SKW-3 and PBMCs in a dose-dependent manner (Figure 1B). The above data indicated that BBR-induced ALL cell death.
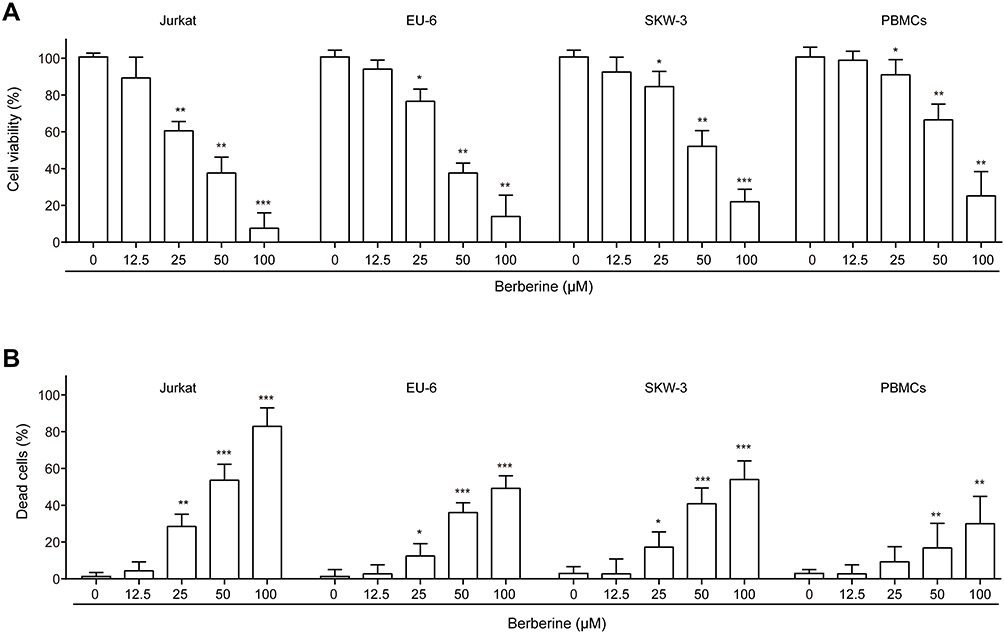 |
Figure 1 BBR inhibits cell viability in ALL cells. ALL Jurkat, EU-6, SKW-3 and peripheral blood-derived mononuclear cells (PBMCs) of ALL patients were treated with BBR at concentrations of 0, 12.5, 25, 50, 100 μM, respectively. (A) Cell viability was determined using CCK-8. (B) Dead cells were detected using trypan blue dye. Data were showed as mean±SD, n=3, *p<0.05, **p<0.01, ***p<0.001. |
BBR Induces Autophagic Death in ALL Cells
The previous study reported BBR promoted autophagic cell death in lung cancer cells.37,38 Next, we investigated whether or not autophagic changes occurred on ALL cell lines and ALL-derived PBMCs after BBR treatment by detecting the autophagic markers light chain 3 (LC3) and Beclin 1. The results showed that BBR treatment increased the percentage of LC3-positive cells in a dosage-dependent manner (Figure 2A and B). Consistently, the expressions of Beclin-1 and LC3-II were all increased by BBR in a dosage-dependent manner (Figure 2C). These findings suggested that BBR treatment induced autophagic death in ALL cells.
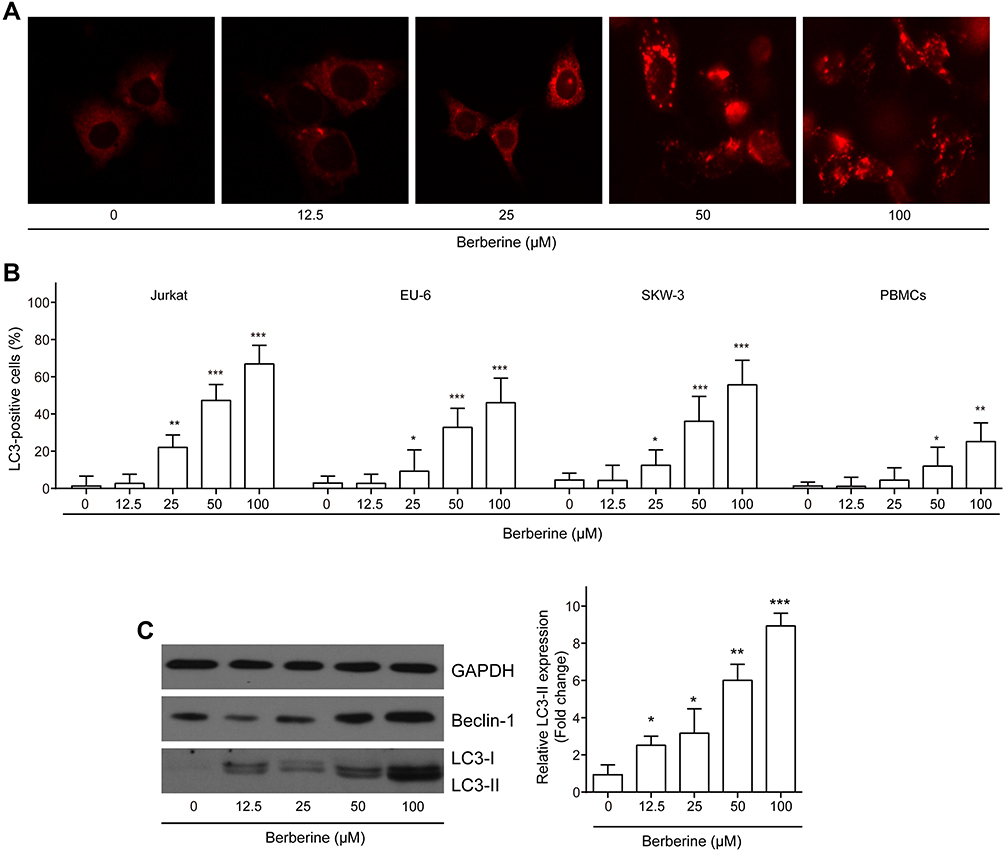 |
Figure 2 BBR promotes cell autophagy in ALL cells. ALL Jurkat, EU-6, SKW-3 and peripheral blood-derived mononuclear cells (PBMCs) of ALL patients were treated with BBR at concentrations of 0, 12.5, 25, 50, 100 μM, respectively. (A and B) The autophagy marker LC3 in EU-6 cells was visualized using immunofluorescence assay. (C) The protein expression of beclin-1, LC3-I and LC3-II in Jurkat, EU-6, SKW-3 and PBMC cells was measured by Western blot. Data were showed as mean±SD, n=3, *p<0.05, **p<0.01, ***p<0.001. |
Inhibition of Autophagy Reverses BBR-Induced Decreased Cell Viability
Next, we chemically verified the autophagic effects of BBR on ALL cells using autophagy inhibitors 3-methyladenine (3-MA) and Bafilomycin A1 (Baf A1). Jurkat, EU-6, SKW-3 and PBMC cells were treated by BBR in the presence or absence of 3-MA or Baf A1, respectively. Intriguingly, both 3-MA and Baf A1 significantly rescued BBR-induced decreased cell viability (Figure 3A). Meanwhile, we genetically verified the autophagic effects using siRNA targeting autophagic mediator ATG5 in Jurkat, EU-6 and SKW-3 (Supplemental Figure 1). The knockdown of ATG5 significantly reversed BBR-induced decreased cell viability and blunted BBR-induced cell autophagy in ALL cells (Figure 3B and C). Taken together, these results indicated that BBR-induced ALL cells into autophagic death.
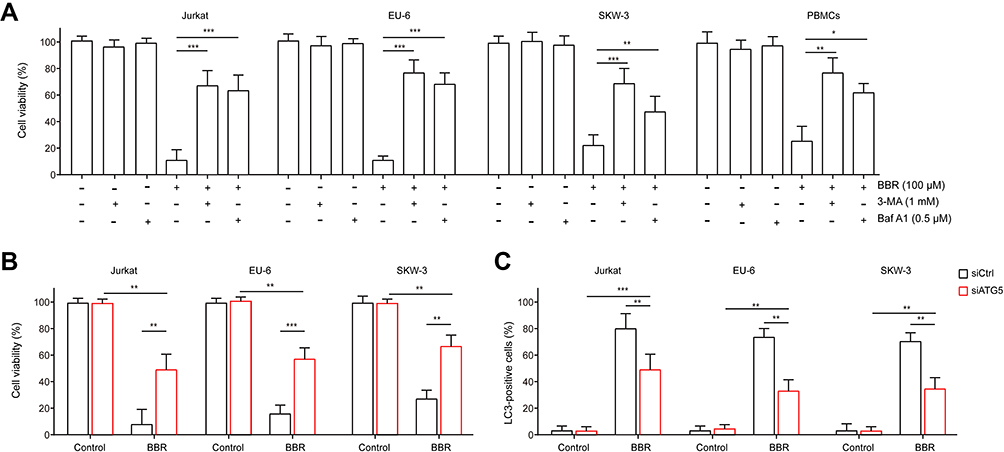 |
Figure 3 Inhibition of autophagy reverses BBR-inhibited cell viability. Jurkat, EU-6, SKW-3 and PBMC cells were treated with BBR in the presence or absence of 3-MA or Baf A1. (A) Cell viability was determined using CCK-8. (B, C) Jurkat, EU-6 or SKW-3 were transfected with siRNA targeting ATG5 or scrambled sequence followed by 100 μM BBR treatment for 48 h. Cell viability and autophagy were determined using CCK-8 and LC3 staining, respectively. Data were showed as mean±SD, n=3, *p<0.05, **p<0.01, ***p<0.001. |
BBR Induces ALL Cell Autophagy by Inactivating AKT/mTORC1 Signaling
Previous studies showed that AKT/mTORC1 signaling mediated cell autophagy in hepatocytes,28 colorectal cancer cells,39 human lung and pancreatic cancer cells.40 Here, we investigated whether AKT/mTORC1 signaling was involved in BBR-induced autophagy in ALL cells. EU-6 cells were treated with different concentrations of BBR, and the protein expressions of AKT/mTORC1 signaling-associated proteins S6, AKT and their phosphorylated proteins (p-S6, p-AKT) were detected. We found BBR treatment markedly inhibited activation of AKT/mTORC1 signaling as showed by the decreased protein expressions of p-S6, p-AKT in a dosage-dependent manner (Figure 4A). To validate AKT/mTORC1 is responsible for the BBR-induced autophagy in ALL cells, EU-6 cells were treated with BBR in the presence or absence of mTORC1 inhibitor rapamycin or activator MHY1485, respectively. It showed that rapamycin exacerbated BBR-induced decreased expressions of p-S6, p-AKT. Conversely, MHY1485 significantly attenuated BBR-induced inhibition of AKT/mTORC1 signaling (Figure 4B). Moreover, Inhibition or activation of AKT/mTORC1 signaling also increased or decreased BBR-induced cell autophagy as showed by the increased or decreased autophagic markers Beclin-3 and LC3-II as well as the percentage of LC3-positive cells, respectively (Figure 4B and C). The results indicated that BBR-induced ALL cell autophagy via inactivating AKT/mTORC1 signaling pathway.
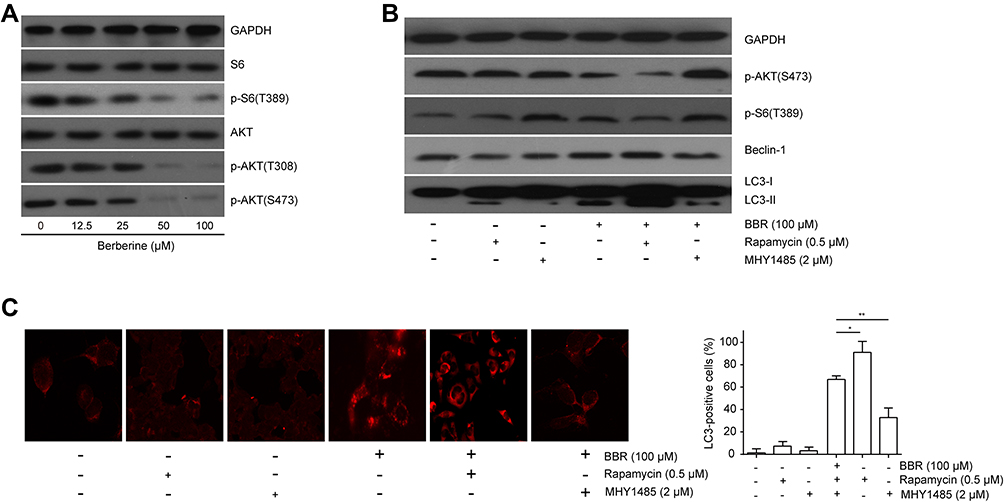 |
Figure 4 AKT/mTORC1 inactivation mediates BBR-induced cell autophagy. (A) EU-6 cells were treated with different concentrations of BBR (0, 12.5, 25, 50, 100 μM), Western blot was performed to detect the proteins of AKT/mTORC1 signaling. (B) EU-6 cells were treated with 100 μM BBR in the presence or absence of rapamycin or MHY1485, Western blot was conducted to determine cell autophagic markers, (C) immunofluorescence assay was performed to detect the LC3-positive cells. Data were showed as mean±SD, n=3, *p<0.05, **p<0.01. |
Inhibition of AKT/mTORC1 Intensifies BBR-Induced Decreased Cell Viability
Next, we confirmed whether inhibition of AKT/mTORC1 intensified BBR-induced decreased cell viability. Jurkat, EU-6, SKW-3 and PBMC cells were treated by BBR in the presence or absence of MHY1485 or rapamycin, respectively. As it is presented in Figure 5A, MHY1485 or rapamycin treatment significantly attenuated or intensified BBR-induced reduced cell viability, respectively. Meanwhile, the increased dead cells induced by BBR were also significantly decreased or enhanced by MHY1485 or rapamycin treatment, respectively (Figure 5B). These data suggested that AKT/mTORC1 signaling is involved in BBR-induced ALL cell death.
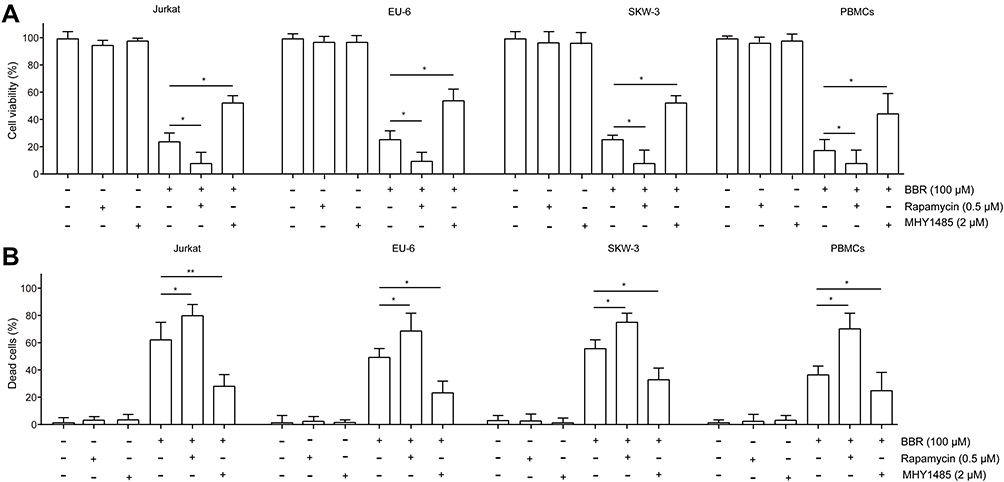 |
Figure 5 Inhibition of AKT/mTORC1 activation intensifies BBR-inhibited cell viability. Jurkat, EU-6, SKW-3 and PBMC cells were treated with 100 μM BBR in the presence or absence of rapamycin or MHY1485, (A) cell viability was determined using CCK-8, (B) dead cells were determined using trypan blue dye. Data were showed as mean±SD, n=3, *p<0.05, **p<0.01. |
BBR Treatment Alleviates Leukemia Condition in ALL Xenograft Mice
Next, the therapeutic effect of BBR on ALL was investigated in vivo. The NOD-SCID mice were intravenously injected with EU-6 cells to establish the ALL xenograft mice. Then, the mice were administrated with BBR or the same volume of solution as the control. It was showed that BBR treatment significantly enhanced the total survival rate and reduced leukemia condition in ALL xenograft mice (Figure 6A and B). Moreover, the numbers of human-derived CD45-positive cells in bone marrow and spleen are reduced in BBR-treated ALL mice as compared with ALL mice (Figure 6C and D). Of note, the levels of red blood cell and hemoglobin are no different in control and BBR-treated mice (Supplemental Figure 2).
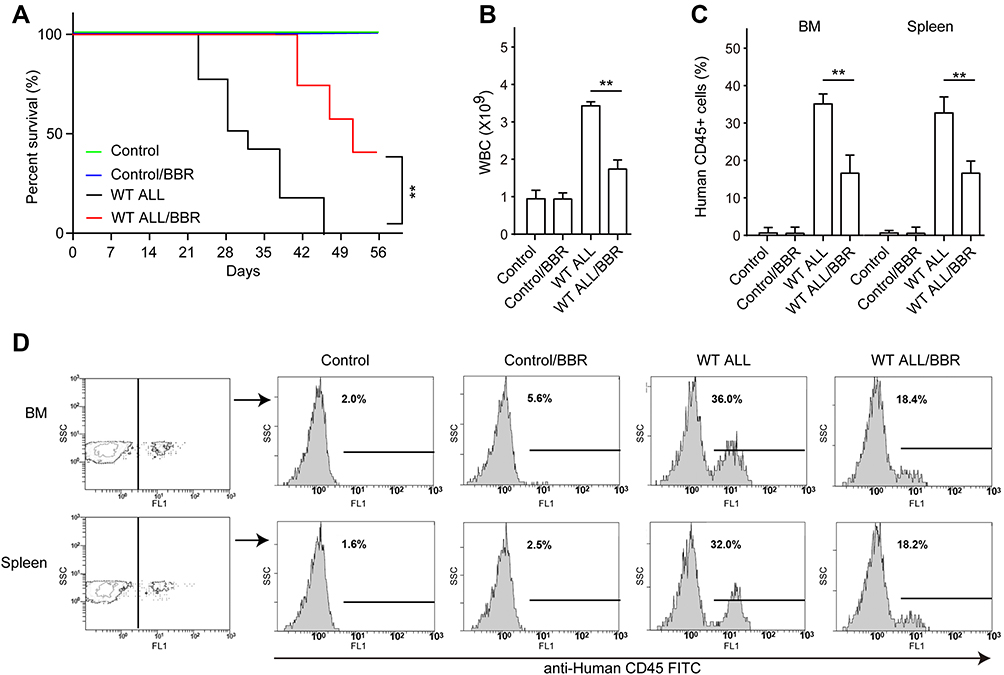 |
Figure 6 BBR treatment alleviates leukemia condition in ALL xenograft mice. NOD-SCID mice were intravenously injected with 1 x 106 EU-6 cells dissolved in 100 μL PBS to generate ALL xenograft mice. Then, the ALL mice and control mice were treated with 10 mg/kg/d BBR or vehicle solution for 2 weeks. (A) The survival rate was detected by Kaplan–Meier analysis in each group. (B) The number of WBC was detected by an automatic blood cell analyzer. (C and D) The percentage of human CD45-positive cells in bone marrow and spleen from each group mice were detected by flow cytometry. Mean±SD, n=3, **p<0.01. |
In vivo Knockdown of ATG5 Results in Aggressive Pathological Condition
To investigate the molecular mechanism underlying the anti-leukemia effect of BBR on ALL in vivo, the ALL xenograft mice bearing ATG5 deficiency were established by injection of EU-6 cells with ATG5 knockdown. Then, the mice were treated with BBR as above. As it is presented in Figure 7A and B, the wild type (WT) ALL and ATG5 deficiency ALL mice (ATG5−/-) exhibited lower survival rate and obvious leukemia condition with a higher level of leukocyte numbers (about 3.62×109, 3.67×109) as compared with healthy mice (about 0.98×109, Figure 6A). BBR treatment significantly increased survival rate and alleviated leukemia condition in both WT and ATG5−/- ALL mice. More importantly, ATG5 knockdown significantly abrogated BBR-caused reversion of survival rate and leukemia condition compared with WT ALL mice. The number of human-derived CD45-positive cells in bone marrow and spleen is increased in ATG5−/- ALL mice as compared in ALL mice after BBR treatment (Figure 7C and D). Meanwhile, either BBR treatment or ATG5 knockdown did not affect the levels of red blood cells and hemoglobin in mice (Supplemental Figure 3). These results demonstrated that BBR alleviated ALL leukemia condition through mediating the autophagy process.
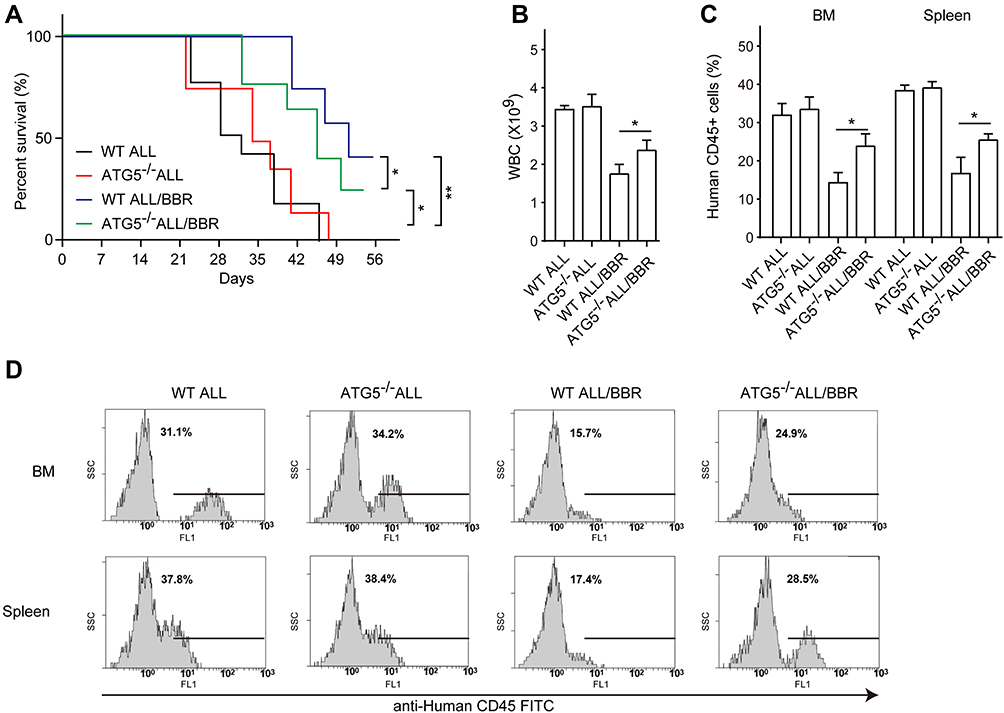 |
Figure 7 In vivo knockdown of ATG5 results in aggressive pathological condition. EU-6 cells were transfected with siRNA targeting ATG5 (ATG5−/-) or scrambled sequence (WT) and then intravenously injected into NOD-SCID mice. The WT and ATG5−/- mice were treated with 10 mg/kg/d BBR or vehicle solution for 2 weeks, respectively. (A) The survival rate was detected by Kaplan–Meier analysis in each group. (B) The number of WBC was detected by an automatic blood cell analyzer. (C and D) The percentage of human CD45-positive cells in bone marrow and spleen from each group mice were detected by flow cytometry. Mean±SD, n=3, *p<0.05, **p<0.01. |
Discussion
In the present study, we investigated the anti-tumor effects and potential mechanism of BBR on ALL. We find that BBR inhibits ALL cell viability and induces autophagy. While autophagy inhibitors 3-MA and Baf A1 could significantly reverse BBR-reduced cell viability. Mechanistic research revealed that AKT/mTORC1 was inactivated by BBR, and inhibition of AKT/mTORC1 activation enhanced BBR-induced cell death. Consistently, the in vivo experiments affirmed that knockdown of ATG5 blunted the ameliorating effects of BBR and resulted in an aggressive pathological condition. This study indicates that BBR served as a preferable agent to efficiently alleviate ALL by promoting autophagic cell death.
Berberine, the widely studied isoquinoline alkaloids, could be used to protect against disorders, such as inflammation, diabetes, hyperlipidemia, parasites, reactive oxygen species, as well as cancers, showed its protective role on ALL in the present study.41 Berberine has a very low oral bioavailability and toxicity. It is widely distributed in liver, kidney and fat and has a round 36 h for plasma concentration maintenance.42 No significant changes in hematological or biochemical parameters were detected in mice treated with berberine.36 It has been reported that autophagy induces cell death and maintains the stability of body by clearing the dysfunctional organelles and incorrect folding proteins.43,44 Recently, autophagy has been implicated in various human diseases and showed an active role against tumors.45,46 For instance, quercetin inhibits cell growth of hepatocellular carcinoma via promoting autophagy in mice.47 Dioscin promotes alveolar macrophage autophagy and further alleviates crystalline silica-induced pulmonary inflammation and fibrosis.48 Accumulating studies also demonstrated that autophagy is involved in the treatment of ALL.49,50 In the present study, we found that BBR-induced cell death by promoting autophagy in ALL cells, indicating the therapeutic role of BBR on ALL. It has been reported that more than 40 autophagy-related genes (ATGs) are involved in the regulation of autophagy,51 among which ATG5 is recognized as a key marker for autophagosome formation. ATG5 can promote autophagy and inhibit endoplasmic reticulum (ER) stress.52 Specifically, microtubule-associated protein 1 light chain 3 (LC3) is considered as a specific marker for autophagosome formation and beclin-1 is widely used in assessing autophagy status. In the current study, we found that BBR treatment significantly increased LC3-I, LC3-II and beclin-1 expression, while ATG5 knockdown abolished the effect of BBR-induced autophagy. The above findings strongly revealed that the anti-ALL role of BBR is attributed to the activation of the autophagic process.
mTORC1 activation is responded to diversity stimuli, such as cytokines, growth factors and antigen receptors.53 And the elevated activity of mTORC1 is observed in various diseases, including neurodegeneration, cancers and metabolic disorders.54 The dysregulated mTORC1 also mediates tumor cell behaviors, for example, mTORC1 regulates cell growth and autophagy in colorectal cancer cells.55 The activation of mTORC1 contributes to cancer cell survival.56 In addition, the hyperactivation of AKT/mTORC1 results in excessive cancer cell proliferation, as well as the impaired autophagy-mediated cell death.40 In the present study, the expression of AKT/mTORC1 markers p-S6 and p-AKT was decreased in BBR-exposed ALL cells. Further, blocking of AKT/mTORC1 by rapamycin significantly promoted autophagy activation and cell death, and in turn suppressed cell viability, and ultimately resulted in an alleviated pathological condition of ALL. The data verified AKT/mTORC1 signaling is involved in BBR-induced cell autophagy in ALL.
In summary, we focused on the pharmacological property of BBR in treating ALL and found that BBR treatment significantly ameliorated ALL condition by promoting tumor cell autophagic death. The further mechanistic study revealed that BBR promoted cell autophagy by inactivation of the AKT/mTORC1 signaling pathway. Our study first identified BBR ameliorates ALL by inducing autophagy and that might be used as a potential agent for ALL treatment.
Abbreviations
BBR, berberine; ALL, acute lymphoblastic leukemia; PDIA6, protein disulfide isomerase family 6; WT, wild type; WBC, white blood cell; RBC, red blood cell; ATGs, autophagy-related genes; ATCC, American Type Culture Collection; HRP, horseradish peroxidase; i.v, intravenously; LC3, light chain 3; ER, endoplasmic reticulum.
Funding
This study was supported by a grant from the National Natural Science Foundation of China (No. 81600133 to J.L.).
Disclosure
The authors declare no conflicts of interest in this work.
References
1. Jabbour E, O’Brien S, Ravandi F, Kantarjian H. Monoclonal antibodies in acute lymphoblastic leukemia. Blood. 2015;125(26):4010–4016. doi:10.1182/blood-2014-08-596403
2. Bhojwani D, Pui CH. Relapsed childhood acute lymphoblastic leukaemia. Lancet Oncol. 2013;14(6):e205–e217. doi:10.1016/S1470-2045(12)70580-6
3. Simioni C, Martelli AM, Zauli G, Melloni E, Neri LM. Targeting mTOR in acute lymphoblastic leukemia. Cells. 2019;8(2):190. doi:10.3390/cells8020190
4. Yilmaz M, Kantarjian H, Jabbour E. Treatment of acute lymphoblastic leukemia in older adults: now and the future. Clin Adv Hematol Oncol. 2017;15(4):266–274.
5. Crazzolara R, Bernhard D. CXCR4 chemokine receptors, histone deacetylase inhibitors and acute lymphoblastic leukemia. Leuk Lymphoma. 2005;46(11):1545–1551. doi:10.1080/10428190500215027
6. Inaba H, Pui CH. Glucocorticoid use in acute lymphoblastic leukaemia. Lancet Oncol. 2010;11(11):1096–1106. doi:10.1016/S1470-2045(10)70114-5
7. Feng X, Sureda A, Jafari S, et al. Berberine in cardiovascular and metabolic diseases: from mechanisms to therapeutics. Theranostics. 2019;9(7):1923–1951. doi:10.7150/thno.30787
8. Kong W, Wei J, Abidi P, et al. Berberine is a novel cholesterol-lowering drug working through a unique mechanism distinct from statins. Nat Med. 2004;10(12):1344–1351. doi:10.1038/nm1135
9. Misik V, Bezakova L, Malekova L, Kostalova D. Lipoxygenase inhibition and antioxidant properties of protoberberine and aporphine alkaloids isolated from mahonia aquifolium. Planta Med. 1995;61(04):372–373. doi:10.1055/s-2006-958107
10. Liang KW, Ting CT, Yin SC, et al. Berberine suppresses MEK/ERK-dependent Egr-1 signaling pathway and inhibits vascular smooth muscle cell regrowth after in vitro mechanical injury. Biochem Pharmacol. 2006;71(6):806–817. doi:10.1016/j.bcp.2005.12.028
11. Hu S, Chen CW, Chen ST, et al. Inhibitory effect of berberine on interleukin-2 secretion from PHA-treated lymphocytic Jurkat cells. Int Immunopharmacol. 2019;66:267–273. doi:10.1016/j.intimp.2018.11.020
12. Lee S, Hallis SP, Jung KA, Ryu D, Kwak MK. Impairment of HIF-1alpha-mediated metabolic adaption by NRF2-silencing in breast cancer cells. Redox Biol. 2019;24:101210. doi:10.1016/j.redox.2019.101210
13. Kuma A, Hatano M, Matsui M, et al. The role of autophagy during the early neonatal starvation period. Nature. 2004;432(7020):1032–1036. doi:10.1038/nature03029
14. Liu X, Zhao P, Wang X, et al. Celastrol mediates autophagy and apoptosis via the ROS/JNK and Akt/mTOR signaling pathways in glioma cells. J Exp Clin Cancer Res. 2019;38(1):184. doi:10.1186/s13046-019-1173-4
15. Bai Y, Liu X, Qi X, et al. PDIA6 modulates apoptosis and autophagy of non-small cell lung cancer cells via the MAP4K1/JNK signaling pathway. EBioMedicine. 2019;42:311–325. doi:10.1016/j.ebiom.2019.03.045
16. Zhang J, Zhang N, Liang S, et al. The amounts and contributions of total drinking fluids and water from food to total water intake of young adults in Baoding, China. Eur J Nutr. 2018;58(7):2669–2677.
17. Wang H, Dong R, Fan WW, Zheng XC, Li AM, Wang WD. Timosaponin AIII induces autophagy of Tcell acute lymphoblastic leukemia Jurkat cells via inhibition of the PI3K/Akt/mTOR pathway. Oncol Rep. 2019;41(5):2937–2944. doi:10.3892/or.2019.7072
18. Li Z, Jiang T, Lu Q, et al. Berberine attenuated the cytotoxicity induced by t-BHP via inhibiting oxidative stress and mitochondria dysfunction in PC-12 cells. Cell Mol Neurobiol. 2019:1–16.
19. Chen Y, Chen Y, Liang Y, Chen H, Ji X, Huang M. Berberine mitigates cognitive decline in an alzheimer’s disease mouse model by targeting both tau hyperphosphorylation and autophagic clearance. Biomed Pharmacother. 2020;121:109670. doi:10.1016/j.biopha.2019.109670
20. Zeng Z, Pan Y, Wu W, et al. Myocardial hypertrophy is improved with berberine treatment via long non-coding RNA MIAT-mediated autophagy. J Pharm Pharmacol. 2019;71(12):1822–1831. doi:10.1111/jphp.13170
21. Lin YS, Chiu YC, Tsai YH, et al. Different mechanisms involved in the berberine-induced antiproliferation effects in triple-negative breast cancer cell lines. J Cell Biochem. 2019;120(8):13531–13544. doi:10.1002/jcb.28628
22. Chen H, Ji Y, Yan X, Su G, Chen L, Xiao J. Berberine attenuates apoptosis in rat retinal Muller cells stimulated with high glucose via enhancing autophagy and the AMPK/mTOR signaling. Biomed Pharmacother. 2018;108:1201–1207. doi:10.1016/j.biopha.2018.09.140
23. Fan X, Wang J, Hou J, et al. Berberine alleviates ox-LDL induced inflammatory factors by up-regulation of autophagy via AMPK/mTOR signaling pathway. J Transl Med. 2015;13(1):92. doi:10.1186/s12967-015-0450-z
24. Silva A, Yunes JA, Cardoso BA, et al. PTEN posttranslational inactivation and hyperactivation of the PI3K/Akt pathway sustain primary T cell leukemia viability. J Clin Invest. 2008;118(11):3762–3774. doi:10.1172/JCI34616
25. Nishioka C, Ikezoe T, Yang J, Koeffler HP, Yokoyama A. Blockade of mTOR signaling potentiates the ability of histone deacetylase inhibitor to induce growth arrest and differentiation of acute myelogenous leukemia cells. Leukemia. 2008;22(12):2159–2168. doi:10.1038/leu.2008.243
26. McCubrey JA, Steelman LS, Abrams SL, et al. Targeting survival cascades induced by activation of Ras/Raf/MEK/ERK, PI3K/PTEN/Akt/mTOR and Jak/STAT pathways for effective leukemia therapy. Leukemia. 2008;22(4):708–722. doi:10.1038/leu.2008.27
27. Steelman LS, Abrams SL, Whelan J, et al. Contributions of the Raf/MEK/ERK, PI3K/PTEN/Akt/mTOR and Jak/STAT pathways to leukemia. Leukemia. 2008;22:686–707.
28. He Y, Mo Q, Luo B, et al. Induction of apoptosis and autophagy via mitochondria- and PI3K/Akt/mTOR-mediated pathways by E. adenophorum in hepatocytes of saanen goat. Oncotarget. 2016;7(34):54537–54548. doi:10.18632/oncotarget.10402
29. Yang J, Chen Q, Tian S, et al. The role of 1,25-dyhydroxyvitamin D3 in mouse liver ischemia reperfusion injury: regulation of autophagy through activation of MEK/ERK signaling and PTEN/PI3K/Akt/mTORC1 signaling. Am J Transl Res. 2015;7(12):2630–2645.
30. Liu Q, Ma H, Sun X, et al. The regulatory ZFAS1/miR-150/ST6GAL1 crosstalk modulates sialylation of EGFR via PI3K/Akt pathway in T-cell acute lymphoblastic leukemia. J Exp Clin Cancer Res. 2019;38(1):199. doi:10.1186/s13046-019-1208-x
31. Tatsumi E, Piontek C, Sugimoto T, Minato K, Minowada J. Selective loss of the expression of OKT-4 or Leu-3A defined antigen by 12-0-tetradecanoylphorbol-13-acetate (TPA) in human lymphoid cells. Am J Hematol. 1984;17(3):287–294. doi:10.1002/ajh.2830170309
32. Serafin V, Capuzzo G, Milani G, et al. Glucocorticoid resistance is reverted by LCK inhibition in pediatric T-cell acute lymphoblastic leukemia. Blood. 2017;130(25):2750–2761. doi:10.1182/blood-2017-05-784603
33. Mohammadi S, Seyedhoseini FS, Asadi J, Yazdani Y. Effects of berberine on the secretion of cytokines and expression of genes involved in cell cycle regulation in THP-1 monocytic cell line. Iran J Basic Med Sci. 2017;20(5):530–537. doi:10.22038/IJBMS.2017.8677
34. Swaminathan S, Klemm L, Park E, et al. Mechanisms of clonal evolution in childhood acute lymphoblastic leukemia. Nat Immunol. 2015;16:766–774.
35. Li H, Miyahara T, Tezuka Y, Tran QL, Seto H, Kadota S. Effect of berberine on bone mineral density in SAMP6 as a senile osteoporosis model. Biol Pharm Bull. 2003;26(1):110–111. doi:10.1248/bpb.26.110
36. Li YH, Zhang M, Fu HB, Xiao HT, Bian ZX. Pre-clinical toxicity of a combination of berberine and 5-aminosalicylic acid in mice. Food Chem Toxicol. 2016;97:150–158.
37. Zhang X, Gu L, Li J, et al. Degradation of MDM2 by the interaction between berberine and DAXX leads to potent apoptosis in MDM2-overexpressing cancer cells. Cancer Res. 2010;70(23):9895–9904. doi:10.1158/0008-5472.CAN-10-1546
38. Peng PL, Kuo WH, Tseng HC, Chou FP. Synergistic tumor-killing effect of radiation and berberine combined treatment in lung cancer: the contribution of autophagic cell death. Int J Radiat Oncol Biol Phys. 2008;70(2):529–542. doi:10.1016/j.ijrobp.2007.08.034
39. Lampada A, O’Prey J, Szabadkai G, Ryan KM, Hochhauser D, Salomoni P. mTORC1-independent autophagy regulates receptor tyrosine kinase phosphorylation in colorectal cancer cells via an mTORC2-mediated mechanism. Cell Death Differ. 2017;24(6):1045–1062. doi:10.1038/cdd.2017.41
40. Erazo T, Lorente M, Lopez-Plana A, et al. The new antitumor drug ABTL0812 inhibits the Akt/mTORC1 axis by upregulating tribbles-3 pseudokinase. Clin Cancer Res. 2016;22(10):2508–2519. doi:10.1158/1078-0432.CCR-15-1808
41. Imanshahidi M, Hosseinzadeh H. Pharmacological and therapeutic effects of Berberis vulgaris and its active constituent, berberine. Phytother Res. 2008;22(8):999–1012. doi:10.1002/ptr.2399
42. Ahmad S, Hussain A, Hussain A, et al. Quantification of berberine in berberis vulgaris l. root extract and its curative and prophylactic role in cisplatin-induced in vivo toxicity and in vitro cytotoxicity. Antioxidants. 2019;8(6):185. doi:10.3390/antiox8060185
43. Nassour J, Radford R, Correia A, et al. Autophagic cell death restricts chromosomal instability during replicative crisis. Nature. 2019;565(7741):659–663. doi:10.1038/s41586-019-0885-0
44. Sehgal AR, Konig H, Johnson DE, et al. You eat what you are: autophagy inhibition as a therapeutic strategy in leukemia. Leukemia. 2015;29(3):517–525. doi:10.1038/leu.2014.349
45. Seton-Rogers S. Eliminating protective autophagy in KRAS-mutant cancers. Nat Rev Cancer. 2019;19(5):247. doi:10.1038/s41568-019-0137-5
46. Vodnala SK, Eil R, Kishton RJ, et al. T cell stemness and dysfunction in tumors are triggered by a common mechanism. Science. 2019;363(6434):eaau0135. doi:10.1126/science.aau0135
47. Ji Y, Li L, Ma YX, et al. Quercetin inhibits growth of hepatocellular carcinoma by apoptosis induction in part via autophagy stimulation in mice. J Nutr Biochem. 2019;69:108–119. doi:10.1016/j.jnutbio.2019.03.018
48. Du S, Li C, Lu Y, et al. Dioscin alleviates crystalline silica-induced pulmonary inflammation and fibrosis through promoting alveolar macrophage autophagy. Theranostics. 2019;9(7):1878–1892. doi:10.7150/thno.29682
49. Dyczynski M, Vesterlund M, Bjorklund AC, et al. Metabolic reprogramming of acute lymphoblastic leukemia cells in response to glucocorticoid treatment. Cell Death Dis. 2018;9(9):846. doi:10.1038/s41419-018-0625-7
50. Evangelisti C, Chiarini F, McCubrey JA, Martelli AM. Therapeutic targeting of mTOR in T-cell acute lymphoblastic leukemia: an update. Int J Mol Sci. 2018;19:1878.
51. Dikic I, Elazar Z. Mechanism and medical implications of mammalian autophagy. Nat Rev Mol Cell Biol. 2018;19(6):349–364. doi:10.1038/s41580-018-0003-4
52. Zheng W, Xie W, Yin D, Luo R, Liu M, Guo F. ATG5 and ATG7 induced autophagy interplays with UPR via PERK signaling. Cell Commun Signal. 2019;17:42.
53. Powell JD, Pollizzi KN, Heikamp EB, Horton MR. Regulation of immune responses by mTOR. Annu Rev Immunol. 2012;30(1):39–68. doi:10.1146/annurev-immunol-020711-075024
54. Jewell JL, Fu V, Hong AW, et al. GPCR signaling inhibits mTORC1 via PKA phosphorylation of Raptor. Elife. 2019;8. doi:10.7554/eLife.43038
55. Silva-Pavez E, Villar P, Trigo C, et al. CK2 inhibition with silmitasertib promotes methuosis-like cell death associated to catastrophic massive vacuolization of colorectal cancer cells. Cell Death Dis. 2019;10(2):73. doi:10.1038/s41419-019-1306-x
56. Martinez-Carreres L, Puyal J, Leal-Esteban LC, et al. CDK4 regulates lysosomal function and mTORC1 activation to promote cancer cell survival. Cancer Res. 2019;79(20):5245–5259. doi:10.1158/0008-5472.CAN-19-0708
 © 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.