")
Back to Journals » Drug Design, Development and Therapy » Volume 16
Berbamine Suppresses the Growth of Gastric Cancer Cells by Inactivating the BRD4/c-MYC Signaling Pathway
Authors Li H, Luo K, Yang Z, Chen M, Yang X, Wang J, Ying Y, Wu D, Wang Q
Received 11 September 2021
Accepted for publication 21 December 2021
Published 11 January 2022 Volume 2022:16 Pages 129—141
DOI https://doi.org/10.2147/DDDT.S338881
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Manfred Ogris
Hongchun Li,1 Kexue Luo,1 Zhuying Yang,2 Miao Chen,3 Xiuyun Yang,1 Jiesheng Wang,1 Yin Ying,4 Dengxuan Wu,5 Qinxian Wang1
1Department of Cadre Health, Tongde Hospital of Zhejiang Province, Hangzhou, 310012, Zhejiang, People’s Republic of China; 2Department of Gastroenterology, Tongde Hospital of Zhejiang Province, Hangzhou, 310012, Zhejiang, People’s Republic of China; 3Department of Oncology, Tongde Hospital of Zhejiang Province, Hangzhou, Zhejiang, 310012, People’s Republic of China; 4Department of Pharmacy, Tongde Hospital of Zhejiang Province, Hangzhou, Zhejiang, 310012, People’s Republic of China; 5Department of Rehabilitation Medicine, Tongde Hospital of Zhejiang Province, Hangzhou, 310012, Zhejiang, People’s Republic of China
Correspondence: Qinxian Wang Tel +86 571-89972437
Email [email protected]
Purpose: Berbamine (Ber), a bioactive constituent extracted from a traditional Chinese medicinal herb, has been shown to exhibit broad inhibitory activity on a panel of cancer cell types. However, its effects and the underlying molecular mechanisms on gastric cancer (GC) remain poorly understood.
Methods: The anti-growth activity of Ber on two GC cell lines and normal gastric epithelial cell line were evaluated using MTS and clone formation assay. Flow cytometry analysis was employed to evaluate the cell cycle distribution and apoptosis of GC cells. Western blot and quantitative PCR (qPCR) analysis were employed to investigate the anti-GC mechanism of Ber. The inhibitory activity and binding affinity of Ber against BRD4 were evaluated by homogeneous time-resolved fluorescence (HTRF) and surface plasmon resonance (SPR) assay, respectively. Molecular docking and molecular simulations were conducted to predict the interaction mode between BRD4 and Ber.
Results: The results demonstrated that Ber reduced the proliferation of GC cell lines SGC-7901 and BGC-823 and induced cell cycle arrest and apoptosis. Mechanistically, Ber was identified as a novel natural-derived BRD4 inhibitor through multiple experimental assay, and its anti-GC activity was probably mediated by BRD4 inhibition. Molecular modeling studies suggested that Ber might bind to BRD4 primarily through hydrophobic interactions.
Conclusion: Our study uncovered the underlying anti-GC activity of Ber in vitro and suggested that Ber holds promise as a potential lead compound in the discovery of novel BRD4 inhibitors.
Keywords: berbamine, BRD4 inhibitor, gastric cancer, molecular modeling
Introduction
Gastric cancer (GC) ranks among the leading causes of cancer deaths around the world.1 Surgical resection plus lymph node dissection is by far the most effective treatment for respectable GC.2 However, distant metastasis and relapses often occur even after curative resection. Tumor progression and metastasis also occur in patients with unresectable GC. One of the biggest challenges in the treatment of GC and the development of novel therapies is to resolve extreme heterogeneity as well as to clarify its histological appearances and molecular basis.
The implementation of chemotherapy for GC, which includes a combination of platinum, fluoropyrimidine, trastuzumab, taxanes, and ramucirumab, has substantially improved the survival rate of advanced GC patients.3 A recent Phase III ATTRACTION-2 trial of nivolumab has also demonstrated the survival benefits of GC.4 However, it was found that the overall remission rate was only 10%, and that almost half the patients showed disease progression. In light of this dire situation, there is an urgent need to discover new GC targets. Another approach to targeted therapy is to widely interfere with cancer growth signals through natural compounds with pleiotropic actions.5 One of the compounds with multiple biological activities is berbamine (Ber), a well-known antineoplastic natural product extracted from Berberis vulgaris, which has been widely used for the treatment of hypertension and arrhythmia in traditional medicine. A growing body of evidence suggests that Ber not only exhibits favorable anti-cancer activities against a wide variety of tumor cells including bladder cancer, hepatocellular carcinoma, leukemia, lung cancer, prostate cancer, and ovarian cancer cells but also exerts potential synergistic effects with many clinical therapies, such as gemcitabine, gefitinib and sorafenib.6–14 Building on the existing findings, it has been further revealed that the anti-cancer effects of Ber might be mediated by its direct inhibition of various oncogenic signaling molecules such as CaMKII, STAT3 and NF-κB.14–16 However, to date, little research has been conducted on the anti-GC effects of Ber and no study has revealed the mechanisms underlying such effects.
In the present study, we evaluated the anti-growth activity of Ber on two GC cell lines and a normal gastric epithelial cell line through MTS and clone formation assay. Our data indicated that Ber dose- and time-dependently suppressed the growth of GC cells (BGC-823 and SGC-7901) but displayed little cytotoxicity against normal GES-1 cells. Furthermore, flow cytometry and Western blot analysis results revealed that Ber induced GC cells cell-cycle arrest and apoptosis via regulation of the expression of key signaling molecules. Mechanistically, our results revealed that Ber directly bound to BRD4 and inhibited its transcription activation activity, which might be a major molecular mechanism of Ber. Molecular modeling results further suggested that Ber might bind to the catalytic-binding pocket of BRD4 through hydrophobic interactions.
Materials and Methods
Reagents
Ber (purity: 99.79%, Figure 1A) was procured from MedChemExpress (Shanghai, China), which was dissolved in Dimethyl sulfoxide (DMSO) as a stock solution (40 mM), and diluted to the final working concentration in the growth medium. CellTiter 96® Aqueous Assay Reagents were purchased from Promega (Madison, USA). Cell Cycle and apoptosis detection kits were purchased from MultiSciences Biotech (Hangzhou, China). The primary antibodies used in this study including β-Actin, BRD4, CDK4, c-Myc, Cleaved-Caspase-3, Cleaved-PARP, Cyclin D1, Phospho-CaMKIIγ (Thr286), Phospho-Rb1 (Ser807/811), Phospho-STAT3 (Tyr705), and Phospho-NF-κB p65 (Ser536) were obtained from Cell Signaling Technology (Danvers, MA). Bcl-2 antibody was obtained from Abcam (Cambridge, USA). The HRP-linked anti-rabbit IgG secondary antibody was procured from Jackson ImmunoResearch Laboratories (West Grove, USA). Recombinant Human BRD4 protein was obtained from Abcam (Cambridge, USA) and its purity was not less than 98% determined by SDS-PAGE.
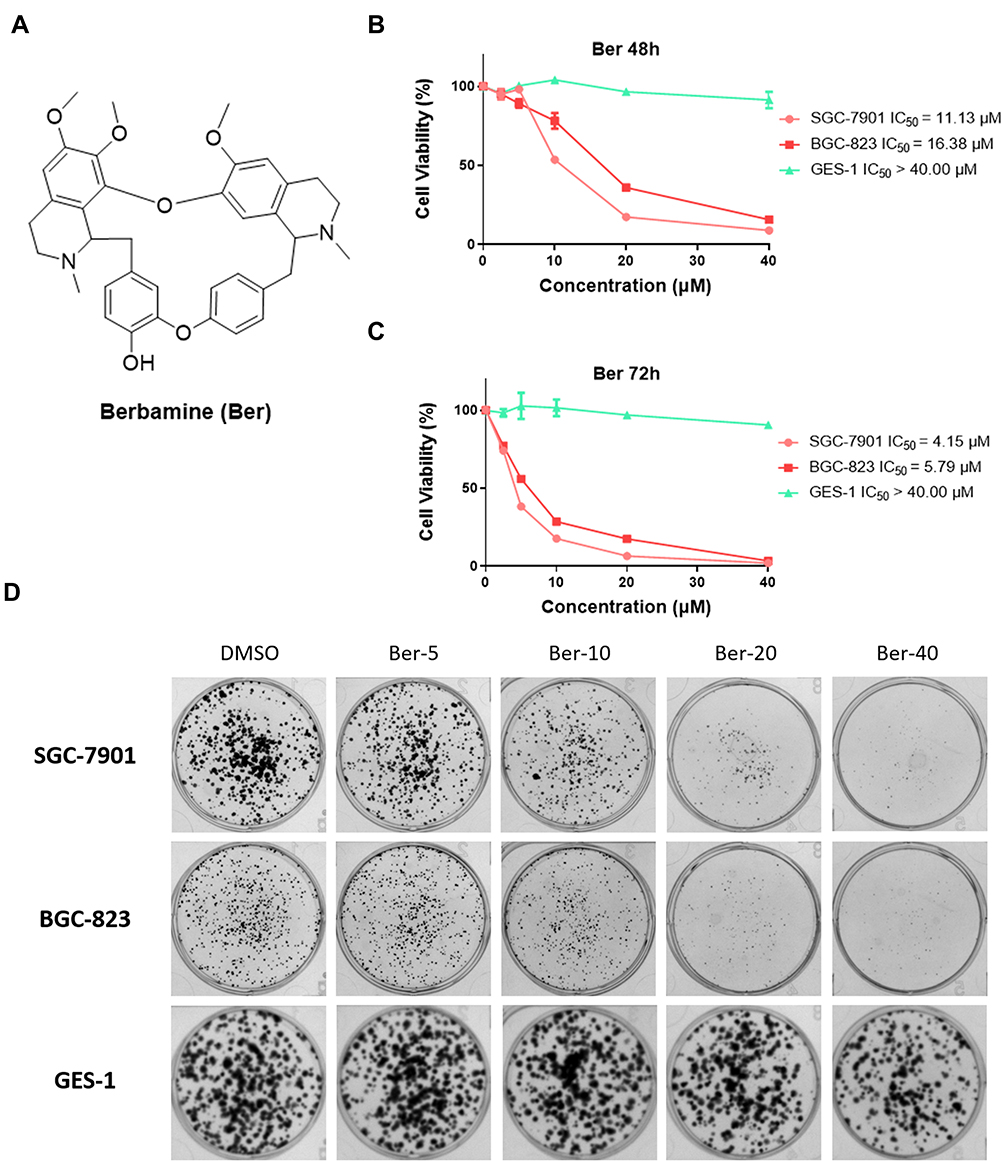 |
Figure 1 Ber selectively suppresses the growth of GC cell lines. (A) Chemical structure of Ber. (B and C) Effects of Ber on the viability of human gastric cancer cell lines SGC-7901, BGC-823, and normal gastric epithelial cell line GES-1. Cells were treated with Ber of increasing concentrations for 48 h (B) and 72 h (C), and cell viability was measured via MTS assay. (D) Effect of varying Ber concentrations on the colony formation of SGC-7901, BGC-823 and GES-1 cells. Cells were incubated with Ber for 24 h and allowed to grow for 10 days. Colonies were stained by crystal violet dye. Each point represented the means ± SDs of triplicates. Each experiment was performed for three times. |
Cell Culture
Normal gastric epithelial cell-line GES-1 was procured from Beyotime Biotechnology (Shanghai, China), and human GC cell line BGC-823 and SGC-7901 was procured from the Institute of Biochemistry and Cell Biology, Chinese Academy of Sciences (Shanghai, China), which was cultured in RPMI-1640 Medium, with 10% heat-inactivated fetal bovine serum (FBS), 100 μg/mL streptomycin and 100 U/mL penicillin (Gibco, Eggenstein, Germany). Then, cells were incubated with 5% CO2 at 37°C.
Cell Viability Assay
BGC-823, GES-1, and SGC-7901 cells were plated at a density of 5000/well on 96-well plates and allowed to adhere overnight. Cells were subjected to treatment with Ber at different concentrations for 48 or 72 h. Then, 100 μL Cell Titer 96 Aqueous Non-Radioactive Cell Proliferation Assay reagent was added per well, and cells were cultured at 37°C for 0.5 h. The formazan product was dissolved through shaking for 20 s and SpectraMax M5 microplate reader (Molecular Devices, USA) was utilized to measure absorbance at 490 nm. The formula of percent inhibition rate was [1 -(treated group/control group)] × 100.
Colony Formation Assay
BGC-823, GES-1, and SGC-7901 cells were seeded at 800 cells/well on 6-well plates and exposed to Ber treatment for 24 h. They were allowed to grow for 10 days in normal growth media. The cells were then fixed in cold methanol for 20 min. Subsequently, they were subjected to crystal violet solution staining (0.5 in 25% methanol) for 25 min. Colonies are defined as clusters of at least 50 cells, and then photographed with the molecular imager Gel Doc XR+ system (Bio-Rad, USA).
Flow Cytometry (FACS) Analysis
FACS analysis was employed to determine cell cycle distribution and cell apoptosis. Cells were trypsinized after incubation with Ber for 24 h and washed twice with cold PBS. For cell cycle detection, the cells were fixed in 75% ice-cold ethanol at 4°C overnight. The next day, the cell pellets were centrifugated, washed with cold PBS, and stained with 50 mg/mL propidium iodide (PI, MultiSciences Biotech, Hangzhou, China) at room temperature for 30 min. For apoptosis detection, the cell pellets were stained with 100 μL 1× binding buffer containing 5 μL FITC Annexin V and PI reagent (MultiSciences Biotech, Hangzhou, China) at room temperature for 15 min. After the samples were diluted with 400 μL 1× binding buffer, they were analyzed with an ACS Calibur flow cytometer (BD Biosciences, USA).
Western Immunoblot Assay
10% SDS-PAGE was used to separate 50 μg of cell lysates. Thereafter, they were electroporated onto PVDF membranes. The membrane was blocked in Tris-buffered saline containing 0.05% Tween 20 and 5% skim milk for 1.5 h. The PVDF membrane was then subjected to incubation with specific primary antibodies. To detect the immunoreactive band, the membrane was incubated with a secondary antibody along with horseradish peroxidase and enhanced chemiluminescence reagent (Bio-Rad, USA). The Image J analysis software version 1.38e was utilized to analyze the amount of protein, after which they were normalized to their respective controls.
Homogeneous Time-Resolved Fluorescence (HTRF) Assay
Authorization of the HTRF was commissioned to ChemPartner (Shanghai, China) after the experimental conditions had been constrained. The detailed operating procedures are described as follows: In accordance with the plate map and with DMSO’s final fraction at 0.1%, the compounds were serially diluted in Echo plate. After that, Echo was utilized to transfer the compounds and DMSO to 384-well assay plate. Subsequently, 2× Protein and Peptide Mix were added to the assay plate, followed by the addition of 2× Detection Mix, which was shaken for 30 s. After the plate was incubated for 1.5 h at room temperature, EnVision (PerkinElmer, UK) was utilized to read the HTRF signals (Excitation wavelength at 340 nm, Emission wavelength at 615 and 665 nm). BRD4-BD1 (RD-11-157) were purchased from Reaction Biology Corp. (PENN, USA). For the purposes of curve fitting, Equation (1) was used to calculate the inhibition values in Excel: Inhibition % = (Max-Signal)/(Max-Min) × 100 (Equation 1), whereas Equation (2) was used to obtain the IC50 values in XL-Fit: Y = Bottom + (Top-Bottom)/(1+(IC50/X) × HillSlope) (Equation 2), where Y refers to inhibition % and X represents the concentration of compound.
Surface Plasmon Resonance (SPR) Assay
The Proteon XPR36 protein interaction array system (Bio-Rad Laboratories, Hercules, USA) was utilized to perform the binding kinetics experiments. Briefly, amine coupling was performed to immobilize recombinant human BRD4 protein to a GLH sensor chip, thereby creating a surface density of up to 8000 RU. Investigation into the kinetics of compound binding was performed in the PBS solution (5 mM, pH 7.4, containing 5%, w/v, DMSO). For the purposes of the kinetics study, a series of compounds at different concentrations that had been diluted in respective buffers were injected over the surface at a flow rate of 30 μL/min, and then a dissociation phase of 120 s was performed in each cycle. To obtain the final chart, the blank sensing map was subtracted from the duplex or quadruple sensor graph. ProteOn manager software was used to perform the data analyses.
Quantitative Real Time PCR (qPCR)
In compliance with the manufacturer’s operating procedures, Trizol reagent (Invitrogen) was used to extract total RNA. NanoDrop 2000 (Nanodrop, USA) was utilized to quantify the RNA concentrations. High-Capacity cDNA Reverse Transcription Kit (Applied Biosystems, USA) was employed to reverse transcribe 2 μg RNA. To determine the mRNA expression level, qPCR was performed on SYBR Green Master Mix Kit and Light Cycler 480 II system (Roche, Shanghai, China).
Primers used for RT-PCR include the following:
c-MYC forward primer, 5ʹ-GCCTCAGAGTGCATCGAC-3ʹ, and reverse primer, 5ʹ-TCCACAGAAACAACATCG-3ʹ;
BCL2 forward primer, 5ʹ-GTGTGTGGAGAGCGTCAACC-3ʹ, and reverse primer, 5ʹ-CTTGTGGCCCAGATAGGCA-3ʹ;
BCL6 forward primer, 5ʹ-TCTCCCTTCCCCACTTCCTT-3ʹ, and reverse primer, 5ʹ-GAGGCCATTTTGTCTTCACCAA-3ʹ;
ACTB forward primer, 5ʹ-ACTCTTCCAGCCTTCCTTCC-3ʹ, and reverse primer, 5ʹ-CGTCATACTCCTGCTTGCTG-3ʹ.
Relative quantities of c-MYC, BCL2 and BCL6 mRNA were normalized against ACTB mRNA.
Molecular Docking and Molecular Dynamics Simulation
The LeDock software was utilized to calculate the binding mode of Ber in the binding site of BRD4.17 The crystal coordinates for human BRD4 (PDB code: 6Z7L) were obtained from the Protein Data Bank (PDB) database.18 The LePro module in the LeDock software was implemented to process the protein structure of BRD4, remove water molecules, and generate grid files. Then, LeDock was performed to generate 100 conformations with the best binding energy conformation and was then submitted for further molecular dynamics (MD) simulation analysis. The restrained electrostatic potential (RESP) method was used to calculate the partial atomic charges of Ber based on the HF/6-13G* basis set. Next, the ff14SB force field and General Amber Force Field were assigned to Ber and BRD4.19,20 Then, the complex was immersed in a water box with TIP3P water model. Ultimately, counter ions were used to neutralize the simulated system.
After the system was ready, 15,000 steps of steepest descent were performed to eliminate the bad contacts in the constructed system, followed by 15,000 steps of conjugate gradient. Thereafter, the constructed system was heated up from 0 to 310 K in 200 ps. After heating, a density procedure with 500 ps at 310 K was performed. Equilibration with 500 ps in the isothermal isobaric (NPT) ensemble was utilized to equilibrate the density and maintain the system temperature. Finally, the prepared system was subjected to a 600 ns MD simulation at a constant temperature of 310 K and pressure of 1 atm, respectively. Coordinates were saved at an interval of 10 ps, and statistical analysis was performed on the CPPTRAJ module in AmberTools 18 package.21 To calculate the binding free energy decomposition, the last 150 ns MD simulation trajectory with 500 snapshots was performed based on the molecular mechanics/generalized Born surface area (MM/GBSA) method.22,23
Statistical Analysis
The results are reported as means ± SD (standard deviation). In order to test whether the differences among the groups are statistically significant, one-way analysis of variance (one-way ANOVA) or two-way ANOVA was performed on GraphPad Prism 8 (GraphPad, San Diego, CA, USA). A statistically significant result was obtained at p<0.05. All the experiments were replicated for a minimum of three times.
Results
Ber Selectively Suppressed the Growth of GC Cells
Previous studies have reported that Ber (Figure 1A) could inhibit the proliferation of a panel of cancer cell lines, including bladder cancer, breast cancer, liver cancer, myeloma, and ovarian cancer cells. However, its inhibitory effect on the proliferation of GC cell lines remains unknown. To determine the potential anti-proliferation activity of Ber against GC cells, we first evaluated the effect of Ber on GC cell viabilities. As is shown in Figure 1B and C, Ber inhibited the viability of SGC-7901 and BGC-823 cells in a dose-dependent and time-dependent fashion (48 h IC50, SGC-7901: 11.13 μM, BGC-823: 16.38 μM; 72 h IC50, SGC-7901: 4.148 μM, BGC-823: 5.788 μM) but had limited influence on the viability of GES-1 cells (48 h IC50 and 72 h IC50 both >40 μM).
In addition to cell viability assay, colony-forming assay was further conducted to evaluate the long-term anti-growth effects of treatment with Ber. SGC-7901 and BGC-823 cells were first exposed to Ber for 24 h before they were cultured in normal growth media for up to 10 days. Crystal violet staining showed reduced colony formation in BGC-823 and SGC-7901 with increasing concentrations of Ber (Figure 1D). Similar to the MTS results, Ber exhibited little inhibition in the clonogenicity of GES-1 cells. Taken together, our results suggested that Ber could be a novel selective anti-GC natural product in vitro.
Ber Induces GC Cell Cycle Arrest at G0/G1 Phase and Cell Apoptosis
To investigate whether the anti-growth effect of Ber is mediated through the induction of cell cycle arrest, flow cytometry was used to analyze the distributions of GC cells at G0/G1, S and G2/M phases after Ber treatment. According to the results, Ber significantly induced GC cell arrest at the G0/G1 phase (Figure 2A). Meanwhile, Western blot results demonstrated that Ber dose-dependently downregulated the expression of G0-G1 phase transition-related proteins, such as CDK4, Cyclin D1, and phosphorylated Rb1 (Figure 2B and C). These findings indicate that Ber could significantly arrest GC cells at the G0/G1 phase, and that this effect is mediated via the reduction of the expression of cell cycle-related proteins.
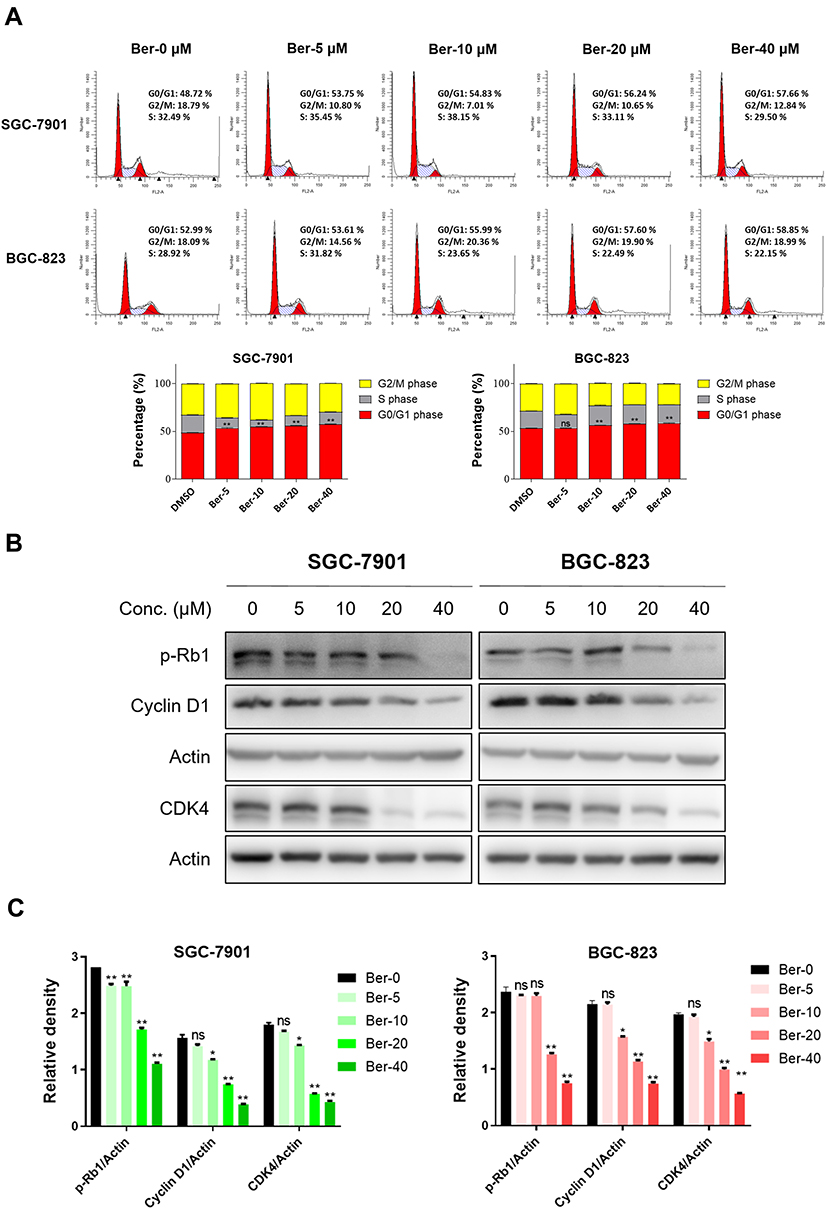 |
Figure 2 Ber induced the cell cycle arrest of GC cells. (A) SGC-7901 and BGC-823 cells were treated with Ber (0, 5, 10, 20 and 40 μM) for 24 h, and the cell cycle distribution was analyzed by flow cytometry. The representative histogram of the cell cycle distribution was shown. Statistics used for reporting include means ± SDs of three experiments conducted independently in triplicate. (B) Western blot analysis of G0-G1 phase transition related proteins p-Rb1 (Ser 807/811), Cyclin D1, CDK4, with Actin as shown a loading control. (C) The relative expression of G0-G1 phase transition related proteins (p-Rb1 (Ser 807/811)/Actin, Cyclin D1/Actin and CDK4/Actin) was quantified by ImageJ and analyzed by GraphPad Prism 8. ns means no significance. *P < 0.05, **P < 0.01 compared to negative control. |
Moreover, flow cytometry was conducted to detect the apoptosis induction effects of Ber on GC cells via Annexin V-FITC/PI double staining assay. According to the results, Ber dose-dependently increased the apoptosis rate of GC cells (Figure 3A). To uncover the mechanisms underlying the pro-apoptosis effects of Ber, Western blot was performed to analyze the expression of the apoptosis-related proteins, including Cleaved-Caspase-3, Cleaved-PARP and Bcl-2. As Figure 3B and C suggest, there is a significant increase in the expression of pro-apoptotic molecules (Cleaved-PARP and Cleaved-Caspase-3), but a decrease in the anti-apoptotic protein Bcl-2.
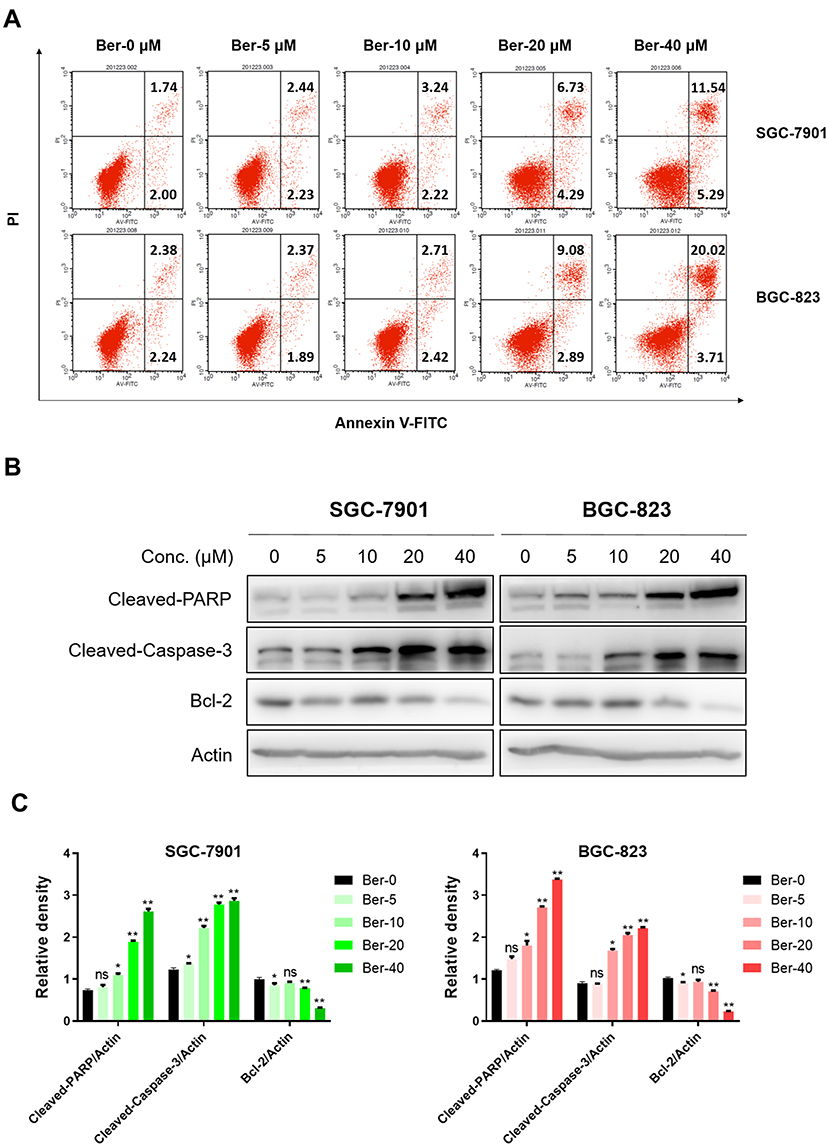 |
Figure 3 Ber induced the apoptosis of GC cells. (A) SGC-7901 and BGC-823 cells were treated with Ber (0, 5, 10, 20 and 40 μM) for 24 h, the apoptosis rates were determined by FITC-Annexin V/ PI double staining assay. Statistics used for reporting include means ± SDs of three experiments conducted independently in triplicate. (B) Western blot analysis of apoptosis-related proteins such as Cleaved-PARP, Cleaved-Caspase 3 and Bcl-2, with Actin as shown a loading control. (C) The relative expression of apoptosis-related proteins (Cleaved-PARP/Actin, Cleaved-Caspase-3/Actin and Bcl-2/Actin) was quantified using the ImageJ and analyzed by GraphPad Prism 8. ns means no significance. *P < 0.05, **P < 0.01 compared to negative control. |
The Identification of Ber as Novel Natural-Derived BRD4 Inhibitor
Several potential molecular targets of Ber have been identified in recent years successively, including CaMKIIγ, STAT3 and NF-κB. To explore whether the anti-GC activity of Ber is involved in the aforementioned signaling pathway, the activation of CaMKIIγ, STAT3 and NF-κB was determined by Western blot. Intriguingly, as shown in Figure 4A, Ber treatment did not inhibit the CaMKIIγ signal, but instead facilitated the activation of STAT3 and NF-κB signals. Considering that the oncogene c-Myc has been reported as one major downstream signal of CaMKIIγ signal in many cancers, we also detected the changes in c-Myc in GC cells. Surprisingly, our results showed that Ber significantly downregulated the expression of c-Myc, even though it had no effect on CaMKIIγ activation. This finding propelled us to hypothesize that Ber might exert anti-GC effects by suppressing a key regulator of c-Myc expression. As a well-studied dominant regulator of c-Myc, Bromodomain-containing protein 4 (BRD4) has been extensively reported as a protumorigenic factor in various cancers, including GC that activates the expression of oncogenes (such as c-MYC and BCL2) through transcription.24 Our results demonstrated that Ber treatment dose-dependently increased the expression of BRD4 in GC cells, indicating that Ber might directly bind to BRD4 protein, thereby elevating the stability of BRD4. To validate the inhibitory activity of Ber on BRD4, HTRF assay was carried out. As shown in Figure 4B, Ber effectively suppressed the binding between BRD4 and substrate peptide with a moderate IC50 = 12.10 μM. In addition, SPR analysis was conducted to determine the binding affinity between BRD4 and Ber. The results revealed that Ber dose-dependently bound to BRD4 protein with a moderate equilibrium dissociation constant (KD) value of 10 μM (Figure 4C). qPCR assay results further confirmed that Ber effectively downregulated the mRNA levels of BRD4 transcriptional targets, such as c-MYC, BCL2 and BCL6, suggesting that Ber is a novel BRD4 inhibitor and that its anti-GC effects are mediated primarily by BRD4 inhibition (Figure 4D).
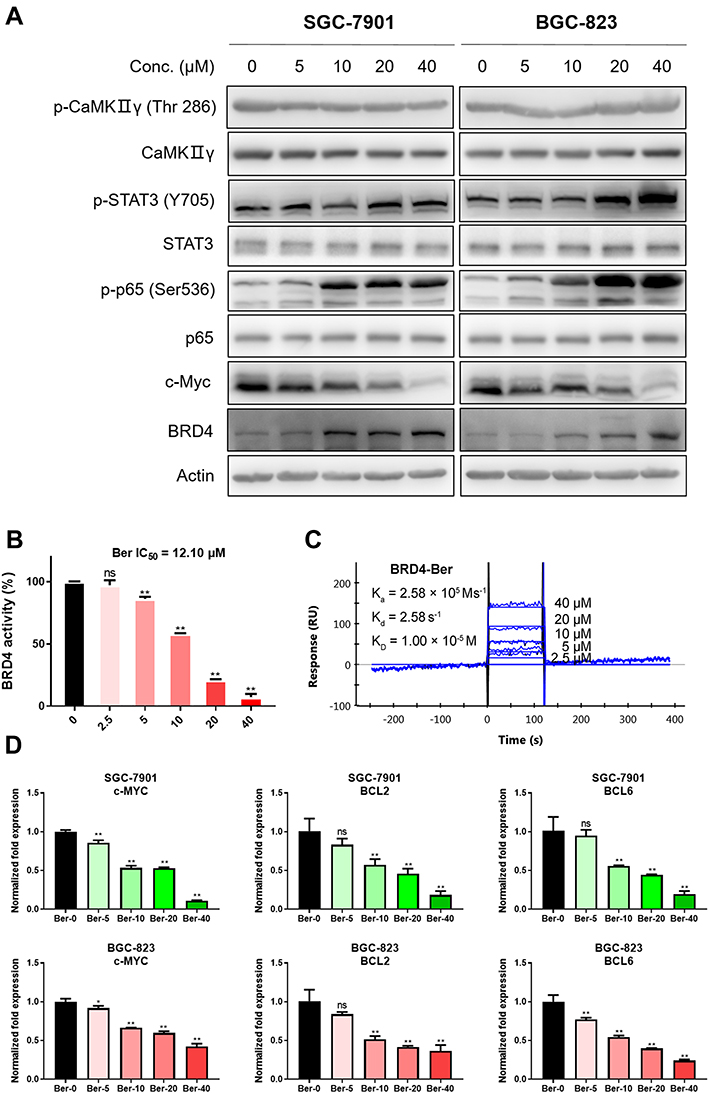 |
Figure 4 Ber was identified as a novel natural-derived BRD4 inhibitor, and suppressed the transcription activation of BRD4-downstream oncogenes in GC cells. (A) SGC-7901 and BGC-823 cells were treated with Ber (0, 5, 10, 20 and 40 μM) for 24 h, and then the expression of p-CaMKIIγ (Thr286), CaMKIIγ, p-STAT3 (Tyr705), STAT3, p-NF-κB p65 (Ser536), p65, c-Myc and BRD4 was detected by Western blot. Actin was used as the loading control. (B) The BRD4 inhibition activity of Ber was measured by HTRF assay. ns means no significance. **P < 0.01 compared to negative control. (C) The binding affinity between Ber and BRD4 was determined by SPR. Kd: dissociation constant, Ka: association constant, KD: equilibrium dissociation constant. (D) qPCR analysis of well-known BRD4 transcriptional targets (c-MYC, BCL2 and BCL6) in SGC-7901 and BGC-823 cells treated with Ber (0, 5, 10, 20 and 40 μM) for 24h. The relative gene expression was normalized to internal control ACTB and analyzed by GraphPad Prism 8. ns means no significance. *P < 0.05, **P < 0.01 compared to negative control. |
Molecular Modeling of Ber Bound to BRD4
Molecular docking was used to construct the initial binding mode between BRD4 and Ber. Then, the predicted binding mode was subjected to 600 ns MD simulations to monitor the dynamic behavior. To analyze the stability of the simulated complex, the root-mean square deviations (RMSDs) of the protein backbone atoms (Cα) of BRD4, and the heavy atoms of Ber were first calculated. As Figure 5A indicates, small fluctuations were observed in the RMSDs of Cα of BRD4 after 180 ns, while Ber remained stable throughout the MD simulation. These observations suggest that we have obtained relatively reasonable binding conformations of the BRD4-Ber complex via MD simulations. Then, the MM/GBSA method was used to decompose the binding free energies into the contributions of each residue to highlight the key residues for BRD4-Ber recognition patterns. As Figure 5B reveals, the residues with the greatest contributions were Trp81, Ile146, Pro82, Asp145 and Met149. Further structural analysis indicated that hydrophobic interactions were the primary interactions (Figure 5C).
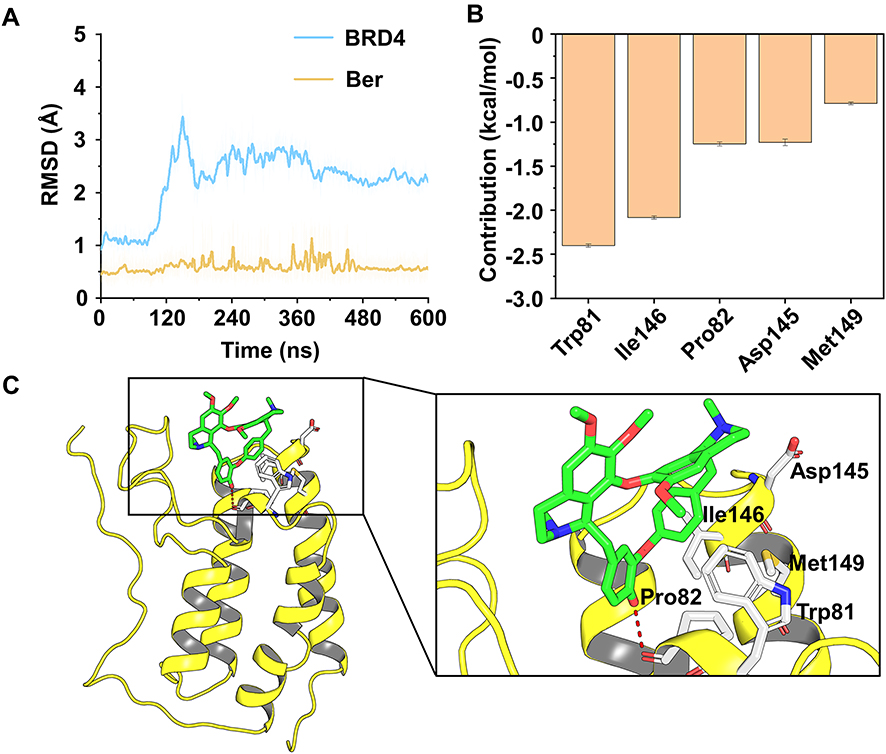 |
Figure 5 Structural and energetic analysis of Ber to the binding pocket of BRD4. (A) RMSD curves for the 600 ns MD simulation. (B) Key residues between BRD4 and Ber. (C) Structural analysis of the key residues of BRD4 to Ber. |
Discussion
In the past few decades, the survival rate of GC patients has improved substantially thanks to improved living standards, reduced chronic H. pylori infection, and better detection and treatment. However, the prognosis of advanced GC patients remains a thorny problem.2 Currently, platinum/fluoropyrimidine-based dual chemotherapy and docetaxel-based triple regimens are the most recommended treatment standards for patients with metastatic and unresectable locally advanced GC, respectively.3 Although the combined treatment of multiple chemotherapeutics has significantly improved their efficacy in clinical practice, it also increases the risk of severe systemic toxicity. Moreover, the emergence of multiple chemotherapy resistance greatly limits the long-term benefits of therapeutic treatments for patients with advanced GC.25 Therefore, there is a pressing need to discover novel and more effective targeted therapies for treating advanced gastric cancer.
Recent research on abnormal signaling pathways in GC has provided many promising therapeutic targets. A representative example is HER2, which is overexpressed in approximately 17–20% of GC patients and is the most common biomarker for advanced GC patient molecular classification.26 Accumulating evidence suggests that oncogenic protein kinases such as VEGFR, FGFR2 and MET could also be potential therapeutic targets for GC treatment.26 Besides, several recent studies further point out that BRD4 is closely associated with the progression and metastasis of GC. Qin et al reported that BRD4 expression was upregulated in human GC tissues and associated with distal metastasis and shorter overall survival.27 Ba et al discovered that BRD4 promoted the growth of GC cells and resistance to cell apoptosis through binding to c-MYC promoter and coordinately activating its transcription.24 Furthermore, BRD4 was reportedly involved in the senescence, stemness and metastasis of GC cells by regulating the expression of non-coding RNAs, such as miR-106b, miR-216a-3p and LncRNA MAGI2-AS3.28–30 In recent years, BRD4 has emerged as a hotspot anti-cancer drug target. As one of the most thoroughly and extensively studied histone H3 and H4 acetylation readers, BRD4 is responsible for recruiting transcription elongation factor b (p-TEFb) complex to phosphorylate RNA polymerase II (RNA pol II), thereby activating a large number of genes, such as oncogenes c-MYC, BCL2 and BCL6. Among these BRD4 downstream oncogenes, c-MYC is the most well characterized. Its expression is estimated to be elevated or deregulated in up to 70% of human cancers. Through the formation of Myc-Max heterodimer, c-Myc controls the transcription of multiple genes involved in cell proliferation, migration and energy metabolism in many cancers. In addition, it has been reported that BRD4 is also involved in DNA damage and telomere regulation in many cancer cells. Therefore, the above evidence suggests that targeting BRD4/c-Myc could be a promising approach for the development of novel GC targeted therapies. Although many powerful and selective BRD4 inhibitors have entered clinical trials, the majority of current BRD4 inhibitors remain in the early stages. For example, the pan-BET inhibitor OTX015 (Birabresib) has now completed Phase 1 clinical trials in patients with multiple myeloma and acute leukaemia.31,32 Another pan-BET inhibitor CPI-0610 (Pelabresib) has also completed Phase 1 trials for progressive lymphoma and multiple myeloma patients.33 Overall, the efficacy and safety of current pan-BET inhibitors is still not very satisfactory. More importantly, none of the above potent BRD4 inhibitors is currently undergoing clinical trials as GC therapies, and there are even fewer studies evaluating the anti-GC activity of BRD4 inhibitors.
Since docetaxel was approved as a mainstream chemotherapeutic drug for GC patients, the discovery of new natural-derived anti-GC small molecules has been a research hotspot in recent years. Several well-known natural products, including celastrol, esculetin and schisantherin A, have been identified as potential anti-GC active compounds.34–36 In recent years, another naturally-derived compound that has been extensively studied is Ber, which has a broad range of anti-cancer effects on breast, lung and liver cancer. However, few studies have investigated the effect of Ber on GC. Herein, our study evaluated the potential anti-GC activity of Ber for the first time. Our results demonstrated that Ber effectively suppressed the growth of GC cell lines, but exerted little cytotoxicity on normal gastric epithelial cell line (GES-1). Moreover, Ber treatment significantly induced cell cycle arrest and apoptosis in GC cells. Overall, our results indicate that Ber holds promise as a potential in vitro anti-GC agent. More importantly, in many Asian countries such as China and Japan, Ber has been widely used as an adjuvant drug for the treatment of chemotherapy-induced leukopenia. This provides additional support for the development of Ber as a new type of anti-GC drug.37
Previous studies have found that Ber exerts its anti-cancer effects on different tumors through different molecular mechanisms. In many cancer types, such as CML, liver cancer and T cell lymphoma, CaMKIIγ has been postulated as the most likely direct target of Ber because of its direct binding affinity to CaMKIIγ and potent inactivation of CaMKIIγ signaling.15,38,39 STAT3 is another Ber interacting molecule that has been confirmed in previous studies and may be a direct target for the gefitinib-sensitizing and sorafenib-sensitizing effects of Ber.13,14 Besides, Liang et al reported that the anti-myeloma effects of Ber may be mediated by the NF-κB signaling pathway blockade.16 In this study, through Western blot, HTRF and SPR assay, we confirmed that the anti-GC effects of Ber might be primarily mediated by BRD4 inhibition, rather than the aforementioned signaling pathway. Western blot and qPCR analysis results suggested that Ber downregulated the expression of key BRD4 downstream oncogenes, such as c-Myc, BCL2 and BCL6. Then, we further carried out molecular docking and MD simulations to study the possible binding modes between Ber and BRD4. Computer simulation results suggested that Ber bound to a hydrophobic pocket in the first bromodomain of BRD4 (BD1 domain) through its oxydibenzene groups to form key hydrophobic interactions with Trp81 and Ile146. Besides, the hydroxyl in Ber formed a key hydrogen bond with Pro82. Based on this finding, several chemical modification strategies were proposed to improve the BRD4 binding affinity and the druggability of Ber. Based on the simulation results, we found that the oxydibenzene core of Ber orientated to the inner side of binding pocket, thereby introducing more hydrogen donors or acceptors into it, which might be related to Ber’s BRD4 binding capacity. Because Ber has a macrocyclic structure and a highly rigid structure, its solubility is poor. The long-chain hydrophilic group should be introduced into the isoquinoline ring toward the outside of the binding pocket, which might increase its water solubility. In addition, we boldly hypothesized that breaking the oxygen bridge bond between the two isoquinoline rings may be another feasible method to increase solubility and BRD4 inhibition of Ber, which may also promote the de novo synthesis of Ber derivatives.
Conclusions
In summary, this study suggests that Ber is a novel natural-derived BRD4 inhibitor that holds promise as a potential therapeutic drug for treating gastric cancer. At the same time, our research provides strong evidence that BRD4 may be a potential carcinogenic driving factor for GC and a promising target for small molecule therapeutic intervention.
Author Contributions
All authors made a significant contribution to the work reported, whether that is in the conception, study design, execution, acquisition of data, analysis and interpretation, or in all these areas; took part in drafting, revising or critically reviewing the article; gave final approval of the version to be published; have agreed on the journal to which the article has been submitted; and agree to be accountable for all aspects of the work.
Funding
Financial support was provided by Zhejiang Province Bureau of Health (2019RC029, 2019KY350) and Zhejiang Province Bureau of Traditional Chinese Medicine (2020ZA024).
Disclosure
The authors declare no conflicts of interest in this work.
References
1. Sung H, Ferlay J, Siegel RL, et al. Global cancer statistics 2020: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin. 2021;71(3):209–249. doi:10.3322/caac.21660
2. Smyth EC, Nilsson M, Grabsch HI, van Grieken NC, Lordick F. Gastric cancer. Lancet. 2020;396(10251):635–648. doi:10.1016/S0140-6736(20)31288-5
3. Salati M, Orsi G, Smyth E, et al. Gastric cancer: translating novels concepts into clinical practice. Cancer Treat Rev. 2019;79:101889. doi:10.1016/j.ctrv.2019.101889
4. Chen LT, Satoh T, Ryu MH, et al. A Phase 3 study of nivolumab in previously treated advanced gastric or gastroesophageal junction cancer (ATTRACTION-2): 2-year update data. Gastric Cancer. 2020;23(3):510–519. doi:10.1007/s10120-019-01034-7
5. Poornima P, Kumar JD, Zhao Q, Blunder M, Efferth T. Network pharmacology of cancer: from understanding of complex interactomes to the design of multi-target specific therapeutics from nature. Pharmacol Res. 2016;111:290–302. doi:10.1016/j.phrs.2016.06.018
6. Xu R, Dong Q, Yu Y, et al. Berbamine: a novel inhibitor of bcr/abl fusion gene with potent anti-leukemia activity. Leuk Res. 2006;30(1):17–23. doi:10.1016/j.leukres.2005.05.023
7. Wang GY, Lv QH, Dong Q, Xu RZ, Dong QH. Berbamine induces Fas-mediated apoptosis in human hepatocellular carcinoma HepG2 cells and inhibits its tumor growth in nude mice. J Asian Nat Prod Res. 2009;11(3):219–228. doi:10.1080/10286020802675076
8. Hou ZB, Lu KJ, Wu XL, Chen C, Huang XE, Yin HT. In vitro and in vivo antitumor evaluation of berbamine for lung cancer treatment. Asian Pac J Cancer Prev. 2014;15(4):1767–1769. doi:10.7314/APJCP.2014.15.4.1767
9. Zhao Y, Lv JJ, Chen J, et al. Berbamine inhibited the growth of prostate cancer cells in vivo and in vitro via triggering intrinsic pathway of apoptosis. Prostate Cancer Prostatic Dis. 2016;19(4):358–366. doi:10.1038/pcan.2016.29
10. Zhang H, Jiao Y, Shi C, et al. Berbamine suppresses cell proliferation and promotes apoptosis in ovarian cancer partially via the inhibition of Wnt/beta-catenin signaling. Acta Biochim Biophys Sin (Shanghai). 2018;50(6):532–539. doi:10.1093/abbs/gmy036
11. Han C, Wang Z, Chen S, et al. Berbamine suppresses the progression of bladder cancer by modulating the ROS/NF-kappaB axis. Oxid Med Cell Longev. 2021;2021:8851763. doi:10.1155/2021/8851763
12. Jin X, Wu Y. Berbamine enhances the antineoplastic activity of gemcitabine in pancreatic cancer cells by activating transforming growth factor-beta/Smad signaling. Anat Rec (Hoboken). 2014;297(5):802–809. doi:10.1002/ar.22897
13. Hu B, Cai H, Yang S, Tu J, Huang X, Chen G. Berbamine enhances the efficacy of gefitinib by suppressing STAT3 signaling in pancreatic cancer cells. Onco Targets Ther. 2019;12:11437–11451. doi:10.2147/OTT.S223242
14. Zhao W, Bai B, Hong Z, Zhang X, Zhou B. Berbamine (BBM), a natural STAT3 inhibitor, synergistically enhances the antigrowth and proapoptotic effects of sorafenib on hepatocellular carcinoma cells. ACS Omega. 2020;5(38):24838–24847. doi:10.1021/acsomega.0c03527
15. Gu Y, Zhang J, Ma X, et al. Stabilization of the c-Myc protein by CAMKIIgamma promotes T cell lymphoma. Cancer Cell. 2017;32(1):115–128 e117. doi:10.1016/j.ccell.2017.06.001
16. Liang Y, Xu RZ, Zhang L, Zhao XY. Berbamine, a novel nuclear factor kappaB inhibitor, inhibits growth and induces apoptosis in human myeloma cells. Acta Pharmacol Sin. 2009;30(12):1659–1665. doi:10.1038/aps.2009.167
17. Zhao H, Caflisch A. Discovery of ZAP70 inhibitors by high-throughput docking into a conformation of its kinase domain generated by molecular dynamics. Bioorg Med Chem Lett. 2013;23(20):5721–5726. doi:10.1016/j.bmcl.2013.08.009
18. Watson RJ, Bamborough P, Barnett H, et al. GSK789: a selective inhibitor of the first bromodomains (BD1) of the bromo and extra terminal domain (BET) proteins. J Med Chem. 2020;63(17):9045–9069. doi:10.1021/acs.jmedchem.0c00614
19. Maier JA, Martinez C, Kasavajhala K, Wickstrom L, Hauser KE, Simmerling C. ff14SB: improving the accuracy of protein side chain and backbone parameters from ff99SB. J Chem Theory Comput. 2015;11(8):3696–3713. doi:10.1021/acs.jctc.5b00255
20. Wang J, Wolf RM, Caldwell JW, Kollman PA, Case DA. Development and testing of a general amber force field. J Comput Chem. 2004;25(9):1157–1174. doi:10.1002/jcc.20035
21. Roe DR, Cheatham TE. PTRAJ and CPPTRAJ: software for processing and analysis of molecular dynamics trajectory data. J Chem Theory Comput. 2013;9(7):3084–3095. doi:10.1021/ct400341p
22. Genheden S, Ryde U. The MM/PBSA and MM/GBSA methods to estimate ligand-binding affinities. Expert Opin Drug Discov. 2015;10(5):449–461. doi:10.1517/17460441.2015.1032936
23. Wang E, Sun H, Wang J, et al. End-point binding free energy calculation with MM/PBSA and MM/GBSA: strategies and applications in drug design. Chem Rev. 2019;119(16):9478–9508. doi:10.1021/acs.chemrev.9b00055
24. Ba M, Long H, Yan Z, et al. BRD4 promotes gastric cancer progression through the transcriptional and epigenetic regulation of c-MYC. J Cell Biochem. 2018;119(1):973–982. doi:10.1002/jcb.26264
25. Ruan T, Liu W, Tao K, Wu C. A review of research progress in multidrug-resistance mechanisms in gastric cancer. Onco Targets Ther. 2020;13:1797–1807. doi:10.2147/OTT.S239336
26. Biagioni A, Skalamera I, Peri S, et al. Update on gastric cancer treatments and gene therapies. Cancer Metastasis Rev. 2019;38(3):537–548. doi:10.1007/s10555-019-09803-7
27. Qin ZY, Wang T, Su S, et al. BRD4 promotes gastric cancer progression and metastasis through acetylation-dependent stabilization of snail. Cancer Res. 2019;79(19):4869–4881. doi:10.1158/0008-5472.CAN-19-0442
28. Dong X, Hu X, Chen J, Hu D, Chen LF. BRD4 regulates cellular senescence in gastric cancer cells via E2F/miR-106b/p21 axis. Cell Death Dis. 2018;9(2):203. doi:10.1038/s41419-017-0181-6
29. Song H, Shi L, Xu Y, et al. BRD4 promotes the stemness of gastric cancer cells via attenuating miR-216a-3p-mediated inhibition of Wnt/beta-catenin signaling. Eur J Pharmacol. 2019;852:189–197. doi:10.1016/j.ejphar.2019.03.018
30. Li D, Wang J, Zhang M, et al. LncRNA MAGI2-AS3 is regulated by BRD4 and promotes gastric cancer progression via maintaining ZEB1 overexpression by sponging miR-141/200a. Mol Ther Nucleic Acids. 2020;19:109–123. doi:10.1016/j.omtn.2019.11.003
31. Amorim S, Stathis A, Gleeson M, et al. Bromodomain inhibitor OTX015 in patients with lymphoma or multiple myeloma: a dose-escalation, open-label, pharmacokinetic, phase 1 study. Lancet Haematol. 2016;3(4):e196–204. doi:10.1016/S2352-3026(16)00021-1
32. Berthon C, Raffoux E, Thomas X, et al. Bromodomain inhibitor OTX015 in patients with acute leukaemia: a dose-escalation, phase 1 study. Lancet Haematol. 2016;3(4):e186–195. doi:10.1016/S2352-3026(15)00247-1
33. Sun Y, Han J, Wang Z, Li X, Sun Y, Hu Z. Safety and efficacy of bromodomain and extra-terminal inhibitors for the treatment of hematological malignancies and solid tumors: a systematic study of clinical trials. Front Pharmacol. 2020;11:621093. doi:10.3389/fphar.2020.621093
34. Guo D, Zhang W, Yang H, et al. Celastrol induces necroptosis and ameliorates inflammation via targeting biglycan in human gastric carcinoma. Int J Mol Sci. 2019;20(22):5716. doi:10.3390/ijms20225716
35. Pan H, Wang BH, Lv W, Jiang Y, He L. Esculetin induces apoptosis in human gastric cancer cells through a cyclophilin D-mediated mitochondrial permeability transition pore associated with ROS. Chem Biol Interact. 2015;242:51–60. doi:10.1016/j.cbi.2015.09.015
36. Wang Z, Yu K, Hu Y, et al. Schisantherin A induces cell apoptosis through ROS/JNK signaling pathway in human gastric cancer cells. Biochem Pharmacol. 2020;173:113673. doi:10.1016/j.bcp.2019.113673
37. Zhao Y, Tan Y, Wu G, et al. Berbamine overcomes imatinib-induced neutropenia and permits cytogenetic responses in Chinese patients with chronic-phase chronic myeloid leukemia. Int J Hematol. 2011;94(2):156–162. doi:10.1007/s12185-011-0887-7
38. Gu Y, Chen T, Meng Z, et al. CaMKII gamma, a critical regulator of CML stem/progenitor cells, is a target of the natural product berbamine. Blood. 2012;120(24):4829–4839. doi:10.1182/blood-2012-06-434894
39. Meng Z, Li T, Ma X, et al. Berbamine inhibits the growth of liver cancer cells and cancer-initiating cells by targeting Ca(2)(+)/calmodulin-dependent protein kinase II. Mol Cancer Ther. 2013;12(10):2067–2077. doi:10.1158/1535-7163.MCT-13-0314
 © 2022 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms.php
and incorporate the Creative Commons Attribution
- Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2022 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms.php
and incorporate the Creative Commons Attribution
- Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.