")
Back to Journals » Neuropsychiatric Disease and Treatment » Volume 14
Beneficial effects of trimetazidine on expression of serotonin and serotonin transporter in rats with myocardial infarction and depression
Authors Liu MY, Wei WL, Stone CR, Zhang LJ, Tian GX, Ding JN
Received 18 November 2017
Accepted for publication 5 February 2018
Published 16 March 2018 Volume 2018:14 Pages 787—797
DOI https://doi.org/10.2147/NDT.S157441
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Wai Kwong Tang
Meiyan Liu,1,* Wanlin Wei,2,* Christopher R Stone,3 Lijun Zhang,1 Guoxiang Tian,4 Jessie N Ding3
1Department of Cardiology, Beijing Anzhen Hospital, Capital Medical University, Beijing, People’s Republic of China; 2Department of Cardiology, PLA Army General Hospital, Beijing, People’s Republic of China; 3Department of Neurosurgery, Wayne State University School of Medicine, Detroit, MI, USA; 4Department of 4th Cadres Ward, PLA Army General Hospital, Beijing, People’s Republic of China
*These authors contributed equally to this work
Background: Trimetazidine is an anti-ischemic drug that can inhibit platelet aggregation and regulate serotonin (5-hydroxytryptamine [5-HT]) release. The purpose of this study was to investigate the therapeutic effects of trimetazidine on 5-HT and serotonin transporter (SERT) expression in experimentally induced myocardial infarction (MI), depression, and MI + depression.
Materials and methods: Eighty Sprague Dawley (SD) rats were randomly divided into a trimetazidine group and a saline group of 40 rats each. The trimetazidine group was given trimetazidine pretreatment for 4 weeks, while the saline group received saline for 4 weeks. Both groups were then subdivided into four subgroups (n=10), which were each subjected to a unique disease condition: sham surgery, MI, depression, or MI + depression. All rats were sacrificed 3 days thereafter, and serum and platelet levels of 5-HT and SERT were assessed. In addition, we experimented with trimetazidine posttreatment. Twenty SD rats underwent MI surgery, and were then randomly divided into a treatment and a saline group (n=10 each). For 4 weeks post-surgery, the trimetazidine group was given trimetazidine, while the saline group received saline. Serum and platelet levels of 5-HT and SERT were assessed.
Results: Pretreatment with trimetazidine: in the nontreatment saline group, MI, depression, and MI + depression showed significant declines (P<0.05) in both serum and platelet 5-HT levels compared to sham. Trimetazidine treatment significantly increased serum and platelet 5-HT levels in the MI, depression, and MI + depression (P<0.05) subgroups compared to their counterparts in the saline group. Results for SERT were heterogeneous between serum and platelets. Trimetazidine treatment significantly decreased serum levels of SERT in the sham surgery subgroup (P<0.05), while significantly increasing levels in depression rats, compared to control (P<0.05). In platelets, trimetazidine significantly decreased SERT in sham surgery, MI, depression, and MI + depression rats, compared to control (P<0.05). This contrast suggests that trimetazidine has opposite effects in serum and platelet SERT levels for the three disease models. Post-surgery trimetazidine: increased serum 5-HT (P<0.05) and serum SERT (P<0.05) were observed, compared to control. In platelets, trimetazidine decreased both 5-HT and SERT compared to control, significantly (P<0.05) for 5-HT, but not significantly for SERT (P>0.05).
Conclusion: Trimetazidine has a regulatory effect on 5-HT and SERT in the serum and platelets. Because of the downstream effects of this regulation on blood vessel function and myocardial protection, trimetazidine may be a therapeutic or preventive agent in several disease processes, including MI, depression, and the comorbidity between these two diseases. Further investigation, aimed at exploring the clinical potential of trimetazidine, is therefore warranted.
Keywords: trimetazidine, MI, depression, serotonin, serotonin transporter
Introduction
In recent years, medical science has seen a veritable surge of research within the field of health psychology. Gone are the days when, in the absence of much concrete data to suggest otherwise, mind–body dualism was the dominant paradigm.1 The epidemiologic modality has since been an important source of paradigm changing data. A study by Russ et al, for example, yielded a dose–response curve between psychological distress and all-cause mortality after adjustment for somatic, behavioral, and socioeconomic covariates.2 Increasingly, the biological modality is also contributing to the pool of mind–body synthetic data. By turning up measurable associations between physiological markers and their proposed psychological causes, these data contribute a mechanistic way to understand the pathophysiological interconnectivity of body and mind.3
One prominent focus of such studies, both epidemiologically and biologically, has been the relationship between coronary heart disease (CHD) and clinical depression. Interest in their comorbidity follows partly from the tremendous burden both diseases exert independently. With an annual mortality rate of 17 million, cardiovascular disease is the number one cause of death worldwide.4
Depression, moreover, is an independent risk factor for heart disease morbidity and mortality. It increases the severity of CHD, seriously affecting prognoses and outcomes.5 This is true both according to outcomes research, and from the perspective of individual patients, it has been shown that, among patients whose CHD is stable, it is depressive symptomology, and not cardiac indicators, that is related to diminished quality of life.6 It is also true epidemiologically. While depression has a population-wide prevalence of about 5%, it is much more prevalent among those who have experienced CHD. Its rate among this population increases four-fold, to 20%.7
Many different biological mechanisms for this comorbidity have been proposed and studied, but it is not within the scope of this work to review them in detail. Instead, this paper will contribute to the depth with which we understand one mechanism: serotonin-mediated platelet dysregulation. This mechanism is of singular interest for two reasons. First, as our study aims to show, it is directly testable through experimentation with the established antianginal drug, trimetazidine. Second, owing to the availability of many modulators of serotonin concentration, such a connection, if properly elucidated, would seem to promise substantial translational potential for the clinical treatment of both depression and CHD.
Serotonin, or 5-hydroxytryptamine (5-HT), was first characterized in 1948 by Rapport et al,8 who extracted it from bovine serum and named it for the effects it exhibited on blood vessels. Subsequently, in 1951, Speeter et al synthesized 5-HT in the laboratory, and confirmed that it was the same chemical found earlier by Rapport.8 5-HT is an intermediate product of tryptophan metabolism, and an important neurotransmitter. It has a wide range of physiological effects,9 which play out mainly in the enterochromaffin cells, platelets, and brain serotonergic neurons,10 and include vasomodulation, mood control, urinary continence, intestinal motility, and regulation of sleep and body temperature. In the body, 95% of 5-HT is synthesized in the enterochromaffin cells;10 other synthetic sites include the raphe nuclei, pineal gland, and pulmonary epithelium.11 After synthesis, 99% of the body’s 5-HT is stored in the dense granules of platelets.12
The effect of serotonin transporter (SERT) must be considered, too. SERT regulates the cytosolic entry and exit of 5-HT according to the intracellular and extracellular levels of 5-HT, as well as the amount of SERT on the plasma membrane.13 SERT levels will change with 5-HT levels to maintain balance. It was once thought that a rise in serum 5-HT led to an increase in plasma membrane SERT to accelerate reuptake, but Nemeroff et al14 found that when serum 5-HT increased platelet plasma membrane SERT levels actually decreased, thus limiting platelet reuptake of serum 5-HT. The specific biochemical mechanism of platelet SERT’s regulation of serum 5-HT is clearly complex. Brenner et al15 showed as much, suggesting that the concentration of 5-HT is related to 5-HT reuptake in an inverse manner via concentration-dependent expression of SERT. That is, when serum 5-HT was low, the SERT density on platelet membranes increased by 35% and the 5-HT reuptake rate increased by 32%. When serum 5-HT was high, 5-HT reuptake decreased by 31%, while platelet membrane SERT density decreased by 30%. Also, SERT function and expression abnormalities have been found in depression.16,17 Mercado et al14 found that depressed patients have reduced platelet SERT binding sites. This may be a consequence of stress from increased consumption of serum 5-HT and reduced release in depression, resulting in platelet dysfunction.
By inhibiting the long-chain 3-ketoacyl coenzyme A thiolase (3-KAT), trimetazidine is likely to have a simultaneous impact on 5-HT and its receptor in myocardial tissue, to thus help protect the myocardium and small blood vessels. Moreover, since 5-HT dysregulation has also been implicated in both CHD and depression, an agent that is cardioprotective because of its regulatory effect on 5-HT physiology has the potential to address CHD and depression comorbidity, as well.
In this study, we investigated the effects of trimetazidine treatment on serum and platelet levels of 5-HT and SERT in myocardial infarction (MI), depression, and MI + depression. Our hypothesis was that this experiment would clarify some of the unknown steps in the syllogism between 5-HT dysregulation and the CHD/depression comorbidity, and thus provide translational potential.
Materials and methods
Subjects
A total of 100 male and female Sprague Dawley (SD) rats, weighing 180–220 g, were purchased from the Experimental Animal Center in Jiangsu Province for use in this study. Rats were housed two per cage with sawdust, in a temperature-controlled room with a 12-hour light–dark cycle (7 pm to 7 am dark cycle). Food and water were available ad libitum. All experimental procedures were approved by the Institutional Animal Investigation Committee of Capital Medical University in accordance with the National Institutes of Health (USA) guidelines for care and use of laboratory animals. All efforts were made to minimize the number of animals used and their suffering.
Groups
In our first experiment (pretreatment), rats were randomly divided into treatment and saline groups of 40 rats each. After 4 weeks, the two groups were further divided into four subgroups to establish four animal disease models of 10 rats each: sham operation, MI, depression, and MI + depression. In a second experiment (posttreatment), 20 additional rats were randomly and equally divided into a trimetazidine group and a saline group after receiving MI. Pharmacological administration was performed at 7 am.
Trimetazidine treatment
Trimetazidine gavage mix at a dosage of 10 mg/kg18,19 dissolved in 2.5 mL saline, which was made fresh daily, was given to each of the rats in the trimetazidine groups (before or after procedures, depending on the experiment) once a day for 4 weeks (on consecutive days). During the same period, the rats in the saline groups received the same amount of normal saline without added trimetazidine.
Pathological induction procedures
Sham-operated group
Rats underwent conventional thoracotomy, but without occlusion of the left anterior artery.
Depression
Depression was induced in rats by a modified forced swimming test (FST) in accordance with the work of Porsolt et al.20 In the light phase, rats were individually plunged into a Pyrex cylinder (21 cm diameter, 46 cm height), with 23°C–25°C water filled to a depth of 30 cm, for 15 minutes of “swimming,” after which they were removed from the pool and allowed to dry in a heated enclosure (32°C) for 15 minutes. They were then returned to their cages. After 24 hours, FST was repeated, but this time with the swimming time reduced to 5 minutes. Throughout the process, rat swimming behavior was captured on video.21
MI
A mixture of 100 mg of ketamine and 10 mg of xylazine in 10 mL saline was administered via intramuscular injection at 0.1 mL/10 g of bodyweight. Rats were then placed in the supine position, and MI was induced with Akbay and Onur’s approach.22 Success of the operation was determined by the following criteria: bright to pale postoperative cardiac infarct zone, cardiac cyanosis, weakened ventricular activity, electrocardiography showing ST segment elevation, and arched T-wave towering or formation of a single curve.
MI combined with depression
MI was induced first as described in the “MI” section. Rats were then treated by FST 3 days thereafter. In the pretreatment experiment, the rates of successful disease model induction – that is, of rats that survived the pathological induction procedure – for the rats that received saline were 8 of 10 (80%) for sham operation, 8 of 10 (80%) for MI, 9 of 10 (90%) for depression, and 8 of 10 (80%) for MI + depression. In rats that received trimetazidine, the success rates were 10 of 10 (100%) for sham operation, 8 of 10 (80%) for MI, 8 of 10 (80%) for depression, and 9 of 10 (90%) for MI + depression. All rats were sacrificed 3 days after disease model induction, and levels of 5-HT and SERT in serum and platelets were measured. In the posttreatment experiment, 20 rats were randomly and equally divided into a trimetazidine group and a saline group after receiving MI which was induced by the same method as above. Eight of 10 (80%) rats receiving saline survived MI induction; 7 of 10 (70%) receiving trimetazidine survived. Table 1 shows the time points of all treatments for all groups.
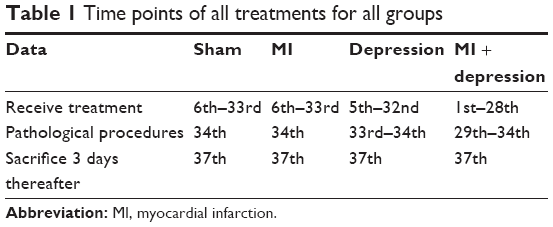 | Table 1 Time points of all treatments for all groups |
Detection of 5-HT and SERT
To stabilize the plasma glucose and avoid fluctuations of the plasma concentrations of tryptophan and other amino acids, all animals were fasted for 12 hours prior to sample collection on the day of sacrifice, which occurred at about the same time for all rats. In the light phase, the rats were anesthetized with pentobarbital and the carotid artery cannulated to collect blood. Blood was drawn through a polythene cannula filled with K2-ethylenediaminetetraacetic acid (K2-EDTA) and collected into plastic tubes with EDTA (final concentration 0.5%–1%). The tubes were then centrifuged and the supernatant collected and stored at −80°C until analyzed.
For analysis of the samples, enzyme-linked immunosorbent assay (ELISA) kits for 5-HT and SERT (product number EFXER 00123) were purchased from Nanjing Yi Fei Xue Biotechnology Ltd (China).
Serum preparation
Blood was coagulated at room temperature for 10–20 minutes, and centrifuged for 20 minutes at 2,000–3,000 rpm. If precipitate was visible, the blood was centrifuged again. The supernatant was then carefully collected.
Platelet preparation
One milliter of blood from the heart was centrifuged in K2-EDTA anticoagulant tubes (Becton, Dickinson and Company, Suzhou, Jiangsu Province, China, item: 367 844) at 2,000 rpm for 15 minutes at room temperature, yielding platelet-rich plasma (PRP). The PRP was further centrifuged at 2,000 rpm for 10 minutes, this time at 4°C. The supernatant was then discarded, and platelets were obtained.
ELISA
5-HT and SERT levels were assessed with double antibody sandwich ELISA. Standard sample, an equal volume of serum, and an equal volume of platelet lysate were all loaded into the microtiter plate (standard and two duplicate wells per sample). They were incubated at 37°C for 30 minutes, washed with PBS (NaCl 137 mmol/L, KCl 2.7 mmol/L, Na2HPO4 10 mmol/L, KH2PO4 2 mmol/L, pH 7.4), and then incubated with enzyme-labeled reagent for 30 minutes. After the coloring solution was added for 10 minutes, the reaction was terminated with dilute sulfuric acid. The concentration of 5-HT and SERT (pg/mL) was determined by comparing the absorbance (OD) of the plate reader to the standard curve at 450 nm.
Statistical analysis
SPSS 17.0 software (SPSS Inc, Chicago, IL, USA) was used for data analysis. Measurements were expressed as  ± s. In the pretreatment experiment, regarding the design of our experiment, the interactions of different models and pretreatments were analyzed by multivariate linear regression analysis. Controlling the variable of the model, the differences of saline and trimetazidine pretreatments in the same model were analyzed by independent t-test. Controlling the variable of pretreatment, the differences of the four models underwent the same pretreatment by analysis of variance univariate analysis, with two groups comparisons using the least significance difference test. In the treatment experiment, the trimetazidine group and saline group were compared using the independent t-test. In all circumstances, P<0.05 was considered statistically significant.
± s. In the pretreatment experiment, regarding the design of our experiment, the interactions of different models and pretreatments were analyzed by multivariate linear regression analysis. Controlling the variable of the model, the differences of saline and trimetazidine pretreatments in the same model were analyzed by independent t-test. Controlling the variable of pretreatment, the differences of the four models underwent the same pretreatment by analysis of variance univariate analysis, with two groups comparisons using the least significance difference test. In the treatment experiment, the trimetazidine group and saline group were compared using the independent t-test. In all circumstances, P<0.05 was considered statistically significant.
Results
Pretreatment experiment
Part one: interaction of different models and pretreatments
Serum 5-HT
Considering the design of our experiment, we regarded sham pretreated with saline as constant, and built the linear equation as follows:
|
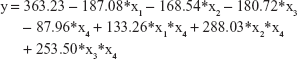
where y: the value of serum 5-HT; x1: MI pretreated with saline; x2: depression pretreated with saline; x3: MI + depression pretreated with saline; x4: pretreated with trimetazidine; x1*x4: the interaction of MI and trimetazidine; x2*x4: the interaction of depression and trimetazidine; x3*x4: the interaction of MI + depression and trimetazidine.
The multivariate linear regression analysis showed that different models including MI, depression, and MI + depression, different pretreatments, and the interaction were all significantly associated with serum 5-HT (all P<0.01). The absolute value of standardized coefficients beta demonstrated that the interaction of depression and trimetazidine had the strongest relationship with serum 5-HT compared to all the other factors (Table 2).
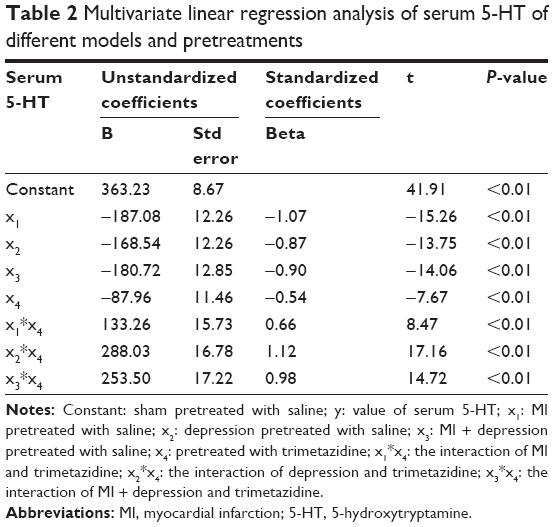 | Table 2 Multivariate linear regression analysis of serum 5-HT of different models and pretreatments |
Serum SERT
Considering the design of our experiment, we regarded sham pretreated with saline as constant, and built the linear equation as follows:
|
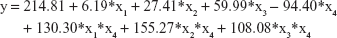
where y: the value of serum SERT; x1: MI pretreated with saline; x2: depression pretreated with saline; x3: MI + depression pretreated with saline; x4: pretreated with trimetazidine; x1*x4: the interaction of MI and trimetazidine; x2*x4: the interaction of depression and trimetazidine; x3*x4: the interaction of MI + depression and trimetazidine.
The multivariate linear regression analysis showed that the model of MI + depression, different pretreatments, and the different interactions were all significantly associated with serum SERT (P=0.00), but there was no significance in the models of MI (P=0.658) and depression (P=0.055). The absolute value of standardized coefficients beta demonstrated that the interaction of depression and trimetazidine had the strongest relationship with serum SERT compared to all the other factors (Table 3).
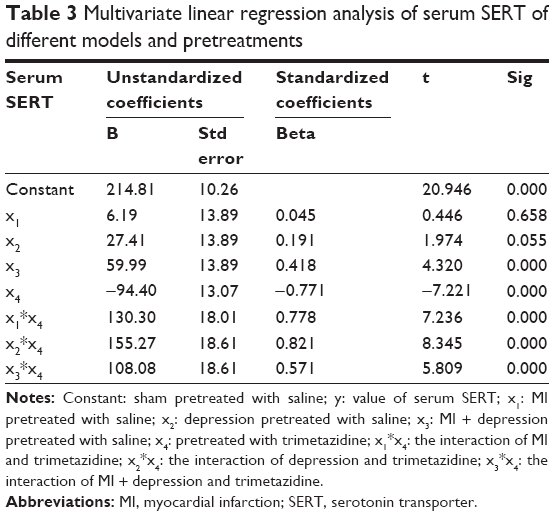 | Table 3 Multivariate linear regression analysis of serum SERT of different models and pretreatments |
Platelet 5-HT
Considering the design of our experiment, we regarded sham pretreated with saline as constant, and built the linear equation as follows:
|
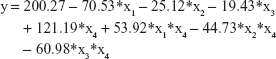
where y: the value of platelet 5-HT; x1: MI pretreated with saline; x2: depression pretreated with saline; x3: MI + depression pretreated with saline; x4: pretreated with trimetazidine; x1*x4: the interaction of MI and trimetazidine; x2*x4: the interaction of depression and trimetazidine; x3*x4: the interaction of MI + depression and trimetazidine.
The multivariate linear regression analysis showed that the models of MI, depression, different pretreatments, and the different interactions were all significantly associated with platelet 5-HT (all P<0.05), but there was no significance in the models of MI + depression (P=0.071). The absolute value of standardized coefficients beta demonstrated that the pretreatment of trimetazidine had the strongest relationship with platelet 5-HT compared to all the other factors (Table 4).
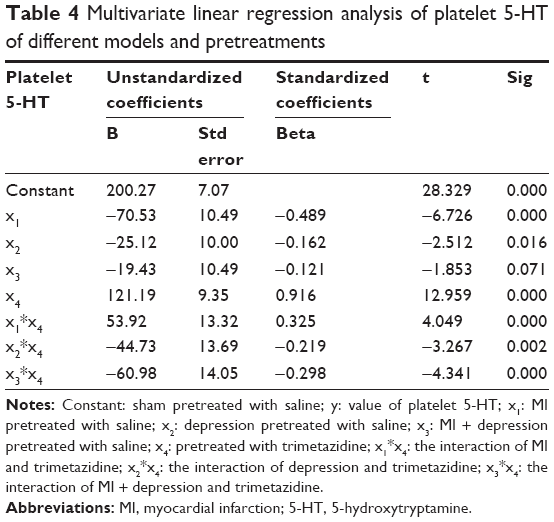 | Table 4 Multivariate linear regression analysis of platelet 5-HT of different models and pretreatments |
Platelet SERT
Considering the design of our experiment, we regarded sham pretreated with saline as constant, and built the linear equation as follows:
|
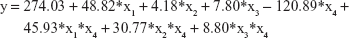
where y: the value of platelet SERT; x1: MI pretreated with saline; x2: depression pretreated with saline; x3: MI + depression pretreated with saline; x4: pretreated with trimetazidine; x1*x4: the interaction of MI and trimetazidine; x2*x4: the interaction of depression and trimetazidine; x3*x4: the interaction of MI + depression and trimetazidine.
The multivariate linear regression analysis showed that the model of MI, different pretreatments, and the interaction of MI and trimetazidine pretreatment were all significantly associated with platelet SERT (all P<0.05), but there was no significance in the models of depression (P=0.734), MI + depression (P=0.527), the interaction of depression and trimetazidine (P=0.073), and the interaction of MI + depression and trimetazidine (P=0.602). The absolute value of standardized coefficients beta demonstrated that the pretreatment of trimetazidine had the strongest relationship with platelet SERT compared to all the other factors (Table 5).
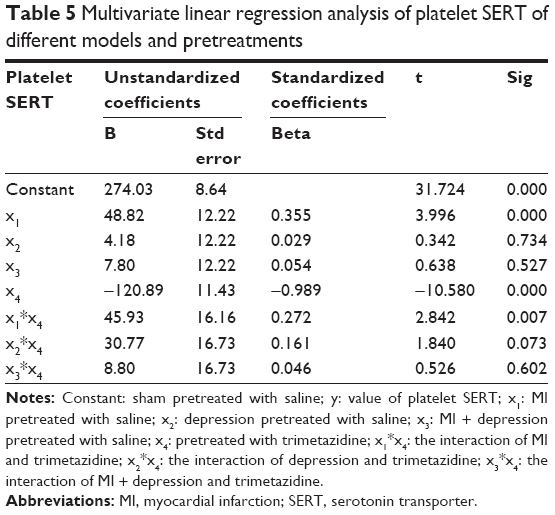 | Table 5 Multivariate linear regression analysis of platelet SERT of different models and pretreatments |
Part two: comparisons of different groups
5-HT levels
Serum 5-HT
In the saline group, the MI, depression, and MI + depression subgroups exhibited levels of serum 5-HT that were significantly lower than sham-operated rat levels (F=141.83, P<0.05). In the trimetazidine group, depression and MI + depression subgroups exhibited serum 5-HT levels that were significantly higher than those in sham operation rats (F=87.20, P<0.05). Trimetazidine treatment significantly decreased serum 5-HT for sham operation rats (F=39.84, t=6.60, df=5.29, P=0.001), and significantly increased serum 5-HT for MI (F=1.64, t=−4.54, df=15, P=0.000), depression (F=19.15, t=−15.40, df=5.26, P=0.000), and MI + depression (F=4.32, t=−12.34, df=9, P=0.000) rats compared with the saline group (Figure 1A).
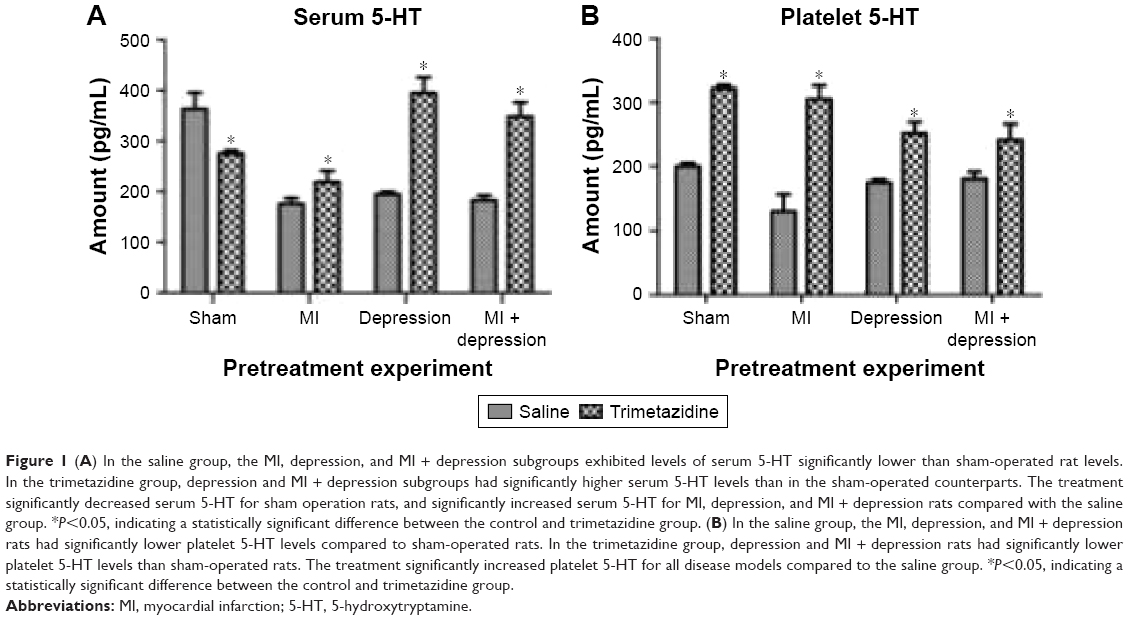 | Figure 1 (A) In the saline group, the MI, depression, and MI + depression subgroups exhibited levels of serum 5-HT significantly lower than sham-operated rat levels. In the trimetazidine group, depression and MI + depression subgroups had significantly higher serum 5-HT levels than in the sham-operated counterparts. The treatment significantly decreased serum 5-HT for sham operation rats, and significantly increased serum 5-HT for MI, depression, and MI + depression rats compared with the saline group. *P<0.05, indicating a statistically significant difference between the control and trimetazidine group. (B) In the saline group, the MI, depression, and MI + depression rats had significantly lower platelet 5-HT levels compared to sham-operated rats. In the trimetazidine group, depression and MI + depression rats had significantly lower platelet 5-HT levels than sham-operated rats. The treatment significantly increased platelet 5-HT for all disease models compared to the saline group. *P<0.05, indicating a statistically significant difference between the control and trimetazidine group. |
Platelet 5-HT
In the saline group, MI, depression, and MI + depression rats had significantly lower platelet 5-HT levels compared with sham-operated rats (F=22.94, P<0.05). In the trimetazidine group, depression and MI + depression rats had significantly lower platelet 5-HT levels than sham-operated rats (F=29.99, P<0.05). Trimetazidine treatment significantly increased platelet 5-HT for sham (F=0.24, t=−38.71, df=12, P=0.000), MI (F=0.91, t=−13.48, df=13, P=0.000), depression (F=8.23, t=−10.12, df=5.69, P=0.000), MI + depression (F=2.91, t=−4.86, df=9, P=0.001) rats compared with the saline group (Figure 1B).
SERT levels
Serum SERT
In the saline group, MI, depression, and MI + depression rats had higher serum SERT than sham-operated rats, but the differences were not statistically significant (F=4.02, P>0.05) except for the MI + depression group (F=4.02, P<0.05). In the trimetazidine group, serum levels of SERT were significantly higher than sham-operated rats in the MI, depression, and MI + depression rats (F=449.40, P<0.05). Compared with the saline group, trimetazidine treatment significantly decreased serum SERT in sham-operated rats (F=7.97, t=6.45, df=4.17, P=0.00), significantly increase serum SERT in depression rats (F=1.36, t=−5.50, df=10, P=0.00), and insignificantly increased serum SERT in MI (F=6.77, t=−2.06, df=5.52, P=0.09) and MI + depression rats (F=4.42, t=−1.06, df=10, P=0.32) (Figure 2A).
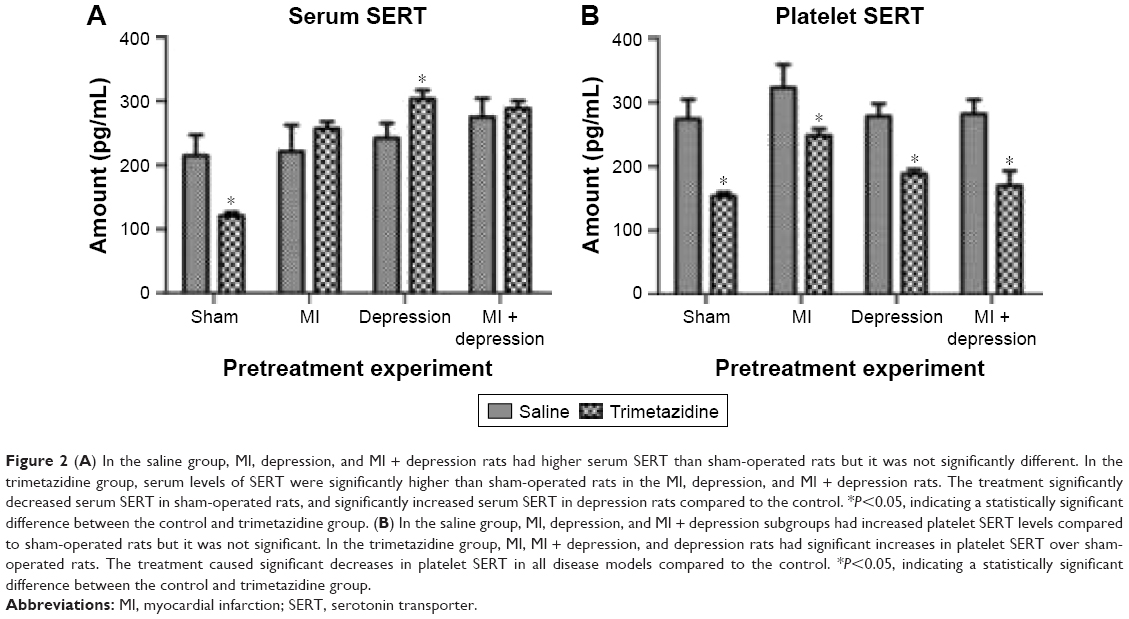 | Figure 2 (A) In the saline group, MI, depression, and MI + depression rats had higher serum SERT than sham-operated rats but it was not significantly different. In the trimetazidine group, serum levels of SERT were significantly higher than sham-operated rats in the MI, depression, and MI + depression rats. The treatment significantly decreased serum SERT in sham-operated rats, and significantly increased serum SERT in depression rats compared to the control. *P<0.05, indicating a statistically significant difference between the control and trimetazidine group. (B) In the saline group, MI, depression, and MI + depression subgroups had increased platelet SERT levels compared to sham-operated rats but it was not significant. In the trimetazidine group, MI, MI + depression, and depression rats had significant increases in platelet SERT over sham-operated rats. The treatment caused significant decreases in platelet SERT in all disease models compared to the control. *P<0.05, indicating a statistically significant difference between the control and trimetazidine group. |
Platelet SERT
In the saline group, MI, depression, and MI + depression subgroups saw increased platelet SERT levels compared with sham-operated rats, but the difference was not statistically significant (F=3.93, P>0.05). In the trimetazidine group, MI, depression, and MI + depression rats had significant increases in platelet SERT over sham-operated rats (F=79.40, P<0.05). Trimetazidine treatment caused significant decreases in platelet SERT in sham (F=181.90, t=9.60, df=5.28, P=0.00), MI (F=20.29, t=4.89, df=5.66, P=0.00), depression (F=5.23, t=10.52, df=6.31, P=0.00), and MI + depression (F=0.00, t=8.52, df=10, P=0.00) models compared with the saline group (Figure 2B).
Posttreatment experiment
5-HT levels
Serum levels of 5-HT were significantly higher in rats that received trimetazidine post-MI than rats that received saline treatment (saline group) post-MI (F=10.30, t=−6.28, df=5.28, P=0.00). Platelet 5-HT was significantly lower in the trimetazidine group than the saline group post-MI (F=0.00, t=16.16, df=14, P=0.00) (Table 6).
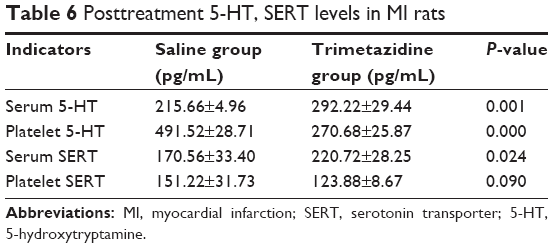 | Table 6 Posttreatment 5-HT, SERT levels in MI rats |
SERT levels
Serum levels of SERT were significantly higher in rats receiving trimetazidine post-MI than rats receiving saline treatment (saline group) post-MI (F=0.40, t=−2.70, df=9, P=0.02). Platelet SERT was lower in the trimetazidine group than in the saline group, but the difference was not statistically significant (F=17.59, t=2.04, df=10, P=0.09) (Table 6).
Discussion
Our results show that both the different models and pretreatments were related with the value of serum 5-HT, serum SERT, platelet 5-HT, and platelet SERT. Compared with saline group, trimetazidine pretreatment significantly increased serum and platelet 5-HT for MI, depression, and MI + depression rats, significantly increased serum SERT in depression rats, and insignificantly increased serum SERT in MI and MI + depression rats. It also significantly decreased platelet SERT in all disease models. Serum levels of 5-HT and SERT were significantly higher in rats receiving trimetazidine post-MI than in the saline group. Platelet 5-HT and SERT were significantly lower in the trimetazidine group than the saline group post-MI.
Platelets have many functional similarities to neurons and are easily accessible to clinical investigation.23 Blood platelets contain a good amount of serotonin as well, which may be an indirect index for functional levels of serotonin in the brain.24 In a previous study,25 we had demonstrated the relationship of CNS 5-HT and periphery 5-HT. In this study, we focused on serum and platelet 5-HT modulation.
From the perspective of its relationship to CHD, the powerful vasomodulatory function exerted by 5-HT is one of its most important. This function can manifest either as constriction or as relaxation of blood vessels, and depends on the interplay of many other physiological variables. The most important of these variables are the integrity of the autonomic nervous system through which 5-HT acts,26 the 5-HT receptors27 whereby its signal is transduced, the integrity of the vascular endothelium,26,28,29 and properties of the 5-HT transporter (SERT). Endothelial integrity is what accounts for the dual vasomodulatory role of 5-HT: on an intact endothelium, 5-HT plays the protective role of vascular relaxation; when, by contrast, the endothelium is missing or dysfunctional, 5-HT initiates a vasoconstrictive signal.28 SERT, with which 5-HT is associated on the blood–brain barrier, regulates 5-HT levels centrally and peripherally, by transporting it through the peripheral blood and central nervous system.30 It is distributed mainly in the heart, blood vessels, brain tissue, adrenal gland and kidneys, and platelets;10 critically, this is the protein through which platelets regulate 5-HT.
Platelets cannot synthesize 5-HT by themselves. They are, however, able to take up, store, and release it. Plasma and platelet 5-HT levels are equilibrated by platelet absorption and release.11 Platelets transport 5-HT by both active transport and passive diffusion, in a similar fashion to 5-HT secretion and reuptake at nerve endings.31 Abnormal platelet 5-HT levels, suggestive of disruption of this process, are involved in a variety of pathological states, including CHD, depression, and neurological diseases.32 In fact, some scholars have reported that 5-HT-catalyzed platelet aggregation is the mechanism of MI and depression comorbidity.33 However, this is not a simple relationship. First, it has been shown that 5-HT cannot cause platelet aggregation unless the plasma 5-HT concentration exceeds a certain threshold. Second, platelet aggregation occurs through synergy with other vasoactive substances (such as thrombin).17 And third, the relationship between depression and 5-HT levels requires the consideration of other variables before it can be related to platelet aggregation. Sanner et al34 found that lower Depression Rating Scale scores correlated with high platelet 5-HT levels in patients, while 5-HT levels in depressed patients generally declined. When 5-HT levels decline, serotonin activity decreases, body exercise tolerance declines, and patients show a depressed state.13 Williams7 has shown that depressed patients show reduced platelet 5-HT levels and decreased SERT binding.
While these relationships (ie, the regulatory role of platelets via SERT over peripheral 5-HT concentration, the dependence of vasomodulatory function of 5-HT on endothelial integrity, the relevance of platelets in CHD pathogenesis, and the relevance of central 5-HT in depressive pathogenesis) are established in the literature, the mechanism that connects them remains to be elucidated. As a consequence of this current lack of depth, feasible, effective interventions that exploit the relationships have so far failed to suggest themselves. Accordingly, it is important at this stage to establish animal models of CHD with depression, and to study these models to understand the comorbid neuroendocrine changes.
In this experiment, we employed trimetazidine in order to do just that. Studies suggest that it exerts its antianginal effects by inhibiting 3-KAT. This causes a metabolic shift away from long-chain fatty acid oxidation and toward glucose oxidation, thus increasing the efficiency of myocardial oxygen utilization. This, in turn, can reduce vascular resistance and increase coronary microcirculatory blood flow.35,36 Moreover, Shirahase et al found that the antianginal effects of trimetazidine may be related to inhibition of platelet aggregation and inhibition of 5-HT release.37 Thus, if experimentation with trimetazidine can confer insight into how 5-HT and platelets are dysregulated in CHD and/or depression, and if trimetazidine can be shown to ameliorate these circumstances, then significant headway would be made toward understanding and treating this comorbidity. There has been some investigation into the comorbidity along these lines. Some authors suggest the relevance of platelet hyper-reactivity to serotonin,38 and our previous studies suggest that the cardiomyocyte and coronary microcirculation injuries seen in the comorbidity are mediated by elevated 5-HT and SERT.39 However, no further studies have investigated the mechanism of action of trimetazidine on the 5-HT system.
Our experiment demonstrated a potential way to inhibit platelet dysfunction resulting from the dysregulation of 5-HT. After trimetazidine pretreatment, serum and platelet 5-HT rose, reversing the declines seen in saline-treated rats. This suggests that trimetazidine has a regulatory effect on 5-HT levels, and that an appropriate increase in serum 5-HT reduces stress on serum 5-HT levels of consumption, increases the storage capacity of platelet 5-HT, and improves the regulatory 5-HT balance inside and outside cells. An appropriate amount of 5-HT elevation therefore not only does not cause platelet aggregation, but also has a protective effect on platelets, demonstrates a protective effect in the tiny blood vessels of the heart, and, since it also induces the inverse of 5-HT trends seen in depression, may prevent MI and depression.
As Brenner et al’s15 study demonstrated, with 5-HT at a low level, SERT transport function was affected, but as serum and platelet SERT density increased, the reuptake of 5-HT also increased to maintain a balance of 5-HT intracellularly. However, with trimetazidine, the rats of the trimetazidine group had higher levels of 5-HT in serum and platelets. At this point, serum and platelet SERT levels appeared to rise. This could have been due to the need for an increased concentration of free serous SERT in order to accommodate transport of the increased 5-HT concentration. Platelet SERT had a downward trend, as the Brenner study suggested it should. With the high 5-HT level, platelet SERT density decreased, and the serum transport of 5-HT also decreased, preventing excessive increase in 5-HT. This maintained the intracellular 5-HT balance, which protects small blood vessels: Hervig and Farstad40 found that platelets with a high endogenous 5-HT content could preserve function better during storage.
In addition, due to the special form of 5-HT and adenosine triphosphate (ATP) in platelets, we speculate that this is related with the pharmacological effect of trimetazidine in treating MI and/or depression. 5-HT forms a chemical complex with ATP and one to two molecules of 5-HT form one molecule of ATP in platelet-dense granules. The levels of ATP in the periphery increased following the release of 5-HT from platelets.41,42 The increased ATP would be a protective factor in maintaining platelet function and coronary artery function, which is consistent with a similar theory that inhibiting the 3-KAT enzyme to increase myocardial ATP in the heart would protect the myocardium. However, we still need further experiments to prove it.
In summary, we hypothesized that pretreatment could increase the 5-HT reserve in advance, and increase the reactivity of platelet 5-HT. This way, the platelets could maintain a high level of 5-HT after MI. In accordance with these expectations, we found that levels of 5-HT in the serum and platelets decreased sharply in injury models, but that serum 5-HT increased after treatment of trimetazidine. This would repair injured blood vessels. Meanwhile, the level of platelet 5-HT in the trimetazidine group was still lower than that of the saline group. A possible mechanistic account of this is as follows: 1) platelets continue to release the right amount of serum 5-HT in order to help the body to recover; 2) platelet SERT and the 5-HT reuptake rate decrease, platelet 5-HT binding increases, and/or platelet 5-HT storage capacity decreases. It seems that, for the purpose of avoiding an increased risk of MI, platelet and plasma 5-HT maintain balance, thereby avoiding an excessive increase in 5-HT-mediated platelet aggregation.
Limitations
There are, however, some limitations to the inductive potential of our experiment. These include the small sample size, short duration, and the homogeneous doses of trimetazidine given. It would be useful to vary these, both to better resemble the temporality of depression and CHD in the clinical setting, as well as to construct dose–response curves for the effects we observed. Also, we need to consider more variables, such as genetic variations of 5-HT and SERT gene expression and polymorphisms, using more advanced methodology. Broadening our account of the mechanism we studied might help to reveal relationships between it and other proposed mechanisms, such as the inflammatory response common to depression and CHD, among others.5,33,43 Finally, our results would benefit from translational follow-up. We aim to provide such follow-up through the study of patients with MI + depression in the clinic. Our hope is that doing so will allow us to discover the potential effect of trimetazidine for treating the CHD and depression comorbidity to fruition in the clinical setting.
Conclusion
Trimetazidine has been used clinically as long-term treatment of myocardial ischemia, but there is currently a lack of in-depth understanding of the mechanism by which it works for this purpose. In this experiment, we made progress toward enhancing this understanding, observing that trimetazidine exerts a regulatory effect on rat serum and platelet 5-HT and SERT. This result suggests that trimetazidine can regulate the 5-HT system, which is associated with protective effects on coronary small blood vessels and microcirculation. Therefore, one important potential implication of our study is that trimetazidine can play a protective role in MI, in both posttreatment and pretreatment settings. Another important implication concerns depression. If the 5-HT system is indeed a link between the observed coincidence of CHD and depression, then the normalizing effect of trimetazidine would presumably diminish the impact of both diseases simultaneously. As the summary of the massive burden imposed by both of these diseases with which we introduced our paper illustrates, such a diminution would reverberate both powerfully and ubiquitously.
Acknowledgments
Thanks go to Yingbin Ge, who made several contributions to animal experiments, and Xiangjun Zhai and Liguo Zhu who contributed to the statistical analysis.
Disclosure
The authors report no conflicts of interest in this work.
References
Miller G, Chen E, Cole SW. Health psychology: developing biologically plausible models linking the social world and physical health. Annu Rev Psychol. 2009;60:501–524. | ||
Russ TC, Stamatakis E, Hamer M, Starr JM, Kivimäki M, Batty GD. Association between psychological distress and mortality: individual participant pooled analysis of 10 prospective cohort studies. BMJ. 2012;345:e4933. | ||
Cohen S, Gianaros PJ, Manuck SB. A stage model of stress and disease. Perspect Psychol Sci. 2016;11:456–463. | ||
World Health Organization. Cardiovascular diseases (CVD) factsheet. Reviewed September 2016. Available from: http://www.who.int/mediacentre/factsheets/fs317/en/. Accessed May, 2017. | ||
Carney RM, Freedland KE, Miller GE, Jaffe AS. Depression as a risk factor for cardiac mortality and morbidity: a review of potential mechanisms. J Psychosom Res. 2002;53:897–902. | ||
Benyamini Y, Roziner I, Goldbourt U, Drory Y, Gerber Y; Israel Study Group on First Acute Myocardial Infarction. Depression and anxiety following myocardial infarction and their inverse associations with future health behaviors and quality of life. Ann Behav Med. 2013;46:310–321. | ||
Williams MS. Platelets and depression in cardiovascular disease: a brief review of the current literature. World J Psychiatry. 2012;2:114–123. | ||
Freyburger WA, Graham BE, Rapport MM, et al. The pharmacology of 5-hydroxytryptamine (serotonin). J Pharmacol Exp Ther. 1952;105:80–86. | ||
Saldanha BD, Kumar MN, Ryali SCV, Srivastava K, Pawar AA. Serum serotonin abnormality in depression. Med J Armed Forces India. 2009;65:108–112. | ||
Ni W, Watts SW. 5-hydroxytryptamine in the cardiovascular system: focus on the serotonin transporter (SERT). Clin Exp Pharmacol Physiol. 2006;33:575–583. | ||
Linder AE, Ni W, Szasz T, et al. A serotonergic system in veins: serotonin transporter-independent uptake. J Pharmacol Exp Ther. 2008;325:714–722. | ||
Bianchi M, Moser C, Lazzarini C, Vecchiato E, Crespi F. Forced swimming test and fluoxetine treatment: in vivo evidence that peripheral 5-HT in rat platelet-rich plasma mirrors cerebral extracellular 5-HT levels, whilst 5-HT in isolated platelets mirrors neuronal 5-HT changes. Exp Brain Res. 2002;143:191–197. | ||
Akbay E, Onur MA. A new modified myocardial infarction animal model. Cardiovasc Surg. 2013;1:69–71. | ||
Nemeroff CB, Knight DL, Franks J, Craighead WE, Krishnan KR. Further studies on platelet serotonin transporter binding in depression. Am J Psychiatry. 1994;151:1623–1625. | ||
Brenner B, Harney JT, Ahmed BA, et al. Plasma serotonin levels and the platelet serotonin transporter. J Neurochem. 2007;102:206–215. | ||
Sauer WH, Berlin JA, Kimmel SE. Effect of antidepressants and their relative affinity for the serotonin transporter on the risk of myocardial infarction. Circulation. 2003;108:32–36. | ||
Artigas F. Future directions for serotonin and antidepressants. ACS Chem Neurosci. 2013;4:5–8. | ||
Wei CY, Huang ZY, Wang LN, et al. Effects of different intervention occasion of trimetazidine on isoproterenol-induced cardiac hypertrophy in rats. Pract Clin Med. 2010;11:4–7. | ||
Nowak P, Zagzi T, Konecki J, et al. Trimetazidine increases [3H]glucose uptake in rat brain. Pharmacol Rep. 2006;58:559–561. | ||
Porsolt RD, Bertin A, Jalfre M. Behavioural despair in mice: a primary screening test for antidepressants. Arch Int Pharmacodyn Ther. 1977;229:327–336. | ||
Carr GV, Lucki I. The role of serotonin receptor subtypes in treating depression: a review of animal studies. Psychopharmacology. 2011;213:265–287. | ||
Akbay E, Onur MA. A new modified myocardial infarction animal model. Cardiovasc Surg. 2013;1:69–71. | ||
Le Quan-Bui KH, Plaisant O, Leboyer M, et al. Reduced platelet serotonin in depression. Psychiatry Res. 1984;13:129–139. | ||
Takahashi S. Reduction of blood platelet serotonin levels in manic and depressed patients. Folia Psychiatr Neurol Jpn. 1976;30:476–486. | ||
Liu MY, Ren YP, Zhang LJ, Ding JY. Pretreatment with ginseng fruit saponins affects serotonin expression in an experimental comorbidity model of myocardial infarction and depression. Aging Dis. 2016;7:680–686. | ||
Saxena PR, Villalla CM. 5-Hydroxytryptamine: a chameleon in the heart. Trends Pharmacol Sci. 1991;12:223–227. | ||
Chester AH, Martin GR, Bodelsson M, et al. 5-Hydroxytryptamine receptor profile in healthy and diseased human epicardial coronary arteries. Circ Res. 1990;24:932–937. | ||
Cocks TM, Angus JA. Endothelium-dependent relaxation of coronary arteries by noradrenaline and serotonin. Nature. 1983;305:627–630. | ||
Golino P, Piscione F, Benedict CR, et al. Local effect of serotonin released during coronary angioplasty. N Engl J Med. 1994;330:523–528. | ||
Young LW, Darios ES, Watts SW. An immunohistochemical analysis of SERT in the blood–brain barrier of the male rat brain. Histochem Cell Biol. 2015;144:321–329. | ||
Meltzer HY, Arora RC, Baber R, Tricou BJ. Serotonin uptake in blood platelets of psychiatric patients. Arch Gen Psychiatry. 1982;38:1322–1326. | ||
Cerrito E, Lazzaro MP, Gaudio E, Arminio P, Aloisi G. 5HT2-receptors and serotonin release: their role in human platelet aggregation. Life Sci. 1993;53:209–215. | ||
Panagiotakos DB, Pitsavos C, Chrysohoou C, et al. Inflammation, coagulation, and depressive symptomatology in cardiovascular disease-free people; the ATTICA study. Eur Heart J. 2004;25:492–499. | ||
Sanner JE, Frazier L, Udtha M. The role of platelet serotonin and depression in the acute coronary syndrome population. Yale J Biol Med. 2013;86:5–13. | ||
Zhou X, Chen J. Is treatment with trimetazidine beneficial in patients with chronic heart failure? PLoS One. 2014;9:e94660. | ||
Kantor PF, Lucien A, Kozak R, Lopaschuk GD. The antianginal drug trimetazidine shifts cardiac energy metabolism from fatty acid oxidation to glucose oxidation by inhibiting mitochondrial long-chain 3-ketoacyl coenzyme A thiolase. Circ Res. 2000;86:580–588. | ||
Shirahase H, Suzuki Y, Osumi S, Kakeya N, Kurahashi K. Inhibitory effects of trimetazidine dihydrochloride on aggregation, serotonin release and malondialdehyde production in rabbit platelets. Jpn J Pharmacol. 1988;47:29–34. | ||
Shimbo D, Child J, Davidson K, et al. Exaggerated serotonin-mediated platelet reactivity as a possible link in depression and acute coronary syndromes. Am J Cardiol. 2002;89:331–333. | ||
Liu MY, Ren YP, Wei WL, Tian GX, Li G. Changes of serotonin (5-HT), 5-HT2A receptor, and 5-HT transporter in the Sprague-Dawley rats of depression, myocardial infarction and myocardial infarction co-exist with depression. Chin Med J (Engl). 2015;128:1905–1909. | ||
Hervig TA, Farstad M. Human blood platelet serotonin studied in vitro: endogenous serotonin may stimulate thrombin-induced serotonin release in stored platelets. Platelets. 1996;7:53–57. | ||
Ruggiero M, Zimmerman TP, Lapetina EG. ATP depletion in human platelets caused by permeabilization with saponin does not prevent serotonin secretion induced by collagen. Biochem Biophys Res Commun. 1985;131:620–627. | ||
Born GV, Gillson RE. Studies on the uptake of 5-hydroxytryptamine by blood platelets. J Physiol. 1959;146:472–491. | ||
Harrison NA, Cooper E, Voon V, Miles K, Critchley HD. Central autonomic network mediates cardiovascular responses to acute inflammation: relevance to increased cardiovascular risk in depression? Brain Behav Immun. 2013;100:189–196. |
 © 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.