")
Back to Journals » Vascular Health and Risk Management » Volume 16
Barriers to Early Diagnosis and Treatment of Familial Hypercholesterolemia: Current Perspectives on Improving Patient Care
Authors Alonso R, Perez de Isla L , Muñiz-Grijalvo O, Mata P
Received 5 November 2019
Accepted for publication 28 December 2019
Published 9 January 2020 Volume 2020:16 Pages 11—25
DOI https://doi.org/10.2147/VHRM.S192401
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Daniel Duprez
Rodrigo Alonso,1,2 Leopoldo Perez de Isla,3 Ovidio Muñiz-Grijalvo,4 Pedro Mata2
1Department of Nutrition, Clínica Las Condes, Santiago, Chile; 2Fundación Hipercolesterolemia Familiar, Madrid, Spain; 3Department of Cardiology, Hospital San Carlos, Madrid, Spain; 4Internal Medicine Department, Hospital Virgen del Rocío, Sevilla, Spain
Correspondence: Pedro Mata
Fundación Hipercolesterolemia Familiar, Gral Alvarez De Castro 14, 1-E, Madrid 28010, Spain
Tel +34 915042206
Email [email protected]
Abstract: Familial hypercholesterolemia (FH) is a frequent disorder associated with premature atherosclerotic cardiovascular disease. Different clinical diagnosis criteria are available, and cost of genetic testing has been reduced in the last years; however, most cases are not diagnosed worldwide. Patients with FH are at high cardiovascular risk and the risk can be reduced with lifelong lifestyle and pharmacological treatment. Statins and ezetimibe are available as generic drugs in most countries reducing the cost of treatment. However, the use of high-intensity statins combined with ezetimibe and PCSK9 inhibitors, if necessary, is low for different reasons that contribute to a high number of patients not reaching LDL-C targets according to guidelines. On the other hand, cardiovascular risk varies greatly in families with FH; therefore, risk stratification strategies including cardiovascular imaging is another element to consider for improving care and management of FH. There are numerous barriers depending on the awareness, knowledge, perception of risk, management and care of patients living with FH that impact in the diagnosis and treatment of the disorder. In this contemporary review, we analyze different barriers in the diagnosis and care of patients to improve patients’ care and prevention of atherosclerotic cardiovascular disease and describe recent advances and strategies to improve the gaps in the care of FH, including global collaboration and advocacy.
Keywords: familial hypercholesterolemia, statins, early detection, iPCSK9, genetic testing, screening strategies, cardiovascular risk
Introduction
Heterozygous familial hypercholesterolemia (FH) is one of the most common monogenic disorders associated with premature atherosclerotic cardiovascular disease (ASCVD), but it remains substantially underdiagnosed.1 If identified and not treated, convey a risk of early onset ASCVD of 3- to 10-fold compared to the general population.2 It is caused by pathogenic variants involving genes related to low-density lipoprotein (LDL) clearance, like the LDL receptor gene (LDLR) in 70–90% of cases, and less frequently in apolipoprotein B-100 (APOB), and proprotein convertase subtilisin/kexin type 9 genes (PCSK9).1
Familial hypercholesterolemia is not a rare disease. The current prevalence is estimated in 1 in 250 individuals in the general population.1,3 In patients with acute coronary syndrome, the prevalence of genetically confirmed FH is 9% in those cases ≤65 years and with LDL-Cholesterol (LDL-C) levels ≥160 mg/dL,4 increasing to 14% in those below 45 years. The estimated prevalence is similar using DNA-based diagnosis or the Dutch Lipid Clinic Network (DLCN) score criteria.5
Due to lifetime exposure to elevated LDL-C levels since birth, patients with FH may suffer from coronary artery disease since the third decade of life. Data from the Spanish FH registry showed that mean age for first CV event was 42 years for men and 51 years for women, being myocardial infarction the most frequent event in males.6 However, most patients are unaware of their FH condition, and the diagnosis is frequently done late in life or after the first cardiovascular event.7
The prognosis for patients with FH has improved in the last 30 years due to the availability of effective lipid-lowering treatments (LLT) with statins and ezetimibe,8 and recently with the development and availability of PCSK9 inhibitors that have proven to be very effective and safe in FH.9,10 Cardiovascular risk was reduced up to 79% in those patients with FH and no history of CAD treated with statins compared to not-treated patients.8
General Barriers
There are several barriers related to early diagnosis and treatment of FH patients (Box 1). Less than 1% of cases has been diagnosed in most countries, being the detection rate higher in those countries that have developed screening programs.1 Current efforts are focused on finding individuals with FH through opportunistic screening in high-risk patients, and cascade screening using LDL-C levels and/or genetic testing.11–13 Most severe FH cases are usually identified in specialist care setting, in coronary or intensive care unit or in lipid clinics, whereas less severe phenotypes are usually seen in primary care. Therefore, knowledge and awareness of the disorder and how to identify a potential FH case is essential in primary care.
 |
Box 1 Some Known Barriers to Early Diagnosis and Management of Familial Hypercholesterolemia |
Many individuals and family members with FH suffering ASCVD have other common risk factors and genetic hypercholesterolemia is not always suspected. Regarding treatment, a significant proportion of patients are treated with low or moderate statin doses and combination treatment is used too sparingly; therefore, many patients do not reach recommended LDL-C targets.14,15 Moreover, patients and physicians must understand that treatment is lifelong and therefore, adherence to chronic LLT recommendations must be improved. Therapy is often started in the late stages of disease when atherosclerosis has already developed as a result of life-long high LDL-C concentrations. Strategies for improving early diagnosis and care of FH among physicians require adequate knowledge, awareness and appropriate practices concerning this condition. Different studies have shown that there are several gaps in the knowledge, perception and practice in FH among general practitioners from different countries. Less than 50% are familiar with the disorder, most of them do not know the prevalence of the disorder and the majority underestimate the increased cardiovascular risk.16–18 Most of the physicians recognize statins to best treat hypercholesterolemia, and between 30% and 70% recognized statin and ezetimibe combination as an alternative to treat severe hypercholesterolemia.16,17 Finally, health care systems are not sufficiently aware of the problem and there is a lack of screening programs.
From the point of view of public health, the best strategy for covering this gap in diagnosis and treatment of FH is the implementation of a family-based cascade screening program. This process consists of diagnosing FH in the family members of an individual, the index case (IC), identified as having FH ideally with genetic confirmation.1,12
Clinical Diagnosis and Genetic Testing
Familial hypercholesterolemia should be suspected in all adult with a total cholesterol level >300 mg/dL or an LDL-C level >190 or 220 mg/dL depending on the guidelines (Figure 1, Table 1), or in those individuals presenting with atherosclerotic cardiovascular events before age 60 years, and/or in those with physical signs like xanthomas or premature arcus cornealis.1,12,19 In children and adolescents, the diagnosis should be suspected when LDL-C is over 150 mg/dL12 or 160 mg/dL.19 Then, family history should be investigated. To make the phenotypic diagnosis of FH, there are different criteria worldwide. The most used are the DLCN criteria assigning a score to different variables like LDL-C levels, family history of hypercholesterolemia in children and adults, history of premature ASCVD in the proband and relatives, and the presence of tendon xanthomas and/or arcus cornealis before age 45 years.20 According to the final score, the diagnosis can be probable (6–7) or certain (≥8). It should be highlighted that the use of the DLCN criteria must be used only in adults and require to know the family history.
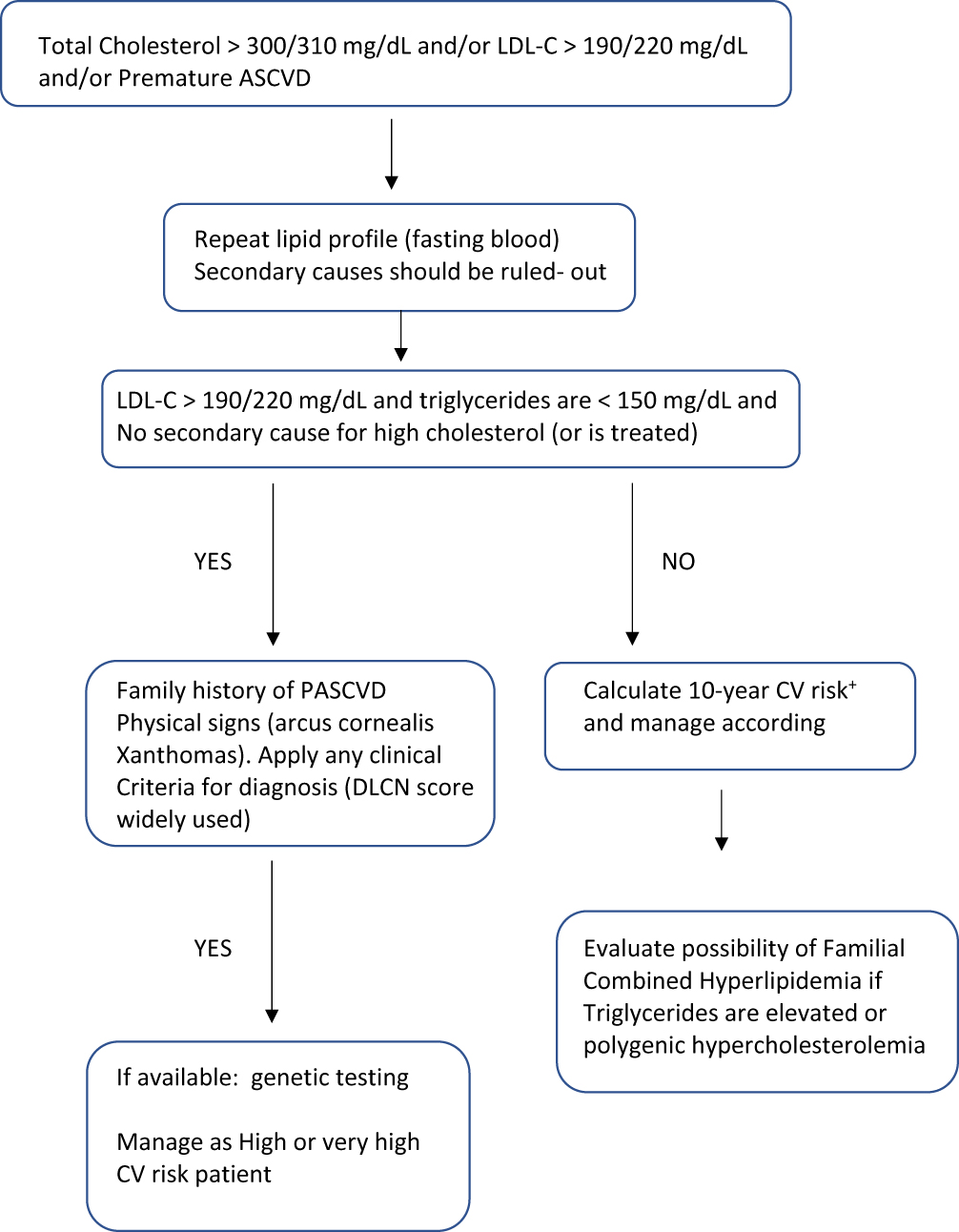 |
Figure 1 Attitude towards an adult with suspected familial Hypercholesterolemia according to different guidelines. |
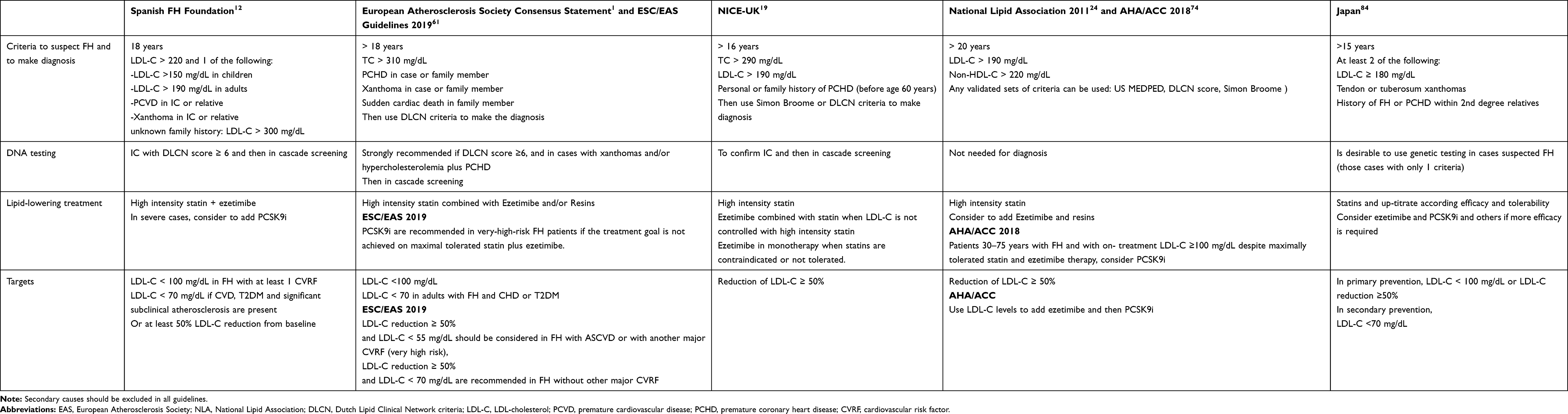 |
Table 1 Criteria for Familial Hypercholesterolemia in Adults, Screening, Lipid-Lowering Treatment and LDL-C Targets According to Different Guidelines or Consensus Panels |
Familial Hypercholesterolemia meets the WHO criteria for systematic disease screening: it is a common disorder, the etiology is well known, it causes premature cardiovascular morbidity, there are criteria for the diagnosis and there are different effective treatments.1
Genetic testing for FH is a Tier 1 genomic application, defined by the Center for disease control and prevention as having significant potential for positive impact on public health based on available evidence-based guidelines and recommendations. Tier 1-related interventions include joint work of public health department with medical providers and payers, in order to screen FH through cascade screening using cholesterol levels with or without DNA testing, identifying new cases and start treatment as early as possible before ASCVD is developed.21 Despite FH is a well-known genetic disorder, genetic testing is underused. Genetic testing provides a definitive diagnosis of the disorder, helps to stratify the risk and improves cascade screening.13 Recently, a scientific expert statement described the limitations and risks of genetic testing highlighting potential discrimination by care insurances in the United States.13
Recently, a systematic review of the literature using a consolidated framework for implementation research (CFIR) identified 26 potential barriers and 15 potential facilitators to FH genetic testing in the United States.22 The cost of FH genetic testing uncovered by insurance payers, stigmatization or discrimination by health insurers, privacy over genetic information among others were prevalent barriers identified in this analysis. This systematic and flexible analysis can guide future research to understand FH genetic testing implementation in diverse clinical settings and identify strategies to reduce the number of barriers and leverage current facilitators for genetic testing.
Screening Strategies
Low awareness and knowledge of FH in specialty and general practice highlight the need to implement different strategies to improve the early detection of FH and ensure adequate treatment to prevent the development of ASCVD, improve quality of life and care of patients with FH.
Universal Screening
Universal screening (US) program means that all individuals in a defined population would be offered a blood test to measure total cholesterol and/or LDL-C levels. Those individuals with cholesterol levels above a defined cut-off level should be considered at high risk of FH and can be referred for a confirmatory diagnostic assessment by a GP or lipid specialist, which may include DNA testing if available. The benefits of this strategy are associated with an increased detection rate in the population, and individuals diagnosed with FH through the program would also represent new index cases, supporting a more effective cascade screening approach to identify other family members. In this sense, universal screening during childhood would maximize the early detection and treatment before atherosclerosis has been established. Universal screening (US) has been proposed to be performed before age 20 years, ideally before puberty. Although the efficacy has been demonstrated in children, the experience is very limited,23 and the yield of the screening in efficacy and cost-effectiveness remains unclear. The National Lipid Association recommended in 2011 that US should be carried out at age 9–11 years when lipid changes associated with puberty are less evident and statin therapy can be started.24 Feasibility of US at age 1–2 years has been demonstrated by Wald et al, through capillary cholesterol measurement during routine immunization in children combined with reverse cascade screening of parents. This has been shown to be a feasible and effective method to detect familial hypercholesterolemia. For every 1000 children screened, 4 children and 4 parents with FH and with high cardiovascular risk can be detected.25 Recently, McKay et al showed that US with cholesterol levels followed by genetic testing and reverse cascade screening is the most cost-effective modelled compared to other strategies at age 1 to 2 years of age in the United Kingdom. Incorporation of lipid profile measurement into routine child healthcare appointment reduces additional healthcare costs and user inconvenience.26 Role of parents in the screening and diagnosis of children is essential because they must consent prior to any strategy. Knowledge and awareness about the importance of early diagnosis and treatment, safety concern of long-term statin treatment started in childhood, potential psychological issues and the right not to know are important factors to consider. In the study of Wald et al,25 84% of parents of 13,097 children invited to participate in a universal screening accepted the offer of screening, suggesting that this approach was acceptable to most families. Regarding psychological effects of genetic diagnosis, the European Consensus Statement recommended that they must be considered, with pretest counselling to parents being essential to the consent/assent procedure, and should take account of parental literacy.27 Other limitations of US include cost and resource consumption, and some ethical issues like insurance implications.28
Opportunistic Screening
Opportunistic detection is another approach that can be implemented in different health care levels. Primary care physicians should be involved actively in the screening and care of FH. One of the barriers to patients’ care is the degree of knowledge that primary care physicians have about the disorder.
The first study of the knowledge and management practices by General practitioners was performed in Australia in 2014.29 Bell et al showed that 62% of GPs rated their familiarity with FH as average or above, and 80% correctly defined FH; however, awareness of national guidelines for diagnosis and management, and knowledge in hereditability, prevalence and diagnostic criteria were suboptimal (<50%). Similar gaps have been described later in other regions of the world and also among cardiologists and other health practitioners like pharmacists.30,31 A survey performed among Cardiologists in 2011 showed that knowledge about FH was also limited, only 10% reported to be very or extremely familiar with the disorder, and most of them (80%) were unaware of the real prevalence, 60% about heritability and all of them did not know about the real high cardiovascular risk.30 A large-scale multinational survey assessing FH knowledge and management gaps was performed in 10 countries from the Asia-Pacific region and results were compared with data from the UK where specific guideline for the care of FH patients exists.17 Important gaps in awareness and knowledge were identified. Only 34% considered to be familiar with FH and less than 50% were aware of national or international guidelines. Knowledge of prevalence (24%), inheritability (41%) and CVD risk (9%) of FH were also suboptimal. However, familiarity and knowledge with statins as an appropriate cholesterol-lowering therapy for FH management was very high (89%). Results of this survey suggest that the implementation of country-specific guidelines and the development of educational and awareness programs are imperative to improve the care of FH in the region.17
Strategies for Opportunistic Screening
Detection of the initial individual of a family with FH (index case) is a crucial step for cascade testing. Different approaches like analysis of cholesterol levels in the laboratory, electronic case-finding programs with alerts in laboratory reports, the addition of interpretative commenting, phone-calls, electronic health records, development of predictive diagnostic algorithm and combination of these strategies has been shown to be useful to detect FH cases in different community settings.32–38
The role of community laboratories, where most of the analysis are requested by GPs, to screen for individuals with potential FH has been demonstrated by Bell et al.33
Considering an LDL-C≥251 mg/dL (99.75 percentile) after the exclusion of secondary causes, prevalence of FH was 1:398 irrespective of age, emphasizing the potential of a community laboratory in the detection of FH. Moreover, the addition of an interpretative comment to the lipid results report raising FH as a possible diagnosis associated with a high risk of ASCVD in those individuals with LDL-C ≥251mg/dL recommending a specialist referral increased the number of cases referred to a lipid clinic and the detection of FH within the community.35 However, the overall referral rate is still low (18%) suggesting that knowledge and awareness of FH among GPs is also low, emphasizing the need for an educational program to improve FH detection.
Large numbers of patients at risk of FH can be systematically identified using electronic methods.36–38 Effectiveness of these methods depends in part in the quality of information recorded in databases, and they should be integrated into clinical practice to consume less time and resources. The use of a simple specific software (FH Audit tool) to identify and flagged all subjects at risk of FH according to LDL-C levels and the subsequent nurse-led review of cases at risk, allowed the identification of 433 new cases of FH and reduced the number of cases at risk and unscreened.36
More sophisticated approaches include the development of predictive algorithm (FAMCAT) including more clinical variables interacting among them to enhance the identification of individuals in primary care. Data from 2,971,562 individuals were analysed using the Clinical Practice Research Datalink in UK, and 5050 documented cases of FH were included in the study.37 FAMCAT model significantly improved discrimination between cases and non-cases when compared to model adapted from variables in the DLCN score, in the Simone-Broome and using only total cholesterol. An important clinical implication is that FAMCAT algorithm could be integrated within electronic health records to rank patients about their probability of having FH rationalizing the use of primary care and specialist resources.37
Another study in Australia showed that the combination of a specific electronic screening tool (TARB-ex) estimating DLCN score with clinical follow-up by GP is an efficient and accurate method of systematically identifying FH risk patients, consuming less time and resources, and that strongly correlates with clinical examination.38 Estimating DLCN score from 3708 medical records by TARB-ex took 10 mins and identified 32 patients at risk of FH (1:116) compared to the 60 hrs that took to a trained GP to identify 22 potential cases (1:168) after manual reviewing of 360 records with high cholesterol. Finally, 10 patients were considered at high risk of FH (31%) and after clinical assessment, clinical diagnosis was confirmed in 6 of 7 patients (86% sensitivity).38
Cascade Screening
Screening programs, using cascade testing of first-degree relatives of identified adults (index case) and also the reverse cascade testing of first-degree relatives of affected children, using cholesterol levels and/or genetic testing (if available), have been reported successfully.12,39 These strategies permit the identification of untreated individuals, which are usually younger and free of ASCVD.25,40,41 The sustainability of cascade testing relies on identifying new unrelated index cases. Different studies have shown that the most cost-effective approach for detecting new cases of FH is cascade screening of relatives of a diagnosed index case using lipid levels and genetic testing.42,43
Detection based on genetic testing can establish the definitive diagnosis if a pathogenic variant is found in any of the genes causative of FH. If DNA-testing is not available, the cascade should be done using country-specific cut-off points of LDL-C plasma levels.1,12,39 However, it should be emphasized that using only cholesterol levels, up to 20% of relatives with LDL-C levels below the 90th percentile, may have a causative mutation in the LDLR gene and will not be diagnosed if genetic testing is not performed.44
Possible FH cases should be considered in those subjects with a history of premature cardiovascular disease or are admitted to a hospital with an acute cardiovascular event before age 60 years.3,4
Receiving a medical diagnosis or genetic confirmation of FH empowered individuals to take control of their condition, providing motivation to initiate or continue lifestyle changes and LLT. The positive influence of diagnosis on medication efficacy beliefs and adherence has been reported.45
Cardiovascular Risk Assessment in FH
Patients with FH are considered to be at high or very high ASCVD risk; however, a considerable number of these vulnerable subjects do not develop cardiovascular events, regardless lifelong exposure to very high LDL-C levels, while others do so despite intensive LLT, suggesting that the ASCVD risk is very variable in this population. Khera et al showed that for any observed LDL-C level, individuals with molecular defined FH have four times increased risk for CAD compared with those individuals without a mutation.46 FH accelerates atherosclerotic coronary disease by 10 to 40 years.38 The analysis of different cohorts has shown a risk for CAD or premature ASCVD 3 to 13 times higher in familial hypercholesterolemia subjects compared with their unaffected relatives or with the general population.47–49 That variability may be due in part to differences in lipid-lowering treatment. However, the individual risk for ASCVD can vary widely in FH patients belonging to the same family or sharing the same mutation,50,51 suggesting that other genetic, environmental or risk factors can modulate the risk. Some approaches have been made to re-stratify the risk in these individuals in order to improve and intensify lipid-lowering therapy with the use of PCSK9i.52,53 Recently, in the SAFEHEART Study (Spanish Familial Hypercholesterolemia Cohort Study), the most comprehensive registry-based prospective study to date, a pragmatic equation was developed using age, sex, history of ASCVD, blood pressure, body-mass index, smoking, LDL-C, and Lipoprotein(a) [Lp(a)] levels that predicts with a high discriminant value of first and recurrent ASCVD events.52 This model was derived from a well-defined, adult cohort already on statin therapy and although requires validation in other populations, it has demonstrated superior in predicting ASCVD in FH patients compared to Framingham and ACC/AHA risk prediction tools.54 This accurate prediction equation (SAFEHEART-RE) provides a global approach to the ASCVD risk and LDL-C remains as an important predictor of the risk, and also Lp(a), confirming the association between high Lp(a) levels and ASCVD in familial hypercholesterolemia.55 Its use in clinical practice in primary and specialist care settings may increase the efficiency in patients care and also optimize the selection of patients for a rational use of the new PCSK9i.56
In recent years, substantial information has emerged regarding the role of Lp(a) as a predictor of risk in FH. Individuals with FH have higher levels of serum Lp(a) compared with their non-affected relatives and an independent association with the type of mutation has been described.55,57 Moreover, Lp(a) is a strong predictor for CAD in FH patients independent of other cardiovascular risk factors and it is recommended to measure Lp(a) once in those individuals with FH.57 Effectiveness of cascade screening for high Lp(a) levels alone has not been assessed and established. However, Lp(a) is an heritable risk factor transmitted in a dominant manner, and the testing of relatives of a patient with very high Lp(a) levels may be useful. It has been shown that systematic testing for elevated Lp(a) during cascade screening for FH in relatives of index cases with FH and high Lp(a) is highly effective identifying 1 new case of elevated Lp(a) for every 2.4 relatives screened.58
Role of Imaging in CV Risk Re-Stratification
In recent years, emerging studies have suggested a large variation in the presence and distribution of coronary artery calcium (CAC), a surrogate marker of atherosclerotic plaque burden in FH. Therefore, assessment of subclinical atherosclerosis may be a valuable clinical tool for determining the ASCVD risk in asymptomatic FH patients.59,60 It has been recommended as a key modifier that allows patients to be re-classified from high to very-high cardiovascular disease risk with subsequent implications in therapy.61
The presence and degree of CAC predicts coronary events in asymptomatic, middle-aged FH patients treated with statins and this is partly mediated by associations with other risk factors, particularly the burden of LDL-C-years.54,55 Moreover, the extent of plaque burden on computed tomography coronary angiography (CTA) in FH is also related to the number of circulating microvesicles.62 Abnormal Coronary CTA findings are associated with favorable changes in lifestyle, intensification of lipid-lowering therapy and patient behavior.63
Therefore, it has been suggested that the screening of subclinical coronary atherosclerosis in FH individuals may be integrated into their overall care, and will permit to identify those cases that will require intensification of lifestyle changes and additional medical therapies.1,63
Carotid intima media thickness (IMT) has also been reported to be significantly thicker in children with FH compared with their unaffected siblings and non-FH children,64 and in adults with FH compared with polygenic hypercholesterolemia.65 Carotid IMT is related to gender, LDL-C levels, age, family history of CVD and the type of mutation. Carotid plaques can also be found in 10% of FH children.64 Therefore, carotid IMT is a feasible method for detecting subclinical atherosclerosis in FH and also to evaluate the impact of statin treatment on progression of carotid atherosclerosis.66
Treatment of Adults Living with FH
LDL-C Targets and Therapeutic Options in FH (Table 1)
In the last years, LDL-C targets in FH have undergone major changes. The consensus statement of the European Atherosclerosis Society in 2013 recommended LDL-C targets below 70 mg/dL for adults with FH and CAD or diabetes, and below 100 mg/dL for those FH cases without CAD or diabetes.1 Other guidelines also recommended a relative reduction in LDL-C levels of at least 50% from baseline values.12,19,39 Recently, the 2019 ESC/EAS guidelines recommended an LDL-C relative reduction of ≥50% from baseline values and an absolute LDL-C goal <55 mg/dL for FH patients with ASCVD (class I, level of evidence A), and the same targets should be considered in primary prevention FH patients with another major CV risk (class IIa, level of evidence C). In those primary prevention FH individuals without other CV risk factor, an LDL-C reduction of ≥50% and an LDL-C goal <70mg/dL are recommended (class I, level of evidence A).61
All patients should be counselled on lifestyle changes, especially dietary modification, promotion of physical activity and avoidance and cessation of smoking. FH patients usually have healthier habits compared to their non-affected relatives. They eat less saturated fats and sugar, and more vegetables and fish. They also do more physical activity and smoke less.67,68
The cornerstone in FH therapy is the use of high-intensity statin and ezetimibe. Both have been demonstrated to be effective and safe in adolescents and adults with FH.69,70
However, in the real-world clinical practice, recommended targets are difficult to achieve in most FH patients with common LLT (statin and ezetimibe) available in most countries as generic drugs. A recent study showed that less than 5% of FH patients with ASCVD receiving LLT to reduce LDL-C levels at least 50% achieved an absolute LDL-C level below 70 mg/dL.71 In this scenario, most of patients with FH could potentially require PCSK9i to achieve the targets. It has been shown that alirocumab and evolocumab are well tolerated and yield an additional 50% to 60% reduction in LDL-C levels in patients with FH under LLT.9,10 However, the high price of PCSK9i has forced to do cost-effectiveness studies, consensus statements, and attempts to identify those patients who will benefit most from these more intensive treatments.56,72,73 In Norway, an economic evaluation study showed that PCSK9i are cost-effective for some groups of molecularly diagnosed FH patients considering age, gender, statin tolerance and history of ASCVD.72 In Spain, it has been shown that using the SAFEHEART risk equation the lowest number necessary to treat to avoid a cardiovascular event (N=12) is among those FH patients receiving high-intensity statin with 5-year risk of ≥5% and an LDL-C ≥160 mg/dL.56
Treatment of Children Living with FH (Table 2)
There is an agreement that LLT should be started at younger ages. The risk of accelerated atherosclerosis in FH is the main reason to start statin treatment in early childhood to avoid atherosclerosis progression. The European Atherosclerosis Society (EAS) consensus panel and the current American College of Cardiology/American Heart Association guidelines for FH advocate initiation of statins between 8 and 10 years old.27,74
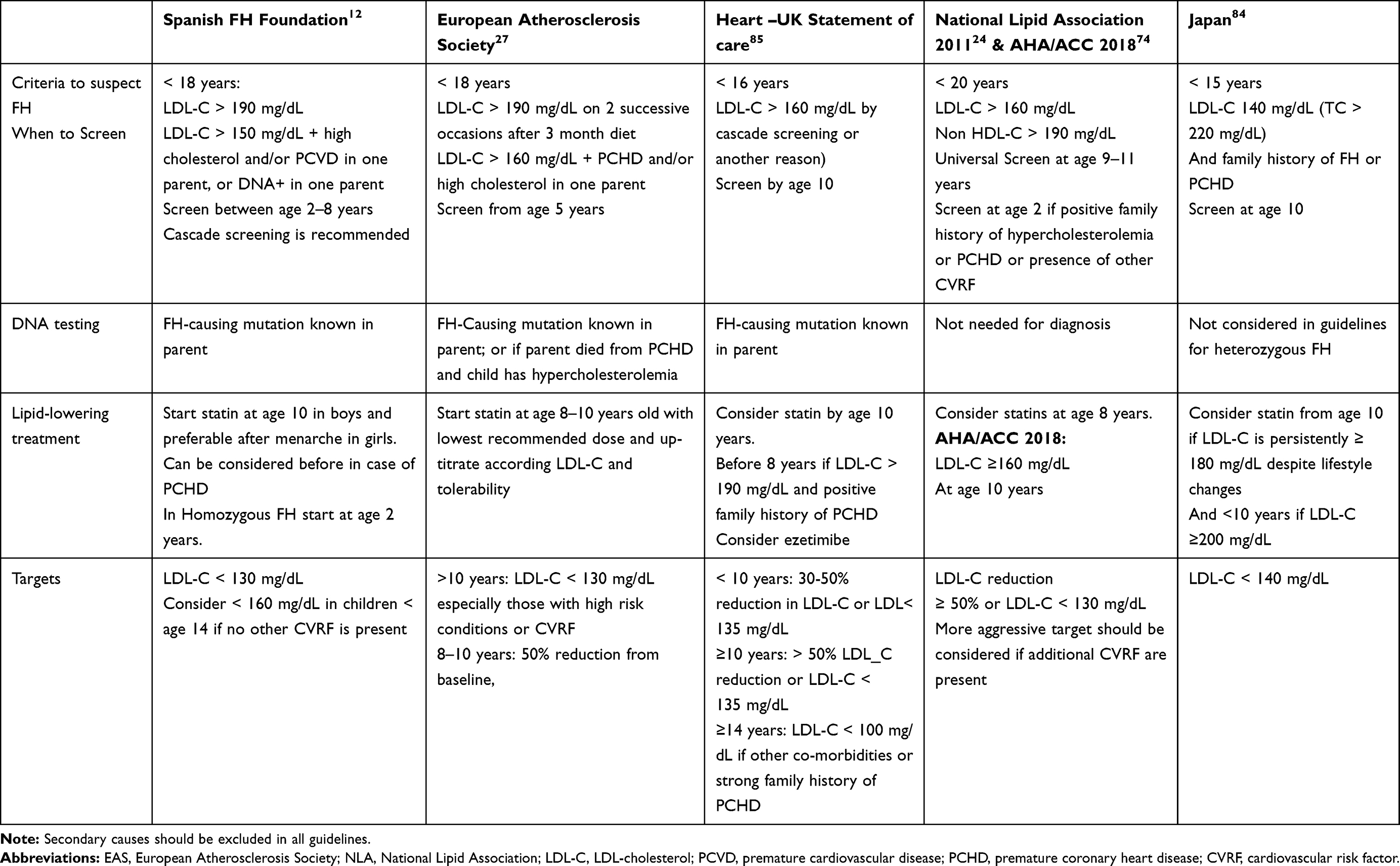 |
Table 2 Criteria for Familial Hypercholesterolemia in Children and Adolescents, Screening, Lipid-Lowering Treatment and LDL-C Targets According to Different Guidelines or Consensus Panels |
Safety and efficacy of statins are well-established in children. There are no concerns regarding the effect of statin on sexual maturity and growth.75,76 On the other hand, the value of initiating statin treatment has been demonstrated initially in carotid intima media thickness (IMT) studies. A mean LDL-C reduction of 41% after 2 years of treatment with rosuvastatin was associated with a significant reduction in the progression of carotid atherosclerosis.77 Moreover, the 10-year follow-up of children with mean age 12 years old treated with pravastatin during 2 years showed that the statin treatment initiated in childhood normalizes the carotid intima media thickness in the young adults (mean age 24 years old).78 Recently, the 20-year follow-up of this population has been published comparing the incidence of ASCVD among these patients that started statins since childhood and their affected parents, assuming that parents started statin treatment later in life. A 32% reduction in LDL-C since baseline in the original trial was observed in this period of time, and carotid IMT in patients was similar to that of their unaffected siblings. Furthermore, the cumulative incidence of fatal and non-fatal CV events at 39 years of age was lower in patients with FH than in their affected parents (1% vs 26%, and 0% vs 7%).79 Therefore, modest and sustained LDL-C reductions since childhood despite not achieving LDL-C targets can have a major impact on reducing ASCVD morbidity and mortality. This is an important issue because only 20% of children reached an LDL-C below 100 mg/dL and most guidelines suggest an LDL-C target below 130 mg/dL or a reduction over 50% from baseline levels (Table 2)
In children and adolescents, the ASCVD risk can be stratified according to the presence and magnitude of risk factors or co-morbidities, such as obesity, hypertension, diabetes, high Lp(a) levels and a family history of premature ASCVD.80 According to the American statement, those children and adolescents with FH are at moderate risk and can be reclassified as high risk if two or more CV risk factors or co-morbidities are present. LDL-C recommended target is <100 mg/dL in the high-risk condition and <130 mg/dL in the moderate-risk condition.80 On the other hand, the 2015 European statement recommended for children aged 8 to 10 years, that LDL-C should be ideally reduced by 50% from pretreatment levels; and for children aged ≥10 years, the target LDL-C should be <130 mg/dL, especially if there are additional cardiovascular risk factors, including elevated Lp(a).27
Patient Networks and Support Groups
Patient support groups and networks may play an important role in FH detection and are essential for advocating value-based health care, especially because healthcare and government policies differ widely across the countries. Sharing experience and networking remain valuable especially for supporting nascent organizations. Beyond providing awareness raising through social media, general education, health professionals counselling, concrete international achievements to date are evidenced by national registries, development of ASCVD risk prediction algorithms, support for new screening strategies and improvements in access to effective therapies. The last role is particularly relevant at a time of access barriers to new and expensive therapies. The value of patient support networks is exemplified by the activities of several organizations like the US FH Foundation, Spanish FH Foundation and the European FH Patient Network.81
Clinical Registries
Clinical registries on FH may improve the knowledge of the disorder and can have an impact on research and precision medicine.82 Registries capture “real world” clinical practice data and these are important not only to raise overall awareness and understanding of FH, but also for gathering information for clinical trials and for health service research, as a mean for improving the quality of patient care and outcomes. Registries of genetically defined patients, such as the SAFEHEART, have generated a wealth of new data that bear on the care of patients with FH.52,55,56 The Familial Hypercholesterolemia Studies Collaboration (FHSC) is an international registry that captures the largest global dataset on FH.83 International registries have provided key information on FH, including the gaps in late diagnosis in those with ASCVD. While the value of statins in diminishing ASCVD has been well emphasized by registry data, there are marked shortfalls in the attainment of treatment targets and in the use of PCSK9 inhibitors. Enrolment in registries may enable attainment of LDL-C targets but significant gaps in treatment persist. The rapid initiation and expansion of clinical registries on FH should take account of expert recommendations on the need to collect high-quality data. Registries may contribute to research for generating new evidence that will inform adaptive models of care and better policy decisions.
Conclusion
Familial Hypercholesterolemia is a frequent and treatable disorder. There is agreement among different guidelines and statements that an early diagnosis when patients are asymptomatic, and adequate treatment is essential to reduce the cardiovascular risk burden and to prevent the development of premature ASCVD. However, there are several barriers on the part of patients, their families, physicians and health systems that must be identified to improve the identification of cases and their long-term treatment and care. Cost and access for medication, long-term adherence to lipid-lowering therapy and lifestyle changes, familiarity of FH and awareness of clinical guidance by general practitioners, low perception of high cardiovascular risk are some of them.
Different detection strategies and models of care have been suggested and implemented in some countries to address the barriers, including patients, their families and physician awareness of FH, highlighting the value of screening for FH and the necessity to follow and adhere to a long-term treatment.
Acknowledgments
The authors thank the Spanish Familial Hypercholesterolemia Foundation team for their great job in improving awareness and knowledge in FH for physicians and families living with familial hypercholesterolemia, and the patients and families participating in the SAFEHEART registry.
Author Contributions
All authors contributed to data analysis, drafting and revising the article, gave final approval of the version to be published, and agree to be accountable for all aspects of the work.
Disclosure
The authors report no conflicts of interest in this work.
References
1. Nordestgaard BG, Chapman MJ, Humphries SE, et al. Familial hypercholesterolaemia is underdiagnosed and undertreated in the general population: guidance for clinicians to prevent coronary heart disease. Eur Heart J. 2013;34:3478–3490. doi:10.1093/eurheartj/eht273
2. Wong B, Kruse G, Kutikova L, Ray K, Mata P, Bruckert E. Cardiovascular disease risk associated with familial hypercholesterolemia: a systematic review of the literature. Clin Ther. 2016;38:1696–1709. doi:10.1016/j.clinthera.2016.05.006
3. Akioyamen LE, Genest J, Shan SD, et al. Estimating the prevalence of heterozygous familial hypercholesterolemia: a systematic review and meta- analysis. BMJ Open. 2017;7:e016461. doi:10.1136/bmjopen-2017-016461
4. Amor-Salamanca A, Castillo S, Gonzlez-Vioque E, et al. Genetically confirmed familial hypercholesterolemia in patients with acute coronary syndrome. J Am Coll Cardiol. 2017;70:1732–1740. doi:10.1016/j.jacc.2017.08.009
5. Kramer AI, Trinder M, Brunham LR. Estimating the prevalence of familial hypercholesterolemia in acute coronary syndrome: a systematic review and meta-analysis. Can J Cardiol. 2019;35:1322–1331. doi:10.1016/j.cjca.2019.06.017
6. Alonso R, Castillo S, Civeria F, et al. Familial Hypercholesterolemia in Spain: description of 819 non-related cases. Med Clin. 2002;118:487–492. doi:10.1016/S0025-7753(02)72428-7
7. deGoma EM, Ahmad ZS, O’Brien EC, et al. Treatment gaps in adults with heterozygous familial hypercholesterolemia in the United States: data from the CASCADE-FH registry, Circ. Cardiovasc Genet. 2016;9:240–249. doi:10.1161/CIRCGENETICS.116.001381
8. Versmissen J, Oosterveer DM, Yazdanpanah M, et al. Efficacy of statins in familial hypercholesterolaemia: a long term cohort study. BMJ. 2008;337:a2423. doi:10.1136/bmj.a2423
9. Hovingh GK, Raal FJ, Dent R, et al. Long-term safety, tolerability and efficacy of evolocumab in patients with heterozygous familial hypercholesterolemia. J Clin Lipidol. 2017;11:1448–1457. doi:10.1016/j.jacl.2017.09.003
10. Kastelein JJ, Hovingh GK, Langslet G, et al. Efficacy and safety of the proprotein convertase subtilisin/kexin type 9 monoclonal antibody alirocumab vs. placebo in patients with heterozygous familial hypercholesterolemia. J Clin Lipidol. 2017;11:195–203. doi:10.1016/j.jacl.2016.12.004
11. Louter L, Defesche J, Roeters van Lennep J. Cascade screening for familial hypercholesterolemia: practical consequences. Atheroscler Suppl. 2017;30:77–85. doi:10.1016/j.atherosclerosissup.2017.05.019
12. Mata P, Alonso R, Ruiz A, et al. Diagnostico y tratamiento de la hipercolesterolemia familiar en Espana: documento de consenso. Aten Primaria. 2014. doi:10.1016/j.aprim.2013.12.015
13. Sturm A, Knowles J, Gidding S, et al. Clinical genetic testing for familial hypercholesterolemia. J Am Coll Cardiol. 2018;72:662–680. doi:10.1016/j.jacc.2018.05.044
14. Hartgers M, Besseling J, Stroes E, et al. Achieved LDL cholesterol levels in patients with heterozygous familial hypercholesterolemia: a model that explores the efficacy of conventional and novel-lipid lowering therapy. J Clin Lipidol. 2018;12:972–980. doi:10.1016/j.jacl.2018.04.002
15. Perez de Isla L, Alonso R, Watts G, et al. Attainment of LDL cholesterol treatment goals in patients with familial hypercholesterolemia: 5-year SAFEHEART registry follow-up. J Am Coll Cardiol. 2016;67:1278–1285. doi:10.1016/j.jacc.2016.01.008
16. Kwok S, Pang J, Adam S, Watts G, Soran H. An online questionnaire survey of UK general practitioners’ knowledge and management of familial hypercholesterolemia. BMJ Open. 2016;6:e012691. doi:10.1136/bmjopen-2016-012691
17. Pang J, Hu M, Lin J, et al. An enquiry based on a standardised questionnaire into knowledge, awareness and preferences concerning the care of familial hypercholesterolaemia among primary care physicians in the Asia-Pacific region: the “Ten Countries Study”. BMJ Open. 2017;7:e017817. doi:10.1136/bmjopen-2017-017817
18. Zimmerman J, Duprez D, McCarthy Veach P, Zierhut H. Barriers to the identification of familial hypercholesterolemia among primary care providers. J Community Genet. 2019;10:229–236. doi:10.1007/s12687-018-0383-3
19. Wierzbicki AS, Humphries SE, Minhas R. Familial hypercholesterolaemia: summary of NICE guidance. BMJ. 2008;337:509–511. doi:10.1136/bmj.a1095
20. World Health Organization. Familial hypercholesterolaemia. Report of a second WHO consultation. Geneva: World Health Organization; 1999.
21. Centers for disease control and prevention, Office of Public Health Genomics. Genomics tests and family health history by levels of evidence: tier 1. Available from: http://www.cdc.gov/genomics/gtesting/tier.htm.
22. Hendrick-Sturrup R, Mazor K, Sturn A, Lu C. Barriers and facilitators to genetic testing for familial hypercholesterolemia in the United States: a review. J Pers Med. 2019;9:32. doi:doi:10.3390/jpm9030032
23. Klančar G, Grošelj U, Kovač J, et al. Universal screening for familial hypercholesterolemia in children. J Am Coll Cardiol. 2015;66:1250–1257. doi:10.1016/j.jacc.2015.07.017
24. Goldberg A, Hopkins P, Toth P, et al. Familial hypercholesterolemia: screening, diagnosis and management of pediatric and adult patients. Clinical guidance from the national lipid association expert panel on familial hypercholesterolemia. Executive summary. J Clin Lipidol. 2011;5:S1–S8. doi:10.1016/j.jacl.2011.04.003
25. Wald D, Bestwick J, Morris J, Whyte K, Jenkins L, Wald N. Child-parent familial hypercholesterolemia screening in primary care. N Engl J Med. 2016;375:1628–1637. doi:10.1056/NEJMoa1602777
26. McKay A, Hogan H, Humphries S, Marks D, Ray K, Miners A. Universal screening at age 1-2 years as an adjunct to cascade testing for familial hypercholesterolemia in the UK: a cost-utility analysis. Atherosclerosis. 2018;275:434–443. doi:10.1016/j.atherosclerosis.2018.05.047
27. Wiegman A, Gidding SS, Watts GF, et al. Familial hypercholesterolaemia in children and adolescents: gaining decades of life by optimizing detection and treatment. Eur Heart J. 2015;36:2425–2437. doi:10.1093/eurheartj/ehv157
28. Kusters DM, de Beaufort C, Widhalm K, et al. Paediatric screening for hypercholesterolaemia in Europe. Arch Dis Child. 2012;97:272–276. doi:10.1136/archdischild-2011-300081
29. Bell D, Garton-Smith J, Vickery A, et al. Familial hypercholesterolaemia in primary care: knowledge and practices among general practitioners in Western Australia. Heart Lung Circ. 2014;23:309–313. doi:10.1016/j.hlc.2013.08.005
30. Foody JM. Familial hypercholesterolemia: an under-recognized but significant concern in cardiology practice. Clin Cardiol. 2014;37:119–125. doi:10.1002/clc.22223
31. Jervis L, Hanson M, Bell D, et al. An audit of pharmacists’ knowledge of familial hypercholesterolaemia: implications for community healthcare. Aust Pharm. 2013;32:73–75.
32. Mirzaee S, Choy KW, Doery JGC, Zaman S, Cameron JD, Nasis A. The tertiary hospital laboratory; a novel avenue of opportunistic screening of familial hypercholesterolemia. Int J Cardiol Heart Vasc. 2019. doi:10.1016/j.ijcha.2019.100354
33. Bell DA, Hooper AJ, Bender R, et al. Opportunistic screening for familial hypercholesterolaemia via a community laboratory. Ann Clin Biochem. 2012;49:534–537. doi:10.1258/acb.2012.012002
34. Bell D, Hooper A, Edwards G, et al. Detecting familial hypercholesterolaemia in the community: impact of a telephone call from a chemical pathologist to the requesting general practitioner. Atherosclerosis. 2014;234:469–472. doi:10.1016/j.atherosclerosis.2014.04.002
35. Bender R, Edwards G, McMahon J, et al. Interpretative comments specifically suggesting specialist referral increase the detection of familial hypercholesterolaemia. Pathology. 2016;48:463–466. doi:10.1016/j.pathol.2016.04.003
36. Green P, Neely D, Humphries S; the Medway FH Audit Steering Committee. Improving detection of familial hypercholesterolemia in primary care using electronic audit and nurse-led clinics. J Eval Clin Pract. 2016;22:341–348. doi:10.1111/jep.12481
37. Weng S, Kai J, Neil HA, Humphries S, Qureshi N. Improving identification of familial hypercholesterolaemia in primary care: derivation and validation of the familial hypercholesterolaemia case ascertainment tool (FAMCAT). Atherosclerosis. 2015;238:336–343. doi:10.1016/j.atherosclerosis.2014.12.034
38. Troeung L, Arnold-Reed D, Chan She Ping-Delfos W, et al. A new electronic screening tool for identifying risk of familial hypercholesterolaemia in general practice. Heart. 2016;102:855–861. doi:10.1136/heartjnl-2015-308824
39. Watts GF, Gidding S, Wierzbicki A, et al. Integrated guidance on the care of familial hypercholesterolaemia from the International FH Foundation. Int J Cardiol. 2014;171:309–325. doi:10.1016/j.ijcard.2013.11.025
40. Rubio-Marín P, Michán-Doña A, Maraver-Delgado J, et al. Cascade screening program for familial hypercholesterolemia. Endocrinol Diabetes Nutr. 2018:
41. Huijigen R, Kindt I, Verhoeven SB, et al. Two years after molecular diagnosis of familial hypercholesterolemia: majority on cholesterol-lowering treatment but a minority reaches treatment goal. PLoS One. 2010;5(2):e9220. doi:10.1371/journal.pone.0009220
42. Ademi Z, Watts GF, Pang J, et al. Cascade screening based on genetic testing is cost effective: evidence for implementation of models of care for familial hypercholesterolemia. J Clin Lipidol. 2014;8(4):390–400. doi:10.1016/j.jacl.2014.05.008
43. Lázaro P, Pérez de Isla L, Watts GF, et al. Cost-effectiveness of a cascade screening program for the early detection of familial hypercholesterolemia. J Clin Lipidol. 2017;11:260–271. doi:10.1016/j.jacl.2017.01.002
44. Damgaard D, Larsen ML, Nissen PH, et al. The relationship of molecular genetic to clinical diagnosis of familial hyper- cholesterolemia in a Danish population. Atherosclerosis. 2005;180:155–160. doi:10.1016/j.atherosclerosis.2004.12.001
45. Kinnear F, Wainwright E, Perry R, et al. Enablers and barriers to treatment adherence in heterozygous familial hypercholesterolemia: a qualitative evidence synthesis. BMJ Open. 2019;9:e030290. doi:10.1136/bmjopen-2019-030290
46. Khera A, Won HH, Peloso G, et al. Diagnostic yield and clinical utility of sequencing familial hypercholesterolemia genes in patients with severe hypercholesterolemia. J Am Coll Cardiol. 2016;67:2578–2589. doi:10.1016/j.jacc.2016.03.520
47. Pérez de Isla L, Alonso R, Mata N, SAFEHEART Investigators, et al. Coronary & heart disease, peripheral arterial disease, and stroke in familial hypercholesterolaemia: insights from the SAFEHEART registry (Spanish familial hypercholesterolaemia Cohort Study). Arterioscler Thromb Vasc Biol. 36;2016:2004–2016. doi:10.1161/ATVBAHA.116.307514
48. Benn M, Watts GF, Tybjaerg-Hansen A, et al. Mutations causative of familial hypercholesterolemia: screening of 98098 individuals from the Copenhagen General Population Study estimated a prevalence of 1 in 217. Eur Heart J. 2016;37:1384–1394. doi:10.1093/eurheartj/ehw028
49. Villa G, Wong B, Kutikova L, et al. Prediction of cardiovascular risk in patients with familial hypercholesterolaemia. Eur Heart J Qual Care Clin Outcomes. 2017;3:274–280. doi:10.1093/ehjqcco/qcx011
50. Umans-Eckenhausen M, Sijbrands E, Kastelein J, Defesche J. Low-density lipoprotein receptor gene mutations and cardiovascular risk in a large genetic cascade screening population. Circulation. 2002;106:3031–3036. doi:10.1161/01.CIR.0000041253.61683.08
51. Mata P, Alonso R, Pérez de Isla L. Atherosclerotic cardiovascular disease risk assessment in familial hypercholesterolemia: does one size fit all? Curr Opin Lipidol. 2018;29:445–452. doi:10.1097/MOL.0000000000000553
52. Pérez de Isla L, Alonso R, Mata N, SAFEHEART investigators, et al. Predicting cardiovascular events in familial hypercholesterolemia: the SAFEHEART registry. Circulation. 135;2017:2133–2144. doi:10.1161/CIRCULATIONAHA.116.024541
53. Santos RD, Gidding SS, Hegele RA, et al. Defining severe familial hypercholesterolaemia and the implications for clinical management: a consensus statement from the International Atherosclerosis Society Severe Familial Hypercholesterolemia Panel. Lancet Diabetes Endocrinol. 2016;4:850–861. doi:10.1016/S2213-8587(16)30041-9
54. Goff DC, Lloyd-Jones DM, Bennett G, et al. 2013 ACC/AHA guideline on the assessment of cardiovascular risk: a report of the American College of Cardiology/American Heart Association Task Force on Practice Guidelines. Circulation. 2014;129:S49–S73. doi:10.1161/01.cir.0000437741.48606.98
55. Alonso R, Andres E, Mata N, SAFEHEART Investigators, et al. Lipoprotein(a) levels in familial hypercholesterolemia: an important predictor of cardiovascular disease independent of the type of LDL receptor mutation. J Am Coll Cardiol. 63;2014:1982–1989. doi:10.1016/j.jacc.2014.01.063
56. Perez de Isla L, Ray KK, Watts G, et al. Potential utility of the SAFEHEART risk equation for rationalizing the use of PCSK9 monoclonal antibodies in adults with heterozygous familial Hypercholesterolemia. Atherosclerosis. 2019;286:40–45. doi:10.1016/j.atherosclerosis.2019.05.003
57. Cegla J, Neely DG, France M, et al. Heart UK consensus statement on Lipoprotein (a)- a call to action. Atherosclerosis. 2019. doi:10.1016/j-atherosclerosis.2019.10.011
58. Ellis K, Pérez de Isla L, Alonso R, et al. Value of measuring Lipoprotein(a) during cascade testing for familial hypercholesterolemia. J Am Coll Cardiol. 2019;73:1029–1039. doi:10.1016/j.jacc.2018.12.037
59. Miname MH, Bittencourt MS, Moraes SR, et al. Coronary artery calcium and cardiovascular events in patients with familial hypercholesterolemia receiving standard lipid-lowering therapy. JACC Cardiovasc Imaging. 2019;12:1797–1804. doi:10.1016/j.jcmg.2018.09.019
60. Pérez de Isla L, Alonso R, Muñiz-Grijalvo O, SAFEHEART investigators, et al. Coronary computed tomographic angiography findings and their therapeutic implications in asymptomatic patients with familial hypercholesterolemia. Lessons from the SAFEHEART study. J Clin Lipidol. 12;2018:948–957. doi:10.1016/j.jacl.2018.04.003
61. Mach F, Baigent C, Catapano A, et al. 2019 ESC/EAS guidelines for the management of dyslipidaemias: lipid modification to reduce cardiovascular risk. Eur Heart J. 2019:ehz455. doi:10.1093/eurheartj/ehz455
62. Suades R, Padró T, Crespo J, et al. Liquid biopsy of extracellular microvesicles predicts future major ischemic events in genetically characterized familial hypercholesterolemia. Arterioscler Thromb Vasc Biol. 2019;39:1172–1181. doi:10.1161/ATVBAHA.119.312420
63. Shapiro M, Fazio S. “Taking a look under the hood”-imaging the phenotypic heterogeneity of familial hypercholesterolemia. J Clin Lipidol. 2018;12:1095–1098. doi:10.1016/j.jacl.2018.05.020
64. Narverud I, Retterstol K, Ole P, et al. Markers of atherosclerotic development in children with familial hypercholesterolemia: a literature review. Atherosclerosis. 2014;235:299–309. doi:10.1016/j.atherosclerosis.2014.05.917
65. Sharifi M, Higginson E, Bos S, et al. Greater preclinical atherosclerosis in treated monogenic familial hypercholesterolemia vs. polygenic hypercholesterolemia. Atherosclerosis. 2017;263:405–411. doi:10.1016/j.atherosclerosis.2017.05.015
66. Vergeer M, Zhou R, Bots ML, et al. Carotid atherosclerosis progression in familial hypercholesterolemia patients: a pooled analysis of the ASAP, ENHANCE, RADIANCE 1, and CAPTIVATE studies. Circ Cardiovasc Imaging. 2010;3:398–404. doi:10.1161/CIRCIMAGING.109.909655
67. Cicero AFG, Fogacci F, Giovannini M, Bove M, Debellis G, Borghi C. Effect of quantitative and qualitative diet prescription on children behavior after diagnosis of heterozygous familial hypercholesterolemia. Int J Cardiol. 2019;293:193–196. doi:10.1016/j.ijcard.2019.05.069
68. Arroyo- Olivares R, Alonso R, Quintana Navarro G, et al. Adults with familial hypercholesterolemia have healthier dietary habits and lifestyle habits compared with their non-affected relatives: the SAFEHEART study. Public Health Nutr. 2019;22:1433–1443. doi:10.1017/S1368980018003853
69. Stein E, Stender S, Mata P, et al. Achieving lipoprotein goals in patients at high risk with severe hypercholesterolemia: efficacy and safety of ezetimibe co-administered with atorvastatin. Am J Cardiol. 2004;148:447–455.
70. van der Graaf A, Cuffie-Jackson C, Vissers MN, et al. Efficacy and safety of coadministration of ezetimibe and simvastatin in adolescents with heterozygous familial hypercholesterolemia. J Am Coll Cardiol. 2008;52:1421–1429. doi:10.1016/j.jacc.2008.09.002
71. Pérez de Isla L, Arroyo-Olivares R, Muñiz-Grijalvo O, et al. Long-term effect of two intensive statin regimens on treatment and incidence of cardiovascular events in familial hypercholesterolemia. The SAFEHEART study. J Clin Lipidol. 2019. doi:10.1016/j.jacl.2019.10.005
72. Wisloff T, Mundal L, Retterstol K, Igland J, Kristiansen IS. Economic evaluation of lipid lowering with PCSK9 inhibitors in patients with familial hypercholesterolemia: methodological aspects. Atherosclerosis. 2019;287:140–146. doi:10.1016/j.atherosclerosis.2019.06.900
73. Landmesser U, Chapman MJ, Stock J, et al. 2017 update of ESC/EAS task force on practical clinical guidance for proprotein convertase subtilisin/kexin type 9 inhibition in patients with atherosclerotic cardiovascular disease or in familial hypercholesterolaemia. Eur Heart J. 2018;39:1131–1143. doi:10.1093/eurheartj/ehx549
74. Grundy SM, Stone NJ, Bailey AL, et al. AHA/ACC/AACVPR/AAPA/ABC/ACPM/ADA/AGS/APhA/ASPC/NLA/PCNA guideline on the management of blood cholesterol: a report of the American College of Cardiology/American Heart Association Task Force on Clinical Practice Guide- lines. Circulation. 2019;139:e1082- e1143. doi:10.1161/CIR.0000000000000625
75. Saltijeral A, Pérez de Isla L, Alonso R, et al. Attainment of LDL cholesterol treatment goals in children and adolescents with familial hypercholesterolemia. The SAFEHEART follow-up registry. Rev Esp Cardiol. 2017;70:444–450. doi:10.1016/j.recesp.2016.10.012
76. Vuorio A, Kuoppala J, Kovanen PT, et al. Statins for children with familial hypercholesterolemia. Cochrane Database Syst Rev. 2017;7:CD006401. doi:10.1002/14651858
77. Braamskamp MJAM, Langslet G, McCrindle BW, et al. Effect of rosuvastatin on carotid intima-media thickness in children with heterozygous familial hypercholesterolemia: the CHARON study (hypercholesterolemia in children and adolescents taking rosuvastatin open label). Circulation. 2017;136:359–366. doi:10.1161/CIRCULATIONAHA.116.025158
78. Kusters DM, Avis HJ, de Groot E, et al. Ten-year follow-up after initiation of statin therapy in children with familial hypercholesterolemia. JAMA. 2014;312:1055–1057. doi:10.1001/jama.2014.8892
79. Luiriniok I, Wiegman A, Kusters DM, et al. 20-year follow-up of statin in children with familial hypercholesterolemia. N Engl J Med. 2019;381:1547–1556. doi:10.1056/NEJMoa1816454
80. de Ferranti S, Steinberger J, Ameduri R, et al. Cardiovascular risk reduction in high-risk pediatric patients: a scientific statement from the American Heart Association. Circulation. 2019;139:e603–e634. doi:10.1161/CIR.0000000000000618
81. Payne J, Williams S, Maxwell D, et al. Familial hypercholesterolaemia patient support groups and advocacy: A multinational perspective. Atherosclerosis. 2018;277:377–382. doi:10.1016/j.atherosclerosis.2018.08.020
82. Kindt I, Mata P, Knowles J. The roe of registries and genetic databases in familial hypercholesterolemia. Curr Opin Lipidol. 2017;28:152–160. doi:10.1097/MOL.0000000000000398
83. Vallejo-Vaz AJ, Akram A, Kondapally Seshasai SR, et al. Pooling and Expanding Registries of Familial Hypercholesterolaemia to Assess Gaps in Care and Improve Disease Management and Outcomes: Rationale and Design of the Global EAS Familial Hypercholesterolaemia Studies Collaboration. Atheroscler Suppl. 2016; 22:1–32.
84. Harada-Shiba M, Arai H, Ishigaki Y, et al. Guidelines for diagnosis and treatment of familial hypercholesterolemia 2017. J Atheroscler Thromb. 2018;25:751–770. doi:10.5551/jat.CR003
85. Ramaswami U, Humphries S, Priestley-Barnham L, et al. Current management of children and young people with heterozygous familial hypercholesterolemia- HEART UK statement of care. Atherosclerosis. 2019;290:1–8. doi:10.1016/j.atherosclerosis.2019.09.005
 © 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms.php
and incorporate the Creative Commons Attribution
- Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms.php
and incorporate the Creative Commons Attribution
- Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.