Back to Journals » Blood and Lymphatic Cancer: Targets and Therapy » Volume 4
B-cell malignancies: capture-sequencing strategies for identification of gene rearrangements and translocations into immunoglobulin gene loci
Authors Boyle EM, Walker BA, Wardell C, Leleu X, Davies F, Morgan G
Received 18 April 2014
Accepted for publication 5 June 2014
Published 30 October 2014 Volume 2014:4 Pages 107—119
DOI https://doi.org/10.2147/BLCTT.S51503
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Eileen M Boyle,1,2 Brian A Walker,1 Christopher P Wardell,1 Xavier Leleu,2 Faith E Davies,1 Gareth J Morgan1
1Centre for Myeloma Research, Institute of Cancer Research, Sutton, Surrey, UK; 2Service des Maladies du Sang, Hôpital Claude Huriez, Centre Hospitalier Universitaire de Lille, Lille, France
Abstract: Aberrant chromosomal translocations involving the immunoglobulin heavy chain (IGH) locus and clonal rearrangements of the variable (V), diversity (D), joining (J) segments (V[D]J) of the immunoglobulin gene are implicated in the initiation of mature B-cell malignancies, including myeloma. Analysis of these events provides information useful for diagnosis and prediction of prognosis, and also provides a useful monitoring strategy for response to treatment in patients with these diseases. Current methods for identification of these events are not generally applicable and give a biased picture of the prognostic significance and clinical relevance of these events. Novel methodologies based on next-generation sequencing are a new and more efficient genetic tool that can be used to screen and characterize these changes at the nucleotide level, offering a deeper and better understanding of the genetic basis of these complex diseases. In this work, we provide a review of the molecular basis of B-cell neoplasms, the methods used to detect them, and how they translate into the clinic.
Keywords: B-cell malignancies, minimal residual disease, molecular, IGH locus, next-generation sequencing
Introduction
Translocations involving the immunoglobulin heavy chain (IGH) locus and clonal rearrangements of the variable (V), diversity (D), joining (J), and constant (C) (V(D)J) segments of the immunoglobulin (Ig) genes are implicated in the initiation of B-cell malignancies, including myeloma. Analysis of these events provides information useful for diagnosing these cancers and for predicting their prognosis after treatment. In this setting, they also provide a tool that can be used to monitor response. Current methods for identification of these events are not generally applicable and give a biased picture of the prognostic significance and clinical relevance of these events. Next-generation sequencing could also be used in a diagnostic setting to identify these changes and may offer additional relevant information for patient management. In this work, we review the molecular basis of B-cell malignancies, the methods used to detect them, and how they might be employed in the clinic.
Immunoglobulin genes and normal B-cell development
The B-cell lineage is derived from lymphoid progenitor cells that differentiate from hematopoietic stem cells. They undergo a complex, highly controlled maturation process leading to expression of a functional Ig formed of two identical heavy chains (coded by the IGH locus) and two identical light chains coded by either of the Ig light chain loci, kappa (IGK) or lambda (IGL).1
The IGH locus is located on chromosome 14 at 14q32. This locus comprises approximately 1.5 Mb, and can be divided into four groups depending on the segments of the heavy chain encoded: V, D, J, and C segments. At the pro-B-cell stage, the DNA undergoes sequential DNA recombinations of single D, J, and V segments, which are controlled by the recombination signal sequences that flank each gene segment. This site-specific DNA recombination process is catalyzed by the proteins encoded by recombination activating genes 1 and 2 (RAG1 and RAG2).2 DNA repair processes such as mismatch repair, base excision repair, and non-homologous end joining,1,3 rejoin them. Junction diversity via random addition of nucleotides by terminal deoxynucleotidyl transferase adds further diversity.4 Once a functional V(D)J rearrangement has been achieved, the other allele is excluded from recombination attempts in a process known as allelic exclusion.5 The final transcript is composed of the V(D)J rearrangement and the constant region immediately 3′ of the J segment, which in a naïve B-cell is the Cμ region that generates an IgM isotype.6 Enhancers (E) have been identified. They can be divided into two groups: Eμ enhancers, located in the intronic region between JH and Cμ, and a series of enhancers located downstream of Cα in a region referred to as the 3′ IGH regulatory region. Eμ is a small 220 bp core element necessary and sufficient to induce transcription, flanked by two matrix attachment regions that contribute positively to Eμ function. They are lineage-specific and are known to drive oncogenesis.7
Following the IGH locus rearrangement, the light chain loci proceed to rearrange. The kappa and lambda light chain loci are located at 2p12 and 22q11.2, respectively. Like the IGH locus, they include V, J, and C segments, but lack D segments. IgK-positive B-cells retain their IGL genes in germline configuration whereas the majority of IgL-positive B-cells bear IGK rearrangements in line with hierarchical IGL chain recombination. First, rearrangements start at the IGK locus where Jκ and Vκ segments rearrange with limited junctional diversity.8 Nonfunctional rearrangements may occur when a downstream element, the kappa-deleting-element, rearranges with the Vκ segment or an intron.9 This leads to inactivation of the kappa locus via deletions of the JκCκ or Cκ regions preventing the expression of that IGK allele and therefore participating in the regulation of allelic and κ/λ isotypic exclusion. If the kappa rearrangement is not functional on both alleles, the IGL locus proceeds to rearrange.10
Finally, pairing of the heavy and light chains results in a functional Ig on the cell surface at the immature B-cell stage, enabling the cell to escape apoptosis and proceed to maturation outside the bone marrow.1
After this initial process where the primary Ig repertoire is created, these immature yet immunocompetent B-cells exit the bone marrow and enter the lymphoid organs through the mantle zone into the germinal center (GC), where they proceed to proliferate and undergo affinity maturation after encounter with an antigen. This maturation or GC reaction requires close interaction between B-cells, antigen-presenting dendritic cells, and T-cells. B-cells carrying an antigen-specific B-cell receptor are selected to survive and proliferate. These positively selected cells, or centroblasts, rapidly divide and expand in the dark zone of the GC. Some of them then enter the light zone, stop dividing, and become centrocytes. Centrocytes can revert to centroblasts or terminally differentiate into either memory B-cells or antibody-secreting cells (plasmablasts or plasmocytes). Plasmablasts are short-lived, cycling, antibody-secreting cells that are found in extrafollicular foci. In a poorly understood process, some of them can return to the GC and undergo further clonal selection.11 The proliferation of naïve lymphocytes occurring during the first encounter with the antigen, the primary immune response, generates IgM or IgD effector B-cells and memory B-cells. The presence of memory B-cells, on subsequent encounters with antigens, enables a quantitative and qualitatively superior response, termed the secondary response. During this GC reaction, two types of DNA modifications occur, class switch recombination and somatic hypermutation. Somatic hypermutation adds point mutations into the variable regions of the Ig genes.12 As the process of somatic hypermutation is repeated several times, populations of B-cells bearing receptors with increasing affinity toward decreasing levels of antigen are selected. This process is referred to as affinity maturation. Class switch recombination occurs when the V(D)J segment is brought into proximity to another constant region (IGHγ1–4, IGHα1-2, or IGHε) by deletion of the intervening DNA between Cμ and the next constant region. Class switch recombination involves large repetitive switch (S) sequences that are located upstream of each CH gene segment. During class switch recombination, the Sμ region recombines with a downstream S region, resulting in a closer proximity of the new CH gene segment to the V(D)J region and the IGH variable region promoter.13 Class switch recombination provides an important means of altering the effector function of the antibodies produced by the B-cells and is central to maturation of the antibody response elicited by natural infections and vaccination (Figure 1). This process occurs during the secondary immune response. The ultimate aim of all these processes is the development of an extensive B-cell repertoire with high affinity antibodies that recognize and optimally bind foreign antigens with effector properties. Although mechanistically different, these two processes are mediated by DNA double-strand breaks induced by transcription-targeted activation-induced cytidine deamination activity. The activation-induced cytidine deamination protein14 belongs to the APOBEC (apolipoprotein B mRNA editing enzyme, catalytic polypeptide-like) family of cytidine deaminases and is capable of deaminating cytidine to uracil in vitro on both single-stranded DNA substrates and single-stranded DNA generated by formation of RNA-DNA hybrids.15,16 Although highly regulated,17 it can induce DNA mutations at a relatively high rate (up to 10−4 to 10−3 mutation per base per division) and is a known oncogene.14
 | Figure 1 Translocations involving the IGH locus in mature B-cell malignancies. |
Upon terminally differentiating, plasma cells home to the bone marrow where they secrete large quantities of antibodies. Expression of surface Ig is downregulated, hence they no longer respond to antigens. By comparison, the recirculating memory B-cells readily respond to subsequent antigenic challenge as they express functional surface Ig and can, on reactivation, terminally differentiate to antibody-secreting cells.1,11 The process of B-cell terminal differentiation is regulated by the coordinated activity of a small group of master regulatory transcription factors. These factors can be divided into those such as Pax5 and Bcl6 that promote and maintain the B-cell program and others such as Blimp1 (B lymphocyte-induced maturation protein) and IRF4 (interferon regulatory factor 4) that control antibodies secreting cells differentiation. There exists mutually antagonistic transcriptional programs in B-cell and antibody secreting cells that ensure the separation of B-cell and antibody secreting cells fate.18 This process is essential for regulating terminal differentiation.
Immunoglobulin events in B-cell malignancies
Overview
Although essential for an effective adaptive immune response, the different processes that lead to B-cell differentiation are implicated in the pathogenesis of B-cell malignancies. B-cell neoplasms are a heterogeneous group of diseases, each with a distinct level of maturation defined in relation to the GC and their normal cellular counterparts. In broad outline, they can be divided into three types based on their relationship with the germinal center:
- pre-GC center neoplasms with unmutated V segments, eg, acute lymphoblastic leukemia (ALL), mantle cell lymphoma (MCL), and chronic lymphocytic lymphoma (CLL)
- GC type with ongoing V gene mutations, eg, follicular lymphoma and diffuse large B-cell (GC-DLBC) tumors
- post-GC neoplasms with stable mutated V genes, eg, activated B-cell lymphoma (ABC-DLBC), marginal zone lymphoma, and myeloma.19,20
These entities are clonal and are characterized by the presence of a clone-specific rearrangement of one of the Ig gene loci.21,22 V(D)J rearrangements occur early in B-cell ontogeny within B-cell precursors located within the bone marrow21 and can be used to define clonality.22 The second key abnormality at the IGH locus is the presence of reciprocal translocations that deregulate target oncogenes by placing them adjacent to the strong enhancers of the Ig genes.23 These translocations rarely give rise to fusion transcripts. Double-strand breaks and abnormal repair mechanisms contribute to translocations in B-cell malignancies. Depending on the stage they occur at, these translocations are believed to be mediated by different genes, such as the RAG complex at the pre-B-cell and AID at the pro B-cell stage.24 In some cases, these translocations are disease-defining (as in follicular lymphoma, MCL, or Burkitt’s lymphoma),25 in some cases, they have prognostic value such as t(4;14) in myeloma26 (Figure 2).
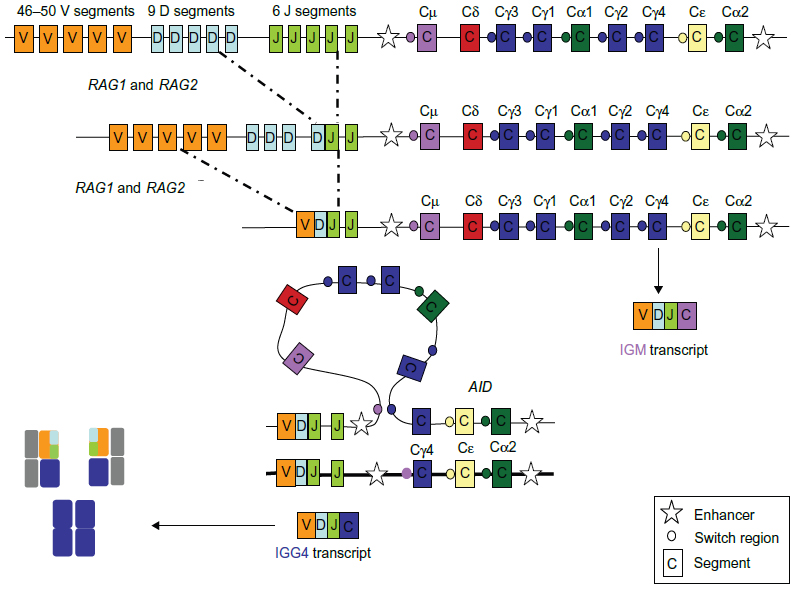 | Figure 2 Heavy chain rearrangement. |
Immunoglobulin events in pre-GC malignancies
ALL is derived from a normal precursor B-cell. Although long believed to be absent, translocations involving the IGH locus have been implicated in approximately 3% of precursor B-cell ALL. The first recurrent translocation described was t(14;19)(q32;q13).27 Unlike observations in mature B-cell malignancies, the breakpoint on chromosome 19 does not involve BCL3 but CEBPA, a gene of the CCAAT/enhancer binding protein (CEBP) gene family of transcription factors.28,29 Other translocations in ALL involving the IGH locus have been identified, such as t(8;14)(q11;q32), t(14;14)(q11;q32)/inv(14)(q11;q32), t(14;20)(q32;p13), and t(14;19)(q32;q13) with an alternative breakpoint. The partners of such translocations consist of other members of the CEBP family, such as CEBPD, CEBPE, and CEBPG.29 A number of other partners have been described, such as ID4 in t(6;14)(p22;q32)30 and EPOR in t(14;19)(q32;p13).31 Although the partners differ, patients show common clinical features with a pre B-immunophenotype and low blood count. As they remain rare events, their prognostic value is uncertain.32,33 IGH rearrangements are present in up to 90% of ALL blasts. Approximately 50% of them also rearrange the IGK locus and the T-cell receptor (TCR) γ/δ loci. A combined study of these four loci enables identification of one or more rearrangements in virtually all cases of ALL.34,35 Therefore, IGH rearrangements may be used to monitor minimal residual disease (MRD) and guide treatment strategies, such as allogeneic transplant.
Burkitt’s lymphoma is an uncommon form of non-Hodgkin lymphoma. A defining feature of Burkitt’s lymphoma is activation of the MYC gene located at 8q24 through translocation with one of the three Ig loci, which introduces a transcriptional enhancer. In 80% of cases, it involves the IGH locus [(t(8;14)(q24;q32)], in 15% the IGK locus [t(2;8)(p12;q24)], and in 5% the IGL locus [t(8;22)(q24;q11.2)]. The breakpoint occurs between the first (non-coding exon) and second exon (where the start codon is located), leading to an enhancer-driven activation via a cryptic promoter (P3). IGH rearrangements are also present in virtually all patients.36
The t(11;14)(q13;q32) translocation that juxtaposes the proto-oncogene CCND1 at 11q13 to the IGH locus at chromosome 14q32 is considered the primary oncogenic event in the development of MCL. It occurs at the pre-B cell stage of differentiation during recombination of the V(D)J segments of the IGH variable region.37 However, the tumor is composed of a specific population of mature B lymphocytes, indicating that the full neoplastic phenotype is acquired at a later stage of differentiation. Historically, naïve B-cells have been considered the normal counterpart to MCL tumor cells based on IgM/IgD and CD5 expression, their location in the mantle zone, and early descriptions of the predominantly unmutated variable region. More recently, comprehensive analysis of the B-cell receptor diversity in MCL has shifted this view to a more complex ontogeny, in which antigen selection plays a role. Recent studies have also shown that the subset of cases that carry somatic hypermutation (15%–40%) also have an indolent phenotype and clinical behavior.38–40
Rearrangements of the IGH locus with oncogenes appear to be rare in CLL. Rare cases of t(14;18)(q32;q21) involving the IGK locus and BCL2, have been described. The t(14;19)(q32;q13) involving the BCL3 gene was found in six of 4,487 cytogenetically analyzed lymphoproliferative disorders. Five of these six cases were eventually classified as CLL.41 Initially, it was thought that the clonal accumulation of CD5+ B-cells in CLL was comprised of naïve B-cells with unmutated IGH variable region. However, recent studies showed the presence of significant somatic mutations in approximately 50% of patients, suggesting the neoplastic cells may correspond to post GC memory B-cells.42–44
Immunoglobulin events in GC malignancies
The hallmark of follicular lymphoma, t(14;18)(q32;q21) results in a constitutive overexpression of BCL2, allowing B-cells to abrogate the default GC apoptotic program. Nonetheless, this translocation is neither necessary nor sufficient for diagnosis, as it is absent in 15% of follicular lymphomas and present in about 20%–30% of GC B-type DLBC lymphomas.20,45 The somatic hypermutation process is ongoing in follicular lymphoma,46 and recent studies suggest that a disruption in this process occurring late in evolution of the disease may select a more aggressive disease and lead to transformation into high-grade lymphoma.47
GC B-type DLBC lymphoma seems to originate from the GC and frequently shows ongoing somatic hypermutation.48 Forty-five percent of patients harbor a t(14;18)(q32;q21) leading to a constitutive activation of BCL2,49 a feature that is absent in ABC-like disease.
Immunoglobulin events in post-GC malignancies
ABC-DLBC lymphoma seems to originate from a B-cell that is in the process of differentiating into plasma cells. Translocations have been described, but unlike GC B-type DLBC lymphoma, they do not harbor any t(14;18)(q32;q21).
The chromosomal translocations t(1;14)(p22;q32), t(14;18)(q32;q21), and t(3;14)(p13;q32) are all known to occur with variable frequencies in mucosa-associated lymphoid tissue (MALT) lymphomas, resulting in IGH-BCL10, IGH-MALT1, and IGH-FOXP1 rearrangements, respectively.50 The t(1;14)(p22;q32) and t(14;18)(q32;q21) occur in a small subgroup of almost exclusively non-gastric MALT lymphomas.50
Myeloma is a late B-cell malignancy where malignant plasma cells expand and accumulate in the bone marrow. As IgM and IgD are rare,51 myeloma is considered a post-class switch disease. Translocations involving the IGH locus occur in approximately 50% of cases and include t(4;14)(p16;q32) (15%), t(11;14)(q13;q32) (20%), t(14;16)(q32;q23) (<5%), t(6;14)(p21;q32) (<5%), t(8;14)(8q24:q32) (<5%), and t(14;20)(q32;q12) (<5%). They are responsible for mediating ectopic expression of specific genes brought under the control of strong enhancers. The t(11;14)(q13;q32) leads to overexpression of CCND1, t(6;14)(p21;q32) deregulates CCND3, and t(4;14)(p16;q32) involves WHSC1 (also known as MMSET ) and FGFR3.52 The majority of the breakpoints are localized within the switch region and are generated through class switch recombination. All t(4;14) are mediated through class switch recombination; however, 21% of t(11;14) and 25% of t(14;20) are generated through DH-JH recombination, with breakpoints within the DH-JH region. These two groups also generate translocations through receptor revision, as determined by the breakpoints in 10% and 50% of t(11;14) and t(14;20) samples, respectively.52 These data suggest that, although similar, they would involve different genes. The RAG complex would be responsible for the translocations occurring through DJ rearrangements via the pro-B-cell stage in the bone marrow or through an aberrant RAG expression in the post-GC. AID would be responsible for the class switch recombination-mediated translocations. In MCL, although the translocation partners are similar, all the t(11;14) are generated through V(D)J rearrangement, suggesting the role of RAG at the pro-B-cell stage.24 Deregulation of MYC by t(8;14)(q24;q32), t(2;8)(p12;q24), and t(8;22)(q24; q11.2) is also present at diagnosis. Similarly, MYC translocations may also involve non-Ig partners. These translocations function in the same way by placing an enhancer from a B cell lineage gene next to MYC, leading to its overexpression. Comparable mechanisms are seen in other B-cell malignancies.49 IGH rearrangements are detectable in most tumor cells in a patient, suggesting they are initiating events. Most plasma cells secrete an Ig coded by a functional hypermutated V(D)J. The rearrangement is often biallelic and the other allele can carry either a complete but non-functional rearrangement (40%) or a single DJ rearrangement in the same frequencies as those observed in normal B-cells, supporting the principle of allelic exclusion. Hypermutation of the rearranged V(D)J is present in most cases. The level of hypermutation ranges from 9% to 23%, which is consistently higher than in other diseases such as CLL, hindering a polymerase chain reaction (PCR)-based molecular diagnosis approach.53 Of note, somatic hypermutations are absent on the non-completely rearranged allele. This allele may therefore be used for PCR. As seen in other B-cell malignancies, the IGK locus is rearranged in kappa myeloma and IGL in the lambda isotype myeloma.54
Clinical value of detecting clonality and translocations
Detection of clonality
The concept of clonality is based on the hallmark of cancer where, once a malignant B-cell has rearranged its Ig genes, every daughter cell will bear the same rearrangement.55 IGH rearrangements are not B-cell-specific, as they can occur in 22% of T-cell ALL, 8%–16% of T-cell lymphomas, and 4%–18% of AML.56 Nevertheless, they are crucial in B-cell malignancies because their identification can be used to confirm the diagnosis and guide follow-up.
Despite the well established morphological and phenotypic diagnostic criteria, definite diagnosis of a suspected lymphoproliferative disease often requires clonality testing.20 Retrospective studies suggest that 5%–15% of suspected lymphoproliferative disease diagnoses cannot be made based on morphology or immunophenotyping alone. This percentage increases in immunocompromised patients.57,58 The ability of demonstrating V(D)J rearrangement is an effective strategy to demonstrate clonality;59 nevertheless it varies among B-cell neoplasms.57,58,60,61 V(D)J rearrangements may also be used to establish the clonal relationship between two coexistent lymphoid malignancies.62–64
Beyond diagnosis, these rearrangements may also be used to monitor MRD. Monitoring MRD is a key requirement for the management of most hematological malignancies as it can be used:
- as an early surrogate marker of treatment efficacy that can therefore be used to guide treatment strategies early on, such as de-escalation strategies leading the way into personalized medicine
- as a prognostic marker that can predict outcome
- as a follow-up marker for remission control that can be used as a trigger for pre-emptive treatment.
Evaluation of MRD by multiparameter flow cytometry requires that neoplastic cells express an antigen profile that differs from that observed in normal hematopoietic cells and that this pattern of expression is not affected by chemotherapy. This immunophenotyping approach was to some extent successful because it was able to predict outcome in diseases such as pediatric ALL,65 myeloma,66 and CLL.67 It was also used to guide treatment approaches, notably in pediatric ALL where MRD could be used to determine requirements for an allogeneic transplant65 or CLL where it can be used in a risk assessment strategy to guide treatment after allogeneic transplant.68 With the introduction of standardized quantitative PCR for the measurement of abnormal rearrangements in ALL,69–72 it appeared that the extent and depth of response based on a molecular approach was even more prognostically informative than that observed by flow cytometry.67,68 With increased sensitivities in comparison with immunophenotyping, in ALL, molecular MRD can distinguish prognostic subgroups.73–77 Although rare cases have a BCR-ABL or a MLL-AFA4 fusion transcript, for technical convenience, the rearrangements more commonly studied for follow-up of MRD are those of TCRγ/δ, IGH, and IGK. MRD technology has more recently been used in the study of Burkitt’s lymphoma78 and MCL.79 In myeloma, there is currently no standard molecular MRD detection as PCR-based assays for V(D)J rearrangements are hindered by somatic hypermutation, given the non-annealing of the PCR primers.54
Detection of somatic hypermutation
This is achieved by sequencing the IGH variable region segment of a clonal population and comparing it to a database of germline sequences. If the difference accounts for more than 2% of the nucleotides, the segment is considered mutated. In fundamental studies, Damle et al and Hamblin et al have shown80,81 that the presence of unmutated IGH variable region genes predicts for an inferior survival in CLL. There is to the extent of our knowledge no equivalent study in myeloma.
Detection of translocation
Translocations in B-cell malignancies can be used to confirm the diagnosis, predict outcome, and maybe monitor MRD. The clinical value of detecting a chromosomal translocation differs according to disease type. They are required for diagnosis in diseases such as MCL [t(11;14)(q13;q32)], follicular lymphoma [t(14;18)(q32;q21)], and Burkitt’s lymphoma [t(8;14)(q24;q32)].
In a clinical setting, apart from diagnosis, the main reason for identifying translocations is to stratify patients and use this information to guide treatment. In myeloma, for instance, some translocations are of prognostic value, such as t(4;14)(p16;q32), and to a lesser extent t(14;16)(q32;q23) or t(14;20)(q32;q12). When used in conjunction with other biologically relevant elements such as beta-2 microglobulin, they can be used to define molecular subgroups with similar outcome that would benefit from specific treatment options.26 Finally, preliminary data suggest that detection of translocations or the fusion transcript that may arise from these balanced translocations can be used to detect MRD in Burkitt’s lymphoma.78
Traditional methods for detecting immunoglobulin loci rearrangement events
Identification of V(D)J rearrangement
V(D)J rearrangements were first assessed by Southern blotting21 with hybridization probes mapping to the V, J, or C segment.59 This is traditionally regarded as a time-consuming, labor-intensive, and technically demanding procedure. It also requires both high quantities and good quality of DNA, so cannot be performed on fixed samples. False positives are rare, and false negatives account for 5% of samples. As it has a lower sensitivity than PCR, this method has gradually been replaced by PCR.
The PCR approach relies on amplification of the IGH locus using conserved sequences. It is believed to be both more rapid and less labor-intensive than Southern blotting. It also requires lower qualities of DNA and can be performed on previously fixed or embedded samples. It is based on the IGH variable region and JH consensus primers FR I, II, III, and IV. Using a single FR yields sensitivity of 50%–75%.57 When combining multiple primers or adding the IGK primers, sensitivities are greater than 90%.58,60 BIOMED-II primers target FR I, II, and III, the incomplete DJ rearrangement, and the IGK locus. Adding the DJ rearrangement primers targets the non-coding allele. This is of particular importance in GC and post-GC malignancies. As it does not undergo somatic hypermutation, primers have a higher probability of binding appropriately. The IGK primers bind the VJ segments as well as the kappa-deleting element. Adjunction of IGL primers does not seem to improve sensitivity.57 False positives will arise in the event of oligo or pseudoclonal populations. This is observed when either the sample or the repertoire is poor (eg, immune-compromised patients or post-treatment samples). False negatives relate to ineffective binding and subsequent lack of amplification. This is chiefly observed in three different circumstances: in the presence of somatic hypermutation (in GC or post-GC malignancies), in the case of a translocation breakpoint involving the consensus region, and in the case of small degraded DNA samples (<200 bp).57,58 PCR is able to yield results in up to 90% of cases of CLL82 and 98% of ALL cases.34 Nevertheless, in post-GC malignancies that have undergone somatic hypermutation, such as DLBC lymphoma, follicular lymphoma, and multiple myeloma, the rates of failure are higher (15% in follicular lymphoma and up to 60% in myeloma).60,61
Identification of translocations
Karyotyping and conventional cytogenetics made their way into the clinic with the development of more effective techniques to culture bone marrow and blood and improved metaphase preparations. More accurate identification of individual chromosomes became possible with banding techniques, first using fluorochromes (Q-banding)83 and then enzymatic treatment strategies (G-banding).84 Although standardization facilitated the description of chromosomal abnormalities, these techniques lacked sensitivity and were not appropriate for tumors with low proliferating indices in vitro. A conventional cytogenetics approach is currently standard for the detection of established chromosomal abnormalities in ALL within a routine clinical environment. Nevertheless, it fails to identify morphologically cryptic or complex translocations. It also has a high failure rate inherent to this technique (requires live cells) and to the tumor characteristic (high failure rates in tumors with low proliferation indices). The latter explains the limitation of such an approach in myeloma. The performances of karyotyping were improved by addition of metaphase fluorescence in situ hybridization (FISH) that helped to identify specific fusions without resolving the technical limitations.
FISH is based on annealing of a DNA probe to a complementary sequence on a target genomic DNA. The use of split signal probes has led to the identification of morphologically cryptic translocations. Interphase FISH on selected cells increased the sensitivity of such approaches. Although sensitivity is increased, this method remains labor-intensive with high failure rates and is inherently biased toward a small selection of translocations.
Whole genome DNA analysis using comparative genomic hybridization was another significant technical development in molecular cytogenetics.85 It can detect abnormalities arising from the gain and loss of DNA. High resolution array comparative genomic hybridization may detect submicroscopic changes in DNA copy number to a resolution <1 Mb.86 This approach enables us to accurately locate the breakpoints involved in such rearrangements.87 A parallel technology utilizing single nucleotide polymorphism arrays has been used to show copy number changes and loss of heterozygosity. These arrays can also detect translocations if they are unbalanced.88
There is a need for a high throughput tool to detect these events at the molecular levels, which could be used to better understand the mechanisms of disease, its heterogeneity, and potential new targets.
High throughput strategies
Here we provide a description of the capture-sequencing method, its role in better characterizing IGH alterations, and its potential applications and limitations in the study of mature B-cell neoplasms.
Principles of next-generation sequencing
The main principle of next-generation sequencing is based on massively parallel sequencing where small DNA fragments are sequenced in parallel (Figure 3). Bioinformatics is used to piece together these fragments by mapping these fragments or reads to the reference genome. Each base of the genome is sequenced multiple times, giving additional depth that translates into accuracy of the data thus generated. This high throughput method can be used on entire genomes, individual genes, or the 25,000 coding genes (whole exome). This method can provide insight into a wide panel of DNA variations, such as missense mutations, small insertions or deletions, and structural changes, such as inversions, translocations, and rearrangements. Copy number changes and microdeletions can also be derived from the data. It does not require any pre-existing knowledge of the gene or its location, and thus offers an unbiased view of the whole exome. The main limits of this approach are the relative error that occurs in the event of a high GC content or a repeat architecture, the infrastructure required to comprehensively analyze the sample, and the cost that limits its implementation in the clinic. Targeted capture approaches based on RNA baits mapping to a specific region offer a methodology to enrich and select specific regions of interest. This has been used in the research setting to define the translocation breakpoints in myeloma52,89 and in DLBC lymphoma.90 As the technique requires small amounts of DNA (50 ng) and can be performed on formalin-fixed, paraffin-embedded extracted DNA, it provides an opportunity for rapid detection of translocation in the clinical setting.
 | Figure 3 Overview of high throughput next-generation sequencing. |
Clonality detection using immunoglobulin gene rearrangements
By defining a capture for the Ig gene loci, high-throughput sequencing methods can be applied to universally amplify these segments and identify all clonal gene rearrangements despite somatic hypermutation. It is also possible to amplify the Ig rearrangements using PCR followed by massively parallel sequencing of the product to detect clonality in B-cells. This provides a more robust and more specific means of detecting subclones emerging through disease progression. Measurements may be repeated at different time points, such as at diagnosis or disease progression. This approach has been tested in ALL where it was able to detect MRD.10 As numerous correlative studies suggest, in ALL the prognostic power of MRD could also increase due to the higher sensitivity of this approach. Studies in CLL suggest it could be a good diagnostic tool and useful for monitoring MRD after transplant.91 In myeloma, 133 newly diagnosed patients with a very good partial response were evaluated using a next-generation sequencing-based MRD test. This approach proved feasible (91% of patients were evaluable) and the median overall survival was proportionate to the depth of response.90 Studies in other B-cell malignancies are encouraging,92 but should nevertheless be confirmed in a prospective setting. The current sequencing costs for rearranged antigen-receptor genes are comparable with those of flow cytometry and PCR. Sequencing data may be complex to interpret, but the current analytic algorithms facilitate detection and quantitation of genetic events.
Detecting chromosomal translocations into immunoglobulin gene loci
The identification of IGH translocations requires firstly the design and fabrication of a targeted capture library directed at the IGH locus. Although not currently available on a routine basis, this technique could be used to detect translocations by detecting the presence of reads mapping to different chromosomes. Exact breakpoints could be identified and could be used to determine PCR-based assays for MRD.52,90
Discussion
Next-generation sequencing-based tests could be implemented to replace the current PCR-based assays for MRD detection. Next-generation sequencing methods yield higher sensitivities in the MRD setting than PCR in most cases of B-cell neoplasms in which they have been tested. When tested in 106 ALL patients and compared with flow cytometry, next-generation sequencing was able to identify all positive patients and an additional ten patients previously considered negative. In the same cohort, when compared with PCR, it was able to identify 97% of positive cases and an additional three patients.93 Similar results were seen in CLL and other B-cell malignancies, thus adding sensitivity and precision to MRD detection. Nevertheless, these data need confirmation in prospective randomized trials in order to determine the exact prognostic significance of next-generation sequencing and how it should be used to guide treatment.91
The capture sequence strategy may also be used to identify translocations. Whole exon sequencing has been used both in myeloma and DLBC lymphoma. New translocation partner chromosomes and exact breakpoints were identified. Furthermore, by providing information on both V(D)J rearrangement and translocations, capture sequencing strategies may also offer additional information by not only determining the breakpoint but also the putative mechanism of the chromosomal event (aberrant V(D)J rearrangement or class switch recombination, for instance). From there it was shown that, in some cases, the initiating events in post-GC-type disease, such as myeloma, occurred earlier in B-cell development (at the pre-GC stage).52 This may be of clinical relevance.
Smaller targeted panels may be used in a diagnostic setting. As the capture sequence strategy is based on DNA rather than viable cells or easily degraded RNA, it would be more reliable and easier to implement, especially in the case of paraffin embedded samples.
Finally, next-generation sequencing may be used to monitor clonal evolution. Firstly, the study of somatic hypermutation and its progression throughout disease could offer further insight into the clonal architecture and evolution of B-cell malignancies, especially those with ongoing somatic mutations, such as follicular lymphoma or myeloma. Secondly, the identification of multiple subclones and their evolution throughout treatment and follow-up may be of additional prognostic value.
Conclusion
The development of molecular diagnostics has significantly improved the detection of Ig events such as translocations and V(D)J rearrangements in B-cell malignancies. From a clinical perspective, this has translated into more accurate diagnosis, better risk stratification, and more precise monitoring of patients. These studies have also given us a better understanding of the ontogeny of B-cell malignancies. The translation of the more novel approaches into the clinic, such as next-generation sequencing, could also benefit patients. Not only are they more sensitive than standard approaches in the detection of these events, but they could also give additional information on the clonal architecture of these malignancies that could translate into the clinic.
Disclosure
The authors have no conflicts of interest in this work.
References
Delves PJ, Roitt IM. The immune system. First of two parts. N Engl J Med. 2000;343(1):37–49. | |
Nishana M, Raghavan SC. Role of recombination activating genes in the generation of antigen receptor diversity and beyond. Immunology. 2012;137(4):271–281. | |
Vuong BQ, Herrick-Reynolds K, Vaidyanathan B, et al. A DNA break- and phosphorylation-dependent positive feedback loop promotes immunoglobulin class-switch recombination. Nat Immunol. 2013;14(11):1183–1189. | |
Victor KD, Capra JD. An apparently common mechanism of generating antibody diversity: length variation of the VL-JL junction. Mol Immunol. 1994;31(1):39–46. | |
Jung D, Giallourakis C, Mostoslavsky R, Alt FW. Mechanism and control of V(D)J recombination at the immunoglobulin heavy chain locus. Annu Rev Immunol. 2006;24:541–570. | |
Xu Z, Zan H, Pone EJ, Mai T, Casali P. Immunoglobulin class-switch DNA recombination: induction, targeting and beyond. Nat Rev Immunol. 2012;12(7):517–531. | |
Roy AL, Sen R, Roeder RG. Enhancer-promoter communication and transcriptional regulation of IgH. Trends Immunol. 2011;32(11):532–539. | |
Victor KD, Vu K, Feeney AJ. Limited junctional diversity in kappa light chains. Junctional sequences from CD43+B220+ early B cell progenitors resemble those from peripheral B cells. J Immunol. 1994;152(7):3467–3475. | |
Langerak AW, van Dongen JJM. Recombination in the human IGK locus. Crit Rev Immunol. 2006;26(1):23–42. | |
Langerak AW, Nadel B, de Torbal A, et al. Unraveling the consecutive recombination events in the human IGK locus. J Immunol. 2004;173(6):3878–3888. | |
Nutt SL, Taubenheim N, Hasbold J, Corcoran LM, Hodgkin PD. The genetic network controlling plasma cell differentiation. Semin Immunol. 2011;23(5):341–349. | |
Fraenkel S, Mostoslavsky R, Novobrantseva TI, et al. Allelic ‘choice’ governs somatic hypermutation in vivo at the immunoglobulin kappa-chain locus. Nat Immunol. 2007;8(7):715–722. | |
Stavnezer J, Guikema JEJ, Schrader CE. Mechanism and regulation of class switch recombination. Annu Rev Immunol. 2008;26:261–292. | |
Muramatsu M, Kinoshita K, Fagarasan S, Yamada S, Shinkai Y, Honjo T. Class switch recombination and hypermutation require activation-induced cytidine deaminase (AID), a potential RNA editing enzyme. Cell. 2000;102(5):553–563. | |
Chaudhuri J, Tian M, Khuong C, Chua K, Pinaud E, Alt FW. Transcription-targeted DNA deamination by the AID antibody diversification enzyme. Nature. 2003;422(6933):726–730. | |
Ramiro AR, Stavropoulos P, Jankovic M, Nussenzweig MC. Transcription enhances AID-mediated cytidine deamination by exposing single-stranded DNA on the nontemplate strand. Nat Immunol. 2003;4(5):452–456. | |
Keim C, Kazadi D, Rothschild G, Basu U. Regulation of AID, the B-cell genome mutator. Genes Dev. 2013;27(1):1–17. | |
Shapiro-Shelef M, Calame K. Regulation of plasma-cell development. Nat Rev Immunol. 2005;5(3):230–242. | |
Lenz G, Staudt LM. Aggressive lymphomas. N Engl J Med. 2010;362(15):1417–1429. | |
Harris NL, Jaffe ES, Diebold J, et al. The World Health Organization classification of neoplastic diseases of the hematopoietic and lymphoid tissues. Report of the Clinical Advisory Committee meeting, Airlie House, Virginia, Nov 1997. Ann Oncol. 1999;10(12):1419–1432. | |
Cleary ML, Chao J, Warnke R, Sklar J. Immunoglobulin gene rearrangement as a diagnostic criterion of B-cell lymphoma. Proc Natl Acad Sci U S A. 1984;81(2):593–597. | |
Aubin J, Davi F, Nguyen-Salomon F, et al. Description of a novel FR1 IgH PCR strategy and its comparison with three other strategies for the detection of clonality in B cell malignancies. Leukemia. 1995;9(3):471–479. | |
Lovén J, Hoke HA, Lin CY, et al. Selective inhibition of tumor oncogenes by disruption of super-enhancers. Cell. 2013;153(2):320–334. | |
Tsai AG, Lieber MR. Mechanisms of chromosomal rearrangement in the human genome. BMC Genomics. 2010;11 Suppl 1:S1. | |
Huret J. Atlas of Genetics and Cytogenetics in Oncology and Haematology. Cancer Net Cytogenet. Available from: http://atlasgeneticsoncology.org/. Accessed September 1, 2014. | |
Boyd KD, Ross FM, Chiecchio L, et al. A novel prognostic model in myeloma based on co-segregating adverse FISH lesions and the ISS: analysis of patients treated in the MRC Myeloma IX trial. Leukemia. 2012;26(2):349–355. | |
Robinson HM, Taylor KE, Jalali GR, Cheung KL, Harrison CJ, Moorman AV. t(14;19)(q32;q13): a recurrent translocation in B-cell precursor acute lymphoblastic leukemia. Genes Chromosomes Cancer. 2004;39(1):88–92. | |
Chapiro E, Radford-Weiss I, Cung HA, et al. Chromosomal translocations involving the IGH@ locus in B-cell precursor acute lymphoblastic leukemia: 29 new cases and a review of the literature. Cancer Genet. 2013;206(5):162–173. | |
Akasaka T, Balasas T, Russell LJ, et al. Five members of the CEBP transcription factor family are targeted by recurrent IGH translocations in B-cell precursor acute lymphoblastic leukemia (BCP-ALL). Blood. 2007;109(8):3451–3461. | |
Russell LJ, Akasaka T, Majid A, et al. t(6;14)(p22;q32): a new recurrent IGH@ translocation involving ID4 in B-cell precursor acute lymphoblastic leukemia (BCP-ALL). Blood. 2008;111(1):387–391. | |
Russell LJ, De Castro DG, Griffiths M, et al. A novel translocation, t(14;19)(q32;p13), involving IGH@ and the cytokine receptor for erythropoietin. Leukemia. 2009;23(3):614–617. | |
Dyer MJS, Akasaka T, Capasso M, et al. Immunoglobulin heavy chain locus chromosomal translocations in B-cell precursor acute lymphoblastic leukemia: rare clinical curios or potent genetic drivers? Blood. 2010;115(8):1490–1499. | |
Russell LJ, Enshaei A, Jones L, et al. IGH@ translocations are prevalent in teenagers and young adults with acute lymphoblastic leukemia and are associated with a poor outcome. J Clin Oncol. 2014;32(14):1453–1462. | |
Macintyre EA, d’Auriol L, Duparc N, Leverger G, Galibert F, Sigaux F. Use of oligonucleotide probes directed against T cell antigen receptor gamma delta variable-(diversity)-joining junctional sequences as a general method for detecting minimal residual disease in acute lymphoblastic leukemias. J Clin Invest. 1990;86(6):2125–2135. | |
Hansen-Hagge TE, Yokota S, Bartram CR. Detection of minimal residual disease in acute lymphoblastic leukemia by in vitro amplification of rearranged T-cell receptor delta chain sequences. Blood. 1989;74(5):1762–1767. | |
Agsalda M, Kusao I, Troelstrup D, Shiramizu B. Screening for residual disease in pediatric Burkitt lymphoma using consensus primer pools. Adv Hematol. 2009;2009:412163. | |
Jares P, Campo E. Advances in the understanding of mantle cell lymphoma. Br J Haematol. 2008;142(2):149–165. | |
Orchard J, Garand R, Davis Z, et al. A subset of t(11;14) lymphoma with mantle cell features displays mutated IgVH genes and includes patients with good prognosis, nonnodal disease. Blood. 2003;101(12):4975–4981. | |
Royo C, Navarro A, Clot G, et al. Non-nodal type of mantle cell lymphoma is a specific biological and clinical subgroup of the disease. Leukemia. 2012;26(8):1895–1898. | |
Del Giudice I, Messina M, Chiaretti S, et al. Behind the scenes of non-nodal MCL: downmodulation of genes involved in actin cytoskeleton organization, cell projection, cell adhesion, tumour invasion, TP53 pathway and mutated status of immunoglobulin heavy chain genes. Br J Haematol. 2012;156(5):601–611. | |
Michaux L, Mecucci C, Stul M, et al. BCL3 rearrangement and t(14;19)(q32;q13) in lymphoproliferative disorders. Genes Chromosomes Cancer. 1996;15(1):38–47. | |
Ritgen M, Lange A, Stilgenbauer S, et al. Unmutated immunoglobulin variable heavy-chain gene status remains an adverse prognostic factor after autologous stem cell transplantation for chronic lymphocytic leukemia. Blood. 2003;101(5):2049–2053. | |
Stilgenbauer S, Bullinger L, Lichter P, Döhner H; German CLL Study Group (GCLLSG). Chronic lymphocytic leukemia. Genetics of chronic lymphocytic leukemia: genomic aberrations and V(H) gene mutation status in pathogenesis and clinical course. Leukemia. 2002;16(6):993–1007. | |
Döhner H, Stilgenbauer S, Benner A, et al. Genomic aberrations and survival in chronic lymphocytic leukemia. N Engl J Med. 2000;343(26):1910–1916. | |
Weiss LM, Warnke RA, Sklar J, Cleary ML. Molecular analysis of the t(14;18) chromosomal translocation in malignant lymphomas. N Engl J Med. 1987;317(19):1185–1189. | |
Bahler DW, Levy R. Clonal evolution of a follicular lymphoma: evidence for antigen selection. Proc Natl Acad Sci U S A. 1992;89(15):6770–6774. | |
Pasqualucci L, Khiabanian H, Fangazio M, et al. Genetics of follicular lymphoma transformation. Cell Rep. 2014;6(1):130–140. | |
Lossos IS, Alizadeh AA, Eisen MB, et al. Ongoing immunoglobulin somatic mutation in germinal center B cell-like but not in activated B cell-like diffuse large cell lymphomas. Proc Natl Acad Sci U S A. 2000;97(18):10209–10213. | |
Rosenwald A, Staudt LM. Clinical translation of gene expression profiling in lymphomas and leukemias. Semin Oncol. 2002;29(3):258–263. | |
Sagaert X, Tousseyn T. Marginal zone B-cell lymphomas. Discov Med. 2010;10(50):79–86. | |
Greipp PR, San Miguel J, Durie BGM, et al. International staging system for multiple myeloma. J Clin Oncol. 2005;23(15):3412–3420. | |
Walker BA, Wardell CP, Johnson DC, et al. Characterization of IGH locus breakpoints in multiple myeloma indicates a subset of translocations appear to occur in pregerminal center B cells. Blood. 2013;121(17):3413–3419. | |
Wagner SD, Martinelli V, Luzzatto L. Similar patterns of V kappa gene usage but different degrees of somatic mutation in hairy cell leukemia, prolymphocytic leukemia, Waldenstrom’s macroglobulinemia, and myeloma. Blood. 1994;83(12):3647–3653. | |
González D, van der Burg M, García-Sanz R, et al. Immunoglobulin gene rearrangements and the pathogenesis of multiple myeloma. Blood. 2007;110(9):3112–3121. | |
Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144(5):646–674. | |
Dupret C, Asnafi V, Leboeuf D, et al. IgH/TCR rearrangements are common in MLL translocated adult AML and suggest an early T/myeloid or B/myeloid maturation arrest, which correlates with the MLL partner. Leukemia. 2005;19(12):2337–2338. | |
Evans PA, Pott CH, Groenen PJ, et al. Significantly improved PCR-based clonality testing in B-cell malignancies by use of multiple immunoglobulin gene targets. Report of the BIOMED-2 Concerted Action BHM4-CT98-3936. Leukemia. 2007;21(2):207–214. | |
Liu H, Bench AJ, Bacon CM, et al. A practical strategy for the routine use of BIOMED-2 PCR assays for detection of B- and T-cell clonality in diagnostic haematopathology. Br J Haematol. 2007;138(1):31–43. | |
Arnold A, Cossman J, Bakhshi A, Jaffe ES, Waldmann TA, Korsmeyer SJ. Immunoglobulin-gene rearrangements as unique clonal markers in human lymphoid neoplasms. N Engl J Med. 1983;309(26):1593–1599. | |
Payne K, Wright P, Grant JW, et al. BIOMED-2 PCR assays for IGK gene rearrangements are essential for B-cell clonality analysis in follicular lymphoma. Br J Haematol. 2011;155(1):84–92. | |
González D, González M, Balanzategui A, et al. Molecular characteristics and gene segment usage in IGH gene rearrangements in multiple myeloma. Haematologica. 2005;90(7):906–913. | |
Konoplev S, Lin P, Qiu X, Medeiros LJ, Yin CC. Clonal relationship of extranodal marginal zone lymphomas of mucosa-associated lymphoid tissue involving different sites. Am J Clin Pathol. 2010;134(1):112–118. | |
Yoshinaga H, Ohashi K, Yamamoto K, et al. Clonal identification of Burkitt’s lymphoma arising from lymphocyte-predominant Hodgkin’s disease. Br J Haematol. 1996;95(2):380–382. | |
Au WY, Srivastava G, Wong KY, et al. Transformation of diffuse large B-cell lymphoma into pre-B acute lymphoblastic leukemia: clinicopathologic features and clonal relationship. Hum Pathol. 2004;35(7):900–903. | |
Rubnitz JE, Inaba H, Dahl G, et al. Minimal residual disease-directed therapy for childhood acute myeloid leukaemia: results of the AML02 multicentre trial. Lancet Oncol. 2010;11(6):543–552. | |
Rawstron AC, Child JA, de Tute RM, et al. Minimal residual disease assessed by multiparameter flow cytometry in multiple myeloma: impact on outcome in the Medical Research Council Myeloma IX Study. J Clin Oncol. 2013;31(20):2540–2457. | |
Rawstron AC, Böttcher S, Letestu R, et al. Improving efficiency and sensitivity: European Research Initiative in CLL (ERIC) update on the international harmonised approach for flow cytometric residual disease monitoring in CLL. Leukemia. 2013;27(1):142–149. | |
Richardson SE, Khan I, Rawstron A, et al. Risk-stratified adoptive cellular therapy following allogeneic hematopoietic stem cell transplantation for advanced chronic lymphocytic leukaemia. Br J Haematol. 2013;160(5):640–648. | |
De Haas V, Verhagen OJ, von dem Borne AE, Kroes W, van den Berg H, van der Schoot CE. Quantification of minimal residual disease in children with oligoclonal B-precursor acute lymphoblastic leukemia indicates that the clones that grow out during relapse already have the slowest rate of reduction during induction therapy. Leukemia. 2001;15(1):134–140. | |
Chen JS, Coustan-Smith E, Suzuki T, et al. Identification of novel markers for monitoring minimal residual disease in acute lymphoblastic leukemia. Blood. 2001;97(7):2115–2220. | |
Moreira I, Papaioannou M, Mortuza FY, et al. Heterogeneity of VH-JH gene rearrangement patterns: an insight into the biology of B cell precursor ALL. Leukemia. 2001;15(10):1527–1536. | |
Mortuza FY, Moreira IM, Papaioannou M, et al. Immunoglobulin heavy-chain gene rearrangement in adult acute lymphoblastic leukemia reveals preferential usage of J(H)-proximal variable gene segments. Blood. 2001;97(9):2716–2726. | |
Eckert C, von Stackelberg A, Seeger K, et al. Minimal residual disease after induction is the strongest predictor of prognosis in intermediate risk relapsed acute lymphoblastic leukaemia – long-term results of trial ALL-REZ BFM P95/96. Eur J Cancer. 2013;49(6):1346–1355. | |
Conter V, Bartram CR, Valsecchi MG, et al. Molecular response to treatment redefines all prognostic factors in children and adolescents with B-cell precursor acute lymphoblastic leukemia: results in 3184 patients of the AIEOP-BFM ALL 2000 study. Blood. 2010;115(16):3206–3214. | |
Bassan R, Spinelli O, Oldani E, et al. Improved risk classification for risk-specific therapy based on the molecular study of minimal residual disease (MRD) in adult acute lymphoblastic leukemia (ALL). Blood. 2009;113(18):4153–4162. | |
Spinelli O, Peruta B, Tosi M, et al. Clearance of minimal residual disease after allogeneic stem cell transplantation and the prediction of the clinical outcome of adult patients with high-risk acute lymphoblastic leukemia. Haematologica. 2007;92(5):612–618. | |
Thörn I, Forestier E, Botling J, et al. Minimal residual disease assessment in childhood acute lymphoblastic leukaemia: a Swedish multi-centre study comparing real-time polymerase chain reaction and multicolour flow cytometry. Br J Haematol. 2011;152(6):743–753. | |
Lovisa F, Mussolin L, Corral L, et al. IGH and IGK gene rearrangements as PCR targets for pediatric Burkitt’s lymphoma and mature B-ALL MRD analysis. Lab Investig. 2009;89(10):1182–1186. | |
Gimenez E, Chauvet M, Rabin L, et al. Cloned IGH VDJ targets as tools for personalized minimal residual disease monitoring in mature lymphoid malignancies; a feasibility study in mantle cell lymphoma by the Groupe Ouest Est d’Etude des Leucémies et Autres Maladies du Sang. Br J Haematol. 2012;158(2):186–197. | |
Damle RN, Wasil T, Fais F, et al. Ig V gene mutation status and CD38 expression as novel prognostic indicators in chronic lymphocytic leukemia. Blood. 1999;94(6):1840–1847. | |
Hamblin TJ, Davis ZA, Oscier DG. Determination of how many immunoglobulin variable region heavy chain mutations are allowable in unmutated chronic lymphocytic leukaemia – long-term follow up of patients with different percentages of mutations. Br J Haematol. 2008;140(3):320–323. | |
Böttcher S, Stilgenbauer S, Busch R, et al. Standardized MRD flow and ASO IGH RQ-PCR for MRD quantification in CLL patients after rituximab-containing immunochemotherapy: a comparative analysis. Leukemia. 2009;23(11):2007–2017. | |
Caspersson T, Zech L, Johansson C, Modest EJ. Identification of human chromosomes by DNA-binding fluorescent agents. Chromosoma. 1970;30(2):215–227. | |
Seabright M. A rapid banding technique for human chromosomes. Lancet. 1971;2(7731):971–972. | |
Kallioniemi A, Kallioniemi OP, Sudar D, et al. Comparative genomic hybridization for molecular cytogenetic analysis of solid tumors. Science. 1992;258(5083):818–821. | |
Barrett MT, Scheffer A, Ben-Dor A, et al. Comparative genomic hybridization using oligonucleotide microarrays and total genomic DNA. Proc Natl Acad Sci U S A. 2004;101(51):17765–17770. | |
Jalali GR, Vorstman JAS, Errami A, et al. Detailed analysis of 22q11.2 with a high density MLPA probe set. Hum Mutat. 2008;29(3):433–440. | |
Mullighan CG, Goorha S, Radtke I, et al. Genome-wide analysis of genetic alterations in acute lymphoblastic leukaemia. Nature. 2007;446(7137):758–764. | |
Walker BA, Wardell CP, Brioli A et al. Translocations at 8q24 juxtapose MYC with genes that harbor superenhancers resulting in overexpression and poor prognosis in myeloma patients. Blood Cancer J. 2014;4:e191. | |
Bouamar H, Abbas S, Lin AP, et al. A capture-sequencing strategy identifies IRF8, EBF1, and APRIL as novel IGH fusion partners in B-cell lymphoma. Blood. 2013;122(5):726–733. | |
Logan AC, Gao H, Wang C, et al. High-throughput VDJ sequencing for quantification of minimal residual disease in chronic lymphocytic leukemia and immune reconstitution assessment. Proc Natl Acad Sci U S A. 2011;108(52):21194–21199. | |
Ladetto M, Brüggemann M, Monitillo L, et al. Next-generation sequencing and real-time quantitative PCR for minimal residual disease detection in B-cell disorders. Leukemia. 2014;28(6):1299–1307. | |
Faham M, Zheng J, Moorhead M, et al. Deep-sequencing approach for minimal residual disease detection in acute lymphoblastic leukemia. Blood. 2012;120(26):5173–5180. |
 © 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.