")
Back to Journals » Drug Design, Development and Therapy » Volume 14
AZD4547 Attenuates Lipopolysaccharide-Induced Acute Kidney Injury by Inhibiting Inflammation: The Role of FGFR1 in Renal Tubular Epithelial Cells
Authors Chen X, Zhang X, Xu J, Zhao Y, Bao J, Zheng Z, Han J
Received 23 July 2019
Accepted for publication 16 February 2020
Published 26 February 2020 Volume 2020:14 Pages 833—844
DOI https://doi.org/10.2147/DDDT.S224343
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 4
Editor who approved publication: Dr Sukesh Voruganti
Xuemei Chen,1,* Xuejiao Zhang,1,* Jiajun Xu,2 Yiqing Zhao,1 Jiachun Bao,1 Zhanxiong Zheng,2 Jibo Han2
1Department of Pharmacy, Affiliated Hospital of Jiangnan University, Wuxi, Jiangsu 214041, People’s Republic of China; 2Department of Cardiology, The Second Affiliated Hospital of Jiaxing University, Jiaxing, Zhejiang 314000, People’s Republic of China
*These authors contributed equally to this work
Correspondence: Jibo Han
Department of Cardiology, The Second Affiliated Hospital of Jiaxing University, Huancheng North Road 1518, Jiaxing, Zhejiang 314000, People’s Republic of China
Tel +86-152 5863 9207
Email [email protected]
Introduction: Inflammation plays an important role in the pathogenesis of acute kidney injury (AKI). Fibroblast growth factor receptor 1 (FGFR1) signaling is implicated in kidney pathology. AZD4547 is a small molecule inhibitor of FGFR1.
Materials and Methods: Here, we investigated whether AZD4547 could mitigate inflammatory responses in AKI. C57BL/6 mice were injected with lipopolysaccharide (LPS) to induce AKI. FGFR1 was blocked using AZD4547 or CRISPR/Cas9 genome editing. After immunofluorescent double-staining of kidney tissues showing that P-FGFR1 was localized to renal tubular epithelial cells, a tubular epithelial cell line (NRK-52E) was used for in vitro analysis.
Results: AZD4547 significantly reduced renal inflammation, cell apoptosis, and kidney dysfunction in AKI mice. In vitro, treatment of NRK-52E cells with AZD4547 attenuated LPS-induced inflammatory responses and was associated with downregulated P-FGFR1 levels. These findings were further confirmed in NRK-52E cells by knocking down the expression of FGFR1.
Conclusion: Our findings provide direct evidence that FGFR1 mediates LPS-induced inflammation leading to renal dysfunction. We also show that AZD4547 is a potential therapeutic agent to reduce inflammatory responses in AKI. Both FGFR1 and AZD4547 may interesting therapeutic options to combat AKI.
Keywords: acute kidney injury, lipopolysaccharide, inflammation, AZD4547, renal tubular epithelial cells
Introduction
Acute kidney injury (AKI), one of the most prominent and clinically critical care syndrome, increases the risk of mortality during the uncontrolled systemic inflammatory response.1,2 In AKI, renal function degrades rapidly, resulting in increased serum creatinine levels and decreased urine output.3 AKI results from various events, including sepsis, organ transplantation, cardiac surgery and rheumatic fever.4–6 Among these various structural and functional events, sepsis seems to be the most important cause of acute renal damage and lipopolysaccharide (LPS) remains the secondary cause of systemic inflammatory response syndrome.1,3 It is estimated that the occurrence of AKI leads to 50% mortality in ICU patients.7 Thus, exploring new and effective therapeutic options to prevent this epidemic is necessary.
AZD4547 is a highly selective, orally bioavailable, small molecule inhibitor of fibroblast growth factor receptor 1 (FGFR1). AZD4547 selectively inhibits FGFR1 phosphorylation and represses the proliferation of cancer cells by inhibiting FGFR1 signaling.8 Several reports have previously implicated FGFR1 signaling in kidney pathology.9–11 Baelde et al observed that FGF1/FGFR1 signaling is downregulated in kidney tissue from diabetic subjects.9 Importantly, with relevance to our study, in normal kidneys, FGFR1 is expressed in the tubular epithelium, mesangial cells and glomerular endothelial cells. The expression of FGFR1 is increased over normal above tubular epithelial cells in inflammatory renal diseases, including lupus nephritis (LN), chronic allograft nephropathy (CAN), and acute interstitial nephritis (AIN).11 Recently, we have shown that FGFR1 antagonism by either AZD4547 or siRNA silencing attenuates LPS- induced activation of hepatic stellate cells via suppressing inflammation.12 These findings suggest that FGFR1 blockage may have the potential to reduce the severity of inflammatory injury in the kidney.
In this study, we investigated the potential activity of AZD4547 against inflammatory responses in AKI. Using the LPS-induced septic mouse model, we show that AZD4547 attenuates indices of inflammatory responses in kidneys and protects against kidney dysfunction. We found that these activities were mediated, at least in part, by modulating FGFR1. We then confirmed the significant contribution of FGFR1 in renal tubular epithelial cells challenged with LPS. Furthermore, we found that AZD4547 modulated the TRAF6/nuclear factor (NF)-κB inflammatory pathway by blocking FGFR1/TRAF6 complex formation.
Methods
Reagents
AZD4547 was purchased from Shanghai Kai Yu Pharmatech Technology Co., Ltd. (Shanghai China). AZD4547 was dissolved in dimethyl sulfoxide (DMSO) for in vitro studies and in 1% sodium carboxymethyl cellulose (CMC-Na) for in vivo experiments. LPS was purchased from Sigma-Aldrich (St. Louis, MO, USA).
Animal Experiments
4-week old male C57BL/6 mice weighing 18–22g were obtained from Wenzhou Medical University Animal Center (Wenzhou, China). The mice were housed at constant room temperature with a 12:12 h light-dark cycle and fed with a standard rodent diet. All animal care and experimental protocols were performed in accordance with the Guidelines for the Care and Use of Laboratory Animals (US National Institutes of Health) and were approved by the Affiliated Hospital of Jiangnan University Animal Policy and Welfare Committee (No. 2019DWLL001). The mice were randomly divided into three weight-matched groups: 1) saline-treated control mice (intragastric administration of 0.9% saline, Ctrl, n=7), 2) mice challenged with LPS (intraperitoneal injection with 15 mg/kg of LPS, LPS, n=7), and 3) mice challenged with LPS and treated with AZD4547 (intragastric administration of AZD4547 at 40 mg/kg13 for 1 hr before LPS treatment, LPS+ AZD, n=7). Twenty-four hours following the initiation of the treatments, the mice were anesthetized by intraperitoneal injection of 1% sodium pentobarbital (40 mg/kg) and sacrificed. Blood and renal tissues were collected. Serum creatinine (Cr) and urea nitrogen (UN) were detected using commercial kits (Nanjing Jiancheng, Jiangsu, China). Kidney tissues were either fixed in 4% paraformaldehyde for histological analysis or flash-frozen in liquid nitrogen for gene and protein analyses.
Histological Assessments
Renal tissues from mice were fixed in 4% paraformaldehyde and embedded in paraffin. Tissues were then cut into 5 μm sections and stained with hematoxylin and eosin (H&E) for histology. Stained sections were viewed under microscope (Nikon, Japan).
Apoptosis was measured by terminal deoxynucleotidyl transferase-mediated dUTP nick end labeling (TUNEL) staining (R&D Systems, Minneapolis, MN). Images were viewed under fluorescence microscope (Nikon, Japan).
For double labeling, the sections were incubated with both P-FGFR1 antibody and an antibody against Wilms tumor 1 (WT-1, Novus Biologicals, Littleton, CO; NBP2-44607, 1:200) or aquaporin 1 (AQO-1, Santa Cruz Biotechnology; sc-32737, 1:200). The slides were then incubated with 2 secondary antibodies (TRITC- labeled secondary, Abcam, ab6786, 1:500 or Alexa Fluor488- labeled secondary, Abcam, ab150077, 1:500) for 1 h at room temperature.
Determination of Cytokine Levels
TNF-α and IL-6 proteins in kidney tissues were determined by using cytokine-specific ELISA kits (eBiosciences Inc, CA, USA).
Cell Culture Studies
Rat tubular epithelial NRK-52E cells were obtained from the Shanghai Institute of Biochemistry and Cell Biology (Shanghai, China). NRK-52E cells were grown in Dulbecco’s modified Eagle’s medium (DMEM) (Gibco, Eggenstein, Germany) containing 5.5 mM D-glucose, 5% fetal bovine serum (FBS), 100 U/mL penicillin, and 100 mg/mL streptomycin.
FGFR1 knockout NRK-52E cells were achieved by CRISPR/Cas9 genome editing according to the protocol described in a previous publication.14 The sgRNA sequence (5′-GCAACCGTACACGCATCACAGGG-3′ for FGFR1) was cloned into the pSpCas9 (BB)-2A-Puro (PX459) vector. Then, the plasmid with sgRNA was transfected into NRK-52E cells using Lipofectamine LTX. Forty-eight hours after transfection, the cells were selected by puromycin dihydrochloride (1 µg/mL). Knockdown was verified by immunoblotting.
The CCK8 assay was used to assess the cell viability of NRK-52E cells according to the manufacturer’s instructions (WST-8, Dojindo, Kumamoto, Japan).
NF-κB Activity Assay
NF-κB activity was detected using the NF-κB-EGFP NRK-52E stable cell line. The NF-κB-EGFP NRK-52E stable cell line was prepared by lentiviral infection as described in our previous publication.15,16 During the experiment, the NFκB-EGFP NRK-52E stable cells were incubated with LPS (0.5 μg/mL) for 6 h. Mean fluorescence intensity was analyzed by flow cytometry.
In vitro Ubiquitination
Ubiquitination reactions were performed in the presence of HEPES buffer (10 mmol/L), E1 (0.067 μL; 30 nM), ubiquitin (0.13 μL; 0.5 μM), UBC13-UEV1A (0.2 μL; 0.4 μM), TRAF6 and AZD4547 (1 μL; 2.5, 5, 10 or 20 μM). The reaction mixtures were incubated on ice for 10 min. ATP was then added, and the samples were incubated at 37°C for 30 min. The reaction was terminated by adding loading buffer and heating the samples at 85°C for 5 min.
Western Blotting and Co-Immunoprecipitation
The general procedure for Western blot analysis was described in our previous publication.17 Antibodies against P-FGFR1, FGFR1, transforming growth factor-β (TGF-β), collagen (col) IV, Bax, Bcl-2, IκB-α, P-TGF-β activated kinase 1 (TAK1), TAK1, GAPDH and the secondary horseradish peroxidase-conjugated antibody were obtained from Santa Cruz Biotechnology (Santa Cruz, CA). Antibody against Ub was obtained from Cell Signaling Technology (CST, CA, USA). Antibody against TNF receptor associated factor-6 (TRAF6) was obtained from Abcam (Cambridge, MA, USA).
For immunoprecipitation experiments, cell extracts were incubated with anti-TRAF6-antibody for 4 h and were then precipitated with protein G-Sepharose beads at 4 °C overnight. FGFR1 and TRAF6 levels were detected by Western blotting using specific antibodies.
Real-Time Quantitative qPCR
The general procedure for RT-qPCR was described in our previous publication.17 Primers for genes including TGF-β, Col-IV, Bax, Bcl-2, TNF-α, IL-6 and β-actin were synthesized by Invitrogen (Invitrogen, Shanghai, China). The primer sequences used are shown in Table S1. Target mRNA was normalized to β-actin.
Statistical Analysis
All in vitro data represent 3 independent experiments and are expressed as the means ± SEM. Statistical analyses were performed using GraphPad Pro Prism 5.0 (GraphPad, San Diego, CA). Student’s t-test, one-way ANOVA followed by multiple comparisons test with Bonferroni correction or two-way ANOVA test was employed to analyze the differences between sets of data. A P value < 0.05 was considered significant.
Results
AZD4547 Protects Against LPS-Induced Renal Dysfunction, Cell Apoptosis and Inflammatory Factor Release
We first investigated the renoprotective effects of AZD4547 in LPS-induced acute kidney injury by treating mice with 40 mg/kg AZD4547.13 AKI mice showed increased serum urea nitrogen (UN) and creatinine (Cr) levels (Figure 1A and B, respectively). These changes were not observed in AKI mice treated with AZD4547. We next analyzed the effects of AZD4547 on kidney tissues by histology. H&E staining of kidney tissues from LPS mice showed renal tubular dilation, distortion and tubular epithelial cell edema (Figure 1C). Administration of AZD4547 normalized these AKI-associated tubule histopathology, indicating that AZD4547 protected kidney function in LPS-treated mice. We then stained the kidney tissues with Masson trichrome to assess fibrotic changes. Our results showed no increased collagen deposition in the interstitial compartments in diabetic mice (Figure S1A). Inconsistent with our histology findings, RT-qPCR analysis of transforming growth factor-β (TGF-β) and collagen IV showed increased transcript levels in kidney tissues of AKI mice, and AZD4547 normalized the levels of collagen IV and TGF-β (Figure 1D–E, respectively).
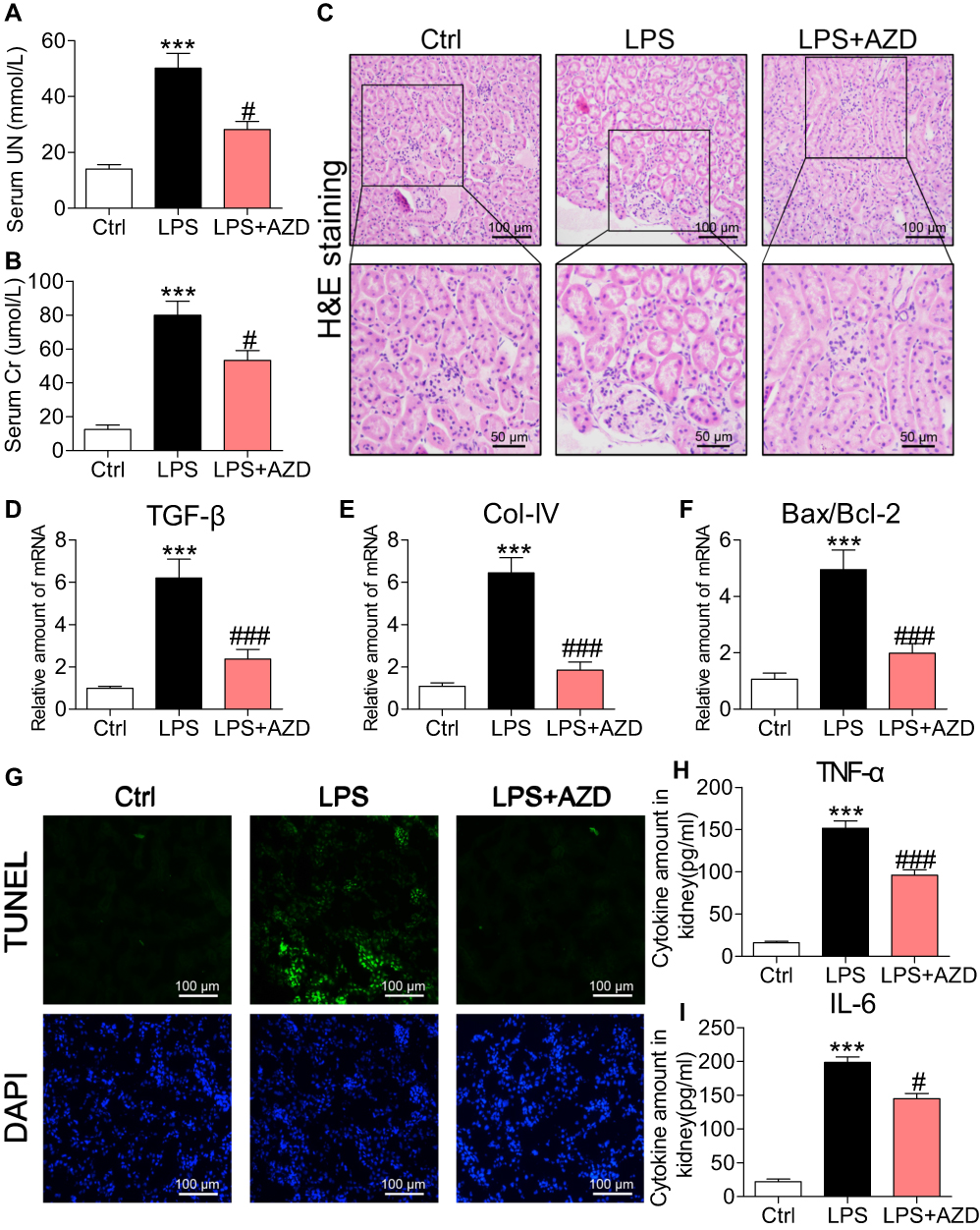 |
Figure 1 AZD4547 normalizes LPS-induced renal dysfunction, cell apoptosis and inflammatory factor release. (A, B) Levels of serum urea nitrogen (UN) and creatinine (Cr) in LPS mice and mice treated with AZD4547[n= 6 per group]. (C) Representative images for H&E staining [scale bar= 100 μm (upper) or 50 μm (below)]. (D–F) mRNA levels of TGF-β, Col-IV and Bax: Bcl-2 ratio in kidney tissues of mice as determined by qPCR [mRNA levels were normalized to β-actin; n= 6 per group]. (G) Apoptotic DNA fragmentation in renal tissues of LPS mice as detected by TUNEL staining (green). Tissues were counterstained with DAPI (blue). [scale bar= 50 μm]. (H, I) Levels of TNF-α and IL-6 proteins in kidney tissues as detected by ELISA [n= 6 per group]. (*, vs Ctrl group; #, vs LPS group; #P<0.05, *** and ###P<0.001.) |
To assess whether the effects of AZD4547 on preserving renal function in AKI are mediated through the modulation of cell apoptosis, we measured the proapoptotic gene Bax and the antiapoptotic gene Bcl-2. RT-qPCR analysis showed an increased Bax: Bcl-2 ratio (Figure 1F). AZD4547 significantly alleviated this LPS-induced change. TUNEL staining of kidney specimens from LPS mice showed increased TUNEL-positive apoptotic cells in renal tubules (Figure 1G). However, treatment with AZD4547 prevented this increase.
We next assessed whether inflammation was involved in the actions of AZD4547. We measured renal TNF-α and IL-6 levels by ELISA and found that these proinflammatory factors were increased in AKI mice (Figure 1H and I, respectively). Such an increase was not observed in LPS mice treated with AZD4547. Renal inflammation is a sum effect of tubular epithelial cells, podocytes and glomeruli cells expressing various proinflammatory cytokines, cell surface molecules, and chemokines that recruit macrophages. Recruited cells substantially increase inflammatory factors in the kidney. Thus, we performed an additional analysis to detect the infiltration of macrophages in the kidney tissues of AKI mice. We stained kidney tissues for CD68 and found increased immunoreactivity in AKI mice compared to that in Ctrl mice (Figure S1B). CD68 immunoreactivity was reduced in kidney tissues of AZD4547-treated mice, suggesting decreased infiltration. These findings strongly suggest that AZD4547 protects against LPS-related renal dysfunction, cell apoptosis and inflammation in mouse kidneys.
FGFR1 Activation Is Upregulated in Renal Tubular Epithelial Cells in LPS-Induced AKI Mice
AZD4547 is a small-molecule inhibitor that specifically inhibits FGFR1 activity.8 To confirm the role of FGFR1 in LPS-related renal inflammation and dysfunction, we examined the activation and localization of FGFR1 in kidney tissues to identify the cellular source. FGFR1 phosphorylation was robustly increased in the kidneys of AKI mice as detected by Western blotting (Figure 2A). Immunofluorescence staining showed P-FGFR1 immunoreactivity in cells expressing AQP-1 implicating renal tubular epithelial cells as the cellular source/target (Figure 2B). P-FGFR1 immunoreactivity, although significantly reduced, was also observed in WT-1 positive podocytes and glomeruli cells (Figure 2C). These findings show the role of FGFR1 in LPS-induced AKI and that renal tubular epithelial cells are perhaps the major contributor to the inflammatory responses.
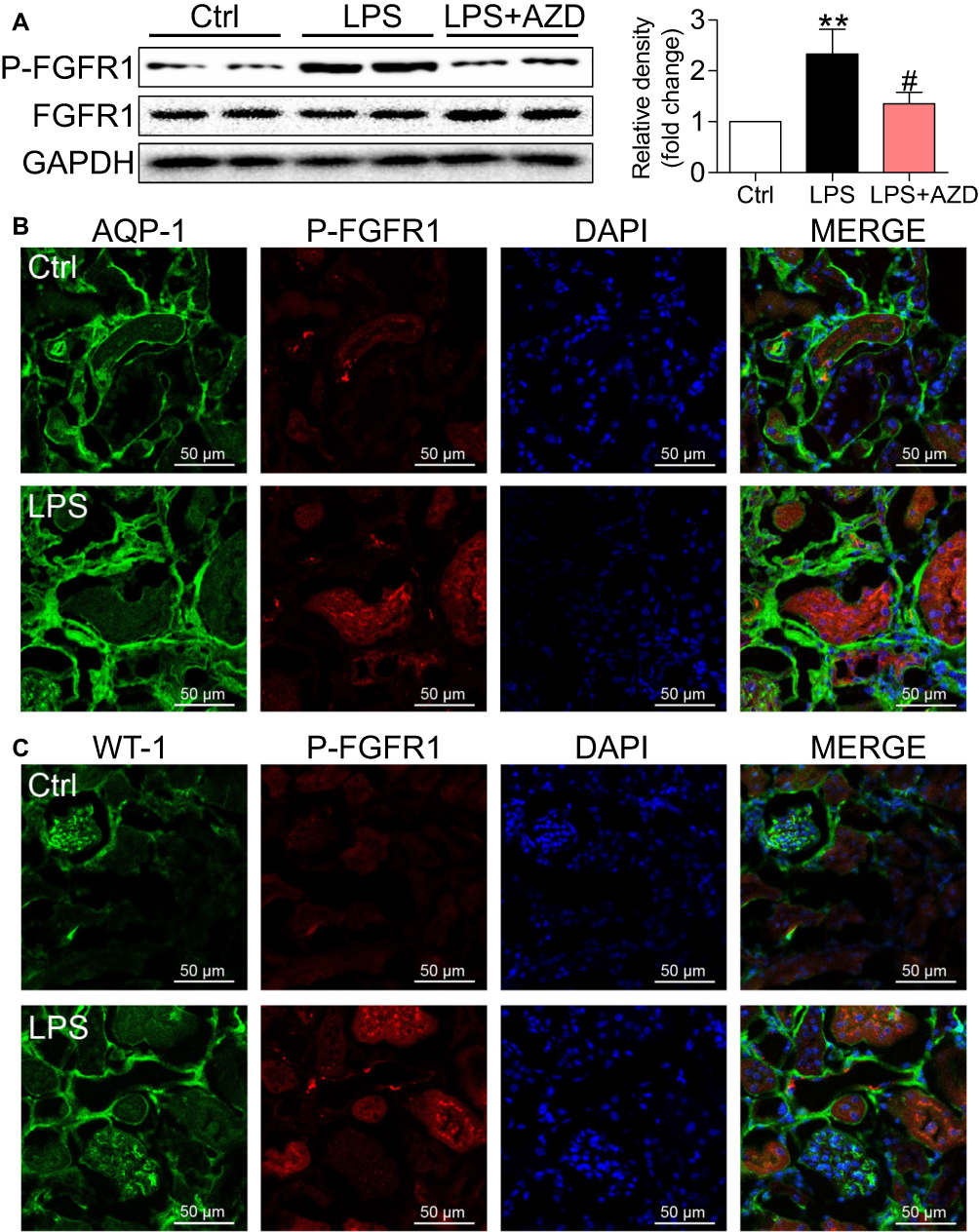 |
Figure 2 FGFR1 activation is upregulated in kidney tissues in LPS mice. (A) Western blot and densitometric analysis of P-FGFR1 and FGFR1 levels in kidney tissues [n=5 per group; 2 Ctrl, 2 LPS and 2 LPS+AZD samples shown in immunoblots]. (B) Immunofluorescence staining for P-FGFR1 (red) and AQO-1 (green) showing immunoreactivity of P-FGFR1 in tubular epithelial cells [scale bar = 50 μm]. Merged images (yellow) showing colocalization. (C) Immunofluorescence staining of kidney tissues for P-FGFR1 (red) and WT-1 (green) [scale bar = 50 μm]. WT-1 was used as a marker of podocytes. (*, vs Ctrl group; #, vs LPS group; #P<0.05, **P<0.01.) |
AZD4547 Inhibits LPS-Induced Inflammatory Deficits in Cultured Renal Tubular Epithelial Cells
To provide mechanistic insights into the role of AZD4547 and FGFR1 in LPS-induced kidney dysfunction, we challenged a cultured rat tubular epithelial cell line (NRK-52E) with LPS to model renal proximal tubular epithelial cells in AKI. As shown in Figure S2, AZD4547 significantly inhibited cell proliferation at a concentration of 20 μM in NRK-52E cells. Thus, we selected 10 μM for our study. NRK-52E cells were pretreated with AZD4547 for 1 h and then stimulated with 0.5 μg/mL LPS for the indicated times. LPS induced a robust increase in FGFR1 phosphorylation that was inhibited by AZD4547 pretreatment (Figure 3A). To evaluate the effect of AZD4547 on LPS-induced inflammatory injury, we determined the cell viability of NRK-52E cells. AZD4547 pretreatment increased cell viability of LPS-stimulated NRK-52E cells (Figure 3B). Upon sustained LPS stimulation for 24 h, the protein expression of Bax increased and Bcl-2 decreased (Figure 3C and D). Pretreatment with AZD4547 reversed these changes. As shown in Figure 3E and F, LPS treatment induced expression of TGF-β and collagen IV, both of which were reduced by AZD4547. We also measured TGF-β, Col-IV, Bax, and Bcl-2 using RT-qPCR in vitro. We have included these data in Figure S2B-D, which showed that under LPS stimulation, the mRNA levels of TGF-β, Col-IV and Bax: Bcl-2 ratio were increased, all these changes were reversed by AZD4547.
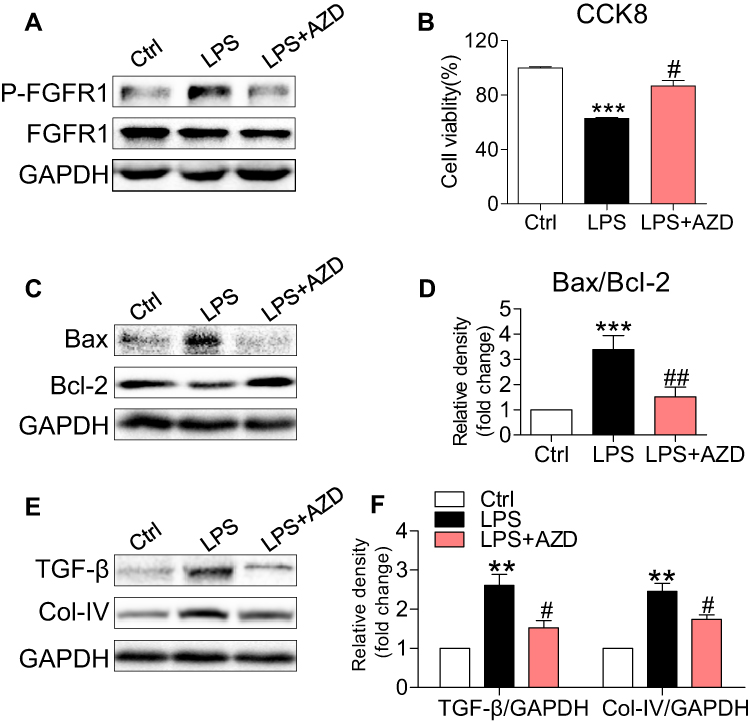 |
Figure 3 AZD4547 inhibits LPS-induced cell apoptosis and fibrosis in NRK-52E cells. NRK-52E cells were pretreated with 10 μM AZD4547 for 1 h, and then exposed to 0.5 μg/mL LPS for the indicated times. (A) Incubation with LPS for 15 min. p-FGFR1 levels was detected by immunoblots. (B) Incubation with LPS for 24 h. The cell viability of NRK-52E cells was detected by CCK8 assay [n= 3 per group]. (C, D) Exposure to LPS for 24 h. The expression of Bax and Bcl-2 in cell lysates were detected by immunoblots [n= 3 per group]. (E, F) Exposure to LPS for 24 h, the levels of TGF-β and Col-IV protein were detected by Western blot [n= 3 per group]. (*, vs Ctrl group; #, vs LPS group; #P<0.05, ## and **P<0.01, ***P<0.001.) |
To confirm the roles of AZD4547 and FGFR1 in LPS-associated renal inflammation, we investigated the levels of proinflammatory factors in NRK-52E cells. AZD4547 reduced LPS-induced TNF-α and IL-6 mRNA levels (Figure 4A and B, respectively). We know from previous studies that LPS-induced activation of NF-κB signaling causes renal inflammation.18,19 To test this hypothesis, we generated a stable NRK-52E cell line expressing the NF-κB reporter (EGFP) and exposed the cells to LPS. AZD4547 diminished LPS-induced NF-κB activity (Figure 4C). Furthermore, we also observed reduced activation of NF-κB by AZD4547, as evidenced by decreased degradation of IκB-α (Figure 4D) in LPS-stimulated NRK-52E cells. TAK1 has been shown to be involved in NF-κB activation.20 Consistent with these findings, AZD4547 inhibited the phosphorylation of TAK1 (Figure 4D). These results indicate that AZD4547 attenuates LPS-induced inflammatory factors by reducing the TAK1/NF-κB signaling pathway in NRK-52E cells.
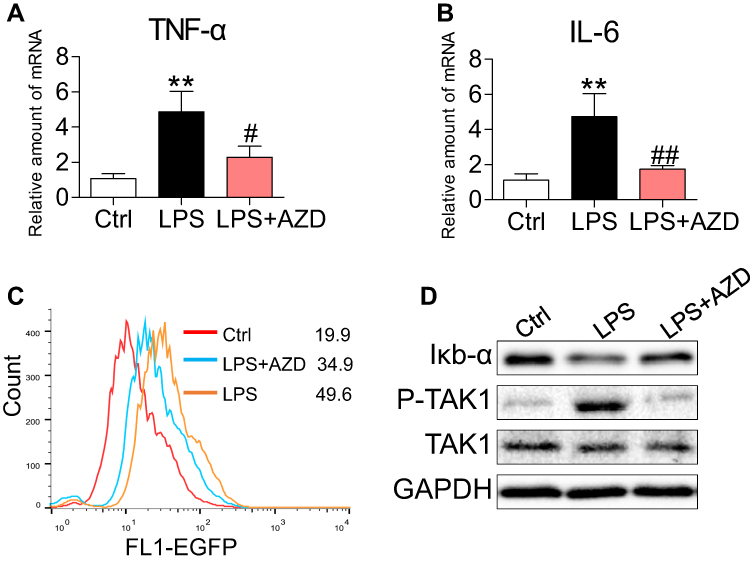 |
Figure 4 AZD4547 attenuates LPS-induced inflammatory factors by reducing TAK1/NF-κB in NRK-52E cells. NRK-52E cells were pretreated with 10 μM AZD4547 for 1 h and then exposed to 0.5 μg/mL LPS for the indicated times. (A, B) Cells were exposed to LPS for 6 h. The inflammatory cytokines TNF-α and IL-6 mRNA levels were detected by RT-qPCR [n= 3 per group]. (C) NF-κB EGFP reporter-expressing NRK-52E cells were treated with AZD4547 (10 μM) for 1 h before challenging the cells with LPS (0.5 μg/mL) for 6 h. NF-κB activity is shown as the mean fluorescence intensity (MFI) detected by flow cytometry. (D) Cells were exposed to LPS for 1 h. Western blot analysis of IκB-α, P-TAK1 and TAK1 levels in NRK-52E cells. (*, vs Ctrl group; #, vs LPS group; #P<0.05, ** and ##P<0.01.) |
FGFR1 Regulates LPS-Related Proinflammatory Pathway Activation in Renal Tubular Epithelial Cells
To confirm the importance of FGFR1 in mediating LPS-induced inflammatory deficits, we knocked out the expression of FGFR1 by using the CRISPR/Cas9 genome editing. Transfection of NRK-52E cells with plasmids targeting FGFR1 led to decreased FGFR1 expression as expected (Figure 5A) and ameliorated the LPS-induced reduction in cell viability (Figure 5B). Similarly, FGFR1 knockout reversed the LPS-induced increases in Bax, TGF-β and collagen IV and decreases in Bcl-2 (Figure 5C). We found that FGFR1 knockout caused decreases in TNF-α and IL-6 mRNA upon LPS stimulation (Figure 5D and E, respectively). We then examined NF-κB activity using a stable NRK-52E cell line expressing an NF-κB reporter (EGFP). FGFR1 knockout diminished LPS-induced NF-κB activity (Figure 5F). Upon sustained LPS stimulation for 1 h, degradation of IκB-α and phosphorylation of TAK1 increased (Figure 5G). FGFR1 knockout reversed these changes. These results suggest that FGFR1 inhibition directly reduces LPS-induced inflammatory responses and cell apoptosis in cultured tubular epithelial cells.
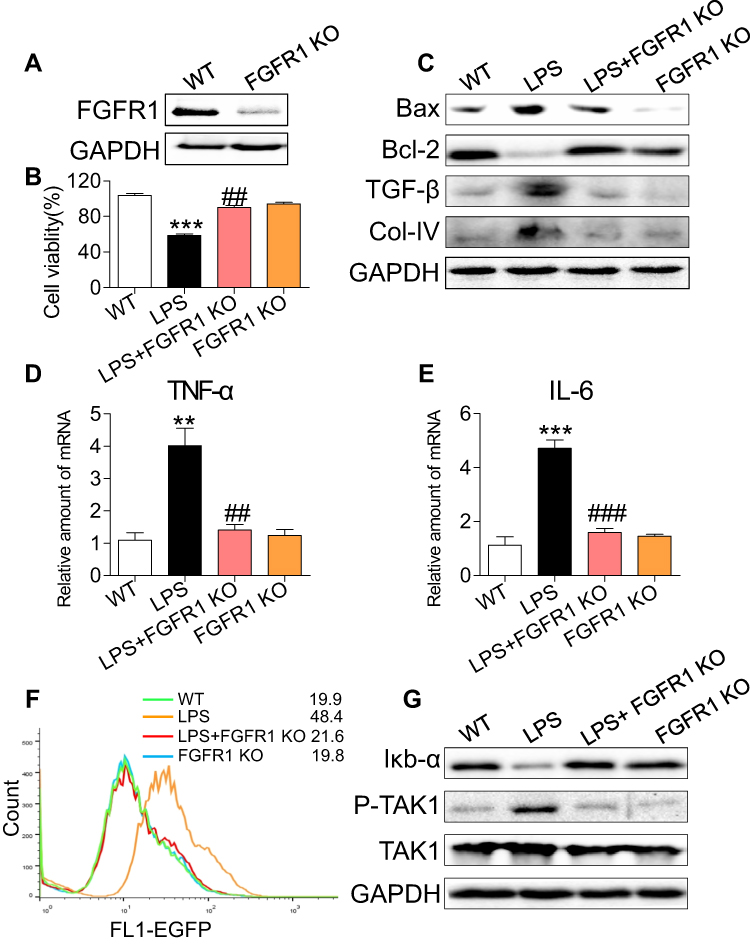 |
Figure 5 FGFR1 knockout decreases LPS-induced inflammatory deficits in NRK-52E cells. (A) Western blot analysis of FGFR1 in lysates generated from wild type (WT) and FGFR1 knockout (KO) NRK-52E cells. WT and FGFR1 KO NRK-52E cells were stimulated with LPS (0.5 μg/mL) for the indicated times. (B) Cells were incubated with LPS for 24 h. Cell viability was detected by CCK8 assay [n= 3 per group]. (C) Cells were exposed to LPS for 24 h. The levels of Bax, Bcl-2, TGF-β, and Col-IV were detected by immunoblots. (D, E) Cells were exposed to LPS for 6 h. TNF-α and IL-6 mRNA levels were detected by RT-qPCR [n= 3 per group]. (F) Cells were incubated with LPS (0.5 μg/mL) for 6 h. NF-κB activity is shown as the mean fluorescence intensity (MFI) detected by flow cytometry. (G) Cells were exposed to LPS for 1 h. Western blot analysis of IκB-α, P-TAK1 and TAK1 levels in WT and FGFR1 KO NRK-52E cells. (*, vs WT group; #, vs LPS group; ** and ##P<0.01, *** and ###P<0.001.) |
AZD4547 Modulates TRAF6-Related Polyubiquitination by Blocking FGFR1/TRAF6 Complex Formation
In LPS-induced TAK1/NF-κB signaling activation, protein ubiquitination of key accessory proteins such as TNF receptor associated factor-6 (TRAF6) may be involved.20 The interaction of TRAF6 with the ubiquitin-conjugating enzymes E1 and Ubc13–Uev2 (E2), catalyzes the formation of polyubiquitin chains.20 Therefore, we reasoned that AZD4547 may modulate TAK1/NF-κB signaling through alteration of TRAF6 ubiquitination. We first investigated the effect of AZD4547 on TRAF6-related ubiquitination in vitro. The ubiquitination reaction was carried out in the presence of E1, Ubc13–Uev2 (E2), TRAF6 (E3) and ubiquitin. Ubc13–Uev2 was activated by adding ATP. Polyubiquitination (polyUb) was significantly inhibited by AZD4547 in a dose-dependent manner (Figure 6A). These data show that AZD4547 blocks TRAF6-related polyUb and possibly inhibits TAK1/NF-κB through this mechanism. Based on our previous research,12 we tested whether LPS could induce the formation of the FGFR1/TRAF6 complex in NRK-52E cells. As shown in Figure 6B, co-immunoprecipitation showed that LPS challenge of NRK-52E cells for 15 to 30 min markedly increased the recruitment of TRAF6 to FGFR1, while the FGFR1 blocker AZD4547 completely prevented the FGFR1/TRAF6 interaction in NRK-52E cells (Figure 6C). These findings show a novel mechanism by which AZD4547 modulates inflammatory deficits in NRK-52E cells that involves LPS-mediated FGFR1/TRAF6 complex formation and protein ubiquitination.
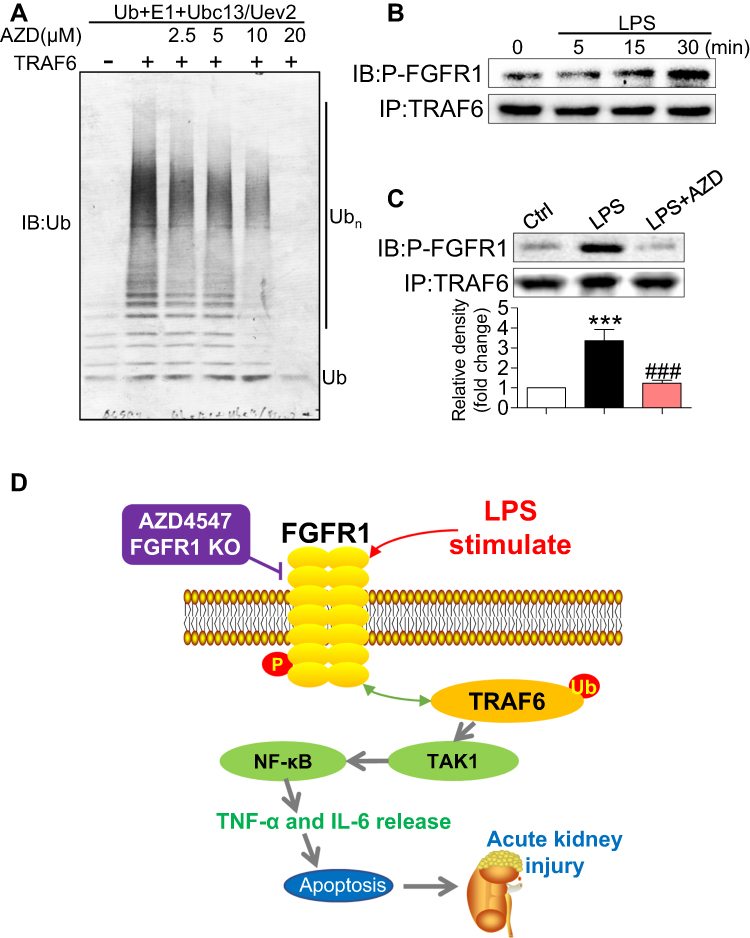 |
Figure 6 AZD4547 modulates TAK1/NF-κB signaling by increasing the interaction between P-FGFR1 and TRAF6. (A) Ubiquitination was carried out in the presence of TRAF6 and blocked by AZD4547 in a dose-dependent manner. Immunoblotting was performed with an anti-Ub antibody. (B) Lysates from LPS-stimulated (indicated times) NRK-52E cells were immunoprecipitated with TRAF6 antibody (IP) and immunoblotted (IB) for P-FGFR1. (C) Immunoprecipitation (IP) of lysates from LPS-stimulated (30 min) NRK-52E cells with AZD4547; IP of TRAF6 and immunoblotting (IB) for P-FGFR1 proteins [n= 3 per group]. (D) Schematic illustration of the potential mechanisms: LPS activates FGFR1 and increases the interaction between P-FGFR1 and TRAF6 to produce TAK1/NF-κB signaling activation and the production of inflammatory cytokines. These inflammatory deficits cause acute kidney injury. (*, vs Ctrl group; #, vs LPS group; *** and ###P<0.001.) |
Discussion
The salient finding of our study includes the demonstration that AZD4547 protects against LPS-associated renal inflammation, cell apoptosis, and functional deficits. We also showed that these activities of AZD4547 are, at least in part, mediated through the suppression of FGFR1. Analysis of kidney tissues showed upregulated activation of FGFR1 in renal tubular epithelial cells of LPS-induced AKI mice. Knocking down the expression of FGFR1 in cultured renal tubular epithelial cells challenged with LPS mirrored the results obtained from AZD4547 treatment. Finally, AZD4547 modulated the TRAF6/NF-κB inflammatory pathway by blocking FGFR1/TRAF6 complex formation (Figure 6D). Collectively, our findings provide evidence that FGFR1 is a potential therapeutic target to normalize inflammation in AKI and that AZD4547 may be a feasible strategy for treating acute renal injury.
FGFR1 expression has been reported in the tubular epithelium, glomerular endothelial cells and mesangial cells in normal human kidneys.11 We found that FGFR1 activation was upregulated in renal tubular epithelial cells in LPS-induced AKI mice (Figure 2). Although studies are needed to examine the contribution of FGFR1 in other renal cells, we do show that suppression of FGFR1 by either AZD4547 (Figures 3 and 4) or CRISPR/Cas9 genome editing (Figure 5) reduces NF-кB activity, and downstream expression of IL-6, TNF-α, as well as molecular markers of fibrosis and apoptosis in renal tubular epithelial cells (NRK-52E cells).
Elevated and sustained inflammation, including increased levels of proinflammatory cytokines IL-6 and TNF-α, is intricately linked to renal injuries.21 It is believed that increased IL-6 and TNF-α play a role in increased permeability, glomerular filtration rate, hemodynamic changes and renal fibrosis.21–23 In addition, inflammatory cytokines may also cause cell apoptosis by inducing the expression of pro-apoptotic factors such as Bax.24 Huang et al showed that AZD4547 improved the survival of cecal ligation and puncture (CLP) model mice and exhibited a protective function against lung injury via inhibiting the expression of the pro-inflammatory factors.25 In the present study, we showed that AZD4547 reduced LPS-induced inflammation, cell apoptosis and renal dysfunction in vivo (Figure 1). These findings suggest that FGFR1 pharmacological inhibition is protective against AKI and that FGFR1 plays an important role in kidney inflammatory responses, and structural and functional characteristics of AKI.
LPS does not directly interact with renal epithelial cells in the AKI model, as no evidence has shown that LPS penetrates the glomerular filtration barrier. LPS enters peripheral circulation and stimulates TNF-α and other proinflammatory cytokines, which induce inflammatory responses in cells. Therefore, we exposed NRK-52E cells to TNF-α with or without AZD4547. Our results show that TNF-α induced FGFR1 phosphorylation and that AZD4547 counteracted the TNF-α-induced increase in mRNA levels of TGF-β, Col-IV, and Bax: Bcl-2 ratio in NRK-52E cells (Figure S3). These results indicate that AZD4547 attenuates TNF-α-induced renal tubular epithelial cell injury.
Studies have shown that overactivation of TRAF6/NF-кB inflammatory signaling leads to inflammation in kidneys and the progression of AKI.18,19,24,26,27 Activation of NF-κB results in increased pro-inflammatory cytokine production in LPS-induced renal damage.18,19 Recent studies have also linked TRAF6 to sepsis complications, including AKI.27 Further implicating this pathway in AKI are studies that show inhibition of inflammation upon TRAF6 deficiency.26 In the Toll-like receptor 4 (TLR4) pathway, TRAF6 serves as the signal transducer that activates IκB kinase (IKK) in response to proinflammatory mediators such as LPS. IKK activation by TRAF6 requires a ubiquitin-conjugating enzyme complex composed of E1 and Ubc13–Uev2 (E2). This complex, together with TRAF6, catalyzes the formation of a polyubiquitin chain that mediates IKK activation.20 LPS-induced phosphorylation of key proteins of the NF-κB/MAPK/STAT3 pathways in RAW264.7 macrophages were inhibited by AZD4547.25 Based on these data, we hypothesized that AZD4547 reduces the polyubiquitination of TRAF6 and suppresses NF-κB activation. Indeed, our results show that AZD4547 reduced TRAF6-related ubiquitination (Figure 6A). The result of this suppression was reduced phosphorylation of TAK1. Furthermore, we identified for the first time that LPS increased the interaction between P-FGFR1 and TRAF6 (Figure 6B), while AZD4547 prevented the recruitment of TRAF6 to FGFR1 (Figure 6C).
In summary, our findings demonstrated that pharmacological inhibition of FGFR1 by AZD4547 and knockdown of FGFR1 attenuates LPS-associated renal inflammation, apoptosis and renal dysfunction. Our results revealed that P-FGFR1 is primarily localized to renal tubular epithelial cells and that inhibiting FGFR1 normalizes LPS-induced inflammatory factor expression by dampening NF-κB activation. Collectively, our study provides evidence that FGFR1 plays a significant role in inflammatory responses in renal tubular epithelial cells. Both FGFR1 and AZD4547 may represent interesting therapeutic options to combat AKI.
Data Sharing Statement
All other data are included within the Article or Supplementary Information or available from the authors on request.
Acknowledgments
This work was supported by the Public Welfare Science and Technology Program of Jiaxing City (grant no. 2018AY32008, 2017AY33032; Jiaxing, China); Wuxi Science and Technology Development Medical and Health Project Guiding Plan (grant no.2014 no.11; Wuxi, China); Youth research project of Wuxi No.3 people’s Hospital (grant no. 2019 no.3; Wuxi, China).
Disclosure
The authors declare no competing financial interests in this work.
References
1. Dellepiane S, Marengo M, Cantaluppi V. Detrimental cross-talk between sepsis and acute kidney injury: new pathogenic mechanisms, early biomarkers and targeted therapies. Crit Care. 2016;20:61. doi:10.1186/s13054-016-1219-3
2. Vaara ST, Pettilä V, Kaukonen K-M, et al. The attributable mortality of acute kidney injury. Crit Care Med. 2014;42(4):878. doi:10.1097/CCM.0000000000000045
3. Harty J. Prevention and management of acute kidney injury. Ulster Med J. 2014;83(3):149.
4. Plataki M, Kashani K, Cabellogarza J, et al. Predictors of acute kidney injury in septic shock patients: an observational cohort study. Clin J Am Soc Nephrol. 2011;6(7):1744. doi:10.2215/CJN.05480610
5. Thiele RH, Isbell JM, Rosner MH. AKI associated with cardiac surgery. Clin J Am Soc Nephrol. 2015;10(3):500. doi:10.2215/CJN.07830814
6. Li PKT, Burdmann EA, Mehta RL, World Kidney Day Steering C. Acute kidney injury: global health alert. Transplantation. 2013;95(5):653–657. doi:10.1097/TP.0b013e31828848bc
7. Uchino S, Kellum JA, Bellomo R, et al. Acute renal failure in critically Ill patients: a multinational, multicenter study. JAMA. 2005;294(7):813–818. doi:10.1001/jama.294.7.813
8. Tucker JA, Klein T, Breed J, et al. Structural insights into FGFR kinase isoform selectivity: diverse binding modes of AZD4547 and ponatinib in complex with FGFR1 and FGFR4. Structure. 2014;22(12):1764–1774. doi:10.1016/j.str.2014.09.019
9. Baelde HJ, Eikmans M, Doran PP, Lappin DW, de Heer E, Bruijn JA. Gene expression profiling in glomeruli from human kidneys with diabetic nephropathy. Am J Kidney Dis. 2004;43(4):636–650. doi:10.1053/j.ajkd.2003.12.028
10. Liang G, Song L, Chen Z, et al. Fibroblast growth factor 1 ameliorates diabetic nephropathy by an anti-inflammatory mechanism. Kidney Int. 2018;93(1):95–109. doi:10.1016/j.kint.2017.05.013
11. Rossini M, Cheunsuchon B, Donnert E, et al. Immunolocalization of fibroblast growth factor-1 (FGF-1), its receptor (FGFR-1), and fibroblast-specific protein-1 (FSP-1) in inflammatory renal disease. Kidney Int. 2005;68(6):2621–2628. doi:10.1111/j.1523-1755.2005.00734.x
12. Lou D, Han J, Zhou L, et al. Fibroblast growth factor receptor 1 antagonism attenuates lipopolysaccharide-induced activation of hepatic stellate cells via suppressing inflammation. Exp Ther Med. 2018;16(4):2909–2916. doi:10.3892/etm.2018.6586
13. Schneider MK, Ioanas HI, Xandry J, Rudin M. An in vivo wound healing model for the characterization of the angiogenic process and its modulation by pharmacological interventions. Sci Rep. 2019;9(1):6004.
14. Ran FA, Hsu PD, Wright J, Agarwala V, Scott DA, Zhang F. Genome engineering using the CRISPR-Cas9 system. Nat Protoc. 2013;8(11):2281–2308. doi:10.1038/nprot.2013.143
15. Chen T, Luo W, Wu G, et al. A novel MyD88 inhibitor LM9 prevents atherosclerosis by regulating inflammatory responses and oxidative stress in macrophages. Toxicol Appl Pharmacol. 2019;370:44–55. doi:10.1016/j.taap.2019.03.012
16. You S, Qian J, Wu G, et al. Schizandrin B attenuates angiotensin II induced endothelial to mesenchymal transition in vascular endothelium by suppressing NF-kappaB activation. Phytomedicine. 2019;62:152955. doi:10.1016/j.phymed.2019.152955
17. Han J, Ye S, Zou C, et al. Angiotensin II causes biphasic STAT3 activation through TLR4 to initiate cardiac remodeling. Hypertension. 2018;72(6):1301–1311. doi:10.1161/HYPERTENSIONAHA.118.11860
18. Amoah-Apraku B, Chandler LJ, Harrison JK, Tang SS, Ingelfinger JR, Guzman NJ. NF-kappa B and transcriptional control of renal epithelial-inducible nitric oxide synthase. Kidney Int. 1995;48(3):674–682. doi:10.1038/ki.1995.337
19. Wang Y, Rangan GK, Goodwin B, Tay YC, Harris DC. Lipopolysaccharide-induced MCP-1 gene expression in rat tubular epithelial cells is nuclear factor-kappaB dependent. Kidney Int. 2000;57(5):2011–2022. doi:10.1046/j.1523-1755.2000.00051.x
20. Wang C, Deng L, Hong M, Akkaraju GR, Inoue J, Chen ZJ. TAK1 is a ubiquitin-dependent kinase of MKK and IKK. Nature. 2001;412(6844):346–351. doi:10.1038/35085597
21. Akcay A, Nguyen Q, Edelstein CL. Mediators of inflammation in acute kidney injury. Mediators Inflamm. 2009;2009:12.
22. Furuichi K, Kaneko S, Wada T. Chemokine/chemokine receptor-mediated inflammation regulates pathologic changes from acute kidney injury to chronic kidney disease. Clin Exp Nephrol. 2009;13(1):9–14. doi:10.1007/s10157-008-0119-5
23. Gomez H, Ince C, De Backer D, et al. A unified theory of sepsis- induced acute kidney injury: inflammation, microcirculatory dysfunction, bioenergetics, and the tubular cell adaptation to injury. Shock. 2014;41(1):3–11. doi:10.1097/SHK.0000000000000052
24. Woods JS, Dieguez-Acuna FJ, Ellis ME, Kushleika J, Simmonds PL. Attenuation of nuclear factor kappa B (NF-kappaB) promotes apoptosis of kidney epithelial cells: a potential mechanism of mercury-induced nephrotoxicity. Environ Health Perspect. 2002;110(Suppl 5):819–822. doi:10.1289/ehp.02110s5819
25. Huang Y, Wang F, Li H, et al. Inhibition of fibroblast growth factor receptor by AZD4547 protects against inflammation in septic mice. Inflammation. 2019;42:1957–1967. doi:10.1007/s10753-019-01056-4
26. Liu S, Lutz J, Chang J, Liu D, Heemann U, Baumann M. TRAF6 knockdown promotes survival and inhibits inflammatory response to lipopolysaccharides in rat primary renal proximal tubule cells. Acta Physiol (Oxf). 2010;199(3):339–346. doi:10.1111/j.1748-1716.2010.02097.x
27. Ma J, Li YT, Zhang SX, Fu SZ, Ye XZ. MiR-590-3p attenuates acute kidney injury by inhibiting tumor necrosis factor receptor-associated factor 6 in septic mice. Inflammation. 2019;42(2):637–649. doi:10.1007/s10753-018-0921-5
 © 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.