Back to Journals » Degenerative Neurological and Neuromuscular Disease » Volume 4
Axonal transport and neurodegenerative disease: vesicle-motor complex formation and their regulation
Authors Anderson E, White II J, Gunawardena S
Received 13 November 2013
Accepted for publication 14 January 2014
Published 10 March 2014 Volume 2014:4 Pages 29—47
DOI https://doi.org/10.2147/DNND.S57502
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Eric N Anderson,* Joseph A White II,* Shermali Gunawardena
Department of Biological Sciences, The State University of New York at Buffalo, Buffalo, NY, USA
*These authors contributed equally to this work
Abstract: The process of axonal transport serves to move components over very long distances on microtubule tracks in order to maintain neuronal viability. Molecular motors – kinesin and dynein – are essential for the movement of neuronal cargoes along these tracks; defects in this pathway have been implicated in the initiation or progression of some neurodegenerative diseases, suggesting that this process may be a key contributor in neuronal dysfunction. Recent work has led to the identification of some of the motor-cargo complexes, adaptor proteins, and their regulatory elements in the context of disease proteins. In this review, we focus on the assembly of the amyloid precursor protein, huntingtin, mitochondria, and the RNA-motor complexes and discuss how these may be regulated during long-distance transport in the context of neurodegenerative disease. As knowledge of these motor-cargo complexes and their involvement in axonal transport expands, insight into how defects in this pathway contribute to the development of neurodegenerative diseases becomes evident. Therefore, a better understanding of how this pathway normally functions has important implications for early diagnosis and treatment of diseases before the onset of disease pathology or behavior.
Keywords: kinesin, dynein, amyloid precursor protein, huntingtin, microtubules
Introduction
Axonal transport within neurons is essential for cell viability, supplying the synapse with important components for survival. Unlike other cell types, neurons are polarized and can extend long distances (up to 1 m in humans). Many proteins that are newly made in the cell body are actively transported down axons along microtubule tracks by molecular motors. Microtubules (MTs) are long, hollow cylinders approximately 25 nm in diameter that are comprised of α- and -tubulin dimers.1 Therefore, all of the transported components synthesized in the cell body must be assembled, associated within complexes, attached to a particular engine or motor, and efficiently transported down the axon. As such, all of these steps must be highly regulated and any misregulation could result in transport deficits leading to neuronal dysfunction.
Experiments on transport kinetics classified two types of axonal transport, as fast and slow.2 Fast axonal transport moves components at rates of 100–400 mm day−1 (1–5 μms−1), while slow transport moves components considerably slower, 0.2–5 mm day−1 (0.01–0.001 μms−1). Fast axonal transport primarily transports vesicular cargoes and membrane-bound organelles, while slow axonal transport transports cytoskeletal components. Current knowledge indicates that the same molecular motors mediate both fast and slow axonal transport.3–5
In this review, we focus on fast axonal transport and its motor-cargo complexes. Given the complexity of axonal transport machinery, deficits in axonal transport can occur by several means, including complications with motor function, problems with cargo complex assembly or cargo attachment to motors, as well as malfunctions in regulatory elements. Recent studies have shown that deficits in these processes can either cause or exacerbate neurodegeneration.6,7 Some proteins involved in neurodegenerative diseases have also been implicated in this pathway with human disease mutations in these proteins causing defects in axonal transport.8–12 Importantly, mutations in motor proteins have also been reported to cause neurodegeneration.13 Furthermore, many of these disease proteins normally form complexes that have functional roles in axonal transport. Alterations in the assembly or defects in the regulation of these protein complexes may result in transport deficits instigating the disease pathology and behavioral defects seen in disease states. Thus, elucidating the mechanisms of axonal transport has important implications for potential early diagnosis of neuronal disease and for the development of therapeutic interventions before the onset of neuronal death.
Motor proteins and regulatory models
Conventional kinesin motor
The kinesin-1 motor protein, also called “conventional kinesin”, is part of a subfamily of motor proteins associated with MTs and move toward their plus ends (Figure 1).14 An important feature of kinesin-1 is its ability to take 8 nm steps as measured using in vitro experiments, performing processive movement.15 Kinesin-1 was first identified in squid axoplasm16,17 and is highly conserved in many species.18–22 Biochemical analyses of kinesin-1 from bovine brains revealed that it is a tetramer consisting of two heavy chains (120 kd) and two light chains (62 kd).23,24
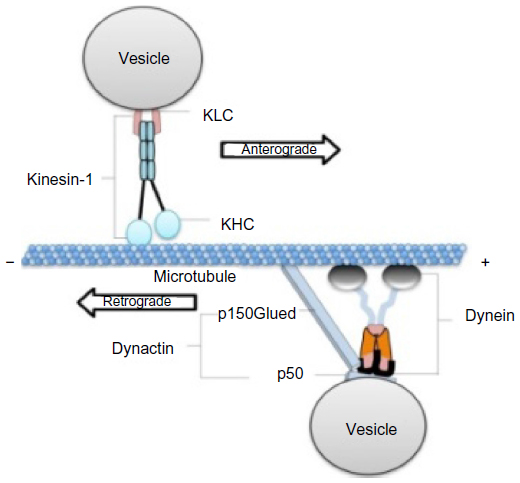 | Figure 1 Conventional kinesin (kinesin-1) and cytoplasmic dynein motor complex on a MT for axonal transport. |
In invertebrates, such as Drosophila, a single gene is responsible for each subunit while, in mammals, kinesin-1 is more complex and requires three genes for kinesin heavy chains (KIF5A, KIF5B, KIF5C) and three for kinesin light chains (KLC1, KLC2, KLC3).20,21
Sequence analysis, expression studies,22 and electron microscopy25,26 revealed that the kinesin heavy chain (KHC) subunit contains three domains: a motor domain; a stalk domain; and a tail domain. The globular motor domain at the N-terminus of KHC contains sites for MT and adenosine triphosphate (ATP) binding. The α-helical coiled-coil stalk domain contains sites where two heavy chains can dimerize, and the C-terminal tail domain interacts with the kinesin light chain (KLC) subunit.22,25–29 Classical studies using truncated fragments of Drosophila KHC expressed in Escherichia coli revealed that KHC is able to exclusively induce movement along MTs.30 Fragments that specifically contained the motor domain were capable of inducing MT motility,30 indicating that the KHC motor domain is required for movement.
Loss of function of KHC in Drosophila showed larval paralysis and axonal accumulations of synaptic vesicles in larval segmental nerves with lethality at early third instar larval stages.31–34 In mice, KIF5B is expressed ubiquitously while KIF5A and KIF5C are neuronal specific. Targeted disruption of the KIF5B or KIF5A gene in mice caused embryonic lethality,35,36 while knockout of the KIF5C gene caused a reduction in brain size with no gross changes in the nervous system. These mice were viable.37 Primary cultures from KIF5B null embryonic cells exhibited striking phenotypes of mitochondrial and lysosomal distribution, indicating that KIF5B may be essential for mitochondrial and lysosomal dispersion.35 A postnatal knockout of KIF5A exhibited seizures and death at approximately 3 weeks of age with neurofilament proteins accumulating in the cell bodies of peripheral sensory neurons, accompanied by a reduction in sensory axon caliber.36 Older animals also developed age-dependent sensory neuron degeneration, accumulation of neurofilaments in cell bodies, reductions in axons, and hind limb paralysis. Recent studies indicated that KIF5A is also essential for gamma-aminobutyric acid receptor transport and that deletion of KIF5A caused epileptic phenotypes.38 Interestingly, a missense mutation in the mammalian KIF5A gene was identified in a family with hereditary spastic paraplegia, an inherited neurodegenerative disease characterized by lower limb spasticity and weakness.39 Thus far, 19 of the 21 hereditary spastic paraplegia (SPG) disease mutations identified in humans are localized to the motor domain of KIF5A,39,40 indicating that these mutations cause disease by perturbing the proper transport of components to the synapse. Indeed, the hereditary spastic paraplegia mutation N256S, which occurs within the KIF5A gene, severely disrupted MT-based transport in Drosophila.13 Furthermore, a KIF5A mutation was also detected in a Charcot–Marie–Tooth type 2 patient belonging to a SPG mutant family.40 Thus, mutations in KHC can lead to neurodegeneration due to deficits in axonal transport.
The KLC subunit contains three domains: an N-terminal coiled-coil domain or heptad repeats which bind KHC; a tetratricopeptide repeat (TPR) domain; and a C-terminal domain. The TPR and C-terminal domains bind cargo and function as a linker between KHC and the transported cargoes.41 The TPR domain, which is a protein–protein interaction domain, has been shown to interact with a variety of adaptor proteins, such as the JNK-interacting proteins (JIP), huntingtin (HTT)-associated protein (HAP) 1,42,43 and amyloid precursor protein (APP).44 Structurally, TPR is composed of tandem repeats of 34 amino acids that each form a helix-turn-helix arrangement and a superhelical conformation of multiple TPR repeats.45 While Drosophila has only one KLC gene, mammals have three KLC genes – KLC1, KLC2, and KLC3. KLC1 is predominantly expressed in neuronal tissues while KLC2 shows a more ubiquitous pattern of expression.46 Removal of functional KLC1 resulted in significantly smaller mutant mice that exhibited pronounced motor disabilities.45 KLC2 was shown to play a role in Na/K-ATPase-containing vesicles in alveolar epithelial cells.47 Interestingly, KLC3 appears to play a unique and specialized role in spermatids.48 Loss of function of the single KLC gene in Drosophila showed paralysis phenotypes, axonal accumulations, and a third instar larval lethality.34 Interestingly, a polymorphism in KLC was found to associate with Alzheimer’s disease (AD),49 indicating that mutations in KLC may also lead to neuronal dysfunction.
Cytoplasmic dynein motor
Dynein, the retrograde motor, (Figure 1), is a multisubunit complex that consists of two catalytic heavy chains (DHC), two intermediate chains (DIC), and several light chains (DLC: Tctex1; roadblock; and LC8 subfamilies).14,50,51 In mammals, DHC is encoded by a single gene (Dync1h1), DIC is encoded by two genes (Dync1i1, Dync1i2), and DLC is encoded by a family of three genes (Dynlt1, Dynlt3, Dynlrb1, Dynlrb2, Dynll1, Dynll2).51,52 The motor domain is localized in the C-terminal region of DHC.53,54 Structurally, the C-terminus of DHC is comprised of six tandemly linked AAA (ATPases Associated with diverse cellular Activities) domains,53–56 with the first four AAA domains essential for dynein movement.57,58 Force production and translocation are achieved through the N-terminal region of DHC, also known as the “stem domain.”59 In vitro studies of the stem domain in Dictyostelium revealed that the dimerization of DHC and the binding of DIC and DLC can be achieved through this region.60 Collectively, the association of the smaller dynein subunits with the N-terminal region (tail domain) of DHC forms the cargo-binding complex for retrograde motility.
Cytoplasmic dynein associates with a protein complex called dynactin (Figure 1) for retrograde motility. Dynactin is composed of: p150Glued; dynamitin (p50); p62; actin-interacting protein (Arp)1 and Arp11; actin; p24; p25; p27; and capZ α and β;61 and plays an important role in dynein’s ability to bind to vesicles.61,62 Dynactin also binds to MTs via p150Glued,63,64 the largest subunit that interacts with both MTs and dynein. The N-terminal of p150Glued binds MTs while the two coiled-coil regions are required for its dimerization and interaction with DIC.63,65,66 Mutations in the MT-binding domain of p150Glued showed decreased affinity for MTs resulting in neuronal abnormalities.67 Binding of the dynein–dynactin complex to vesicles is mediated by the Arp1 filament, which has been shown to bind to the spectrin matrix on organelles.68–71
Both dynein and dynactin are required for viability. Using targeted disruption mice homozygous for loss of DHC died early, while heterozygous mice showed no abnormalities.72 However, in vitro cultures of DHC–/– blastocysts showed defects in Golgi vesiculation and lysosome dispersion, suggesting that DHC is required for cell proliferation and proper distribution of endosomes and lysosomes.73
In flies, dynein was also essential for survival. Loss of function of DHC or DLC caused lethality while partial loss of function mutations caused pleiotropic morphogenetic defects in bristle and wing development and resulted in female sterility.74–76 Most of these defects were associated with loss of cellular shape and structure due to a disorganization of the cytoskeleton.75 Functions for DLC in the regulation of axonogenesis have also been shown, indicating a role of cellular motors in growth cone guidance.77 Interestingly, missense point mutations in DHC resulted in decreased retrograde transport and progressive motor neuron degeneration accompanied by the formation of Lewy-like inclusion bodies.7 Two of these missense point mutations, legs at odd angles (Loa) and cramping 1 (Cra1) occurred in the DIC binding site and in the homo dimerization site of DHC.
Mutations in p150Glued are also linked to neurodegeneration. Mutations in the p150Glued dynactin subunit were identified in familiar forms of motor neuron disease, including amyotrophic lateral sclerosis (ALS) and distal spinal and bulbar muscular atrophy.78–83 A mutation in p150Glued was also identified in Perry syndrome, a rare atypical form of Parkinson’s disease resistant to L-3,4-dihydroxyphenylalanine (L-DOPA).84 Interestingly, postnatal overexpression of the p50 dynactin subunit induced late onset motor neuron disease in mice.78 Motor nerves from specimens obtained from ALS patients showed decreased transport of organelles, such as mitochondria.85 Interestingly, in vivo studies in intact sciatic nerves from the G93A superoxide dismutase 1 (SOD1) transgenic ALS mouse model showed decreased retrograde transport, which preceded visible disease symptoms.86,87 Collectively, these observations indicate that defects in dynein/dynactin function can lead to progressive neurodegeneration.
Regulatory models of bidirectional movement
Since both kinesin-1 and dynein motors are found together on mobile vesicles,88,89 experimental emphasis has been placed on elucidating the mechanisms of how these motors coordinate to achieve bidirectional motility.90 Thus far, three models have been proposed to explain bidirectional transport – the exclusionary, the tug-of-war, and the coordination models.
The exclusionary model of transport suggests that there is only one type of motor or motor complex attached to a vesicle at a given time, and so these vesicles move unidirectionally. In this model, different motors would have to bind/unbind to achieve the bidirectional movement seen with many cargoes. Alternatively, the entire complex could hop onto opposite polarity MTs in areas where MTs are not unipolar, such as dendrites.91 However, recent in vivo evidence has shown that there are, in fact, stable populations of both kinesin-1 and dynein motors on APP vesicles,92 suggesting that this model is incorrect, at least for APP vesicles under physiological conditions. Furthermore, in retrogradely moving early endosomes in Ustilago maydis, dynein-containing vesicles also contained the kinesin-3 complex, while kinesin-3 was located on vesicles moving in both directions.93 Therefore, although more than one type of kinesin together with dynein could be associated with cargo during motility under physiological conditions, only one motor might be active at a given time.
The tug-of-war model suggests that both kinesin-1 and dynein motors are simultaneously active on vesicles and that the motors producing the highest net force ultimately move vesicles in their respective direction,94 similar to a tug-of-war, with back-and-forth motility, ultimately resulting in the net movement in one direction. Much of the evidence for this model comes from studies done in vitro using purified motors. In contrast, the coordination model of bidirectional motility proposes that opposite-polarity motors work in regulated concert to achieve the observed complex vesicle motility rather than a simple tug-of-war mechanism. In this context, at one particular time, one motor is active for movement while the other motor is inactive. While the active motor dictates net movement in one direction, there is little back-and-forth motility.
Part of the reasoning behind the tug-of-war model lies in the observation that many transported vesicles will often change directions quickly in a process called “reversing”.95 In 2009, Soppina et al96 using Dictyostelium cells showed that endosomes experience elongation and slowed velocities during reversals, which is consistent with a tug-of-war mechanism. However, these results do not preclude the possibility of a transient tug-of-war mechanism that could result from delayed inactivation of externally regulated motors. Several studies have used various mathematical models, such as Monte Carlo simulations and models based on a lipid droplet system97,98 to show that the tug-of-war model is not only cooperative, but it can also be used to explain the dynamics of bidirectional movement, such as reversing.97,98
Research using origami DNA99 showed that opposite-polarity motors work to stall synthetic DNA cargoes and seem to follow a cooperative tug-of-war model. Furthermore, the motility of purified vesicles reconstituted in vitro closely resembled the movement of LysoTracker-positive vesicles in primary neurons, where bidirectional motility was interrupted with frequent directional switches, diffusional movement, and pauses. Quantitative analysis indicated that these vesicles copurified with a low number of stably bound motors, suggesting that a small complement of tightly bound motors, and small changes in the number of engaged motors can mediate large changes in the motility of cargo. These observations are consistent with predictions for a motor coordination via a stochastic tug-of-war model, where transport could be driven by the force-dependent kinetics of opposing motors in the absence of external regulation.100
Recently, many researchers have leaned toward a motor coordination model with motor regulation to explain bidirectional motility in vivo. This is, in part, due to the relatively high velocities of vesicle motility observed during in vivo analysis, which contradicts with the in vitro velocities. Since bidirectional motility must occur in a crowded environment, many cargoes must maneuver in both directions on a single microtubule track. In vivo studies of peroxisomes in Drosophila S2 cells indicated that peroxisomes can move at very fast velocities, reaching speeds much faster than those observed during in vitro experiments that contained only one type of motor polarity (ie, [+] ended motors, such as kinesin-1).101 In vivo analysis of APP vesicles in Drosophila larvae showed that APP velocities were dependent on the amount of kinesin available, as 50% reduction of KHC resulted in decreased velocities.92
Reduction of dynein affected both anterograde and retrograde velocities,92,128 indicating that the functions of kinesin-1 and dynein are interdependent and that loss of either motor can lead to the impairment of bidirectional transport. Indeed, several studies support this proposal. Barkus et al showed that mutations in Drosophila Unc-104, a kinesin-3 protein, also inhibited retrograde movement.102 Likewise, in axonal neurofilaments, knockouts of kinesin-1 in mice and reductions of dynein motors inhibited both anterograde and retrograde transport.103 Pilling et al showed that mitochondria are not only transported by kinesin-1, but that kinesin-1 mutants drastically reduced retrograde transport.89 Furthermore, replacement of either kinesin-1 or dynein motors with motors of similar polarity (such as Unc-104 for kinesin-1) was able to activate cargo movement in the opposite direction.104 However, mutated motors with active heads but lacking motility failed to initiate retrograde transport. As a result, it was suggested that the opposing force generated by the anterograde motor could activate retrograde movement. Furthermore, a complete and stable kinesin-1 holenzyme not only mediated anterograde movement of prion protein-containing vesicles, but it also activated its retrograde movement.105
Together, these studies indicate an interdependence of opposite-polarity motors on their activation and motor function and suggest that precise regulation is needed to maintain complex motility in axons. Perhaps activation of both kinesin-1 and dynein/dynactin via phosphorylation may be an important mechanism by which coordination of these motors occur under physiological conditions.108,208 Indeed, both KHC and KLC subunits are known to be phosphorylated and contain sites for several kinases.245,256 While little is known about dynein phosphorylation, putative phosphorylation sequences are seen in dynein subunits.208 While binding of dynactin to dynein activates retrograde motility,106,107 evidence also suggests that kinesin-1 can mechanically interact with dynein to coordinate motor activity and movement.88,92,105,128 Therefore, several mechanisms of motor regulation and coordination must exist in vivo for proper bidirectional motility of several cargos on a single microtubule track.
Motor-cargo complexes in neurodegenerative disease
Recent work suggests that several motor-cargo complexes are transported on a single microtubule track. Several of these motor-cargo complexes contain proteins involved in neurodegenerative disease (Table 1). Here we will discuss a few of these motor-cargo complexes with an emphasis on the normal function of these disease proteins to determine how defects in vesicle motility result in neuronal dysfunction.
 | Table 1 Vesicle-motor complexes associated with neurodegenerative diseases |
APP-cargo complex
APP, the AD protein, is found in many tissues, but it is highly enriched in the brain.109,110 Although the function of APP remains unclear, there is evidence that it can function as a receptor for kinesin-1 during axonal transport. While APP can bind to components of the extracellular matrix, the structural and sequence analysis of APP gives insight into several possible functional domains, including domains for protease inhibition.111 Further, APP can function as an extracellular receptor112,113 much like Notch because of its similarity in structure and the ability of proteases, such as secretases, to cleave full-length APP into fragments. Similarly, APP has also been implicated in cell adhesion through the binding of extracellular matrix proteins.114,115 There is also evidence for APP to function as a regulator of neuronal outgrowth and aptogenesis during differentiation116–118 and neuronal repair following trauma.119
Concurrent with roles in neurite growth and synaptogenesis, APP is transported by fast axonal transport via KLC, either directly since the C-terminus of APP has been found to bind to KLC120–122 or indirectly via an interaction between KLC and JNK interacting protein 1 (JIP1).121,247 Biophysical evidence indicated that 15aa of the C-terminus of APP was sufficient to promote the transport of APP in squid axons122 and that APP beads had a high-binding affinity for kinesin-1.123 In addition, genetic evidence showed that excess human APP or Drosophila APP-like (APPL) and the loss of function of Drosophila APPL resulted in phenotypes similar to motor protein mutants, indicating a function for APP in axonal transport.124
Interestingly, the C-terminal deletions of human APP or APPL (including the kinesin-binding region) disrupted APP transport and failed to interact functionally with kinesin-1.124 JIP1 has also been shown to interact preferentially with APP that is phosphorylated at threonine 668 (Thr668) and affects the transport of phosphorylated (p)APP vesicles125 (Figure 2A). Recently, subpixel colocalization analysis suggested that APP vesicles contain both kinesin-1 and dynein,126 indicating that APP utilizes both kinesin-1 and dynein motors for bidirectional motility within axons. Furthermore, studies have also shown that the aberrant processing of APP by either β or γ secretase/presenilin can lead to the impaired transport of APP along axons which, in turn, may alter its normal function, leading to synaptic degeneration.127,128
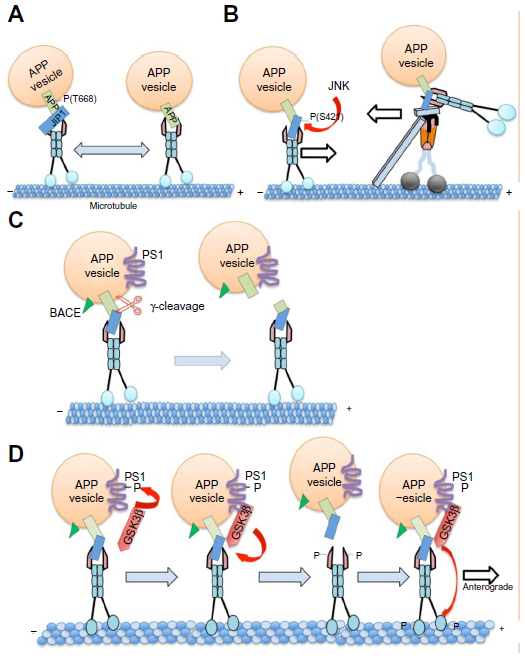 | Figure 2 Models of APP-cargo complexes and their regulation during transport. |
During axonal transport, APP is thought to be contained within a subclass of vesicles that contain the proteins: presenilin 1 (PS1), the catalytic component of γ-secretase); beta-site APP cleaving enzyme 1 (BACE1); synapsin 1; tyrosine kinase receptor type A (Trk A); and growth-associated protein 43 (GAP43).120 In addition, APP functionally interacts with PS1, BACE1, KLC, and c-Jun-amino-terminal kinase-interacting protein 1 (JIP1),125,128–130 and some of these proteins have also been shown to have enzymatic activity on APP itself, illustrating the importance of APP as an axonal protein. Evidence that APP is not transported in vesicles containing synaptotagmin120,128 further solidifies APP as a marker for a distinct subset of fast axonal transport vesicles. APP has also been shown to be cotransported with calsyntenin-1, which is a ligand for KLC.131 Further, reduction of calsyntenin-1 impaired APP transport and caused accumulations of Aβ,131 perhaps due to the fact that BACE1 and PS1 form a complex with APP.129 Although a study using fluorescent imaging failed to find colocalization of APP and BACE1 in retinal ganglion cells,132 BACE1 activity has also been observed in axons.132 While enhanced β-secretase processing altered APP axonal transport,127 a recent study showed that BACE1 and APP vesicles converge via an endocytosis dependent pathway in neurons.133 In cultured hippocampal neurons, dendritic APP/BACE1-containing vesicles were largely segregated in physiologic states, where BACE1 was sorted into acidic recycling endosomes, and APP was conveyed in Golgi-derived vesicles. However, upon activity induction, a known trigger of the amyloidogenic pathway, APP was routed into BACE1-positive recycling endosomes via a clathrin-dependent mechanism,133 suggesting that BACE1 and APP can be together in a particular functional pathway.
Recent work has demonstrated that APP vesicle motility is regulated through several different mechanisms. Anterograde velocities of APP vesicles are dependent on kinesin-1 concentration, and DIC may play a role in the coordinated regulation of APP motility.92 APP is phosphorylated and, although the role of phosphorylation is still uncertain, it may play a role in the regulation of APP vesicles during axonal transport. APP is phosphorylated at residue Thr668, although it is unclear which kinase is responsible for this phosphorylation. Previous studies have shown that cyclin dependent kinase (CDK5), and c-Jun amino-terminal kinase (JNK), are potential kinases responsible for APP phosphorylation at Thr668.134,135
Further work has shown that JIP1 prefers to interact with pAPP, such that reductions in JIP1 impaired transport of pAPP, but not APP.125 More recently,136 JIP1 has been shown to potentially regulate the anterograde/retrograde movements of APP. In vitro experiments showed that JNK can phosphorylate JIP1 at S421 to promote anterograde movement and that lack of JIP1 phosphorylation caused retrograde movement via interactions with dynein (Figure 2B). However, short interfering RNA experiments in cultured rat cortical neurons showed no changes in APP transport with reductions of JIP1.137
Recent in vivo experiments have shown that GSK-3β can negatively regulate APP transport via kinesin-1 by regulating the numbers of active motors on these vesicles.138 GSK-3β is thought to phosphorylate KLC, causing vesicles to release from motors.108 (Figure 2C). Alternatively, GSK-3β can phosphorylate KHC to regulate vesicle motility.208 Evidence has also shown that GSK-3β-mediated activities on APP may be via an action on presenilin,128,208 which is found on APP-containing vesicles129 and is known to cleave APP.139,140 In vivo imaging showed that presenilin also negatively regulates APP motility, similar to GSK-3β.128 Loss-of-function presenilin mutants resulted in increased APP vesicle velocities in both the anterograde and retrograde directions along MTs.128 In contrast to cell culture data, in vivo analysis showed that presenilin can increase the activity of GSK-3β, which may lead to increased kinesin-1 and dynein binding to APP vesicles, causing uncoordinated motility.208 Perhaps PS1 and GSK-3β may form a complex with motors to regulate motility in vivo. Indeed, PS1 associates with GSK-3β141 and can act as a potential scaffold to bring GSK-3β within close proximity of β-catenin (Figure 2D). Similarly, perhaps PS1 can also act as a scaffold that brings GSK-3β into close proximity of kinesin-1 and dynein, thereby regulating the motility of APP vesicles. Alternatively, since PS1 is the catalytic component of the gamma secretase complex, perhaps PS1 may cleave APP vesicles from motors139,140 (Figure 2C) and may serve as another form of regulation of APP motility. Indeed, high levels of APP peptides were shown to impair transport in squid axons,122 which may be a mechanism of disrupting transport in some forms of familial Alzheimer’s disease.
Since the transport of APP and its associated proteins may be regulated through several mechanisms, it is not surprising that misregulation of these complexes can lead to the impairment of axonal transport, which may be an early trigger in disease pathology. Indeed, axonopathy and transport deficits were observed in mouse models of AD that preceded known disease-related pathology by more than a year and in the early stages of AD in humans.142 Furthermore, in Down syndrome, where an increased expression of APP is seen, in the context of trisomy, abnormal transport of nerve growth factor and cholinergic neurodegeneration was observed,143 and these transport defects were thought to be independent of Aβ production.144
Taken together, these studies suggest that defects in axonal transport may be an early event in the progression of AD and may result, in part, due to the misregulation of axonal transport.
HTT cargo complex
HTT, the protein involved in Huntington’s disease (HD), is a ubiquitously expressed protein found in many cell types, but it is concentrated in neurons. HTT is comprised of two expanded regions of polyglutamine (polyQ) and polyproline, at the N-terminus and approximately 28–36 HEAT repeat domain motifs,145 which likely form a superhelical structure146 that is involved in protein–protein interactions. The polyQ tract can expand to longer lengths of CAG repeats (normally, approximately 35 repeats) that tend to correspond to the age of HD onset. X-ray crystallography of the N-terminus of HTT147 has given insight into the structure of the polyQ region, showing various confirmations depending on the surrounding proteins that could potentially affect its binding capabilities. In addition, the predicted structure of pathogenic and nonpathogenic N-terminal fragments of HTT aligned with this X-ray structure suggests an aggregation model for the pathogenesis of HD.148
Like APP, the primary function of HTT is still unclear. However, HTT is essential for development, since HTT null mice are embryonic lethal and heterozygous mice contain a myriad of defects, including cognitive deficits and neurodegeneration.149 Despite the lack of clarity on function, HTT interacts with a variety of different proteins, including huntingtin-interacting proteins and HAPs. These proteins appear to have various functions, including regulation of transcription, cell signaling, and cargo trafficking.150 The structure of HTT and the fact that HTT interacts with many different proteins implies that HTT may act as a scaffolding protein that is essential for cellular trafficking.
HTT has also been shown to have important functions in axonal transport and is transported along MTs.151 Both biochemical and genetic evidence suggest that HTT interacts directly with both kinesin9,152 and the dynein/dynactin complex.9,153 HAP1, which binds to HTT,154,155 has also been shown to interact with p150Glued156 and KLC.43 These findings indicate that HAP1 may act to tether HTT (and its cargo) to the antero- and retrograde motors. Furthermore, HTT has been shown to transport vesicles containing brain-derived neurotrophic factor (BDNF) (Figure 3A) through its interaction with both HAP1 and P150Glued.157 Interestingly, HAP1 has also recently been shown to associate with APP, since deletion of HAP1 resulted in an increased in motionless APP vesicles158 and impaired the anterograde transport of APP.43 Furthermore, HTT may regulate organelle dynamics by influencing different Rab proteins (Figure 3A). Indeed, Pal et al showed that HTT can mediate the transport of Rab5–HAP40–HTT containing early endosomes on actin filaments.159 HTT was also linked to Rab8 through FIP2 (family interacting protein 2), and this interaction regulated cell polarization and morphogenesis.160 Further, impairment of Rab11 caused the defective formation of vesicles in HD models,161,162 membrane binding of Rab11 was decreased with knockout of HD,163 and the axonal transport of Rab11 was perturbed with reduction of HTT,164 indicating that HTT may mediate the transport of Rab11 vesicles on MTs and defects in Rab11 transport may contribute to the early stages of the synaptic dysfunction in HD. Consistent with this proposal, early synaptic dysfunction,165 dendritic spine loss, and neurodegeneration166 observed in HD models were reversed by overexpression of Rab11.
 | Figure 3 Models of HTT-cargo complexes and their regulation during transport. |
Much like APP, phosphorylation is also associated with the regulation of HTT. Akt has been shown to act as a molecular switch by phosphorylating HTT at serine 421.167 This modification caused HTT to recruit kinesin to the BDNF vesicles, and possibly to dynactin, resulting in increased anterograde transport of BDNF vesicles in cultured cortical neurons. Conversely, lack of HTT phosphorylation caused kinesin to release from MTs and to promote retrograde transport.168 As a result, perhaps pHTT may stabilize the interaction between kinesin and dynactin to promote anterograde transport. Similarly, HAP1 is thought to be regulated via phosphorylation since protein kinase A (PKA) can phosphorylate HAP1 at T598, causing a decrease in the association of HAP1 with dynactin and KLC.169 Interestingly, this study also showed that nerve growth factor stimulation led to an increase in activated Akt and enrichment of HAP1 at neurites, indicating an increase in HAP1 anterograde transport which is consistent with Zala et al.168 Together, these findings suggest a mechanism by which HTT and HAP1 work together to coordinate vesicle directionality through two competing kinase pathways (PKA and Akt) (Figure 3B).
More recently, HTT has been implicated as a scaffold for vesicular glycolysis machinery that regulates vesicle transport by providing onboard energy for the transport of a subset of vesicles.170 They showed that HTT can tether glycolytic proteins, such as glyceraldehyde 3-phosphate dehydrogenase (GAPDH), onto BDNF vesicles,170 suggesting that the HTT-mediated glycolytic machinery provides the ATP that is used by BDNF vesicles for axonal transport. These observations suggest that HTT is not only important for carrying cargoes to target regions, but that HTT is also required for the proper transport of potentially any vesicle that relies on ATP generated by glycolysis.
Since HTT influences both kinesin-1 and dynein motors via a series of different complexes that include Rab proteins164 and BDNF,157 it is not surprising that misregulation of these HTT vesicular complexes could also lead to an impairment of axonal transport, which may be an early trigger in disease pathology. Expression of mutant HTT also caused neuritic aggregates, which blocked protein transport in neurites and caused neuritic degeneration before nuclear DNA fragmentation occurred. Simultaneously, expression of cytoplasmic polyQ proteins resulted in axonal blockages, retinal degeneration, and larval lethality in a Drosophila model of HD.9 Together, these studies suggest that the early neuropathology of HD can originate from axonal dysfunction mediated by defects in axonal transport. Indeed, mutant HTT can activate JNK3 (a neuronal specific c-Jun kinase) that phosphorylates the kinesin-1 motor, resulting in the disassociation of kinesin from MTs172 (Figure 3C) perturbing transport.
Mitochondria complex
Distal compartments, such as synapses, depend on mitochondrial transport to supply ATP for cellular processes like synaptic transmission.173 Mitochondria is known to undergo both anterograde and retrograde transport89,174,175 by utilizing kinesin-1 and cytoplasmic dynein motors.89,176–179 Evidence suggests that a specific protein complex is involved in anchoring mitochondria to KHC. Genetic screens in Drosophila identified a Rho guanosine triphosphate (GTP)ase, Miro, and trafficking protein Milton, which form a complex for mitochondrial transport.173,177,180 (Figure 4A). While Drosophila contains only one Milton gene,173,180 mammals have two orthologs named TRAK1/OIP106/Milton1 and GRIF1/Milton2.177 Biochemical evidence showed interactions between Miro and Milton.173,181
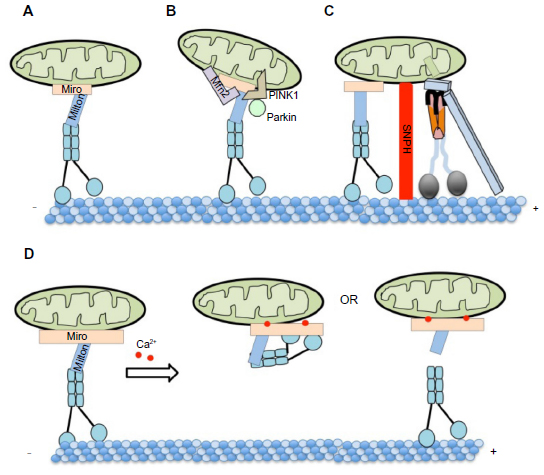 | Figure 4 Proposed models of mitochondrial cargo complexes and their regulation during transport. |
While the overexpression of Miro recruited Milton to mitochondria and regulated its trafficking in hippocampal neurons,181 the loss of Miro impaired the kinesin-mediated transport of mitochondria.182 Evidence using yeast two-hybrid interaction assays, coimmunoprecipitation, and colocalization indicated that GRIF1/Milton2 directly interacts with KHC through its C-terminal cargo-binding domain.183 Furthermore, the Milton interaction with KHC is independent of KLC.173 Therefore, Milton functions as an adaptor that links KHC to Miro-mediating mitochondrial transport.
Since mitochondria also undergo retrograde transport in axons, a Miro complex could exist with dynein. Loss or overexpression of Miro in Drosophila reduced retrograde transport of mitochondria by impairing both kinesin and dynein-mediated movement.181 Fractionation studies in Drosophila suggested a physical association between cytoplasmic dynein and mitochondria, and mutations in DHC-altered mitochondrial retrograde transport.89 Furthermore, dynein colocalized with mitochondria in both anterograde and retrograde directions.184 Although a physical association between dynein and Miro is unknown, together, these studies suggest that dynein is a key player in the retrograde transport of mitochondria. Furthermore, the Drosophila homolog of JIP1, APP-like protein interacting protein 1 (APLIP1), has also been implicated in mitochondrial transport.185 While a mutation in APLIP1 caused larval paralysis, swelling in axons, and reduced both anterograde and retrograde vesicle transport,185 only the retrograde transport of mitochondria was decreased, suggesting an inhibition of the dynein motor. However, it remains unclear whether APLP1 acts as an adaptor for the dynein-mediated retrograde transport of mitochondria. Perhaps several adaptor proteins may link mitochondria to dynein.186
Several proteins have been implicated in the regulation of mitochondrial movement187–190 (Figure 4B). Mitofusins (Mfn1 and Mfn2), which are located on the outer membrane of mitochondria187 and are essential for mitochondria fusion,191,192 are thought to be involved in mitochondrial transport.187 Biochemical evidence suggested that Mfn1/Mfn2 interacts with the Milton/Miro complex.187 Furthermore, defects in anterograde and retrograde mitochondrial transport were seen in neurons from mice with a knockout of Mfn2 or mice with a Charcot–Marie–Tooth (CMT2a) disease mutant in Mfn2 (R94Q),187 suggesting that Mfn has a role in mitochondrial motility. Another mitochondrial-associated protein, PINK1, has been proposed to form a multiprotein complex with Milton/Miro.193 Biochemical and genetic evidence indicated that Miro/Milton and PINK1 could interact with each other.185 Furthermore, PINK1 can regulate mitochondrial motility in motor neurons.194 Overexpression of PINK1 decreased mitochondrial flux and velocities in both directions, while the loss of PINK1 had the opposite effect. Altering PINK1 activity with RNA interference affected the distribution of mitochondria in motor neuron axons.194 Interestingly, Wang et al suggested that PINK1 can phosphorylate Miro on Ser156, and this can activate the proteasomal degradation of Miro in a parkin-dependent manner.189 Perhaps removal of Miro via proteasomal degradation is a potential mechanism for the detachment of kinesin motors from mitochondria, targeting dysfunctional mitochondria for mitophagy.189 Additionally, syntaphilin (SNPH), an axonally targeted protein, was identified as a mitochondria-docking receptor to MTs (Figure 4C). Deletion of SNPH increased the percentage of motile mitochondria, while overexpression abolished motility.195 Furthermore, neuronal activity and Ca2+ promoted the recruitment of SNPH to mitochondria and was necessary to maintain the balance between stationary and mobile mitochondria.196 Together, these observations propose an engine-switch- and-brake model, in which SNPH may function as an engine-off switch by sensing Miro-Ca2+ and as a brake by anchoring mitochondria to MTs.196
While little is known about the regulatory mechanisms of mitochondrial motility, work has shown that Ca2+ may be important. Miro contains two EF-hand Ca2+ binding domains and is a Ca2+ sensor that regulates mitochondrial motility.176,186,197–199 While the assembly of the KHC-Milton-Miro protein complex increased mitochondrial motility along axons, elevated cytosolic Ca2+, as a result of neuronal activity inhibited mitochondrial motility. The inhibition of mitochondrial motility in neurons with Miro mutations in the two EF hands that cannot bind Ca2+197,198 suggests that Miro-mediated Ca2+ levels play a key role in the regulation of mitochondrial motility. Perhaps a Miro-Ca2+ sensing mechanism exists whereby the binding of Ca2+ to Miro releases KHC from mitochondria181,186,197 (Figure 4D).
Interestingly, impairment in mitochondrial transport has been observed in many neurodegenerative diseases, such as AD.200–202 Impaired mitochondrial motility is observed in Aβ-rich environments,203–206,208 implicating axonal mitochondrial changes in synaptic dysfunction in AD neurons. Interestingly, in vitro studies in hippocampal neurons indicate a potential role for PKA and GSK-3β on Aβ and its action on mitochondrial transport.207 While the inhibition of PKA had no effect on Aβ-mediated mitochondrial transport, activation via forskolin completely eliminated mitochondrial transport defects induced by Aβ. On the other hand, GSK-3β inhibition via lithium also completely abolished Aβ-induced mitochondrial defects,208 while excess of GSK-3β inhibited the bidirectional movement of mitochondria in vivo.208 Similarly, Aβ oligomers impaired mitochondrial motility in hippocampal neurons, which appeared to be mediated by N-methyl-D-aspartate receptors.203
Considering that PKA inhibits GSK-3β via phosphorylation,207 and GSK-3β affects the phosphorylation state of KHC,209 a potential mechanism may exist whereby PKA suppresses Aβ-induced mitochondrial transport defects by modulating GSK-3β activity.
Work has also shown mitochondrial transport defects in HD.210–212 Mitochondria were progressively immobilized with frequent stops in neurons from HD transgenic animals, and these defects occurred early during development prior to the onset of measurable neurological or mitochondrial abnormalities.210 Further, changes in the levels of electron transport chain genes and mitochondrial structural genes were thought to be responsible for the abnormal mitochondrial dynamics observed in the cortices of HD patients.211 Moreover, mutant HTT associated with the mitochondrial protein Drp1 influenced axonal transport.212 Perhaps mutant HTT may induce aberrant associations that impair mitochondrial dynamics, decreasing mitochondrial function in HD.
RNA cargo complex
Several lines of evidence suggest that molecular motors are responsible for the spatial localization of RNA, which regulates protein production at specific sites within a cell.213 In Drosophila oocytes, kinesin-1 is responsible for the transport of Oskar messenger RNA (mRNA) to the posterior pole,214,215 while dynein mediates the anterior-dorsal localization of gurken mRNA and anterior localization of bicoid mRNA.216,217 Similarly, intracellular mRNA localization is also a conserved mechanism for spatially regulating protein production in polarized neurons since local translation of mRNAs is thought to be essential for many neuronal functions, such as the regulation of axon growth, axon maintenance, and regeneration after axonal injury.
Recent imaging studies have shown that distinct mRNA-containing complexes, including granules and mitochondrial mRNA, are transported long distances within neuronal projections.218 Neuronal RNA granules are essentially ribonucleoprotein particles whose roles involve transporting mRNA along MTs and modulating local protein synthesis in response to synaptic activity.219–221 Further, the mRNA encoding the translational repressor Nanos (nos) forms ribonucleoprotein (RNP) particles that are dendritically localized in dendritic arborization neurons in Drosophila.222
Interestingly, the RNA-binding protein Rumpelstiltskin and the germ plasm protein Oskar, also functions in dopaminergic neurons to assemble and to transport nosRNP particles mediated by dynein.222 Although the exact mechanisms of how these RNP complexes associate with kinesin-1 or dynein for localization is still unclear, these studies show that localization factors are adaptable and can regulate and target transcripts in different cellular contexts.
Biochemical, imaging, and immunofluorescence studies on the Fragile X mental retardation (FMR) mRNA complex have provided evidence for its involvement in axonal transport. FMR mRNA, which encodes the Fragile X mental retardation protein (FMRP), forms granules containing its own RNA and moves along neurites.223 Interestingly, FMRP was found in the same complex as KHC in mouse homogenates.224 The Drosophila homolog of FMRP, dFMRP, was shown to associate with both KHC and DHC and the RNAi knockdown of either motor blocked transport of FMRP-containing granules.225 Moreover, while one study found that the FMRP interaction with kinesin-1 was specific to KHC and not KLC,225 another study showed that the C-terminal domain of FMRP can directly interact with KLC.226 Together, these studies indicate that both motors are required for the transport of FMRP within axons. Furthermore, FMRP is thought to negatively regulate the number of mitochondria in axons and in neuromuscular junction synapses, affecting mitochondrial flux and processivity.227
Work on the La RNA chaperone protein exposed an interesting mechanism for RNA transport.228 In dorsal root ganglion neurons, sumoylation of La at lysine 41 was essential for its interaction with dynein but not kinesin.228 Furthermore, sumoylated La moved retrogradely in ligated sciatic nerves from adult rats causing distal accumulations of sumoylated La.228 This study suggests that modifying protein–protein interactions may control the directionality of RNA transport via sumoylation and may constitute an important regulatory mechanism. However, whether this is the central mechanism that governs all RNA transport is unclear, given that only a few RNA-binding proteins have been shown to undergo sumoylation.229 Thus, it is becoming evident that long-distance mRNA transport requires microtubule-dependent motors, but the molecular mechanisms underlying the sorting and trafficking of mRNAs into axons or dendrites remains elusive. While the roles of RNPs are still unclear, their sequestration into transported RNP particles and their specific localization is likely the result of specific targeting rather than passive diffusion. Therefore, local translation of RNA must be a rapid response mechanism to signals at the synapses to mediate synaptic plasticity230–232 during development and regeneration.
Other cargo complexes
Many more potential disease protein complexes are emerging that may rely on axonal transport to maintain their normal functions in neurons. Two of these are briefly discussed.
Potential spinal muscular atrophy (SMA)-cargo complex
The survival motor neuron (SMN) protein is vital for the maturation of small nuclear ribonucleoproteins233 and, possibly, the functional localization of messenger RNPs.234 Lack of SMN due to SMN deletion is one cause of SMA. Although it is unclear what complexes SMN is a part of, SMN has been found in axonal termini and growth cones.235 Furthermore, SMN granules were observed to be transported in cultured primary neurons with speeds that were consistent with fast axonal transport (1.8 μms–1).236 Since associations between SMN and mRNA binding proteins HuD240 and FMRP241 have been shown, perhaps SMN functions in RNA motility. Interestingly, several mutations in the tail domain of DHC (DYNC1H1) that disrupt dynein function have been found in patients with SMA.237 More recently, two other mutations in the neck and motor domain of dynein were associated with SMA.238 Interestingly, pharmacological compounds known to rescue kinesin-mediated larval locomotion in Drosophila stimulated neurite growth in rat spinal neurons and rescued motor neuron development in a Zebra fish model of SMA.239 Taken together, these observations suggest that SMN has a role in axonal transport and may form the unique cargo complex that interacts with MT motors.
Potential ALS-cargo complex
ALS is a motor neuron disease caused by mutations in several genes, including SOD1 and TDP-43. Recent work has shown that mutant superoxide dismutase (SOD1) can induce defects in axonal transport.249 KAP3 (kinesin-associated protein 3), a subunit of the kinesin 2 family, was shown to bind misfolded-mutant SOD1.244 KAP3 is known to bind to choline acetyltransferase (ChAT), which requires kinesin-2 for transport in axons.242,243 Tateno et al suggested that misfolded SOD1 was able to sequester kinesin-2 motors and impair ChAT transport.244 Indeed, a similar mechanism, the impairment of axonal transport via sequestration of motors, was previously proposed in a Drosophila model of HD9 and was recently theorized to occur in ALS through computational analysis.245 Computational analysis indicated that defects in axonal transport in SOD1 models of ALS result in protein aggregation and motor depletion. Furthermore, it was also shown that misfolded SOD1 impaired acetylcholine release, which was rescued via overexpression of KAP3.244 Together, these results suggest that disrupting the transport of acetylcholine- and ChAT-containing vesicles may lead to aberrant protein accumulations, leading to the progression of ALS, postulating a ChAT/acetylcholine cargo complex in the context of ALS.
Alternatively, similar to SMA, disruption in RNA transport could also lead to the progression of ALS. A pathological hallmark of ALS is the cytoplasmic inclusions containing hyperphosphorylated and ubiquitinated TDP-43.251 TDP-43 is a ubiquitous DNA/RNA-binding protein with a nuclear role in pre-mRNA splicing, and mutations have recently been linked to the development of ALS.250 Recent work has shown that TDP-43 is localized and actively transported in motor neuron axons and colocalizes with well-studied axonal mRNA-binding proteins, such as FMRP, HuD, and SMN.250
Perhaps similar to SMA, a TDP-43-RNA complex also exists. These observations suggest that TDP-43 has an important role in the regulation of axonal growth, and that impairment in the posttranscriptional regulation of mRNAs in the cytoplasm of motor neurons may be a major factor in the development of ALS.
Summary
It is intriguing that a vast majority of disease-related proteins have been shown to interact with molecular motors or to be present on transported cargo complexes. Many of these disease-related proteins also have roles – either during axonal transport or they are dependent on axonal transport for their proper localization and function. Further, the motility of these cargo complexes appears to be highly regulated at many levels; regulation of the motors themselves and the regulation of the association of these complexes with motors by a myriad of adaptor proteins and/or post translational modifications. Moreover, in some diseases where more than one gene or protein is involved in causing the disease, all of the disease proteins appear to converge in the axonal transport pathway. Either they are contained in the same vesicle or they are in different types of vesicle or RNA granules. Thus highlighting the importance of the axonal transport pathway and implicating perturbations in axonal transport in the initiation and progression of many neurodegenerative diseases. Therefore, it is not surprising that mutations in motor proteins cause neurodegeneration defects in transport have been shown to precede behavioral defects and neuropathology in many human neurodegenerative diseases. Thus, unraveling the identity of these motor-cargo complexes, the mechanisms of transport and how normal disease protein complexes function are all critical in understanding how problems in transport contribute to disease states. Such analysis has important implications for potential early diagnosis of neuronal disease and for the development of therapeutic interventions before the onset of neuronal death.
Acknowledgments
The authors regret that space limitations restricted some work from being cited. The authors thank members of the Gunawardena Laboratory at the State University of New York at Buffalo, NY, USA for helpful discussions. SG thanks Priyantha Karunaratne for constant support. SG is funded by the John R Oishei Foundation, Buffalo, NY, USA, and the National Institutes of Health/National Institute of Neurological Disorders and Stroke (R03NS084386).
Disclosure
The authors report no conflicts of interest in this work.
References
Weisenberg RC. Microtubule formation in vitro in solutions containing low calcium concentrations. Science. 1972;177(4054):1104–1105. | |
Tytell M, Black MM, Garner JA, Lasek RJ. Axonal transport: each major component reflects the movement of distinct macromolecular complexes. Science. 1981;214(4517):179–181. | |
Wang L, Ho CL, Sun D, Liem RK, Brown A. Rapid movement of axonal neurofilaments interrupted by prolonged pauses. Nat Cell Biol. 2000;2(3):137–141. | |
Roy S, Coffee P, Smith G, Liem RKH, Brady ST, Black MM. Neurofilaments are transported rapidly but intermittently in axons: implications for slow axonal transport. J Neurosci. 2000;20(18):6849–6861. | |
Tang Y, Das U, Scott DA, Roy S. The slow transport of alpha-synuclein – mechanistic commonalities amongst diverse cytosolic cargoes. Cytoskeleton (Hoboken). 2012;69(7):506–513. | |
Perlson E, Maday S, Fu MM, Moughamian AJ, Holzbaur EL. Retrograde axonal transport: pathways to cell death? Trends Neurosci. 2010;33(7):335–344. | |
Hafezparast M, Klocke R, Ruhrberg C, et al. Mutations in dynein link motor neuron degeneration to defects in retrograde transport. Science. 2003;300(5620):808–812. | |
Koo EH, Sisodia SS, Archer DR, et al. Precursor of amyloid protein in Alzheimer disease undergoes fast anterograde axonal transport. Proc Natl Acad Sci U S A. 1990;87(4):1561–1565. | |
Gunawardena S, Her LS, Brusch RG, et al. Disruption of axonal transport by loss of huntingtin or expression of pathogenic polyQ proteins in Drosophila. Neuron. 2003;40(1):25–40. | |
Zala D, Hinckelmann MV, Saudou F. Huntingtin’s function in axonal transport is conserved in Drosophila melanogaster. PLoS ONE. 2013;8(3):e60162. | |
Saha AR, Hill J, Utton MA, et al. Parkinson’s disease alpha-synuclein mutations exhibit defective axonal transport in cultured neurons. J Cell Sci. 2004;117(Pt 7):1017–1024. | |
Liu S, Sawada T, Lee S, et al. Parkinson’s disease-associated kinase PINK1 regulates Miro protein level and axonal transport of mitochondria. PLoS Genet. 2012;8(3):e1002537. | |
Füger P, Sreekumar V, Schüler R, et al. Spastic paraplegia mutation N256S in the neuronal microtubule motor KIF5A disrupts axonal transport in a Drosophila HSP model. PLoS Genet. 2012;8(11):e1003066. | |
Hirokawa N, Niwa S, Tanaka Y. Molecular motors in neurons: transport mechanisms and roles in brain function, development, and disease. Neuron. 2010;68(4):610–638. | |
Howard J, Hudspeth AJ, Vale RD. Movement of microtubules by single kinesin molecules. Nature. 1989;342(6246):154–158. | |
Vale RD, Reese TS, Sheetz MP. Identification of a novel force-generating protein, kinesin, involved in microtubule-based motility. Cell. 1985;42(1):39–50. | |
Brady ST. A novel brain ATPase with properties expected for the fast axonal transport motor. Nature. 1985;317(6032):73–75. | |
Scholey JM, Porter ME, Grissom PM, McIntosh JR. Identification of kinesin in sea urchin eggs, and evidence for it localization in the mitotic spindle. Nature. 1985;318(6045):483–486. | |
Saxton WM, Porter ME, Cohn SA, Scholey JM, Raff EC, McIntosh JR. Drosophila kinesin: characterization of microtubule motility and ATPase. Proc Natl Acad Sci U S A. 1988;85(4):1109–1113. | |
Xia CH, Rahman A, Yang Z, Goldstein LS. Chromosomal localization reveals three kinesin heavy chain genes in mouse. Genomics. 1998;52(2):209–213. | |
Rahman A, Friedman DS, Goldstein LS. Two kinesin light chain genes in mice. Identification and characterization of the encoded proteins. J Biol Chem. 1998;273(25):15395–15403. | |
Yang JT, Laymon RA, Goldstein LS. A three-domain structure of kinesin heavy chain revealed by DNA sequence and microtubule binding analysis. Cell. 1989;56(5):879–889. | |
Kuznetsov SA, Vaisberg EA, Shanina NA, Magretova NN, Chernyak VY, Gelfand VI. The quaternary structure of bovine brain kinesin. EMBO J. 1988;7(2):353–356. | |
Bloom GS, Wagner MC, Pfister KK, Brady ST. Native structure and physical properties of bovine brain kinesin and identifcation of the ATP-binding subunit polypeptide. Biochemistry. 1988;27(9):3409–3416. | |
Hirokawa N, Pfister KK, Yorifuji H, Wagner MC, Brady ST, Bloom GS. Submolecular domains of bovine brain kinesin identified by electron microscopy and monoclonal antibody decoration. Cell. 1989;56(5):867–878. | |
Scholey JM, Heuser J, Yang JT, Goldstein LS. Identification of globular mechanochemical heads of kinesin. Nature. 1989;338(6213):355–357. | |
McDonald HB, Goldstein LS. Identification and characterization of a gene encoding a kinesin-like protein in Drosophila. Cell. 1990;61(6):991–1000. | |
Gilbert SP, Johnson KA. Expression, purification, and characterization of the Drosophila kinesin motor domain produced in Escherichia coli. Biochemistry. 1993;32(17):4677–4684. | |
Ma YZ, Taylor EW. Kinetic mechanism of the kinesin motor domain. Biochemistry. 1995;34(40):13233–13241. | |
Yang JT, Saxton WM, Stewart RJ, Raff EC, Goldstein LS. Evidence that the head of kinesin is sufficient for force generation and motility in vitro. Science. 1990;249(4964):42–47. | |
Saxton WM, Hicks J, Goldstein LS, Raff EC. Kinesin heavy chain is essential for viability and neuromuscular functions in Drosophila, but mutants show no defects in mitosis. Cell. 1991;64(6):1093–1102. | |
Gho M, McDonald K, Ganetzky B, Saxton WM. Effects of kinesin mutations on neuronal functions. Science. 1992;258(5080):313–316. | |
Hurd DD, Saxton WM. Kinesin mutations cause motor neuron disease phenotypes by disrupting fast axonal transport in Drosophila. Genetics. 1996;144(3):1075–1085. | |
Gindhart JG Jr, Desai CJ, Beushausen S, Zinn K, Goldstein LS. Kinesin light chains are essential for axonal transport in Drosophila. J Cell Biol. 1998;141(2):443–454. | |
Tanaka Y, Kanai Y, Okada Y, et al. Targeted disruption of mouse conventional kinesin heavy chain, kif5B, results in abnormal perinuclear clustering of mitochondria. Cell. 1998;93(7):1147–1158. | |
Xia CH, Roberts EA, Her LS, et al. Abnormal neurofilament transport caused by targeted disruption of neuronal kinesin heavy chain KIF5A. J Cell Biol. 2003;161(1):55–66. | |
Kanai Y, Okada Y, Tanaka Y, Harada A, Terada S, Hirokawa N. KIF5C, a novel neuronal kinesin enriched in motor neurons. J Neurosci. 2000;20(17):6374–6384. | |
Nakajima K, Yin X, Takei Y, Seog DH, Homma N, Hirokawa N. Molecular motor KIF5A is essential for GABA(A) receptor transport, and KIF5A deletion causes epilepsy. Neuron. 2012;76(5):945–961. | |
Reid E, Kloos M, Ashley-Koch A, et al. A kinesin heavy chain (KIF5A) mutation in spastic paraplegia (SPG10). Am J Hum Genet. 2002;71(5):1189–1194. | |
Crimella C, Baschirotto C, Arnoldi A, et al. Mutations in the motor and stalk domains of KIF5A in spastic paraplegia type 10 and in axonal Charcot–Marie–Tooth type 2. Clin Genet. 2012;82(2):157–164. | |
Kamal A, Goldstein LS. Principles of cargo attachment to cytoplasmic motor proteins. Curr Opin Cell Biol. 2002;14(1):63–68. | |
Verhey KJ, Meyer D, Deehan R, et al. Cargo of kinesin identified as JIP scaffolding proteins and associated signaling molecules. J Cell Biol. 2001;152(5):959–970. | |
McGuire JR, Rong J, Li SH, Li XJ. Interaction of Huntingtin-associated protein-1 with kinesin light chain: implications in intracellular trafficking in neurons. J Biol Chem. 2006;281(6):3552–3559. | |
Kamal A, Stokin GB, Yang Z, Xia CH, Goldstein LS. Axonal transport of amyloid precursor protein is mediated by direct binding of the kinesin light chain subunit of kinesin-1. Neuron. 2000;28(2):449–459. | |
D’Andrea LD, Regan L. TPR proteins: the versatile helix. Trends Biochem Sci. 2003;28(12):655–662. | |
Rahman A, Kamal A, Roberts EA, Goldstein LS. Defective kinesin heavy chain behavior in mouse kinesin light chain mutants. J Cell Biol. 1999;146(6):1277–1288. | |
Trejo HE, Lecuona E, Grillo D, et al. Role of kinesin light chain-2 of kinesin-1 in the traffic of Na,K-ATPase-containing vesicles in alveolar epithelial cells. FASEB J. 2010;24(2):374–382. | |
Junco A, Bhullar B, Tarnasky HA, van der Hoorn FA. Kinesin light-chain KLC3 expression in testis is restricted to spermatids. Biol Reprod. 2001;64(5):1320–1330. | |
Dhaenens CM, Van Brussel E, Schraen-Maschke S, Pasquier F, Delacourte A, Sablonnière B. Association study of three polymorphisms of kinesin light-chain 1 gene with Alzheimer’s disease. Neurosci Lett. 2004;368(3):290–292. | |
Karki S, Holzbaur EL. Cytoplasmic dynein and dynactin in cell division and intracellular transport. Curr Opin Cell Biol. 1999;11(1):45–53. | |
Pfister KK, Fisher EM, Gibbons IR, et al. Cytoplasmic dynein nomenclature. J Cell Biol. 2005;171(3):411–413. | |
Pfister KK, Shah PR, Hummerich H, et al. Genetic analysis of the cytoplasmic dynein subunit families. PLoS Genet. 2006;2(1):e1. | |
Samsó M, Radermacher M, Frank J, Koonce MP. Structural characterization of dynein motor domain. J Mol Biol. 1998;276(5):927–937. | |
Samsó M, Koonce MP. 25 Angstrom resolution structure of a cytoplasmic dynein motor reveals a seven-member planar ring. J Mol Biol. 2004;340(5):1059–1072. | |
Neuwald AF, Aravind L, Spouge JL, Koonin EV. AAA+: A class of chaperone-like ATPases associated with the assembly, operation, and disassembly of protein complexes. Genome Res. 1999;9(1):27–43. | |
Mitchell DR, Brown KS. Sequence analysis of the Chlamydomonas alpha and beta dynein heavy chain genes. J Cell Sci. 1994;107(Pt 3):635–644. | |
Kon T, Nishiura M, Ohkura R, Toyoshima YY, Sutoh K. Distinct functions of nucleotide binding/hydrolysis sites in the four AAA modules of cytoplasmic dynein. Biochemistry. 2004;43(35):11266–11274. | |
Takahashi Y, Edamatsu M, Toyoshima YY. Multiple ATP-hydrolyzing sites that potentially function in cytoplasmic dynein. Proc Natl Acad Sci U S A. 2004;101(35):12865–12869. | |
Burgess SA, Walker ML, Sakakibara H, Knight PJ, Oiwa K. Dynein structure and power stroke. Nature. 2003;421(6924):715–718. | |
Habura A, Tikhonenko I, Chisholm RL, Koonce MP. Interaction mapping of a dynein heavy chain. Identification of dimerization and intermediate-chain binding domain. J Biol Chem. 1999;274(22):15447–15553. | |
Schroer TA. Dynactin. Annu Rev Cell Dev Biol. 2004;20:759–779. | |
Holleran EA, Karki S, Holzbaur EL. The role of the dynactin complex in intracellular motility. Int Rev Cytol. 1998;182:69–109. | |
Karki S, Holzbaur EL. Affinity chromatography demonstrates a direct binding between cytoplasmic dynein and the dynactin complex. J Biol Chem. 1995;270(48):28806–28811. | |
Vaughan KT. Microtubule plus ends, motors, and traffic of Golgi membranes. Biochim Biophys Acta. 2005;1744(3):316–324. | |
Vaughan KT, Vallee RB. Cytoplasmic dynein binds dynactin through a direct interaction between the intermediate chain and p150Glued. J Cell Biol. 1995;131(6 Pt 1):1507–1516. | |
Zhapparova ON, Bryantseva SA, Dergunova LV, et al. Dynactin subunit p150Glued isoforms notable for differential interaction with microtubules. Traffic. 2009;10(11):1635–1646. | |
Chevalier-Larsen ES, Wallace KE, Pennise CR, Holzbaur EL. Lysosomal proliferation and distal degeneration in motor neurons expressing the G59S mutation in the p150Glued subunit of dynactin. Hum Mol Genet. 2008;17(13):1946–1955. | |
Holleran EA, Tokito MK, Karki S, Holzbaur EL. Centractin (ARP1) associates with spectrin revealing a potential mechanism to link dynactin to intracellular organelles. J Cell Biol. 1996;135(6 Pt 2):1815–1829. | |
Holleran EA, Ligon LA, Tokito M, Stankewich MC, Morrow JS, Holzbaur EL. beta III spectrin binds to the Arp1 subunit of dynactin. J Biol Chem. 2001;276(39):36598–36605. | |
Muresan V, Stankewich MC, Steffen W, Morrow JS, Holzbaur EL, Schnapp BJ. Dynactin-dependent, dynein-driven vesicle transport in the absence of membrane proteins: a role for spectrin and acidic phospholipids. Mol Cell. 2001;7(1):173–183. | |
Fath KR, Trimbur GM, Burgess DR. Molecular motors and a spectrin matrix associate with Golgi membranes in vitro. J Cell Biol. 1997; 139(5):1169–1181. | |
Eschbach J, Dupuis L. Cytoplasmic dynein in neurodegeneration. Pharmacol Ther. 2011;130(3):348–363. | |
Harada A, Takei Y, Kanai Y, Tanaka Y, Nonaka S, Hirokawa N. Golgi vesiculation and lysosome dispersion in cells lacking cytoplasmic dynein. J Cell Biol. 1998;141(1):51–59. | |
Bowman AB, Patel-King RS, Benashski SE, McCaffery JM, Goldstein LS, King SM. Drosophila roadblock and Chlamydomonas LC7: a conserved family of dynein-associated proteins involved in axonal transport, flagellar motility, and mitosis. J Cell Biol. 1999;146(1):165–180. | |
Dick T, Ray K, Salz HK, Chia W. Cytoplasmic dynein (ddlc1) mutations cause morphogenetic defects and apoptotic cell death in Drosophila melanogaster. Mol Cell Biol. 1996;16(5):1966–1977. | |
Hays TS, Porter ME, McGrail M, et al. A cytoplasmic dynein motor in Drosophila: identification and localization during embryogenesis. J Cell Sci. 1994;107(Pt 6):1557–1569. | |
Phillis R, Statton D, Caruccio P, Murphey RK. Mutations in the 8 kDa dynein light chain gene disrupt sensory axon projections in the Drosophila imaginal CNS. Development. 1996;122(10):2955–2963. | |
LaMonte BH, Wallace KE, Holloway BA, et al. Disruption of dynein/dynactin inhibits axonal transport in motor neurons causing late-onset progressive degeneration. Neuron. 2002;34(5):715–727. | |
Puls I, Jonnakuty C, LaMonte BH, et al. Mutant dynactin in motor neuron disease. Nat Genet. 2003;33(4):455–456. | |
Lai C, Lin X, Chandran J, Shim H, Yang WJ, Cai H. The G59S mutation in p150(glued) causes dysfunction of dynactin in mice. J Neurosci. 2007;27(51):13982–13990. | |
Laird FM, Farah MH, Ackerley S, et al. Motor neuron disease occurring in a mutant dynactin mouse model is characterized by defects in vesicular trafficking. J Neurosci. 2008;28(9):1997–2005. | |
Münch C, Sedlmeier R, Meyer T, et al. Point mutations of the p150 subunit of dynactin (DCTN1) gene in ALS. Neurology. 2004;63(4):724–726. | |
Münch C, Rosenbohm A, Sperfeld AD, et al. Heterozygous R1101K mutation of the DCTN1 gene in a family with ALS and FTD. Ann Neurol. 2005;58(5):777–780. | |
Farrer MJ, Hulihan MM, Kachergus JM, et al. DCTN1 mutations in Perry syndrome. Nat Genet. 2009;41(2):163–165. | |
Breuer AC, Lynn MP, Atkinson MB, et al. Fast axonal transport in amyotrophic lateral sclerosis: an intra-axonal organelle traffic analysis. Neurology. 1987;37(5):738–748. | |
Kieran D, Hafezparast M, Bohnert S, et al. A mutation in dynein rescues axonal transport defects and extends the life span of ALS mice. J Cell Biol. 2005;169(4):561–567. | |
Bilsland LG, Sahai E, Kelly G, Golding M, Greensmith L, Schiavo G. Defects in axonal transport precede ALS symptoms in vivo. Proc Natl Acad Sci U S A. 2010;107(47):20523–20528. | |
Ligon LA, Tokito M, Finklestein JM, Grossman FE, Holzbaur EL. A direct interaction between cytoplasmic dynein and kinesin 1 may coordinate motor activity. J Biol Chem. 2004;279(18):19201–19208. | |
Pilling AD, Horiuchi D, Lively CM, Saxton WM. Kinesin-1 and Dynein are the primary motors for fast transport of mitochondria in Drosophila motor axons. Mol Biol Cell. 2006;17(4):2057–2068. | |
Welte MA. Bidirectional transport along microtubules. Curr Biol. 2004;14(13):R525–R537. | |
Kapitein LC, Schlager MA, Kuijpers M, et al. Mixed microtubules steer dynein-driven cargo transport into dendrites. Curr Biol. 2010;20(4):290–299. | |
Reis GF, Yang G, Szpankowski C, et al. Molecular motor function in axonal transport in vivo probed by genetic and computational analysis in Drosophila. Mol Biol Cell. 2012;23(9):1700–1714. | |
Schuster M, Kilaru S, Fink G, Collemare J, Roger Y, Steinberg G. Kinesin-3 and dynein cooperate in long-range retrograde endosome motility along a nonuniform microtubule array. Mol Biol Cell. 2011;22(19):3645–3657. | |
Gross SP, Welte MA, Block SM, Wieschaus EF. Coordination of opposite-polarity microtubule motors. J Cell Biol. 2002;156(4):715–724. | |
Gross SP. Hither and yon: a review of bi-directional microtubule-based transport. Phys Biol. 2004;1(1–2):R1–R11. | |
Soppina V, Rai AK, Ramaiya AJ, Barak P, Mallik R. Tug-of-war between dissimilar teams of microtubule motor regulates transport and fission of endosomes. Proc Natl Acad Sci U S A. 2009;106(46):19381–19386. | |
Müller MJ, Klumpp S, Lipowsky R. Motility states of molecular motors engaged in a stochastic tug-of-war. J Stat Phys. 2008;133(6):1059–1081. | |
Müller MJ, Klumpp S, Lipowsky R. Tug-of-war as a cooperative mechanism for bidirectional cargo transport by molecular motors. Proc Natl Acad Sci U S A. 2008;105(12):4609–4614. | |
Derr ND, Goodman BS, Jungmann R, Leschziner AE, Shih WM, Reck-Peterson SL. Tug-of-war in motor protein ensembles revealed with a programmable DNA origami scaffold. Science. 2012;338(6107):662–665. | |
Hendricks AG, Perlson E, Ross JL, Schroeder HW 3rd, Tokito M, Holzbaur EL. Motor coordination via a tug-of-war mechanism drives bidirectional vesicle transport. Curr Biol. 2010;20(8):697–702. | |
Kural C, Kim H, Syed S, Goshima G, Gelfand VI, Selvin PR. Kinesin and dynein move a peroxisome in vivo: a tug-of-war or coordinated movement? Science. 2005;308(5727):1469–1472. | |
Barkus RV, Klyachko O, Horiuchi D, Dickson BJ, Saxton WM. Identification of an axonal kinesin-3 motor for fast anterograde vesicle transport that facilitates retrograde transport of neuropeptides. Mol Biol Cell. 2008;19(1):274–283. | |
Uchida A, Alami NH, Brown A. Tight functional coupling of kinesin-1A and dynein motors in the bidirectional transport of neurofilaments. Mol Biol Cell. 2009;20(23):4997–5006. | |
Ally S, Larson AG, Barlan K, Rice SE, Gelfand VI. Opposite-polarity motors activate one another to trigger cargo transport in live cells. J Cell Biol. 2009;187(7):1071–1082. | |
Encalada SE, Szpankowski L, Xia CH, Goldstein LS. Stable kinesin and dynein assemblies drive the axonal transport of mammalian prion protein vesicles. Cell. 2011;144(4):551–565. | |
King SJ, Schroer TA. Dynactin increase the processivity of the cytoplasmic dynein motor. Nat Cell Biol. 2000;2(1):20–24. | |
Vaughan PS, Leszyk JD, Vaughan KT. Cytoplasmic dynein intermediate chain phosphorylation regulates binding to dynactin. J Biol Chem. 2001;276(28):26171–26179. | |
Morfini G, Szebenyi G, Elluru R, Ratner N, Brady ST. Glycogen synthase kinase 3 phosphorylates kinesin light chains and negatively regulates kinesin-based motility. EMBO J. 2002;21(3):281–293. | |
Martin-Morris LE, White K. The Drosophila transcript encoded by the beta-amyloid protein precursor-like gene is restricted to the nervous system. Development. 1990;110(1):185–195. | |
Lorent K, Overbergh L, Moechars D, De Strooper B, Van Leuven F, Van den Berghe H. Expression in mouse embryos and in adult mouse brain of three members of the amyloid precursor protein family, of the alpha-2-macroglobulin receptor/low density lipoprotein receptor-related protein and of its ligands apolipoprotein E, lipoprotein lipase, alpha-2-macroglobulin and the 40,000 molecular weight receptor-associated protein. Neuroscience. 1995;65(4):1009–1025. | |
Small DH, Nurcombe V, Reed G, et al. A heparin-binding domain in the amyloid protein precursor of Alzheimer’s disease is involved in the regulation of neurite outgrowth. J Neurosci. 1994;14(4):2117–2127. | |
Ho A, Südhof TC. Binding of F-spondin to amyloid-beta precursor protein: a candidate amyloid-beta precursor protein ligand that modulates amyloid-beta precursor protein cleavage. Proc Natl Acad Sci U S A. 2004;101(8):2548–2553. | |
Park JH, Gimbel DA, GrandPre T, et al. Alzheimer precursor protein interaction with the Nogo-66 receptor reduces amyloid-beta plaque deposition. J Neurosci. 2006;26(5):1386–1395. | |
Soba P, Eggert S, Wagner K, et al. Homo- and heterodimerization of APP family members promotes intercellular adhesion. EMBO J. 2005;24(20):3624–3634. | |
Yamazaki T, Koo EH, Selkoe DJ. Cell surface amyloid beta-protein precursor colocalizes with beta 1 intergrins at substrate contact sites in neural cells. J Neurosci. 1997;17(3):1004–1010. | |
Hung AY, Koo EH, Haass C, Selkoe DJ. Increased expression of beta-amyloid precursor protein during neuronal differentiation is not accompanied by secretory cleavage. Proc Natl Acad Sci U S A. 1992;89(20):9439–9443. | |
Moya KL, Benowitz LI, Schneider E, Allinquant B. The amyloid precursor protein is developmentally regulated and correlated with synaptogenesis. Dev Biol. 1994;161(2):597–603. | |
Priller C, Bauer T, Mitteregger G, Krebs B, Kretzschmar HA, Herms J. Synapse formation and function is modulated by the amyloid precursor protein. J Neurosci. 2006;26(27):7212–7221. | |
Van den Heuvel C, Blumbergs PC, Finnie JW, et al. Upregulation of amyloid precursor protein messenger RNA in response to traumatic brain injury: an ovine head impact model. Exp Neurol. 1999;159(2):441–450. | |
Kamal A, Stokin GB, Yang Z, Xia CH, Goldstein LS. Axonal transport of amyloid precursor protein is mediated by direct binding of the kinesin light chain subunit of kinesin-1. Neuron. 2000;28(2):449–459. | |
Inomata H, Nakamura Y, Hayakawa A, et al. A scaffold protein JIP-1b enhances amyloid precursor protein phosphorylation by JNK and its association with kinesin light chain 1. J Biol Chem. 2003;278(25):22946–22955. | |
Satpute-Krishnan P, DeGiorgis JA, Conley MP, Jang M, Bearer EL. A peptide zipcode sufficient for anterograde transport within amyloid precursor protein. Proc Natl Acad Sci U S A. 2006;103(44):16532–16537. | |
Seamster PE, Loewenberg M, Pascal J, et al. Quantitative measurements and modeling of cargo-motor interactions during fast transport in the living axon. Phys Biol. 2012;9(5):055005. | |
Gunawardena S, Goldstein LS. Disruption of axonal transport and neuronal viability by amyloid precursor protein mutations in Drosophila. Neuron. 2001;32(3):389–401. | |
Muresan Z, Muresan V. Coordinated transport of phosphorylated amyloid-beta precursor protein and c-Jun NH2-terminal kinase-interacting protein-1. J Cell Biol. 2005;171(4):615–625. | |
Szpankowski L, Encalada SE, Goldstein LS. Subpixel colocalization reveals amyloid precursor protein-dependent kinesin-1 and dynein association with axonal vesicles. Proc Natl Acad Sci U S A. 2012;109(22):8582–8587. | |
Rodrigues EM, Weissmiller AM, Goldstein LS. Enhanced β-secretase processing alters APP axonal transport and leads to axonal defects. Hum Mol Genet. 2012;21(21):4587–4601. | |
Gunawardena S, Yang G, Goldstein LS. Presenilin controls kinesin-1 and dynein function during APP-vesicle transport in vivo. Hum Mol Genet. 2013;22(19):3828–3843. | |
Kamal A, Almenar-Queralt A, LeBlanc JF, Roberts EA, Goldstein LS. Kinesin-mediated axonal transport of a membrane compartment containing beta-secretase and presenilin-1 requires APP. Nature. 2001;414(6864):643–648. | |
Lazarov O, Morfini GA, Lee EB, et al. Axonal transport, amyloid precursor protein, kinesin-1, and the processing apparatus: revisited. J Neurosci. 2005;25(9):2386–2395. | |
Vagnoni A, Perkinton MS, Gray EH, Francis PT, Noble W, Miller CC. Calsyntenin-1 mediates axonal transport of the amyloid precursor protein and regulates Aβ production. Hum Mol Genet. 2012;21(13):2845–2854. | |
Goldsbury C, Mocanu MM, Thies E, et al. Inhibition of APP trafficking by tau protein does not increase the generation of amyloid-beta peptides. Traffic. 2006;7(7):873–888. | |
Das U, Scott DA, Ganguly A, Koo EH, Tang Y, Roy S. Activity-induced convergence of APP and BACE-1 in acidic microdomains via an endocytosis-dependent pathway. Neuron. 2013;79(3):447–460. | |
Iijima KI, Ando K, Takeda S, et al. Neuron-specific phosphorylation of Alzheimer’s beta-amyloid precursor protein by cyclin-dependent kinase 5. J Neurochem. 2000;75(3):1085–1091. | |
Standen CL, Brownlees J, Grierson AJ, et al. Phosphorylation of thr(668) in the cytoplasmic domain of the Alzheimer’s disease amyloid precursor protein by stress-activated protein kinase 1b (Jun N-terminal kinase-3). J Neurochem. 2001;76(1):316–320. | |
Fu MM, Holzbaur EL. JIP1 regulates the directionality of APP axonal transport by coordinating kinesin and dynein motors. J Cell Biol. 2013;202(3):495–508. | |
Vagnoni A, Glennon EB, Perkinton MS, Gray EH, Noble W, Miller CC. Loss of c-Jun N-terminal kinase-interacting protein-1 does not affect axonal transport of the amyloid precursor protein or Aβ production. Hum Mol Genet. 2013;22(22):4646–4652. | |
Weaver C, Leidel C, Szpankowski L, Farley NM, Shubeita GT, Goldstein LS. Endogenous GSK-3/shaggy regulates bidirectional axonal transport of the amyloid precursor protein. Traffic. 2013;14(3):295–308. | |
Zhang Z, Nadeau P, Song W, et al. Presenilins are required for gamma-secretase cleavage of beta-APP and transmembrane cleavage of Notch-1. Nat Cell Biol. 2000;2(7):463–465. | |
De Strooper B, Saftig P, Craessaerts K, et al. Deficiency of presenilin-1 inhibits the normal cleavage of amyloid precursor protein. Nature. 1998;391(6665):387–390. | |
Takashima A, Murayama M, Murayama O, Kohno T, et al. Presenilin 1 associates with glycogen synthase kinase-3beta and its substrate tau. Proc Natl Acad Sci U S A. 1998;95(16):9637–9641. | |
Stokin GB, Lillo C, Falzone TL, et al. Axonopathy and transport deficits early in the pathogenesis of Alzheimer’s disease. Science. 2005;307(5713):1282–1288. | |
Salehi A, Delcroix JD, Belichenko PV, et al. Increased App expression in a mouse model of Down’s syndrome disrupts NGF transport and causes cholinergic neuron degeneration. Neuron. 2006;51(1):29–42. | |
Stokin GB, Almenar-Queralt A, Gunawardena S, et al. Amyloid precursor protein-induced axonopathies are independent of amyloid-beta peptides. Hum Mol Genet. 2008;17(22):3474–3486. | |
Takano H, Gusella JF. The predominantly HEAT-like motif structure of huntingtin and its association and coincident nuclear entry with dorsal, an NF-κB/Rel/dorsal family transcription factor. BMC Neurosci. 2002;3:15. | |
Li W, Serpell LC, Carter WJ, Rubinsztein DC, Huntington JA. Expression and characterization of full-length human huntingtin, an elongated HEAT repeat protein. 2006. J Biol Chem. 2006;281(23):15916–15922. | |
Kim MW, Chelliah Y, Kim SW, Otwinowski Z, Bezprozvanny I. Secondary structure of Huntingtin amino-terminal region. Structure. 2009;17(9):1205–1212. | |
Dlugosz M, Trylska J. Secondary structures of native and pathogenic huntingtin N-terminal fragments. J Phys Chem B. 2011;115(40):11597–11608. | |
Nasir J, Floresco SB, O’Kusky JR, et al. Targeted disruption of the Huntington’s disease gene results in embryonic lethality and behavioral and morphological changes in heterozygotes. Cell. 1995;81(5):811–823. | |
Harjes P, Wanker EE. The hunt for huntingtin function: interaction partners tell many different stories. Trends Biochem Sci. 2003;28(8):425–433. | |
Block-Galarza J, Chase KO, Sapp E, et al. Fast transport and retrograde movement of huntingtin and HAP1 in axons. Neuroreport. 1997;8(9–10):2247–2251. | |
Her LS, Goldstein LS. Enhanced sensitivity of striatal neurons to axonal transport defects induced by mutant huntingtin. J Neurosci. 2008;28(50):13662–13672. | |
Caviston JP, Ross JL, Antony SM, Tokito M, Holzbaur EL. Huntingtin facilitates dynein/dynactin-mediated vesicle transport. Proc Natl Acad Sci U S A. 2007;104(24):10045–10050. | |
Li XJ, Li SH, Sharp AH, et al. A huntingtin-associated protein enriched in brain with implications for pathology. Nature. 1995; 378(6555):398–402. | |
Li SH, Hosseini SH, Gutekunst CA, Hersch SM, Ferrante RJ, Li XJ. A human HAP1 homologue. Cloning, expression, and interaction with huntingtin. J Biol Chem. 1998;273(30):19220–19227. | |
Li SH, Gutekunst CA, Hersch SM, Li XJ. Interaction of huntingtin-associated protein with dynactin P150Glued. J Neurosci. 1998;18(4):1261–1269. | |
Gauthier LR, Charrin BC, Borrell-Pagès M, et al. Huntingtin controls neurotrophic support and survival of neurons by enhancing BDNF vesicular transport along microtubules. Cell. 2004;118(1):127–138. | |
Yang GZ, Yang M, Lim Y, et al. Huntingtin associated protein 1 regulates trafficking of the amyloid precursor protein and modulates amyloid beta levels in neurons. J Neurochem. 2012;122(5):1010–1022. | |
Pal A, Severin F, Lommer B, Shevchenko A, Zerial M. Huntingtin-HAP40 complex is a novel Rab5 effector that regulates early endosome motility and is up-regulated in Huntington’s disease. J Cell Biol. 2006;172(4):605–618. | |
Hattula K, Peränen J. FIP-2, a coiled-coil protein, links Huntingtin to Rab8 and modulates cellular morphogenesis. Curr Biol. 2000;10(24):1603–1606.. | |
Li X, Standley C, Sapp E, et al. Mutant huntingtin impairs vesicle formation from recycling endosomes by interfering with Rab11 activity. Mol Cell Biol. 2009;29(22):6106–6116. | |
Li X, Sapp E, Chase K, et al. Disruption of Rab11 activity in a knock-in mouse model of Huntington’s disease. Neurobiol Dis. 2009;36(2):374–383. | |
Li X, Sapp E, Valencia A, et al. A function of huntingtin in guanine nucleotide exchange on Rab11. Neuroreport. 2008;19(16):1643–1647. | |
Power D, Srinivasan S, Gunawardena S. In-vivo evidence for the disruption of Rab11 vesicle transport by loss of huntingtin. Neuroreport. 2012;23(16):970–977. | |
Richards P, Didszun C, Campesan S, et al. Dendritic spine loss and neurodegeneration is rescued by Rab11 in models of Huntington’s disease. Cell Death Differ. 2011;18(2):191–200. | |
Steinert JR, Campesan S, Richards P, Kyriacou CP, Forsythe ID, Giorgini F. Rab11 rescues synaptic dysfunction and behavioural deficits in a Drosophila model of Huntington’s disease. Hum Mol Genet. 2012;21(13):2912–2922. | |
Colin E, Zala D, Liot G, et al. Huntingtin phosphorylation acts as a molecular switch for anterograde/retrograde transport in neurons. EMBO J. 2008;27(15):2124–2134. | |
Zala D, Colin E, Rangone H, Liot G, Humbert S, Saudou F. Phosphorylation of mutant huntingtin at S421 restores anterograde and retrograde transport in neurons. Hum Mol Genet. 2008;17(24):3837–3846. | |
Rong J, McGuire JR, Fang ZH, et al. Regulation of intracellular trafficking of huntingtin-associated protein-1 is critical for TrkA protein levels and neurite outgrowth. J Neurosci. 2006;26(22):6019–6030. | |
Zala D, Hinckelmann MV, Yu H, et al. Vesicular glycolysis provides on-board energy for fast axonal transport. Cell. 2013;152(3):479–491. | |
Li H, Li SH, Yu ZX, Shelbourne P, Li XJ. Huntingtin aggregate-associated axonal degeneration is an early pathological event in Huntington’s disease mice. J Neurosci. 2001;21(21):8473–8481. | |
Morfini GA, You YM, Pollema SL, et al. Pathogenic huntingtin inhibits fast axonal transport by activating JNK3 and phosphorylating kinesin. Nat Neurosci. 2009;12(7):864–871. | |
Stowers RS, Megeath LJ, Górska-Andrzejak J, Meinertzhagen IA, Schwarz TL. Axonal transport of mitochondria to synapses depends on milton, a novel Drosophila protein. Neuron. 2002;36(6):1063–1077. | |
Tanaka K, Sugiura Y, Ichishita R, Mihara K, Oka T. KLP6: a newly identified kinesin that regulates the morphology and transport of mitochondria in neuronal cells. J Cell Sci. 2011;124(Pt 14):2457–2465. | |
Hollenbeck PJ, Saxton WM. The axonal transport of mitochondria. J Cell Sci. 2005;118(Pt 23):5411–5419. | |
Fransson S, Ruusala A, Aspenström P. The atypical Rho GTPases Miro-1 and Miro-2 have essential roles in mitochondrial trafficking. Biochem Biophys Res Commun. 2006;344(2):500–510. | |
Brickley K, Smith SJ, Beck M, Stephenson FA. GRIF-1 and OIP106, members of a novel gene family of coiled-coil domain protein. J Biol Chem. 2005;280(15):14723–14732. | |
Brickley K, Stephenson FA. Trafficking kinesin protein (TRAK)-mediated transport of mitochondria in axons of hippocampal neurons. J Biol Chem. 2011;286(20):18079–18092. | |
Brickley K, Pozo K, Stephenson FA. N-acetylglucosamine transferase is an integral component of a kinesin-directed mitochondrial trafficking complex. Biochim Biophys Acta. 2011;1813(1):269–281. | |
Glater EE, Megeath LJ, Stowers RS, Schwarz TL. Axonal transport of mitochondria requires milton to recruit kinesin heavy chain and is light chain independent. J Cell Biol. 2006;173(4):545–557. | |
MacAskill AF, Brickley K, Stephenson FA, Kittler JT. GTPase dependent recruitment of Grif-1 by Miro1 regulates microtubules mitochondrial trafficking in hippocampal neurons. Mol Cell Neurosci. 2009;40(3):301–312. | |
Russo GJ, Louie K, Wellington A, et al. Drosophila Miro is required for both anterograde and retrograde axonal mitochondrial transport. J Neurosci. 2009;29(17):5443–5455. | |
Smith MJ, Pozo K, Brickely K, Stephenson FA. Mapping the GRIF-1 binding domain in the kinesin, KIF5C, substantiates a role for GRIF-1 as an adaptor protein in the anterograde trafficking of cargoes. J Biol Chem. 2006;281(37):27216–27228. | |
Hirokawa N, Sato-Yoshitake R, Yoshida T, Kawashima T. Brain dynein (MAP1C) localizes on both anterogradely and retrogradely transported membranous organelles in vivo. J Cell Biol. 1990;111(3):1027–1037. | |
Horiuchi D, Barkus RV, Pilling AD, Gassman A, Saxton WM. APLIP1, a kinesin binding JIP-1/JNK scaffold protein, influences the axonal transport of both vesicles and mitochondria in Drosophila. Curr Biol. 2005;15(23):2137–2141. | |
Sheng ZH, Cai Q. Mitochondrial transport in neurons: impact on synaptic homeostasis and neurodegeneration. Nat Rev Neurosci. 2012;13(2):77–93. | |
Misko A, Jiang S, Wegorzewska I, Milbrandt J, Baloh RH. Mitofusin 2 is necessary for transport of axonal mitochondria and interacts with the Miro/Milton complex. J Neurosci. 2010;30(12):4232–4240. | |
Baloh RH, Schmidt RE, Pestronk A, Milbrandt J. Altered axonal mitochondrial transport in the pathogenesis of Charcot–Marie–Tooth disease from mitofusin 2 mutations. J Neurosci. 2007;27(2):422–430. | |
Wang X, Winter D, Ashrafi G, et al. PINK1 and Parkin target Miro for phosphorylation and degradation to arrest mitochondrial motility. Cell. 2011;147(4):893–906. | |
Chen Y, Sheng ZH. (2013, Kinesin-1-syntaphilin coupling mediates activity-dependent regulation of axonal mitochondrial transport. J Cell Biol. 202(2):351–364. | |
Rojo M, Legros F, Chateau D, Lombès A. Membrane topology and mitochondrial targeting of mitofusins, ubiquitous mammalian homologs of the transmembrane GTPase Fzo. J Cell Sci. 2002;115(Pt 8):1663–1674. | |
Chen H, Detmer SA, Ewald AJ, Griffin EE, Fraser SE, Chan DC. Mitofusins Mfn1 and Mfn2 coordinately regulate mitochondrial fusion and are essential for embryonic development. J Cell Biol. 2003;160(2):189–200. | |
Weihofen A, Thomas KJ, Ostaszewski BL, Cookson MR, Selkoe DJ. Pink1 forms a multiprotein complex wth Miro and Milton, linking Pink1 function to mitochondrial trafficking. Biochemistry. 2009;48(9):2045–2052. | |
Chapman AL, Bennett EJ, Ramesh TM, De vos KJ, Grierson AJ. Axonal Transport Defects in a Mitofusin 2 Loss of Function Model of Charcot-Marie-Tooth Disease in Zebrafish. PLoS ONE. 2013;8(6):e67276. | |
Kang JS, Tian JH, Pan PY, et al. Docking of axonal mitochondria by syntaphilin controls their mobility and affects short-term facilitation. Cell. 2008;132(1):137–148. | |
Chen Y, Sheng ZH. Kinesin-1-syntaphilin coupling mediates activity-dependent regulation of axonal mitochondria transport. J Cell Biol. 2013;202(2):351–364. | |
MacAskill AF, Rinholm JE, Twelvetrees AE, et al. Miro 1 is a calcium sensor for glutamate receptor-dependent localization of mitochondria at synapses. Neuron. 2009;61(4):541–555. | |
Wang X, Schwartz TL. The mechanism of Ca2+-dependent regulation of kinesin-mediated mitochondrial motility. Cell. 2009;136(1):163–174. | |
Saotome M, Safiulina D, Szabadkai G, et al. Bidirectional Ca2+-dependent control of mitochondrial dynamics by the Miro GTPase. Proc Natl Acad Sci U S A. 2008;105(52):20728–20733. | |
Anandatheerthavarada HK, Biswas G, Robin MA, Avadhani NG. Mitochondrial targeting and a novel transmembrane arrest of Alzheimer’s amyloid precursor protein impairs mitochondrial function in neuronal cells. J Cell Biol. 2003;161(1):41–54. | |
Lustbader JW, Cirilli M, Lin C, et al. ABAD directly links Abeta to mitochondrial toxicity in Alzheimer’s disease. Science. 2004;304(5669):448–452. | |
de Moura MB, dos Santos LS, Van Houten B. Mitochondrial dysfunction in neurodegenerative diseases and cancer. Environ Mol Mutagen. 2010;51(5):391–405. | |
Decker H, Lo KY, Unger SM, Ferreira ST, Silverman MA. Amyloid-beta peptide oligomers disrupt axonal transport through an NMDA receptor-dependent mechanism that is mediated by glycogen synthase kinase 3beta in primary cultured hippocampal neurons. J Neurosci. 2010;30:9166–9171. | |
Calkins MJ, Manczak M, Mao P, Shirendeb U, Reddy PH. Impaired mitochondrial biogenesis, defective axonal transport of mitochondria, abnormal mitochondrial dynamics and synaptic degeneration in a mouse model of Alzheimer’s disease. Hum Mol Genet. 2011;20(23):4515–4529. | |
Calkins MJ, Reddy PH. Amyloid beta impairs mitochondrial anterograde transport and degenerates synapses in Alzheimer’s disease neurons. Biochim Biophys Acta. 2011;1812(4):507–513. | |
Wang X, Perry G, Smith MA, Zhu X. Amyloid-beta-derived diffusible ligands cause impaired axonal transport of mitochondria in neurons. Neurodegener Dis. 2010;7(1–3):56–59. | |
Rui Y, Tiwari P, Xie Z, Zheng JQ. Acute impairment of mitochondrial trafficking by beta-amyloid peptides in hippocampal neurons. J Neurosci. 2006;26(41):10480–10487. | |
Rintoul GL, Filiano AJ, Brocard JB, Kress GJ, Reynolds IJ. Glutamate decrease mitochondrial size and movement in primary forebrain neurons. J Neurosci. 2003;23(21):7881–7888. | |
Dolma K, Iacobucci GJ, Hong Zheng K, et al. Presenilin influences glycogen synthase kinase-3β (GSK-3β) for kinesin-1 and dynein function during axonal transport. Hum Mol Genet. Epub October 18, 2013. | |
Trushina E, Dyer RB, Badger JD 2nd, et al. Mutant huntingtin impairs axonal trafficking in mammalian neurons in vivo and in vitro. Mol Cell Biol. 2004;24(18):8195–8209. | |
Shirendeb U, Reddy AP, Manczak M, et al. Abnormal mitochondrial dynamics, mitochondrial loss and mutant huntingtin oligomers in Huntington’s disease: implications for selective neuronal damage. Hum Mol Genet. 2011;20(7):1438–1455. | |
Shirendeb UP, Calkins MJ, Manczak M, et al. Mutant huntingtin’s interaction with mitochondrial protein Drp1 impairs mitochondrial biogenesis and causes defective axonal transport and synaptic degeneration in Huntington’s disease. Hum Mol Genet. 2012;21(2):406–420. | |
Gumy LF, Katrukha EA, Kapitein LC, Hoogenraad CC. New insights into mRNA trafficking in axons. Dev Neurobiol. Epub August 19, 2013. | |
Cha BJ, Serbus LR, Koppetsch BS, Theurkauf WE. Kinesin 1-dependent cortical exclusion restricts pole plasm to the oocyte posterior. Nat Cell Biol. 2002;4(8):592–598. | |
Brendza RP, Serbus LR, Duffy JB, Saxton WM. A function for kinesin 1 in posterior transport of oskar mRNA and Staufen protein. Science. 2000;289(5487):2120–2122. | |
MacDougall N, Clark A, MacDougall E, Davis I. Drosophila gurken (TGFalpha) mRNA localizes as particles that move within the oocyte in two dynein-dependent steps. Dev Cell. 2003;4(3):307–319. | |
Schnorrer F, Bohmann K, Nüsslein-Volhard C. The molecular motor dynein is involved in targeting swallow and bicoid RNA to the anterior pole of Drosophila oocytes. Nat Cell Biol. 2000;2(4):185–190. | |
Pathak GK, Love JM, Chetta J, Shah SB. A comparative quantitative assessment of axonal and dendritic mRNA transport in maturing hippocampal neurons. PLoS One. 2013;8(7):e65917. | |
Savas JN, Ma B, Deinhardt K, et al. A role for huntington disease protein in dendritic RNA granules. J Biol Chem. 2010;285(17):13142–13153. | |
Huber KM, Gallagher SM, Warren ST, Bear MF. Altered synaptic plasticity in a mouse model of fragile X mental retardation. Proc Natl Acad Sci U S A. 2002;99(11):7746–7750. | |
Kang H, Schuman EM. A requirement for local protein synthesis in neurotrophin-induced hippocampal synaptic plasticity. Science. 1996;273(5280):1402–1406. | |
Xu X, Brechbiel JL, Gavis ER. Dynein-dependent transport of nanos RNA in Drosophila sensory neurons requires Rumpelstiltskin and the germ plasm organizer Oskar. J Neurosci. 2013;33(37):14791–14800. | |
De Diego Otero Y, Severijnen LA, van Cappellen G, Schrier M, Oostra B, Willemsen R. Transport of fragile X mental retardation protein via granules in neuritis of PC12 cells. Mol Cell Biol. 2002;22(23):8332–8341. | |
Kanai Y, Dohmae N, Hirokawa N. Kinesin transports RNA: isolation and characterization of an RNA-transporting granule. Neuron. 2004;43(4):513–525. | |
Ling SC, Fahrner PS, Greenough WT, Gelfand VI. Transport of Drosophila fragile X mental retardation protein-containing ribonucleoprotein granules by kinesin-1 and cytoplasmic dynein. Proc Natl Acad Sci U S A. 2004;101(50):17428–17433. | |
Dictenberg JB, Swanger SA, Antar LN, Singer RH, Bassell GJ. A direct role for FMRP in activity-dependent dendritic mRNA transport links filopodial-spine morphogenesis to fragile X syndrome. Dev Cell. 2008;14(6):926–939. | |
Yao A, Jin S, Li X. Drosophila FMRP regulates microtubule network formation and axonal transport of mitochondria. Hum Mol Genet. 2011;20(1):51–63. | |
van Niekerk EA, Willis DE, Chang JH, Reumann K, Heise T, Twiss JL. Sumoylation in axons triggers retrograde transport of the RNA-binding protein La. Proc Natl Acad Sci U S A. 2007;104(31):12913–12918. | |
Flight MH. Axonal transport: SUMmOned back to the nucleus. Nat Rev Neurosci. 2007;8:656–656. | |
Gagnon JA, Mowry KL. Molecular motors: directing traffic during RNA localization. Crit Rev Biochem Mol Biol. 2011;46(3):229–239. | |
Wang DO, Kim SM, Zhao Y, et al. Synapse- and stimulus-specific local translation during long-term neuronal plasticity. Science. 2009;324(5934):1536–1540. | |
Wang DO, Martin KC, Zukin RS. Spatially restricting gene expression by local translation at synapses. Trends Neurosci. 2010;33(4):173–182. | |
Lefebvre S, Burglen L, Reboullet S, et al. Identification and characterization of a spinal muscular atrophy-determining gene. Cell. 1995;80(1):155–165. | |
Sleeman J. Small nuclear RNA and mRNAs: linking RNA processing and transport to spinal muscular atrophy. Biochem Soc Trans. 2013;41(4):871–875. | |
Fan L, Simard LR. Survival motor neuron (SMN) protein: role in neurite outgrowth and neuromuscular maturation during neuronal differentiation and development. Hum Mol Genet. 2002;11(14):1605–1614. | |
Fallini C, Bassell GJ, Rossoll W. High-efficiency transfection of cultured primary motor neurons to study protein localization, trafficking, and function. Mol Neurodegener. 2010;5:17. | |
Harms MB, Ori-McKenney KM, Scoto M, et al. Mutations in the tail domain of DYNC1H1 cause dominant spinal muscular atrophy. Neurology. 2012;78(22):1714–1720. | |
Fiorillo C, Moro F, Yi J, et al. Novel Dynein DYNC1H1 Neck and Motor Domain Mutations Link Distal Spinal Muscular Atrophy and Abnormal Cortical Development. Hum Mutat. Epub December 4, 2013. | |
Gassman A, Hao le T, Bhoite L, et al. Small molecule suppressors of Drosophila kinesin deficiency rescue motor axon development in a zebrafish model of spinal muscular atrophy. PLoS One. 2013;8(9):e74325. | |
Fallini C, Zhang H, Su Y, et al. The survival of motor neuron (SMN) protein interacts with the mRNA-binding protein HuD and regulates localization of poly(A) mRNA in primary motor neuron axons. J Neurosci. 2011;31(10):3914–3925. | |
Piazzon N, Rage F, Schlotter F, Moine H, Branlant C, Massenet S. In vitro and in cellulo evidences for association of the survival of motor neuron complex with the fragile X mental retardation protein. J Biol Chem. 2008;283(9):5598–5610. | |
Ray K, Perez SE, Yang Z, et al. Kinesin-II is required for axonal transport of choline acetyltransferase in Drosophila. J Cell Biol. 1999;147(3):507–518. | |
Baqri R, Charan R, Schimmelpfeng K, Chavan S, Ray K. Kinesin-2 differentially regulates the anterograde axonal transports of acetylcholinesterase and choline acetyltransferase in Drosophila. J Neurobiol. 2006;66(4):378–392. | |
Tateno M, Kato S, Sakurai T, Nukina N, Takahashi R, Araki T. Mutant SOD1 impairs axonal transport of choline acetyltransferase and acetylcholine release by sequestering KAP3. Hum Mol Genet. 2009;18(5):942–955. | |
Mitchell CS, Lee RH. Cargo distributions differentiate pathological axonal transport impairments. J Theor Biol. 2012;300:277–291. | |
Hollenbeck PJ. Phosphorylation of neuronal kinesin heavy and light chains in vivo. J Neurochem. 1993(6);60:2265–2275. | |
McDonald A, Fogarty S, Leclerc I, Hill EV, Hardie DG, Rutter GA. Cell-wide analysis of secretory granule dynamics in three dimensions in living pancreatic beta-cells: evidence against a role for AMPK-dependent phosphorylation of KLC1 at Ser517/Ser520 in glucose-stimulated insulin granule movement. Biochem Soc Trans. 2010; 38(Pt 1):205–208. | |
Fu MM, Holzbaur EL. JIP1 regulates the directionality of APP axonal transport by coordinating kinesin and dynein motors. J Cell Biol. 2013;202(3):495–508. | |
Shi P, Ström AL, Gal J, Zhu H. Effects of ALS-related SOD1 mutants on dynein- and KIF5-mediated retrograde and anterograde axonal transport. Biochim Biophys Acta. 2010;1802(9):707–716. | |
Mackenzie IR, Rademakers R, Neumann M. TDP-43 and FUS in amyotrophic lateral sclerosis and frontotemporal dementia. Lancet Neurol. 2010;9(10):995–1007. | |
Neumann M, Sampathu DM, Kwong LK, et al. Ubiquitinated TDP-43 in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Science. 2006;314(5796):130–133. |
 © 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.