Back to Archived Journals » Advances in Genomics and Genetics » Volume 4
Autosomal dominant polycystic kidney disease: genetics, epidemiology, and treatment
Authors Reed-Gitomer B
Received 18 June 2014
Accepted for publication 4 September 2014
Published 31 October 2014 Volume 2014:4 Pages 173—183
DOI https://doi.org/10.2147/AGG.S53161
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 5
Editor who approved publication: Dr John Martignetti
Berenice Reed-Gitomer
Division of Renal Diseases and Hypertension, Department of Medicine, University of Colorado Anschutz Medical Campus, Aurora, CO, USA
Abstract: Autosomal dominant polycystic kidney disease (ADPKD) is the most common potentially lethal genetic disorder, accounting for 2%–8% of end-stage renal disease worldwide. While development of renal cysts is the major characteristic of ADPKD, several disease-related complications contribute significantly to morbidity and mortality. Since introduction of renal replacement therapies, cardiovascular disease is the leading cause of death among ADPKD patients. Loss of renal function requiring renal replacement therapy occurs in 50% of patients by the age of 60 years. The results of recent large epidemiological studies in ADPKD have shown little progress in delaying onset of renal failure despite an improvement in patient mortality. Significant progress has been made over the past decade in elucidating the genetics of the disorder. Genetic testing is now available in most countries, and development of more reliable methods for prenatal diagnosis of ADPKD has increased the sensitivity of testing. While there are no approved drugs for treatment of ADPKD within the USA, the first agent targeting renal disease progression in this disorder was recently approved for clinical use in Japan. Furthermore, several additional drugs for treatment of ADPKD are currently under clinical investigation. Overall, this presents an optimistic future for new therapeutic interventions in this disease. This paper reviews the current knowledge related to the epidemiology, genetics, and treatment of ADPKD.
Keywords: autosomal dominant polycystic kidney disease, hereditary renal disease, renal cyst, genetics
Introduction
Autosomal dominant polycystic kidney disease (ADPKD) results from a mutation in either of two genes, ie, PKD1 encoding the protein polycystin 1 (PC1) or PKD2 encoding polycystin 2 (PC2).1–3 The key feature of the disease, namely, development of multiple fluid-filled cysts within the kidneys, results from focal proliferation of single tubular epithelial cells, as shown in the sequential steps depicted in Figure 1A–C.4,5 Continued cell proliferation and fluid secretion results in cyst growth throughout the lifetime, causing displacement and loss of the normal renal parenchyma, with increasing impairment of renal function (Figure 2). Polycystin proteins are expressed in several tissues, including the biliary ducts of the liver, pancreas, endothelial cells, and myocardial smooth muscle cells.6,7 Polycystins interact through their carboxyl-terminal tails to form a complex with both receptor and ion channel functions.8 At a cellular level, PC1 is expressed in the plasma lateral membrane of renal tubular cells, at apical junctions, and in the primary cilium.9–11 Within these locations, PC1 forms a complex with PC2 and other intracellular adhesion molecules. In addition, both of the polycystin proteins in their cleaved forms have been found in urinary exosome-like vehicles that interact with the primary cilium.12 This has been suggested as a mechanism to explain the localization of PC1 at disparate cellular locations. However, despite significant progress toward understanding the pathophysiology of ADPKD, the precise function of the polycystins, and their role in cystogenesis is incompletely understood.
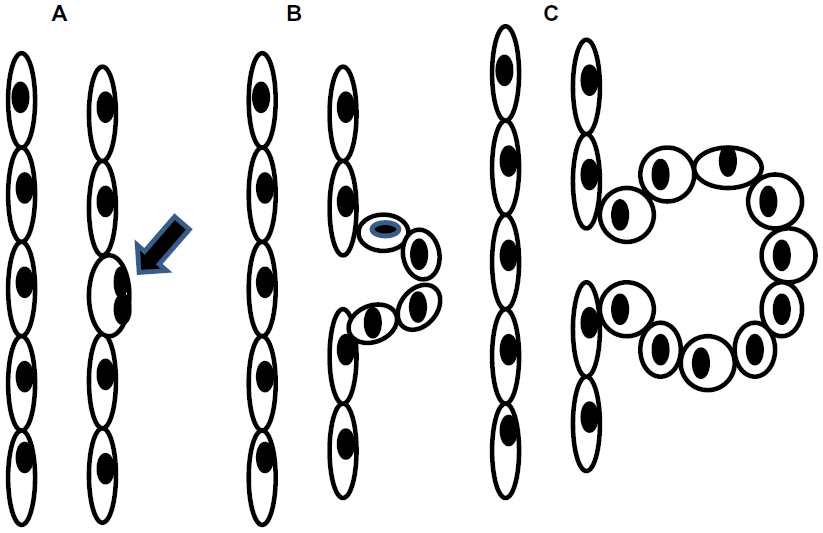 | Figure 1 Schematic of cyst growth progression. |
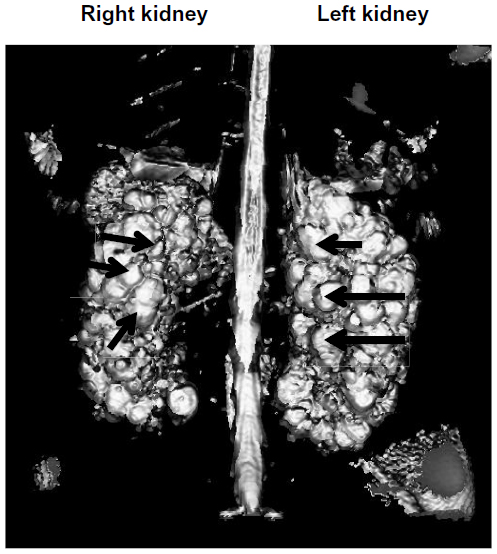 | Figure 2 Magnetic resonance image of ADPKD kidneys in a 14-year-old male. The left and right kidneys are indicated. The arrows indicate a few of the multiple cysts of varying size that completely fill both kidneys. The parenchyma is completely displaced by cysts. |
Consistent with the broad cellular and tissue expression of the polycystins, ADPKD is a systemic disorder. Cysts occur in multiple organs, most commonly the liver and pancreas. Liver cysts, which develop from cholangiocytes lining the biliary ducts, are more common in women, suggesting a role for estrogen in liver cyst growth.13 It is significant that the leading cause of death among affected patients is cardiovascular complications.14,15 Hypertension occurs early in ADPKD, and is an important risk factor associated with rapid decline in kidney function and development of left ventricular hypertrophy and consequent cardiovascular morbidity.16–18 Renal cyst growth results in compression of the renal vasculature, and the resulting intrarenal ischemia has been implicated in activation of the renin–angiotensin–aldosterone system (RAAS).19 RAAS stimulation occurs early in ADPKD before decline in renal function, even prior to development of hypertension, and contributes to left ventricular hypertrophy and atherosclerosis.19,20 Moreover, consistent with the vascular expression of the polycystins, endothelial dysfunction also occurs early in ADPKD, and has been detected in normotensive patients with preserved kidney function.21 A key mechanism contributing to endothelial dysfunction in the earlier stages of ADPKD is increased oxidative stress.22,23 Further extrarenal manifestations of ADPKD include intracranial aneurysms, cardiac valvular defects, abdominal wall hernias, and nephrolithiasis. Pain and hematuria are common complications and frequent reasons leading to diagnosis.
Epidemiology
ADPKD affects an estimated 600,000 individuals in the USA, with an incidence rate of approximately 1/500 to 1/1,000 individuals. The disease affects both sexes equally and all races and ethnicities. In population-based studies, male sex has been associated with both earlier age at diagnosis of hypertension and earlier age at onset of end-stage renal disease.24–26 Up to 50% of patients with ADPKD require renal replacement therapy by the age of 60 years. Based on the 2013 US Renal Data System report, cystic kidney disease was the fourth most common cause for patients initiating renal replacement therapy in the USA between 2009–2011.27 A recent study based on the UK Renal Registry reported an incidence rate of 6.8% for adult ADPKD patients commencing renal replacement therapy between 2000 and 2011.28 A report from the European Renal Association European Dialyses and Transplant Registry based on data collected between 1991 and 2010 demonstrated little apparent improvement in postponing age at onset of end-stage renal disease over the past two decades.29 However, a slight improvement in ADPKD patient mortality was indicated by a UK General Practice Research Database study that showed a trend toward decreased mortality associated with increasing coverage and intensity of antihypertensive therapy.30
Genetics of ADPKD
Genes
ADPKD results from a mutation in either of two genes, ie, PKD1 located on chromosome 16p13.31 or PKD2 located on chromosome 4q21.2,31 While there have been reports of a potential third PKD locus,32,33 these have not been substantiated by reanalysis of PKD3 families.34 However, in animals, mutation in several other genes, including Anks6 in the Han Sprague Dawley rat and Bicc1 in the Jcpk mouse, has been associated with slowly progressive disease that has similar characteristics to ADPKD.35,36 A comprehensive overview of genetic models of polycystic kidney disease has previously been presented by Torres and Harris.37 The PKD1 gene is large, comprising 46 exons that encode a 12,909 base pair transcript.3 Sequence analysis of PKD1 is complicated by duplication of exons 1–33 in six pseudo genes which share approximately 98% identity with the gene.38 PKD2 is a smaller gene, with 15 exons coding a 2,904 base pair transcript.2 Mutation of PKD1 accounts for approximately 85% of all disease cases, with PKD2 mutation accounting for the remaining 15%. More recently, a higher incidence of PKD2 mutations has been reported in the Canadian and US populations (26% and 36%, respectively).39,40 There is significant heterogeneity in disease phenotype between affected individuals. PKD1 mutation is associated with more severe and rapidly progressive disease, with onset of end-stage renal disease occurring by the mid to late 50s. Disease associated with PKD2 mutation is milder, with the onset of end-stage renal disease typically delayed by 10–20 years compared with PKD1. Results from the Consortium of Radiologic Imaging Study of PKD indicated that kidneys of PKD1-affected individuals were significantly larger than those with PKD2, with more cysts developing at an earlier age, while renal growth rate was similar for PKD1 and PKD2.41
Sequence analysis of PKD1 is complex due to the similarities between PKD1 and the pseudo genes (PKD1P1–PKD1P6). However, strategies have been developed that exploit the slight sequence differences between PKD1 and the pseudogenes, allowing unique amplification of the target.42 In contrast, as PKD2 is a nonduplicated gene sequence, analysis by conventional sequencing strategies is possible for mutation identification. More recently, next-generation sequencing has been successfully applied to sequence analysis of both PKD1 and PKD2.43
Allelic heterogeneity
To date, the Mayo Clinic PKD mutation database (http://pkdb.mayo.edu/) lists 1,272 potentially pathogenic mutations in PKD1 and 202 in PKD2. Several studies have reported an association between mutation position or mutation type with severity of ADPKD phenotype. A more 5’ mutation location in PKD1 has been associated with earlier age at onset of end-stage renal disease and a vascular phenotype.44,45 Similarly, in a European study, PKD1 mutation type was shown to influence renal disease severity.46 In PKD2, while mutation position and type have been suggested to influence disease severity, the association is less well delineated over normal disease variability.47
Hypomorphic alleles and mosaicism
Presentation of unusual cases of severe ADPKD that manifest as onset of symptomatic disease in early childhood have been associated with coinheritance of an incompletely penetrant (hypomorphic) allele in trans with either a PKD1 truncating mutation or a second PKD1 missense mutation.48,49 When occurring alone, hypomorphic alleles are usually associated with mild disease; however, when homozygous or presenting as a compound heterozygote, they may manifest as a severe phenotype. Several hypomorphic alleles have been described for PKD1, including p.R3227C, p.Y528C,48,50 and T2250M.51
Genetic mosaicism, where cells carrying a mutation and nonmutated cells are present in the same individual, has been described in ADPKD.52 Mosaicism may result from either a germline or a somatic mutation, and may complicate genetic diagnosis. In the case of a somatic mutation, different levels of the mutated allele may be present in different tissues.
Genetic testing
Mapping of PKD1 and PKD2 and flanking microsatellite markers has allowed genetic diagnosis by linkage analysis.53,54 Determination of the disease-associated haplotype allows ascertainment of at-risk family members and also identification of the disease-associated causative gene. Linkage analysis requires a definitive diagnosis in at least one family member, and several other family members must be available and willing to participate in testing.55 Existence of mosaicism may also complicate diagnosis. Since development of reliable methods for sequencing PKD1,42 diagnosis by mutation identification is possible and available in several countries, including the USA. However, the pathogenicity of certain missense variants may be difficult to establish even with available predictive tools. Preimplantation diagnosis (PGD) with in vitro fertilization presently has limited availability for ADPKD diagnosis. While PGD has been successfully applied in cases of other severe hereditary diseases, testing is more complex in ADPKD due to the presence of the large duplicated region in PKD1.56 Introduction of a single cell duplex-polymerase chain reaction method for PGD that includes an intragenic and extragenic marker for PKD1,57 coupled with more reliable cell sampling techniques for PGD, has increased the reliability of testing in ADPKD.58
Second hit and potential third hit?
The fact that cyst formation is focal, with only a small number of tubular epithelial cells forming cysts within the nephron, has led to the hypothesis that a “second hit” resulting in loss of heterozygosity is necessary for cyst formation.59,60 Thus, the disease could be viewed as recessive at the tissue level, requiring a second somatic mutation in addition to the germline mutation for initiation of cystogenesis. The continued development of cysts throughout life supports this hypothesis. Several studies have identified second somatic mutations in addition to the germline mutation in individual cysts from the kidney or liver in patients where the primary mutation affected PKD1 or PKD2.59,61,62 Studies in animal models of PKD further support a second-hit mechanism.63 In human disease, the mechanism leading to loss of heterozygosity is unknown. Whether the presence of a germline mutation directly conveys a degree of instability to the normal gene copy or whether a potential third hit is necessary remains to be tested. However, there are similarities between growth of a benign tumor and cyst growth, such that clues may be obtained from the cancer paradigm. Chromosomal instability is a hallmark of cancer cells. Interestingly, multiple cytogenetic aberrations have been found in individual ADPKD cyst cells by comparative genomic hybridization and loss of heterozygosity analyses.64 Moreover, a recent study indicated potential genomic instability in peripheral blood lymphocyte DNA isolated from ADPKD patients.65 Chronic inflammation and oxidative stress occur in ADPKD, and both processes have been linked to molecular mechanisms associated with DNA damage and chromosomal instability leading to cancer initiation.66
There is also strong evidence suggesting that gene dosage alone, whether associated with increased or decreased expression of Pkd1 or Pkd2, is sufficient for cyst development.67–69 Likewise, analyses of families with hypomorphic alleles similarly support an effect of gene dosage on phenotype development.48
Modifier genes
Significant phenotypic variability is apparent even among members of the same family, all of whom carry the same germline mutation. This has been partly attributed to the expressed effects of genes that modify disease severity (modifier genes). Based on study of PKD1 families, heritability analyses confirmed that one or more modifier genes in the genetic background accounted for a significant fraction of the phenotypic variability seen in patients with a PKD1 mutation.70 While several individual genes have been examined as candidate modifiers of ADPKD severity, the results of these studies have been varied and confounded by small study populations and lack of patient stratification. A larger study that included 794 PKD1 patients examined 173 candidate genes and found suggestive evidence for an association between the Dickkopf 3 gene and estimated glomerular filtration rate.71 In a few pedigrees with cases of severe very early-onset disease, in addition to the germline PKD mutation, HNF-1β or PKDH1 mutations have been described in the severely affected patients.72 This suggests that these genes may modify disease expression. The results from a large ongoing genome-wide association study funded by the National Institutes of Health (U01 DK079856) will provide further insight into the identity of genes that modify ADPKD severity.
Extracellular matrix gene expression
Abnormal expression of extracellular matrix proteins has been described in ADPKD, and is thought to contribute to proliferation of cyst epithelial cells and cyst growth. Overproduction of extracellular matrix proteins has been reported in both ADPKD cell and animal studies.73,74 Moreover, upregulated gene expression of several matrix protein-related messenger RNAs (mRNAs) has been described in human cystic tissue.75 Mutations in several cell adhesion and matrix protein-related genes, including laminin-5, have been shown to result in a cystic phenotype.76 In a more recent study, increased type 1 collagen and matrix metalloproteinase expression has been suggested to induce cyst formation and expansion in both in vivo and in vitro models.77 The role of polycystins, focal adhesions, and extracellular matrix adhesions in progression of cystic disease has recently been reviewed by Drummond.78
Epigenetics
Epigenetic modulation including histone modification, DNA methylation, or noncoding RNA expression is an important mechanism of cellular regulation. Several recent studies have examined potential epigenome changes in ADPKD. Histone deacetylation is controlled by histone deacetylases (HDACs). Loss of function mutations in HDAC5 have been shown to decrease cyst formation in a Pkd2−/− mouse model, suggesting a role for HDACs in cystogenesis.79 Administration of a HDAC inhibitor to pregnant Pkd2+/− female mice resulted in significant cyst reduction in kidneys of Pkd2−/− embryos.79 Similar results have been reported in Pkd1 mice following HDAC inhibition.80 Significantly, in renal epithelial cells, HDAC5 expression is regulated by flow-mediated shear stress in a polycystin-dependent manner.79
Hypermethylation of PKD1 and other genes related to ion transport and cell adhesion has been identified in ADPKD renal tissue.81 Oxidative stress and inflammatory cytokines have both been implicated in epigenetic instability.82 Both oxidative stress and inflammation play significant roles in the pathophysiology of ADPKD, and it is therefore intriguing to postulate a role for epigenetic modulation in ADPKD.
Micro RNA
Micro RNAs (miRNAs) are short noncoding nucleotides approximately 20 nucleotides in length that play a significant role in post-transcriptional regulation of gene expression. Several differentially expressed miRNAs have been described in animal models of ADPKD.83,84 In particular, the oncogenic cluster miRNA17–92 is upregulated in mouse models of PKD while inactivation of miRNA17–92 retards cyst growth.85 Furthermore, members of the miRNA-17 family have been shown to post-transcriptionally regulate PKD2.86 The RNA binding protein, bicaudal 1, regulates PC2 by interfering with miR-17 family binding, enhancing PKD2 translation.87 In contrast, in non-PKD disease models, loss of bicaudal 1 expression results in a PKD-like phenotype.88 Dysregulated expression of several miRNAs has been found in the urine of human ADPKD patients when compared with urine from patients with chronic kidney disease, including miRNA-1 and miRNA-133, previously implicated as tumor suppressors, and miRNA-223/miRNA-199.89 Thus, a role for miRNAs in the pathogenesis of ADPKD is suggested, and modulation of miRNA levels may have future therapeutic benefits. A summary of the various genetic influences on ADPKD progression is shown in Figure 3.
 | Figure 3 Genetic and epigenetic factors that affect progression of ADPKD. |
Treatment
There is currently no approved therapy to slow progression of ADPKD. Treatment of hypertension or disease-related symptoms are the mainstays of management for ADPKD patients. However, over the past 5 years, significant advances have been made regarding potential new interventions in ADPKD. ClinicalTrials.gov (https://clinicaltrials.gov) currently lists 20 active and five additional trials with unknown status in ADPKD.
Blood pressure management
Early activation of the RAAS in ADPKD is an important factor contributing to the development of hypertension. Studies in adults and children have indicated that hypertension is associated with larger kidney volumes, increased left ventricular mass index, and a risk for left ventricular hypertrophy.17,19 Rigorous blood pressure control has been shown to reduce left ventricular mass index in hypertensive patients with ADPKD.90 Also, some studies have reported an improvement in kidney survival in more recent years, associated with increased use of drugs targeting the RAAS.26,91 Similarly, data from the UK General Practice Database demonstrated a significant trend toward decreased mortality among ADPKD patients, associated with increased use of antihypertensive agents and number of agents prescribed.30 However, a recent large study of 3,598 ADPKD patients from the UK Renal Registry between 2000 and 2011 failed to show any improvement in age at onset of renal replacement therapy.28 The multicenter HALT-Progression of Polycystic Kidney Disease clinical trial is testing the effect of blood pressure control and the effect of angiotensin-converting enzyme inhibitor (ACEI) monotherapy compared with a combination of an ACEI and an angiotensin receptor blocker on renal outcomes.92 The results of this study will provide guidance regarding optimum blood pressure targets and RAAS intervention in ADPKD.
Bosutinib
Bosutinib (SKI-606) is a tyrosine kinase inhibitor with action on Bcr-Abl and Src kinase family members. Activation of protooncogene c-Src occurs by autophosphorylation at tyrosine (Y)418 and is associated with increased proliferation and matrix adhesion in tumors.93 In ADPKD, pY(418)-Src is increased in cyst-lining cells, while inhibition of protooncogene c-Src with SKI-606 attenuates hyperproliferation and reduces renal epithelial cell-matrix adhesion.94 Similarly, SKI-606 retarded renal cyst formation in animal models.95 Preclinical results indicate that bosutinib may be beneficial in ADPKD, and the results of an ongoing clinical trial will provide further information on these benefits. However, significant side effects have been noted in cancer studies, which may affect the long-term use of the drug.
Mineralocorticoid inhibition
Patients with ADPKD demonstrate impaired endothelium-dependent dilation,21,96,97 as well as large elastic artery stiffening indicated by increased aortic pulse wave velocity.98 Activation of the RAAS occurs early in ADPKD and in several other pathological conditions, and aldosterone excess has been implicated in the development of endothelial dysfunction even in the general population (Figure 4).19,99,100 In contrast, aldosterone antagonists have been shown to improve arterial dysfunction and decrease mortality in patients with heart failure receiving ACEI.101,102 Similarly, spironolactone reduced inflammation and preserved endothelial function in elderly hypertensive patients treated with calcium antagonists and angiotensin blockers.103 This indicates that spironolactone may be an effective adjunctive therapy in ADPKD patients taking an ACEI for prevention of endothelial dysfunction and reduction of inflammation. The benefits of spironolactone therapy in ADPKD will be revealed by the outcome of an ongoing clinical trial (ClinicalTrials.gov identifier NCT01853553).
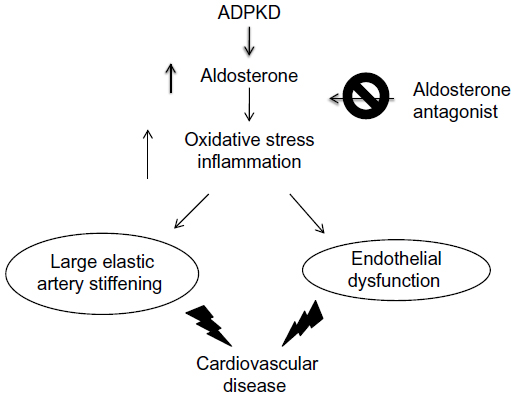 | Figure 4 Relationship between aldosterone and vascular disease in ADPKD. |
Inhibition of mTOR
Mammalian target of rapamycin (mTOR) is a serine/threonine kinase that regulates cell growth, proliferation, and survival.104 The mTOR pathway is regulated by PC1, and thus plays a significant role in ADPKD.105 Rapamycin effectively decreases cyst growth in animal models of ADPKD.106,107 However, results from human studies using either sirolimus (rapamycin) or everolimus have not been as impressive. To date, the results of six clinical trials, five using sirolimus and one using everolimus, have been published.108–113 The trial durations were short, with the longest trial being 24 months. Only one small study showed an improvement in renal function in patients treated with sirolimus.112 The effects on kidney volume were modest. However, the short trial duration and low patient number in some trials may have influenced the ability to detect a change in total kidney volume. There were significant side effects associated with therapy in all trials. A meta-analysis that included four of the above studies indicated that short-duration mTOR inhibitor therapy is relatively safe when used to slow down the increase in kidney volume in patients with early-stage disease, but had limited impact on slowing the decrease in renal function.114
Niacinamide
The nicotinamide adenine dinucleotide-dependent protein deacetylase sirtuin 1 (SIRT1) is upregulated in renal epithelial cells of a Pkd-mutant mouse.115 In a Pkd1 and Sirt1 double knockout mouse model cyst formation is exacerbated over that found in the Pkd mutant mouse indicating a potential role for SIRT1 in cystogenesis.115 Treatment of the Pkd1 knockout mouse with nicotinamide (niacinamide), a pan-sirtuin inhibitor, delayed cyst growth. Thus, a potential therapeutic role of niacinamide in ADPKD is suggested. An ongoing Phase II pilot study (ClinicalTrials.gov identifier NCT02140814) is currently evaluating niacinamide in ADPKD.
Somatostatin
Disruption of intracellular calcium homeostasis occurs in ADPKD and is linked with increased tissue levels of cyclic adenosine monophosphate (cAMP).116 Enhanced cAMP and protein kinase A signaling may impact tubulogenesis and epithelial cell proliferation, thereby facilitating cystogenesis. Somatostatin has been shown to decrease cAMP production and fluid secretion in both renal tubular cells and liver cholangiocytes.117 This observation has led to clinical trials testing the effectiveness of the somatostatin analogs, octreotide and lanreotide, in human ADPKD.118–123 To date, results from four trials have been published. Three of these trials tested octreotide LAR, a long-acting formulation of octreotide, and one lanreotide. While the primary endpoint in three of the studies assessed structural changes in the liver, a small effect on total kidney volume was observed.118,120,122 However, no study has demonstrated a significant sustained beneficial effect on kidney growth with longer treatment duration.119,121,123 The effect of slowing the increase in liver volume has been more promising insofar as sustained beneficial effects on liver growth have been observed. Somatostatin analogs hold promise for the treatment of polycystic liver disease, but larger trials of longer duration are needed to fully assess the benefits of this therapy.
Statins
Statins are hydroxyl-methyl glutaryl coenzyme A inhibitors that reduce cholesterol levels. Statins also have pleiotropic effects, including an ability to reduce inflammation.124 Several studies have shown that statins improve hemodynamic parameters in chronic kidney disease.125,126 This suggests a potential beneficial action in ADPKD. A reduction in cyst burden and improvement of renal function has been observed with statin treatment in animal models of ADPKD.127,128 To date, three clinical studies have assessed the benefit of statins in ADPKD. In a small study, simvastatin improved glomerular filtration rate and increased effective renal plasma flow when compared with placebo treatment.126 However, a study in 49 patients did not show any improvement in renal function parameters with pravastatin treatment.129 Both of these studies were relatively small, and therefore possibly underpowered to show an effect. The results of a recent larger, placebo-controlled study of pravastatin in children and young adults with ADPKD were more promising.130 Over a 3-year duration, treatment with pravastatin lowered the incremental increase in height-corrected total kidney volume when compared with placebo treatment. In this study, all subjects were also treated with an ACEI. Future large studies of adequate duration are needed to assess the benefits of statin therapy in the adult ADPKD population.
Tolvaptan
Vasopressin is an adenylyl cyclase antagonist and its activity is mediated by binding to the V2 receptor. In cell and animal studies, V2 receptor antagonists, including tolvaptan, have been shown to lower cAMP levels, blocking vasopressin-mediated fluid secretion into cysts, thereby ameliorating the cyst burden in animal models of PKD.117 The results of a 3-year, randomized, double-blind, placebo-controlled clinical trial with tolvaptan that included 1,445 participants have shown promise for ADPKD.131 A significantly smaller increase in total kidney volume over 3 years (2.8% versus 5.5% per year) and a slower decline in renal function was observed in the tolvaptan-treated patients when compared with controls. This effect was greatest during the first year of treatment. Fewer patients in the tolvaptan-treated group reported PKD-related complications, including pain and hematuria. Elevated liver enzyme levels experienced by some patients mean that monitoring of liver function is necessary during treatment. Tolvaptan has recently been approved in Japan for the treatment of ADPKD, while additional clinical studies are ongoing in the USA (Otsuka Pharmaceutical Development and Commercialization, Inc).
Triptolide
Triptolide, a diterpene derived from the herb Tripterygium wilfordii, induces apoptosis and has anti-inflammatory benefits.132 In animal models of ADPKD, triptolide restores calcium signaling, attenuates cyst formation, and improves renal function.133–135 In a small uncontrolled clinical study of nine proteinuric ADPKD patients treated with triptolide for 6 months, a decrease in proteinuria, inhibition of increase in total kidney volume, and stabilization of renal function were observed.136 A larger randomized, double-blind, placebo-controlled clinical trial (ClinicalTrials.gov identifier NCT02115659) of triptolide is currently ongoing and will provide insight into the benefits of triptolide in ADPKD.
Conclusion
In conclusion, studies of the natural history of ADPKD have improved our understanding of the course of disease and identified several factors that impact disease progression. Significant strides have been made in elucidating the genetics of the disorder, thereby furthering our understanding of the etiology of phenotypic variability. Genetic testing by mutation identification for both PKD1 and PKD2 is now feasible, and preimplantation genetic diagnosis is available in some countries.
While an improvement in mortality for patients with ADPKD has been reported over the past two decades, progress in prolonging renal function has been disappointing. However, based on the outcomes of recent and ongoing clinical trials, the first drugs to slow progression of renal disease in ADPKD should become available within the next few years. Cardiovascular disease remains the leading cause of mortality in affected patients, so there is an urgent need to establish the efficacy of interventions to decrease cardiovascular risk. The anticipated results of the HALT-PKD trial should better inform regarding optimal blood pressure management and therapy in ADPKD patients with regard to both renal and cardiac outcomes. An ongoing mineralocorticoid antagonism study may identify an intervention to ameliorate endothelial dysfunction in this at-risk population. New therapies to treat cystic disease in the liver may soon become available. Thus, the next decade should herald significant improvement for patient outcomes in ADPKD.
Disclosure
The author has served on an advisory panel for Otsuka Pharmaceutical Co, USA.
References
Hughes J, Ward CJ, Peral B, et al. The polycystic kidney disease 1 (PKD1) gene encodes a novel protein with multiple cell recognition domains. Nat Genet. 1995;10(2):151–160. | |
Mochizuki T, Wu G, Hayashi T, et al. PKD2, a gene for polycystic kidney disease that encodes an integral membrane protein. Science. 1996;272(5266):1339–1342. | |
European Polycystic Kidney Disease Consortium. The polycystic kidney disease gene encodes a 14 kb transcript and lies within a duplicated region of chromosome 16. Cell. 1994;77(6):881–894. | |
Fedeles SV, Gallagher AR, Somlo S. Polycystin-1: a master regulator of intersecting cystic pathways. Trends Mol Med. 2014;20(5):251–260. | |
Grantham JJ, Mulamalla S, Swenson-Fields KI. Why kidneys fail in autosomal dominant polycystic kidney disease. Nat Rev Nephrol. 2011;7(10):556–566. | |
Griffin MD, Torres VE, Grande JP, Kumar R. Immunolocalization of polycystin in human cultured cells. Proc Assoc Am Physicians. 1996;108(3):185–197. | |
Ong AC, Ward CJ, Butler RJ, et al. Coorrdinate expression of the autosomal dominant polycystic kidney disease proteins, polycystin-2 and polycystin-1, in normal and cystic tissue. Am J Pathol. 1999;154(6):1721–1729. | |
Hanaoka K, Guggino WB. Co-assembly of polycystin-1 and 2 produces unique cation-permeable currents. J Am Soc Nephrol. 2000;11(7):1179–1187. | |
Foggensteiner L, Bevan AP, Thomas R, et al. Cellular and subcellular distribution of polycystin-2, the product of the PKD2 gene. J Am Soc Nephrol. 2000;11(5):814–827. | |
Yoder BK, Hou X, Guay-Woodford LM. The polycystic kidney disease proteins, polycystin-1, polycystin-2, polaris, and cystin, are co-localized in renal cilia. J Am Soc Nephrol. 2002;13(10):2508–2516. | |
Nauli SM, Alenghat FJ, Luo Y, et al. Polycystins 1 and 2 mediate mechanosensation in the primary cilium of kidney cells. Nat Genet. 2003;33(2):129–137. | |
Hogan MC, Manganelli L, Wollard JR, et al. Characterization of PKD protein-positive exosome-like vesicles. J Am Soc Nephrol. 2009;20(2):278–288. | |
Gabow PA, Johnson AM, Kaehny WD, Manco-Johnson ML, Duley IT, Everson GT. Risk factors for the development of hepatic cysts in autosomal dominant polycystic kidney disease. Hepatology. 1990;11(6):1033–1037. | |
Fick GM, Johnson AM, Hammond WS, Gabow PA. Causes of death in autosomal dominant polycystic kidney disease. J Am Soc Nephrol. 1995;5(12):2048–2056. | |
Rahman E, Naiz FA, Al-Suwaida A, et al. Analysis of causes of mortality in patients with autosomal dominant polycystic kidney disease: a single center study. Saudi J Kidney Dis Transpl. 2009;20(5):806–810. | |
Gabow PA, Johnson AM, Kaehny WD, et al. Factors affecting the progression of renal disease in autosomal-dominant polycystic kidney disease. Kidney Int. 1992;41(5):1311–1319. | |
Cadnapaphornchai MA, McFann K, Strain JD, Masoumi A, Schrier RW. Increased left ventricular mass in children with autosomal dominant polycystic kidney disease and borderline hypertension. Kidney Int. 2008;74(9):1192–1196. | |
Ecder T. Cardiovascular complications in autosomal dominant polycystic kidney disease. Curr Hypertens Rev. 2013;9(1):2–11. | |
Schrier RW. Renal volume, renin-angiotensin-aldosterone system, hypertension, and left ventricular hypertrophy in patients with autosomal dominant polycystic kidney disease. J Am Soc Nephrol. 2009;20(9):1888–1893. | |
Ecder T, Schrier RW. Hypertension in autosomal-dominant polycystic kidney disease: early occurrence and unique aspects. J Am Soc Nephrol. 2001;12(1):194–200. | |
Wang D, Iversen J, Strandgaard S. Endothelium-dependent relaxation of small resistance vessels is impaired in patients with autosomal dominant polycystic kidney disease. J Am Soc Nephrol. 2000;11(8):1371–1376. | |
Wang D, Strandgaard S, Borresen ML, et al. Asymmetric dimethylarginine and lipid peroxidation products in early autosomal dominant polycystic kidney disease. Am J Kidney Dis. 2008;51(2):184–191. | |
Menon V, Rudym D, Chandra P, Miskulin D, Perrone R, Sarnak M. Inflammation, oxidative stress, and insulin resistance in polycystic kidney disease. Clin J Am Soc Nephrol. 2011;6(1):7–13. | |
Choukroun G, Itakura Y, Albouze G, et al. Factors influencing renal failure in autosomal dominant polycystic kidney disease. J Am Soc Nephrol. 1995;6(6):1634–1642. | |
Kelleher CL, McFann KK, Johnson AM, Schrier RW. Characteristics of hypertension in young adults with autosomal dominant polycystic kidney disease. Am J Hypertens. 2004;17(11 Pt 1):1029–1034. | |
Schrier RW, McFann KK, Johnson AM. Epidemiological study of kidney survival in autosomal dominant polycystic kidney disease. Kidney Int. 2003;63(2):678–685. | |
United States Renal Data System. 2013 Annual Data Report: Atlas of Chronic Kidney Disease and End-Stage Renal Disease in the United States. Bethesda, MD, USA: National Institutes of Health, National Institute of Diabetes and Digestive and Kidney Diseases; 2013. Available from: http://www.usrds.org/atlas.aspx. Accessed September 14, 2014. | |
Shaw C, Simms RJ, Pitcher D, Sandford R. Epidemiology of patients in England and Wales with autosomal dominant polycystic kidney disease and end-stage renal failure. Nephrol Dial Transplant. April 15, 2014. [Epub ahead of print.] | |
Spithoven EM, Kramer A, Meijer E, et al. Analysis of data from the ERA-EDTA Registry indicates that conventional treatments for chronic kidney disease do not reduce the need for renal replacement therapy in autosomal dominant polycystic kidney disease. Kidney Int. 2005;12(3):300–311. | |
Patch C, Charlton J, Roderick PJ, Gulliford MC. Use of antihypertensive medications and mortality of patients with autosomal dominant polycystic kidney disease: a population-based study. Am J Kidney Dis. 2011;57(6):856–862. | |
Kimberling WJ, Kumar S, Gabow PA, Kenyon JB, Connolly CJ, Somlo S. Autosomal dominant polycystic kidney disease: localization of the second gene to chromosome 4q13-q23. Genomics. 1993;18(3):467–472. | |
de Almeida S, de Almeida E, Peters D, et al. Autosomal dominant polycystic kidney disease: evidence for the existence of a third locus in a Portuguese family. Hum Genet. 1995;96(1):83–88. | |
Bogdanova N, Dworniczak B, Dragova D, et al. Genetic heterogeneity of polycystic kidney disease in Bulgaria. Hum Genet. 1995;95(6):645–650. | |
Paul BM, Consugar MB, Ryan Lee M, et al. Evidence of a third ADPKD locus is not supported by re-analysis of designated PKD3 families. Kidney Int. 2014;85(2):383–392. | |
Bihoreau MT, Ceccherini I, Browne J, et al. Location of the first locus, PKDr1, controlling autosomal dominant polycystic kidney disease in Han:SPRD cy/+ rat. Hum Mol Genet. 1997;6(4):609–613. | |
Cogswell C, Price SJ, Hou X, Guay-Woodford LM, Flaherty L, Bryda EC. Positional cloning of jcpk/bpk locus of the mouse. Mamm Genome. 2003;14(4):242–249. | |
Torres VE, Harris PC. Polycystic kidney disease: genes, proteins, animal models, disease mechanisms and therapeutic opportunities. J Intern Med. 2007;261(1):17–31. | |
Harris PC, Rossetti S. Molecular diagnostics for autosomal dominant polycystic kidney disease. Nat Rev Nephrol. 2010;6(4):197–206. | |
Barua M, Cil O, Paterson AD, et al. Family history of renal disease severity predicts the mutated gene in ADPKD. J Am Soc Nephrol. 2009;20(8):1833–1838. | |
Rossetti S, Consugar MB, Chapman AB, et al. Comprehensive molecular diagnostics in autosomal dominant polycystic kidney disease. J Am Soc Nephrol. 2007;18(7):2143–2160. | |
Harris PC, Bae KT, Rossetti S, et al. Cyst number but not the rate of cystic growth is associated with the mutated gene in autosomal dominant polycystic kidney disease. J Am Soc Nephrol. 2006;17(11):3013–3019. | |
Rossetti S, Chauveau D, Walker D, et al. A complete mutation screen of the ADPKD genes by DHPLC. Kidney Int. 2002;61(5):1588–1599. | |
Rossetti S, Hopp K, Sikkink RA, et al. Identification of gene mutations in autosomal dominant polycystic kidney disease through targeted resequencing. J Am Soc Nephrol. 2012;23(5):915–933. | |
Rossetti S, Burton S, Strmecki L, et al. The position of the polycystic kidney disease 1 (PKD1) gene mutation correlates with the severity of renal disease. J Am Soc Nephrol. 2002;13(5):1230–1237. | |
Rossetti S, Chauveau D, Kubly V, et al. Association of mutation position in polycystic kidney disease 1 (PKD1) gene and development of a vascular phenotype. Lancet. 2003;361(9376):2196–2201. | |
Cornec-Le Gall E, Audrezet MP, Chen JM, et al. Type of PKD1 mutation influences renal outcome in ADPKD. J Am Soc Nephrol. 2013; 24(6):1006–1013. | |
Magistroni R, He N, Wang K, et al. Genotype-renal function correlation in type 2 autosomal dominant polycystic kidney disease. J Am Soc Nephrol. 2003;14(5):1164–1174. | |
Rossetti S, Kubly VJ, Consugar MB, et al. Incompletely penetrant PKD1 alleles suggest a role for gene dosage in cyst initiation in polycystic kidney disease. Kidney Int. 2009;75(8):848–855. | |
Vujic M, Heyer CM, Ars E, et al. Incompletely penetrant PKD1 alleles mimic the renal manifestations of ARPKD. J Am Soc Nephrol. 2010;21(7):1097–1102. | |
Pei Y, Lan Z, Wang K, et al. A missense mutation in PKD1 attenuates the severity of renal disease. Kidney Int. 2012;81(4):412–417. | |
Reiterova J, Stekrova J, Merta M, et al. Autosomal dominant polycystic kidney disease in a family with mosaicism and hypomorphic allele. BMC Nephrol. 2013;14:59. | |
Torra Balcells R, Ars Criach E. Molecular diagnosis of autosomal dominant polycystic kidney disease. Nefrologia. 2011;31(1):35–43. | |
Coto E, Sanz de Castro S, Aguado S, et al. DNA microsatellite analysis of families with autosomal dominant polycystic kidney diseasetypes 1 and 2: evaluation of clinical heterogeneity between both forms of the disease. J Med Genet. 1995;32(6):442–445. | |
Torra R, Badenas C, Darnell A, et al. Linkage, clinical features, and prognosis of autosomal dominant polycystic kidney disease types 1 and 2. J Am Soc Nephrol. 1996;7(10):2142–2151. | |
Pei Y. Diagnositic approach in autosomal dominant polycystic kidney disease. Clin J Am Soc Nephrol. 2006;1(5):1108–1114. | |
Verlinsky Y, Rechitsky S, Verlinsky O, Ozen S, Beck R, Kuliev A. Preimplantation genetic diagnosis for polycystic kidney disease. Fertil Steril. 2004;82(4):926–929. | |
De Rycke M, Georgiou I, Sermon K, et al. PGD for autosomal dominant polycystic kidney disease type 1. Mol Hum Reprod. 2005;11(1):65–71. | |
Collins SC. Preimplantation genetic diagnosis: technical advances and expanding applications. Curr Opin Obstet Gynecol. 2013;25(3):201–206. | |
Qian F, Watnick TJ, Onuchic LF, Germino GG. The molecular basis of focal cyst formation in human autosomal dominant polycystic kidney disease type I. Cell. 1996;87(6):979–987. | |
Brasier JL, Henske EP. Loss of the polycystic kidney disease (PKD1) region of chromosome 16p13 in renal cyst cells supports a loss-of-function model for cyst pathogenesis. J Clin Invest. 1997;99(2):194–199. | |
Koptides M, Hadjimichael C, Koupepidou P, Pierides A, Constantinou Deltas C. Germinal and somatic mutations in the PKD2 gene of renal cysts in autosomal dominant polycystic kidney disease. Hum Mol Genet. 1999;8(3):509–513. | |
Pei Y, Watnick T, He N, et al. Somatic PKD2 mutations in individual kidney and liver cysts support a “two-hit” model of cystogenesis in type 2 autosomal dominant polycystic kidney disease. J Am Soc Nephrol. 1999;10(7):1524–1529. | |
Wu G, D’Agati V, Cai Y, et al. Somatic inactivation of Pkd2 results in polycystic kidney disease. Cell. 1998;93(2):177–188. | |
Gogusev J, Murakami I, Doussau M, et al. Molecular cytogenetic aberrations in autosomal dominant polycystic kidney disease tissue. J Am Soc Nephrol. 2003;14(2):359–366. | |
Li M, Qin S, Wang L, Zhou J. Genomic instability in patients with autosomal-dominant polycystic kidney disease. J Int Med Res. 2013;41(1):169–175. | |
Wu Y, Antony S, Meitzler JL, Doroshow JH. Molecular mechanisms underlying chronic inflammation-associated cancers. Cancer Lett. 2014;345(2):164–173. | |
Lantinga-van Leeuwen IS, Dauwerse JG, Baelde HJ, et al. Lowering of Pkd1 expression is sufficient to cause polycystic kidney disease. Hum Mol Genet. 2004;13(24):3069–3077. | |
Park EY, Sung YH, Yang MH, et al. Cyst formation in kidney via B-Raf signaling in the PKD2 transgenic mice. J Biol Chem. 2009;284(11):7214–7222. | |
Eccles MR, Stayner CA. Polycystic kidney disease – where gene dosage counts. F1000Prime Rep. 2014;6:24. | |
Fain PR, McFann KK, Taylor MR, et al. Modifier genes play a significant role in the phenotypic expression of PKD1. Kidney Int. 2005;67(4):1256–1267. | |
Liu M, Shi S, Senthilnathan S, et al. Genetic variation of DKK3 may modify renal disease severity in ADPKD. J Am Soc Nephrol. 2010;21(9):1510–1520. | |
Bergmann C, von Bothmer J, Ortiz Brüchle N, et al. Mutations in multiple PKD genes may explain early and severe polycystic kidney disease. J Am Soc Nephrol. 2011;22(11):2047–2056. | |
Wilson PD, Hreniuk D, Gabow PA. Abnormal extracellular matrix and excessive growth of human adult polycystic kidney disease epithelia. J Cell Physiol. 1992;150(2):360–369. | |
Ebihara I, Nakamura T, Takahashi T, et al. Altered extracellular matrix component gene expression in murine polycystic kidney. Ren Physiol Biochem. 1995;18(2):73–80. | |
Lee JE, Park MH, Park JH. The gene expression profile of cyst epithelial cells in autosomal dominant polycystic kidney disease patients. J Biochem Mol Biol. 2004;37(5):612–617. | |
Shannon MB, Patton BL, Harvey SJ, Miner JH. A hypomorphic mutation in the mouse laminin alpha5 gene causes polycystic kidney disease. J Am Soc Nephrol. 2006;17(7):1913–1922. | |
Liu B, Chenghai L, Liu Z, Dai Z, Tao Y. Increasing extracellular matrix collagen level and MMP activity induces cyst development in polycystic kidney disease. BMC Nephrol. 2012;13:109. | |
Drummond I. Polycystins, focal adhesions and extracellular matrix interactions. Biochim Biophys Acta. 2011;1812(10):1322–1326. | |
Xia S, Li X, Johnson T, Seidel C, Wallace DP, Li R. Polycystin-dependent fluid flow sensing targets histone deacetylase 5 to prevent the development of renal cysts. Development. 2010;137(7):1075–1084. | |
Fan LX, Li X, Magenheimer B, Calvet JP, Li X. Inhibition of histone deacetylases targets the transcription regulator Id2 to attenuate cystic epithelial cell proliferation. Kidney Int. 2012;81(1):76–85. | |
Woo YM, Bae JB, Oh YH, et al. Genome-wide methylation profiling of ADPKD identified epigenetically regulated genes associated with renal cyst development. Hum Genet. 2014;133(3):281–297. | |
Nishida N, Kudo M. Oxidative stress and epigenetic instability in human hepatocarcinogenesis. Dig Dis. 2013;31(5–6):447–453. | |
Pandey P, Brors B, Srivastava PK, et al. Microarray-based approach identifies microRNAs and their target functional patterns in polycystic kidney disease. BMC Genomics. 2008;9:624. | |
Pandey P, Qin S, Ho J, Zhou J, Kreidberg JA. Systems biology approach to identify transcriptome reprogramming and candidate microRNA targets during the progression of polycystic kidney disease. BMC Syst Biol. 2011;5:56. | |
Patel V, Williams D, Hajarnis S, et al. miR-17~92 miRNA cluster promotes kidney cyst growth in polycystic kidney disease. Proc Nat Acad Sci U S A. 2013;110(26):10765–10770. | |
Sun H, Li QW, Lv XY, et al. MicroRNA-17 post-transcriptionally regulates polycystic kidney disease-2 gene and promotes cell proliferation. Mol Biol Rep. 2010;37(6):2951–2958. | |
Tran U, Zakin L, Schweickert A, et al. The RNA-binding protein bicaudal C regulates polycystin 2 in the kidney by antagonizing miR-17 activity. Development. 2010;137(7):1107–1116. | |
Tran U, Pickney LM, Ozpolat BD, Wessely O. Xenopus Bicaudal-C is required for the differentiation of the amphibian pronephros. Dev Biol. 2007;307(1):152–164. | |
Ben-Dov IZ, Tan YC, Morozov P, et al. Urine microRNA as potential biomarkers of autosomal dominant polycystic kidney disease progression: description of miRNA profiles at baseline. PLoS One. 2014;9(1):e86856. | |
Schrier R, McFann K, Johnson A, et al. Cardiac and renal effects of standard versus rigorous blood pressure control in autosomal-dominant polycystic kidney disease: results of a seven-year prospective randomized study. J Am Soc Nephrol. 2002;13(7):1733–1739. | |
Orskov B, Romming Sorensen V, Feldt-Rasmussen B, Strandgaard S. Improved prognosis in patients with autosomal dominant polycystic kidney disease in Denmark. Clin J Am Soc Nephrol. 2010;5(11):2034–2039. | |
Chapman AB, Torres VE, Perrone RD, et al. The HALT polycystic kidney disease trials: design and implementation. Clin J Am Soc Nephrol. 2010;5(1):102–109. | |
Frame MC. Newest findings on the oldest oncogene; how activated src does it. J Cell Sci. 2004;117 Pt 7:989–998. | |
Elliott J, Zheleznova NN, Wilson PD. c-Src inactivation reduces renal epithelial cell-matrix adhesion, proliferation, and cyst formation. Am J Physiol Cell Physiol. 2011;301(2):C522–C529. | |
Sweeney WE Jr, von Vigier RO, Frost P, Avner ED. Src inhibition ameliorates polycystic kidney disease. J Am Soc Nephrol. 2008;19(7):1331–1341. | |
Kocaman O, Oflaz H, Yekeler E, et al. Endothelial dysfunction and increased carotid intima-media thickness in patients with autosomal dominant polycystic kidney disease. Am J Kidney Dis. 2004;43(5):854–860. | |
Wang D, Iversen J, Strandgaard S. Contractility and endothelium-dependent relaxation of resistance vessels in polycystic kidney disease rats. J Vasc Res. 1999;36(6):502–509. | |
Borresen ML, Wang D, Strandgaard S. Pulse wave reflection is amplified in normotensive patients with autosomal-dominant polycystic kidney disease and normal renal function. Am J Nephrol. 2007;27(3):240–246. | |
Tsuchiya K, Yoshimoto T, Hirata Y. Endothelial dysfunction is related to aldosterone excess and raised blood pressure. Endocr J. 2009;56(4):553–559. | |
Nishizaka MK, Zaman MA, Green SA, Renfroe KY, Calhoun DA. Impaired endothelium-dependent flow-mediated vasodilation in hypertensive subjects with hyperaldosteronism. Circulation. 2004;109(23):2857–2861. | |
Pitt B, Zannad F, Remme WJ, et al. The effect of spironolactone on morbidity and mortality in patients with severe heart failure. Randomized Aldactone Evaluation Study Investigators. N Engl J Med. 1999;341(10):709–717. | |
Abiose AK, Mansoor GA, Barry M, Soucier R, Nair CK, Hager D. Effect of spironolactone on endothelial function in patients with congestive heart failure on conventional medical therapy. Am J Cardiol. 2004;93(12):1564–1566. | |
Yamanari H, Nakamura K, Miura D, Yamanari S, Ohe T. Spironolactone and chlorthalidone in uncontrolled elderly hypertensive patients treated with calcium antagonists and angiotensin II receptor-blocker: effects on endothelial function, inflammation, and oxidative stress. Clin Exp Hypertens. 2009;31(7):585–594. | |
Lieberthal W, Levine JS. Mammalian target of rapamycin and the kidney. II. Pathophysiology and therapeutic implications. Am J Physiol Renal Physiol. 2012;303(2):F180–F191. | |
Shillingford JM, Murcia NS, Larson CH, et al. The mTOR pathway is regulated by polycystin-1, and its inhibition reverses renal cystogenesis in polycystic kidney disease. Proc Nat Acad Sci U S A. 2006;103(14):5466–5471. | |
Wahl PR, Serra AL, Le Hir M, Molle KD, Hall MN, Wuthrich RP. Inhibition of mTOR with sirolimus slows disease progression in Han:SPRD rats with autosomal dominant polycystic kidney disease (ADPKD). Nephrol Dial Transplant. 2006;21(3):598–604. | |
Shillingford JM, Piontek KB, Germino GG, Weimbs T. Rapamycin ameliorates PKD resulting from conditional inactivation of Pkd1. J Am Soc Nephrol. 2010;21(3):489–497. | |
Soliman AR, Ismail E, Zamil S, Lotfy A. Sirolimus therapy for patients with adult polycystic kidney disease: a pilot study. Transplant Proc. 2009;41(9):3639–3641. | |
Perico N, Antiga L, Caroli A, et al. Sirolimus therapy to halt the progression of ADPKD. J Am Soc Nephrol. 2010;21(6):1031–1040. | |
Serra AL, Poster D, Kistler AD, et al. Sirolimus and kidney growth in autosomal dominant polycystic kidney disease. N Engl J Med. 2010;363(9):820–829. | |
Stallone G, Infante B, Grandaliano G, et al. Rapamycin for treatment of type I autosomal dominant polycystic kidney disease (RAPYD-study): a randomized, controlled study. Nephrol Dial Transplant. 2012;27(9):3560–3567. | |
Braun WE, Schold JD, Stephany BR, Spirko RA, Herts BR. Low-dose rapamycin (sirolimus) effects in autosomal dominant polycystic kidney disease: an open-label randomized controlled pilot study. Clin J Am Soc Nephrol. 2014;9(5):881–888. | |
Walz G, Budde K, Mannaa M, et al. Everolimus in patients with autosomal dominant polycystic kidney disease. N Engl J Med. 2010;363(9):830–840. | |
He Q, Lin C, Ji S, Chen J. Efficacy and safety of mTOR inhibitor therapy in patients with early-stage autosomal dominant polycystic kidney disease: a meta-analysis of randomized controlled trials. Am J Med Sci. 2012;344(6):491–497. | |
Zhou X, Fan LX, Sweeney WE Jr, Denu JM, Avner ED, Li X. Sirtuin 1 inhibition delays cyst formation in autosomal-dominant polycystic kidney disease. J Clin Invest. 2013;123(7):3084–3098. | |
Yamaguchi T, Nagao S, Kasahara M, Takahashi H, Grantham JJ. Renal accumulation and excretion of cyclic adenosine monophosphate in a murine model of slowly progressive polycystic kidney disease. Am J Kidney Dis. 1997;30(5):703–709. | |
Torres VE, Harris PC. Strategies targeting cAMP signaling in the treatment of polycystic kidney disease. J Am Soc Nephrol. 2014;25(1):18–32. | |
Ruggenenti P, Remuzzi A, Ondei P, et al. Safety and efficacy of long-acting somatostatin treatment in autosomal-dominant polycystic kidney disease. Kidney Int. 2005;68(1):206–216. | |
Caroli A, Perico N, Perna A, et al. Effect of long-acting somatostatin analogue on kidney and cyst growth in autosomal dominant polycystic kidney disease (ALADIN): a randomised, placebo-controlled, multicentre trial. Lancet. 2013;382(9903):1485–1495. | |
Hogan MC, Masyuk TV, Page LJ, et al. Randomized clinical trial of long-acting somatostatin for autosomal dominant polycystic kidney and liver disease. J Am Soc Nephrol. 2010;21(6):1052–1061. | |
Hogan MC, Masyuk TV, Page L, et al. Somatostatin analog therapy for severe polycystic liver disease: results after 2 years. Nephrol Dial Transplant. 2012;27(9):3532–3539. | |
van Keimpema L, Nevens F, Vanslembrouck R, et al. Lanreotide reduces the volume of polycystic liver: a randomized, double-blind, placebo-controlled trial. Gastroenterology. 2009;137(5):1661–1668. | |
Chrispijn M, Nevens F, Gevers TJ, et al. The long-term outcome of patients with polycystic liver disease treated with lanreotide. Aliment Pharmacol Ther. 2012;35(2):266–274. | |
McFarlane SI, Muniyappa R, Francisco R, Sowers JR. Clinical review 145: Pleiotropic effects of statins: lipid reduction and beyond. J Clin Endocrinol Metab. 2002;87(4):1451–1458. | |
Hafez KS, Inman SR, Stowe NT, Novick AC. Renal hemodynamic effects of lovastatin in a renal ablation model. Urology. 1996;48(6):862–867. | |
van Dijk MA, Kamper AM, van Veen S, Souverijn JH, Blauw GJ. Effect of simvastatin on renal function in autosomal dominant polycystic kidney disease. Nephrol Dial Transplant. 2001;16(11):2152–2157. | |
Gile RD, Cowley BD Jr, Gattone VH 2nd, O’Donnell MP, Swan SK, Grantham JJ. Effect of lovastatin on the development of polycystic kidney disease in the Han:SPRD rat. Am J Kidney Dis. 1995;26(3):501–507. | |
Zafar I, Tao Y, Falk S, McFann K, Schrier RW, Edelstein CL. Effect of statin and angiotensin-converting enzyme inhibition on structural and hemodynamic alterations in autosomal dominant polycystic kidney disease model. Am J Physiol Renal Physiol. 2007;293(3):F854–F859. | |
Fassett RG, Coombes JS, Packham D, Fairley KF, Kincaid-Smith P. Effect of pravastatin on kidney function and urinary protein excretion in autosomal dominant polycystic kidney disease. Scand J Urol Nephrol. 2010;44(1):56–61. | |
Cadnapaphornchai MA, George DM, McFann K, et al. Effect of pravastatin on total kidney volume, left ventricular mass index, and microalbuminuria in pediatric autosomal. 2011;301(5):F1005–F1013. | |
Torres VE, Chapman AB, Devuyst O, et al. Tolvaptan in patients with autosomal dominant polycystic kidney disease. N Engl J Med. 2012;367(25):2407–2418. | |
Kiviharju TM, Lecane PS, Sellers RG, Peehl DM. Antiproliferative and proapoptotic activities of triptolide (PG490), a natural product entering clinical trials, on primary cultures of human prostatic epithelial cells. Clin Cancer Res. 2002;8(8):2666–2674. | |
Leuenroth SJ, Okuhara D, Shotwell JD, et al. Triptolide is a traditional Chinese medicine-derived inhibitor of polycystic kidney disease. Proc Nat Acad Sci U S A. 2007;104(11):4389–4394. | |
Leuenroth SJ, Bencivenga N, Igarashi P, Somlo S, Crews CM. Triptolide reduces cystogenesis in a model of ADPKD. J Am Soc Nephrol. 2008;19(9):1659–1662. | |
Leuenroth SJ, Bencivenga N, Chahboune H, Hyder F, Crews CM. Triptolide reduces cyst formation in a neonatal to adult transition Pkd1 model of ADPKD. Nephrol Dial Transplant. 2010;25(7):2187–2194. | |
Chen D, Ma Y, Wang X, et al. Triptolide-containing formulation in patients with autosomal dominant polycystic kidney disease and proteinuria: an uncontrolled trial. Am J Kidney Dis. 2014;63(6):1070–1072. |
 © 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.