Back to Journals » International Journal of Chronic Obstructive Pulmonary Disease » Volume 13
Autophagy, selective autophagy, and necroptosis in COPD
Authors Mizumura K ![]() , Maruoka S
, Maruoka S ![]() , Shimizu T, Gon Y
, Shimizu T, Gon Y ![]()
Received 30 May 2018
Accepted for publication 1 August 2018
Published 9 October 2018 Volume 2018:13 Pages 3165—3172
DOI https://doi.org/10.2147/COPD.S175830
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Prof. Dr. Richard Russell
Kenji Mizumura, Shuichiro Maruoka, Tetsuo Shimizu, Yasuhiro Gon
Division of Respiratory Medicine, Department of Internal Medicine, Nihon University School of Medicine, Tokyo, Japan
Abstract: COPD is characterized by persistent respiratory symptoms and airflow limitation, caused by a mixture of small airway disease and pulmonary emphysema. Programmed cell death has drawn the attention of COPD researchers because emphysema is thought to result from epithelial cell death caused by smoking. Although apoptosis has long been thought to be the sole form of programmed cell death, recent studies have reported the existence of a genetically programmed and regulated form of necrosis called necroptosis. Autophagy was also previously considered a form of programmed cell death, but this has been reconsidered. However, recent studies have revealed that autophagy can regulate programmed cell death, including apoptosis and necroptosis. It is also becoming clear that autophagy can selectively degrade specific proteins, organelles, and invading bacteria by a process termed “selective autophagy” and that this process is related to the pathogenesis of human diseases. In this review, we outline the most recent studies implicating autophagy, selective autophagy, and necroptosis in COPD. Strategies targeting these pathways may yield novel therapies for COPD.
Keywords: ciliophagy, mitophagy, programmed cell death, pulmonary emphysema
Introduction
COPD involves persistent respiratory symptoms and airflow limitation. The chronic airflow limitation is caused by a mixture of small airway disease and pulmonary emphysema, usually due to significant exposure to noxious particles or gases. Cigarette smoke (CS) is the most common identifiable risk factor for COPD, with smokers known to have a greater COPD mortality rate than non-smokers.1 Pulmonary emphysema is thought to result from epithelial cell death caused by smoking; therefore, COPD researchers have devoted considerable attention to programmed cell death.
Apoptosis was previously recognized as the sole form of programmed cell death, whereas necrosis was considered as uncontrolled cell death induced by extreme physical or chemical stress. However, emerging studies have demonstrated the existence of a genetically programmed and regulated form of necrosis, termed “necroptosis,” defined as necrotic cell death dependent on the receptor-interacting protein kinase 3 (RIPK3).2–4 RIPK3, RIPK1, and mixed-lineage kinase domain-like protein (MLKL) form a multiprotein complex called the “necrosome,” in which MLKL is an essential necroptosis inducer that acts downstream of RIPK3.5,6 Unlike apoptosis, which is thought to be a weak inducer of inflammation with little release of damage-associated molecular patterns (DAMPs) from dying cells, necroptosis is considered a strong inducer of inflammation that releases massive amounts of DAMPs.7
Autophagy is a highly conserved process through which cells can recycle organelles and proteins by degrading them in lysosomes.8 Autophagy proceeds through sequential steps beginning with the generation of autophagosomes from an isolation membrane, followed by elongation to form a mature autophagosome that captures cytosolic cargo. While the term “autophagic cell death” has long been used to refer to a type of cell death associated with excessive cytoplasmic vacuolization,9 autophagy is considered to primarily act as a protective mechanism that may prevent cell death by maintaining cellular integrity through regenerating metabolic precursors and clearing subcellular debris.10 Thus, currently, autophagic cell death is not considered as a form of programmed cell death,11 but is ascribed to cell death with, rather than by, autophagy. In contrast, it is becoming clear that autophagy can regulate other cell death pathways. In cells of childhood acute lymphoblastic leukemia, induction of autophagy-dependent necroptosis is required to overcome glucocorticoid resistance.12 In endothelial cells, autophagy inhibition rescues palmitic acid-induced necroptosis.13 Thus, accumulating evidence suggests that autophagy may regulate necroptosis in the pathogenesis of human diseases.
While autophagy has long been considered as simply a non-specific homeostatic cellular process, increasing evidence suggests that it represents a more selective process than originally anticipated.14 Selective autophagy delivers a wide range of autophagic cargo from protein aggregates to whole organelles and even intracellular microbes to the lysosome for degradation. It is thought that ubiquitin-positive substrates, such as protein aggregates, mitochondria, and invading bacteria not dealt with the proteasome system, are selectively degraded by autophagy.15 Ubiquitination seems to function as a general tag for selective autophagy in mammalian cells. Various selective autophagy subtypes have been recognized and named after their specific targets, for example, aggregated proteins: aggrephagy,16 mitochondria: mitophagy,17 pathogens: xenophagy,18 and cilia: ciliophagy.19 The study of selective autophagy is an emerging field that is expected to provide new insights into the pathogenesis of human lung diseases.14 In this review, we examine the growing evidence favoring the contribution of autophagy, selective autophagy (Table 1), and necroptosis (Table 2) to COPD pathogenesis and discuss the dual nature of these processes in the lungs. A better understanding of the protective and injurious effects of these processes in disease pathogenesis will help design personalized therapies for COPD treatment.
 | Table 1 Roles of selective autophagy in COPD |
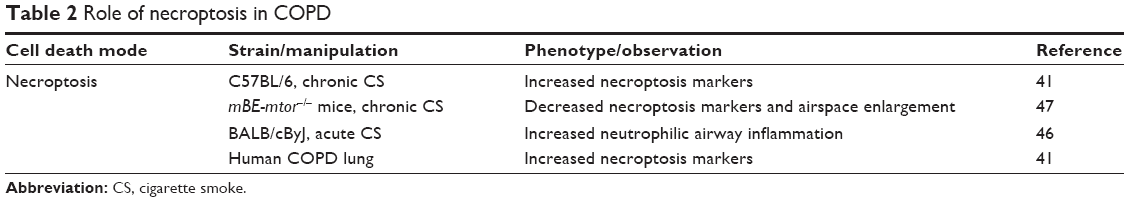 | Table 2 Role of necroptosis in COPD |
Molecular mechanisms of autophagy, selective autophagy, and necroptosis
Autophagy is an evolutionarily conserved catabolic process by which cytoplasmic materials are delivered to and degraded in the lysosome.10 Often referred to simply as autophagy, macroautophagy is the best characterized form of autophagy, involving the engulfment of cytoplasmic contents and organelles through a complex reorganization of subcellular membranes to form a new organelle: the autophagosome. In the 1990s, genetic studies in yeast identified a series of autophagy-related (ATG) genes that regulate the macroautophagic process.20,21 Autophagosome elongation requires two ubiquitin-like conjugation systems, the ATG5-12 conjugation system and the ATG8 conjugation system, which are regulated by various ATG proteins. Microtubule-associated protein light chain 3 (LC3), a homolog of yeast ATG8, is one of the most well-known ATG proteins and is often used as a specific marker to evaluate autophagy in vitro and in vivo.22
Selective autophagy serves to selectively degrade mitochondria and other specific organelles, bacteria, and protein aggregates using the autophagic machinery.23 To evaluate the inclusive list of molecular mechanisms involved in selective autophagy currently in the literature is beyond the scope of this review; therefore, we focus on mitophagy, the autophagy-dependent elimination of mitochondria, in this review. The proposed model for mitophagy is that damaged and depolarized mitochondria stabilize PTEN-induced putative kinase protein 1 (PINK1), which in turn recruits the E3 ubiquitin ligase Parkin. Subsequently, Parkin ubiquitinylates various mitochondrial outer membrane proteins, including the mitofusins MFN1 and MFN2,24 voltage-dependent anion channels (VDACs),25 and mitochondrial rho GTPase (MIRO),26 and induces mitophagy by recruiting autophagic receptors such as p62.
A multiprotein complex, the necrosome, is formed by RIPK3, RIPK1, and MLKL and regulates necroptosis. Among these proteins, MLKL is an essential necroptosis inducer that acts downstream of RIPK3. Oligomerization and intramolecular autophosphorylation of RIPK3 lead to the recruitment and phosphorylation of MLKL, which exposes a 4-helical bundle domain.27 Recent studies suggest two functions for MLKL: it serves as a platform at the plasma membrane for the recruitment of Ca2+ or Na+ ion channels28,29 and serves as a direct pore-forming complex recruited by the binding of the amino terminus of its four-helical bundle domain to negatively charged phosphatidylinositolphosphates.30–32
Autophagy: regulation and function in COPD
Ning et al33 examined comprehensive gene expression profiles in GOLD-2 vs GOLD-0 smokers, which suggested that the autophagy-related protein ATG8/microtubule-associated protein-1 LC3 was a candidate of molecular target in COPD. Further investigation demonstrated pivotal functional roles for autophagy proteins in CS-induced emphysema.34,35 In COPD lung tissues, autophagic vacuoles (autophagosomes/autolysosomes) were increased compared to those in control tissues as observed by electron microscopy, a gold-standard method for autophagy determination, whereas little vacuole formation was evident in control tissues.35 Expression of the active form of LC3B, LC3B-II, as well as Atg4B, Atg5, Atg12, and Atg7 was significantly increased in COPD lungs.35 Genetic depletion of the essential autophagy mediators, LC3B and Beclin 1, ameliorated CS extract (CSE)-induced epithelial cell death.34 To determine whether increased autophagosome formation was correlated with autophagic activity in the lungs of CS-treated mice, we conducted in vivo autophagic flux assays.19,36 Analysis of LC3B steady-state levels in a lysosome-enriched fraction of lung homogenates revealed a time-dependent increase in leupeptin-sensitive LC3B degradation in vivo, which persisted until 24 hours after CS exposure, supporting the conclusion that CS causes a cumulative increase in autophagic flux in lung tissues.19 These data suggest that the CS-induced autophagic pathway may regulate epithelial cell death associated with emphysematous airspace enlargement in chronic CS-exposed mice and in patients with COPD.
While CS induces autophagy in pulmonary epithelial cells, Monick et al37 reported defective autophagy in CS-exposed macrophages. Such a deficit in autophagy was also found in the alveolar macrophages of smokers, suggesting that impaired delivery of bacteria to lysosomes may lead to recurrent infections in patients with COPD.37 Moreover, Fujii et al38 evaluated autophagy-regulated senescence in bronchial epithelial cells treated with CSE. While 3-methyladenine, an autophagy inhibitor, enhanced CSE-induced senescence in primary human bronchial epithelial cells (HBECs), Torin-1, an autophagy inducer, suppressed CSE-induced HBEC senescence.38 The authors found that baseline autophagic activity in HBECs from patients with COPD was significantly higher than that in HBECs from non-smokers and non-COPD smokers; however, autophagy induction in HBECs in response to CSE exposure was significantly lower in COPD patients than in non-smokers and non-COPD smokers.38 These findings suggest that the autophagic response is insufficient in the lungs of patients with COPD, which leads to accelerated epithelial cell senescence.
Interestingly, mTOR signaling also has been linked to CS-induced COPD/emphysema.39 mTOR is an evolutionarily conserved serine-threonine kinase that acts as a sensor of environmental and cellular nutrition and energy status that also plays an important role in regulating autophagy.40 Rtp801, a stress-related protein triggered by adverse environmental conditions, was overexpressed in human emphysematous lungs and in lungs of mice exposed to CS.39 Rtp801 stabilized the assembly of the mTOR inhibitory complex TSC1-TSC2, resulting in exacerbation of oxidative stress-induced cell death.39 The mTOR inhibitor rapamycin reduced alveolar inflammation in wild-type mice exposed to CS, whereas it increased the number of apoptotic and inflammatory cells in room air-exposed wild-type mice and abrogated the protective effects of Rtp801 knockout in mice exposed to CS. These studies highlight that the timing and lung cell targets of mTOR inhibition may be essential to define its beneficial and pathological roles in COPD.
Thus, accumulating evidence demonstrates that autophagy plays previously unforeseen roles in COPD pathogenesis and that it can have both protective and injurious effects on COPD progression. Although there is no unifying explanation for the discrepancies between the results of various studies, the timing and lung cell targets for autophagy may be essential to define its beneficial vs pathologic roles. A better understanding of the balance between cytoprotective and pro-death functions of autophagy in response to CS will be required for the therapeutic targeting of this process in COPD.
Selective autophagy: mitophagy and ciliophagy in COPD
Recently, we reported that mitophagy regulates necroptosis, which contributes to the pathogenesis of COPD (Figure 1).41 Mitophagy selectively eliminates mitochondria by employing the autophagic machinery.42 Parkin and PINK1 are key regulators of mitophagy.42 CSE causes significant mitochondrial depolarization and induces mitophagy in lung epithelial cells.41 We demonstrated that the mitochondrial division/mitophagy inhibitor Mdivi-1 protected against CS-induced cell death by reducing the phosphorylation of MLKL, a substrate for RIP3 in the necroptosis pathway.41 Mice genetically deficient in PINK1 were protected against mitochondrial dysfunction, airspace enlargement, and mucociliary clearance disruption during CS exposure.41 Our results suggest that CS-activated mitophagy may alter mitochondrial membrane integrity and induce mitophagy and necroptosis in pulmonary epithelial cells. Furthermore, recent studies have suggested that CS-induced mitophagy may regulate cellular senescence in COPD pathogenesis.43 Genetic blocking of mitophagy resulted in enhanced CS-induced mitochondrial ROS production and cellular senescence in HBECs. The precise mechanism by which a cell “decides” to undergo either mitophagy-induced necroptosis or senescence remains unclear. One hypothesis is that mitophagy may lead to either senescence or necroptosis depending on the degree of cellular injury.43
 | Figure 1 Mitophagy and necroptosis in COPD. |
We also reported that ciliophagy, the consumption of cilia components by autophagy, regulates cilia length during CS exposure (Figure 2).19 We showed that autophagy-impaired (Becn1+/− or Map1lc3B−/−) mice, as well as tracheal epithelial cells isolated from these mice, display reduced CS-induced cilia shortening.19 We identified cytosolic deacetylase histone deacetylase 6 (HDAC6) as a critical regulator of ciliophagy during CS exposure.19 Importantly, analysis of human COPD specimens demonstrated epigenetic deregulation of HDAC6 by hypomethylation and increased protein expression in the airway.19 These data suggest that ciliophagy, an HDAC6-dependent selective autophagy pathway, may represent a novel pathway critical to cilia homeostasis in response to CS exposure.
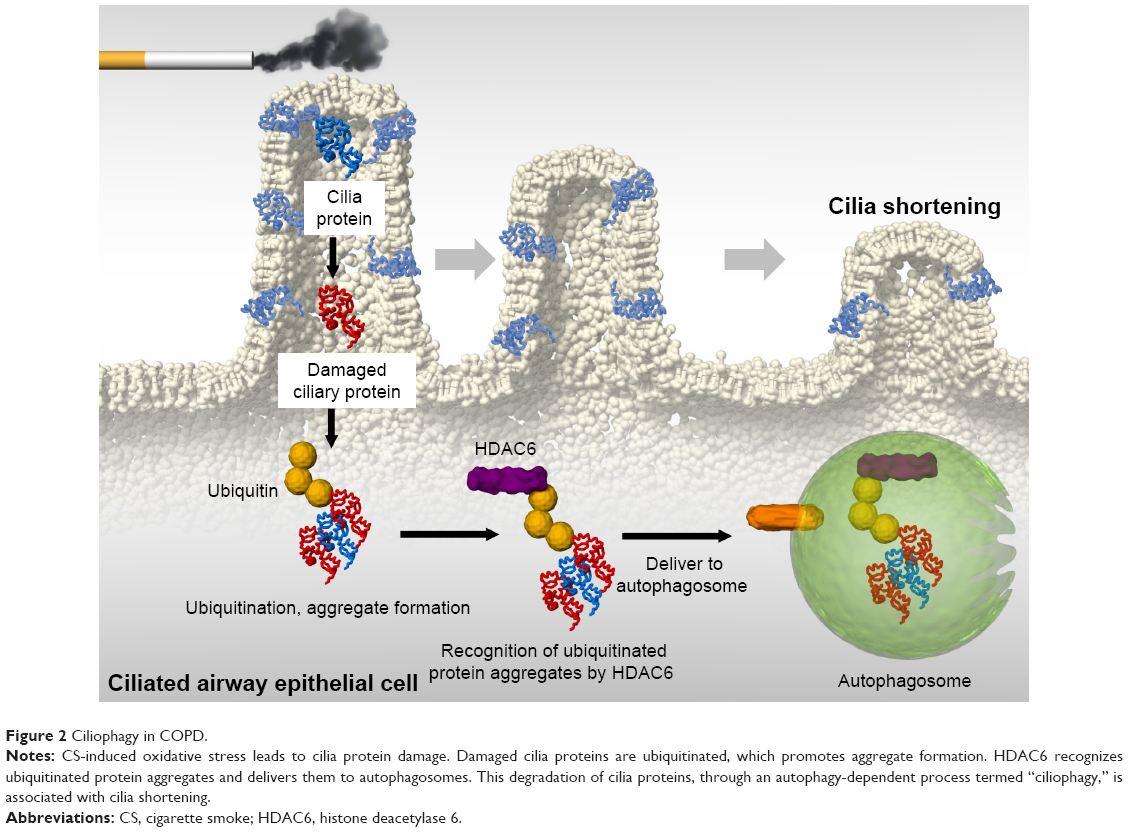 | Figure 2 Ciliophagy in COPD. |
It remains unclear how cells utilize non-selective and selective autophagy pathways in response to CS exposure. We speculate that CS induces non-selective autophagy coincident with injury to organelles (eg, mitochondria, cilia), which would be degraded by selective autophagy. Thus, non-selective and selective autophagy may likely proceed simultaneously in a single cell. In addition, either process may be impacted by cell type and stimulus intensity. Thus, future studies are needed to elucidate the relationship between both types of autophagy in response to CS exposure.
Necroptosis: a critical regulator of cell death in COPD
It is becoming clear that autophagy and selective autophagy can regulate apoptosis and necroptosis in COPD.34,41 Chen et al34 showed that dynamic interactions of the autophagy protein LC3B with Cav-1 and Fas regulate CS-induced lung epithelial cell apoptosis, leading to emphysematous airspace enlargement. Recently, we reported that CS-induced mitophagy may alter mitochondrial membrane integrity, leading to necroptosis induction (Figure 1).41 CSE-induced cell death in pulmonary epithelial cells was effectively reduced by the treatment with necrox-5, a necrosis inhibitor with antioxidant activity that localizes primarily in the mitochondria, or with necrostatin-1, a necroptosis inhibitor. CSE treatment resulted in increased phosphorylation of MLKL, a substrate for RIP3 in the necroptosis pathway, which was decreased in PINK1-knockdown cells.41 Similarly, we observed that the mitochondrial division/mitophagy inhibitor Mdivi-1 inhibited MLKL phosphorylation induced by CSE in pulmonary epithelial cells. These results suggest that mitophagy regulates CSE-induced necroptosis in pulmonary epithelial cells. Furthermore, we detected high levels of RIP3 near emphysematous regions in the lungs of mice exposed to CS for 3 months.41 Importantly, we observed stronger PINK1 and RIP3 expression in the epithelial cells of patients with COPD than in that of control subjects. Confocal imaging confirmed higher and coincident expression of PINK1 and RIP3 in COPD compared to that in healthy lungs. These observations in human clinical samples strongly suggest that our experimental data associating mitophagy with necroptosis are relevant to COPD. Moreover, we have reported that sphingolipids regulate lung epithelial cell mitophagy and necroptosis during CS exposure.44 Inhibition of ceramide-generating acid sphingomyelinase reduces both CS-induced PINK1 phosphorylation and necroptosis.44 Our data provide a mechanistic explanation for how CS induces mitophagy-driven necroptosis and further support the fact that the dysregulated sphingolipid metabolism is implicated in the pathogenesis of structural cell injury in COPD.
COPD is also characterized by chronic inflammation of the airways, lung tissue, and pulmonary blood vessels as a result of exposure to CS. COPD patients show chronic neutrophilic inflammation in the airways, accompanied by aberrant tissue repair and remodeling.45 While apoptosis has often been described as a major physiological process for emphysema, apoptosis is not generally accompanied by inflammation because of no or limited release of DAMPs. Therefore, it has been proposed that additional mechanisms underlie CS-induced apoptosis leading to airway inflammation in COPD. In contrast, there is a general consensus that necroptosis directly triggers inflammation through a massive release of DAMPs from the disintegrating cells.7 Pouwels et al46 revealed that CS-induced necroptosis and the release of DAMPs trigger neutrophilic airway inflammation in mice. Exposure to CS increased the levels of DAMPs and numbers of neutrophils in bronchoalveolar lavage fluid in mice, and this effect was statistically reduced on treatment with the necroptosis inhibitor necrostatin-1.46 More recently, Wang et al47,48 reported a novel regulatory mechanism of necroptosis-mediated inflammation. Endoplasmic reticulum chaperone GRP78 promoted a CSE-induced inflammatory response and mucus hyperproduction in airway epithelial cells, likely through the upregulation of necroptosis and subsequent activation of the nuclear factor-κB and activator protein-1 pathways.48 In contrast, the mTOR suppresses the CS-induced inflammatory cytokines interleukin-6 and interleukin-8 through the nuclear factor-kB pathway, likely through the modulation of autophagy, apoptosis, and necroptosis.47 These results suggest that necroptosis might be a promising therapeutic target for emphysema and inflammation.
Conclusion
Accumulating evidence demonstrates that autophagy, selective autophagy, and necroptosis exert previously unknown functions during COPD pathogenesis. While autophagic cell death is not currently considered a form of programmed cell death, autophagy proteins can regulate programmed cell death, including necroptosis and apoptosis, in a context-specific fashion. Furthermore, the autophagic pathway shares a number of signal molecules with programmed cell death pathways. In COPD, cell death has been mainly evaluated in terms of clinical phenotypes of emphysema (eg, loss of alveolar surface area); however, it is now predicted that cell death can regulate further biological processes, such as inflammation. In particular, necroptosis is a promising target for regulating COPD inflammation, as it directly triggers inflammation through a massive release of DAMPs. Therefore, careful consideration and further research are needed for designing strategies to manipulate these pathways as valid therapeutic interventions.
Acknowledgment
The authors are grateful to Augustine MK Choi for collaboration and suggesting the topic treated in this article.
Author contributions
KM, SM, TS, and YG designed the strategy and goal of this review. KM drafted the article. All authors read and approved the final manuscript. All authors contributed to data analysis, drafting and revising the article, gave final approval of the version to be published, and agree to be accountable for all aspects of the work.
Disclosure
The authors report no conflicts of interest in this work.
References
Kohansal R, Martinez-Camblor P, Agustí A, Buist AS, Mannino DM, Soriano JB. The natural history of chronic airflow obstruction revisited: an analysis of the Framingham offspring cohort. Am J Respir Crit Care Med. 2009;180(1):3–10. | ||
Cho YS, Challa S, Moquin D, et al. Phosphorylation-driven assembly of the RIP1-RIP3 complex regulates programmed necrosis and virus-induced inflammation. Cell. 2009;137(6):1112–1123. | ||
He S, Wang L, Miao L, et al. Receptor interacting protein kinase-3 determines cellular necrotic response to TNF-alpha. Cell. 2009;137(6):1100–1111. | ||
Zhang DW, Shao J, Lin J, et al. RIP3, an energy metabolism regulator that switches TNF-induced cell death from apoptosis to necrosis. Science. 2009;325(5938):332–336. | ||
Sun L, Wang H, Wang Z, et al. Mixed lineage kinase domain-like protein mediates necrosis signaling downstream of RIP3 kinase. Cell. 2012;148(1–2):213–227. | ||
Zhao J, Jitkaew S, Cai Z, et al. Mixed lineage kinase domain-like is a key receptor interacting protein 3 downstream component of TNF-induced necrosis. Proc Natl Acad Sci U S A. 2012;109(14):5322–5327. | ||
Pasparakis M, Vandenabeele P. Necroptosis and its role in inflammation. Nature. 2015;517(7534):311–320. | ||
Klionsky DJ, Emr SD. Autophagy as a regulated pathway of cellular degradation. Science. 2000;290(5497):1717–1721. | ||
Schweichel JU, Merker HJ. The morphology of various types of cell death in prenatal tissues. Teratology. 1973;7(3):253–266. | ||
Mizushima N, Komatsu M. Autophagy: renovation of cells and tissues. Cell. 2011;147(4):728–741. | ||
Kroemer G, Levine B. Autophagic cell death: the story of a misnomer. Nat Rev Mol Cell Biol. 2008;9(12):1004–1010. | ||
Bonapace L, Bornhauser BC, Schmitz M, et al. Induction of autophagy-dependent necroptosis is required for childhood acute lymphoblastic leukemia cells to overcome glucocorticoid resistance. J Clin Invest. 2010;120(4):1310–1323. | ||
Khan MJ, Rizwan Alam M, Waldeck-Weiermair M, et al. Inhibition of autophagy rescues palmitic acid-induced necroptosis of endothelial cells. J Biol Chem. 2012;287(25):21110–21120. | ||
Mizumura K, Choi AM, Ryter SW. Emerging role of selective autophagy in human diseases. Front Pharmacol. 2014;5:244. | ||
Kirkin V, Mcewan DG, Novak I, Dikic I. A role for ubiquitin in selective autophagy. Mol Cell. 2009;34(3):259–269. | ||
Yamamoto A, Simonsen A. The elimination of accumulated and aggregated proteins: a role for aggrephagy in neurodegeneration. Neurobiol Dis. 2011;43(1):17–28. | ||
Lemasters JJ. Selective mitochondrial autophagy, or mitophagy, as a targeted defense against oxidative stress, mitochondrial dysfunction, and aging. Rejuvenation Res. 2005;8(1):3–5. | ||
Levine B. Eating oneself and uninvited guests: autophagy-related pathways in cellular defense. Cell. 2005;120(2):159–162. | ||
Lam HC, Cloonan SM, Bhashyam AR, et al. Histone deacetylase 6-mediated selective autophagy regulates COPD-associated cilia dysfunction. J Clin Invest. 2013;123(12):5212–5230. | ||
Tsukada M, Ohsumi Y. Isolation and characterization of autophagy-defective mutants of Saccharomyces cerevisiae. FEBS Lett. 1993;333(1–2):169–174. | ||
Klionsky DJ, Cregg JM, Dunn WA, et al. A unified nomenclature for yeast autophagy-related genes. Dev Cell. 2003;5(4):539–545. | ||
Kabeya Y, Mizushima N, Yamamoto A, Oshitani-Okamoto S, Ohsumi Y, Yoshimori T. LC3, GABARAP and GATE16 localize to autophagosomal membrane depending on form-II formation. J Cell Sci. 2004;117(Pt 13):2805–2812. | ||
Stolz A, Ernst A, Dikic I. Cargo recognition and trafficking in selective autophagy. Nat Cell Biol. 2014;16(6):495–501. | ||
Gegg ME, Cooper JM, Chau KY, Rojo M, Schapira AH, Taanman JW. Mitofusin 1 and mitofusin 2 are ubiquitinated in a PINK1/parkin-dependent manner upon induction of mitophagy. Hum Mol Genet. 2010;19(24):4861–4870. | ||
Geisler S, Holmström KM, Skujat D, et al. PINK1/Parkin-mediated mitophagy is dependent on VDAC1 and p62/SQSTM1. Nat Cell Biol. 2010;12(2):119–131. | ||
Wang X, Winter D, Ashrafi G, et al. PINK1 and Parkin target Miro for phosphorylation and degradation to arrest mitochondrial motility. Cell. 2011;147(4):893–906. | ||
Hildebrand JM, Tanzer MC, Lucet IS, et al. Activation of the pseudokinase MLKL unleashes the four-helix bundle domain to induce membrane localization and necroptotic cell death. Proc Natl Acad Sci U S A. 2014;111(42):15072–15077. | ||
Cai Z, Jitkaew S, Zhao J, et al. Plasma membrane translocation of trimerized MLKL protein is required for TNF-induced necroptosis. Nat Cell Biol. 2014;16(1):55–65. | ||
Chen X, Li W, Ren J, et al. Translocation of mixed lineage kinase domain-like protein to plasma membrane leads to necrotic cell death. Cell Res. 2014;24(1):105–121. | ||
Su L, Quade B, Wang H, Sun L, Wang X, Rizo J. A plug release mechanism for membrane permeation by MLKL. Structure. 2014;22(10):1489–1500. | ||
Wang H, Sun L, Su L, et al. Mixed lineage kinase domain-like protein MLKL causes necrotic membrane disruption upon phosphorylation by RIP3. Mol Cell. 2014;54(1):133–146. | ||
Dondelinger Y, Declercq W, Montessuit S, et al. MLKL compromises plasma membrane integrity by binding to phosphatidylinositol phosphates. Cell Rep. 2014;7(4):971–981. | ||
Ning W, Li CJ, Kaminski N, et al. Comprehensive gene expression profiles reveal pathways related to the pathogenesis of chronic obstructive pulmonary disease. Proc Natl Acad Sci U S A. 2004;101(41):14895–14900. | ||
Chen ZH, Lam HC, Jin Y, et al. Autophagy protein microtubule-associated protein 1 light chain-3B (LC3B) activates extrinsic apoptosis during cigarette smoke-induced emphysema. Proc Natl Acad Sci U S A. 2010;107(44):18880–18885. | ||
Chen ZH, Kim HP, Sciurba FC, et al. Egr-1 regulates autophagy in cigarette smoke-induced chronic obstructive pulmonary disease. PLoS One. 2008;3(10):e3316. | ||
Haspel J, Shaik RS, Ifedigbo E, et al. Characterization of macroautophagic flux in vivo using a leupeptin-based assay. Autophagy. 2011;7(6):629–642. | ||
Monick MM, Powers LS, Walters K, et al. Identification of an autophagy defect in smokers’ alveolar macrophages. J Immunol. 2010;185(9):5425–5435. | ||
Fujii S, Hara H, Araya J, et al. Insufficient autophagy promotes bronchial epithelial cell senescence in chronic obstructive pulmonary disease. Oncoimmunology. 2012;1(5):630–641. | ||
Yoshida T, Mett I, Bhunia AK, et al. Rtp801, a suppressor of mTOR signaling, is an essential mediator of cigarette smoke-induced pulmonary injury and emphysema. Nat Med. 2010;16(7):767–773. | ||
Kim YC, Guan KL. mTOR: a pharmacologic target for autophagy regulation. J Clin Invest. 2015;125(1):25–32. | ||
Mizumura K, Cloonan SM, Nakahira K, et al. Mitophagy-dependent necroptosis contributes to the pathogenesis of COPD. J Clin Invest. 2014;124(9):3987–4003. | ||
Youle RJ, Narendra DP. Mechanisms of mitophagy. Nat Rev Mol Cell Biol. 2011;12(1):9–14. | ||
Ito S, Araya J, Kurita Y, et al. PARK2-mediated mitophagy is involved in regulation of HBEC senescence in COPD pathogenesis. Autophagy. 2015;11(3):547–559. | ||
Mizumura K, Justice MJ, Schweitzer KS, et al. Sphingolipid regulation of lung epithelial cell mitophagy and necroptosis during cigarette smoke exposure. Faseb J. 2018;32(4):1880–1890. | ||
Curtis JL, Freeman CM, Hogg JC. The immunopathogenesis of chronic obstructive pulmonary disease: insights from recent research. Proc Am Thorac Soc. 2007;4(7):512–521. | ||
Pouwels SD, Zijlstra GJ, van der Toorn M, et al. Cigarette smoke-induced necroptosis and DAMP release trigger neutrophilic airway inflammation in mice. Am J Physiol Lung Cell Mol Physiol. 2016;310(4):L377–L386. | ||
Wang Y, Liu J, Zhou JS, et al. MTOR suppresses cigarette smoke-induced epithelial cell death and airway inflammation in chronic obstructive pulmonary disease. J Immunol. 2018;200(8):2571–2580. | ||
Wang Y, Zhou JS, Xu XC, et al. Endoplasmic reticulum chaperone GRP78 mediates cigarette smoke-induced necroptosis and injury in bronchial epithelium. Int J Chron Obstruct Pulmon Dis. 2018;13:571–581. |
 © 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.