Back to Journals » Neuropsychiatric Disease and Treatment » Volume 11
Autophagy dysfunction upregulates beta-amyloid peptides via enhancing the activity of γ-secretase complex
Authors Cai Z, Zhou Y, Liu Z, Ke Z, Zhao B, He X, Tian F, Yan N
Received 17 March 2015
Accepted for publication 21 April 2015
Published 17 August 2015 Volume 2015:11 Pages 2091—2099
DOI https://doi.org/10.2147/NDT.S84755
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 6
Editor who approved publication: Professor Wai Kwong Tang
Zhiyou Cai,1 Yingjun Zhou,1 Zhou Liu,2,3 Zunyu Ke,1 Bin Zhao2,3
1Department of Neurology, Renmin Hospital, Hubei University of Medicine, Shiyan, Hubei Province, 2Department of Neurology, The Affiliated Hospital of Guangdong Medical College, 3Institute of Neurology, Guangdong Medical College, Zhanjiang, Guangdong Province, People’s Republic of China
Abstract: Numerous studies have shown that autophagy failure plays a critical role in the pathogenesis of Alzheimer’s disease, including increased expression of beta-amyloid (Aβ) protein and the dysfunction of Aβ clearance. To further evaluate the role of autophagy in Alzheimer’s disease, the present study was implemented to investigate the effects of autophagy on α-secretase, β-secretase, or γ-secretase, and observe the effects of autophagy on autophagic clearance markers. These results showed that both autophagy inhibitor and inducer enhanced the activity of α-, β-, and γ-secretases, and Aβ production. Autophagy inhibitor may more activate γ-secretase and promote Aβ production and accumulation than its inducer. Both autophagy inhibitor and inducer had no influence on Aβ clearance. Hence, autophagy inhibitor may activate γ-secretase and promote Aβ production and accumulation, but has no influence on Aβ clearance.
Keywords: Alzheimer’s disease, autophagy, beta-amyloid, secretases
Introduction
Alzheimer’s disease (AD) is a progressive neurodegenerative disease, of which cognitive impairment gradually worsens over time.1–3 As AD advances through the brain, it eventually affects all aspects of a person’s life including mental abilities, emotions and moods, behavior, and the ability to carry out daily activities like eating and grooming.4,5 It is well known that there are three consistent neuro-pathological hallmarks associated with AD, including extracellular amyloid-rich senile plaques, intracellular neurofibrillary tangles and neuronal degeneration of basal forebrain cholinergic neurons that innervate the hippocampus and the cortex.6–8
Autophagy is one of the major degradation pathway characterized by a ubiquitous cellular process responsible for the bulk degradation of long-lived proteins and organelles through an autophagosome-lysosomal pathway.9,10 The word autophagy is derived from the Greek words auto (meaning “self”) and phage (meaning “eat”), of which the main function of autophagy refers to clear abnormal or obsolete cellular proteins.11 There are at least three processes by which intracellular constituents enter lysosomes for degradation distinguishable by their mechanisms: macro-autophagy (the most prevalent form), micro-autophagy, and chaperone-mediated autophagy. Autophagy exists in both normal cellular homeostasis and disease states. Increasing findings have demonstrated that autophagosome-lysosomal dysfunction contributes to severe neurodegenerative disorders related to accumulations of lysosomes and autophagic vacuoles (AVs).11 Compelling research studies have supported that the pivotal role of autophagy in the clearance of aggregate-prone proteins is responsible for several neurodegenerative disorders,12,13 which are implicated in the pathogenesis of AD,13,14 Parkinson’s disease,15,16 Huntington’s disease,17,18 and other related disorders.
An autophagosome, a spherical structure with double layer membranes, is a cellular vesicle that ingests cellular debris and transports the debris to lysosomes. Growing evidence indicates that the rate of autophagosome formation and maturation and the efficiency of autophagosome/lysosome fusion decline in neurodegenerative diseases with age.19–21 A growing number of studies have shown that dysfunction of autophagy plays a critical role in the pathogenesis of AD, including senile plaques, neurofibrillary tangles, and neuronal degeneration.13,22 Moreover, it has been found that immature AVs accumulate during the early evolution of pathology in a dendrite in the PS1-APP (amyloid protein precursor) mouse model of AD while pathological AVs’ accumulation is associated with inhibited retrograde AVs’ transport and impaired autophagosome/lysosome fusion.23–25 Furthermore, a link between autophagy dysfunction and beta-amyloid (Aβ) generation and clearance has been reported to occur in AD.26,27 A number of papers have investigated the precise role of autophagy in the Aβ generation and clearance. However, understanding the exact mechanism may help to design more effective therapeutic strategies to prevent neuronal degeneration and death. It is well known that Aβ production and deposition represent a key feature and is thought as the classic pathological hallmarks in AD. Aβ is generated from APP by the sequential actions of two proteolytic enzymes: β-secretase (beta-site APP cleavage enzyme, BACE) and γ-secretase complex.28,29 In addition, APP undergoes another cleavage: the non-amyloidogenic processing by α-secretase and γ-secretase complex to release membrane-anchored carboxy-terminal fragments that may be associated with apoptosis.30,31 Therefore, it is possible that autophagy regulated Aβ generation via controlling the activity of α-, β-, or γ-secretases. The present study was implemented to investigate the effects of autophagy on α-, β-, and γ-secretase, and the level of Aβ, and to observe the effects of autophagy on autophagic clearance markers. The aim is to further evaluate the role of autophagy in the neurodegenerative process of AD.
These results noted that the both autophagy inhibitor and inducer enhanced Aβ1–42 expression while the level of Aβ1–42 peptide was more remarkably increased by the autophagy inhibitor than by the autophagy inducer. Both autophagy inhibitor and inducer increased the activity of α-, β-, and γ-secretases while the components of the γ-secretase complex (Presenilin 1, Nicastrin, and presenilin enhancer 2 [Pen-2]) were more activated by autophagy inhibitor, compared with the inducer treatment. However, this study revealed that there was no difference between the treatment of the autophagy inhibitor and autophagy inducer. Our study suggests that autophagy inhibitor may activate γ-secretase and promote Aβ accumulation, but has no influence on Aβ clearance.
Materials and methods
Cell culture
SH-SY5Y, a human-derived neuroblastoma cell line, is thrice-cloned originally from SK-N-SH and widely used in the scientific research of neurodegenerative disorders.32 SH-SY5Y was grown in Dulbecco’s Modified Eagle’s Medium (Thermo Fisher Scientific, Waltham, MA, USA) supplemented with 10% fetal bovine serum (HyClone, Logan, UT, USA) and 1% penicillin/streptomycin. Cells were maintained in a humidified atmosphere at 37°C with 5% CO2. The autophagy inhibitor (3-methyladenine, 3-MA, Santa Cruz Biotechnology, Dallas, TX, USA) and inducer (STF-62247, Selleck Chemicals, Houston, TX, USA) were dissolved in dimethyl sulfoxide and used at the following concentrations: the inhibitor (3-MA), 10 mM; the inducer (STF-62247), 10 μM. The cells were treated with the autophagy inhibitor and inducer without fetal bovine serum for 24 hours.
RNA extraction and real-time quantitative PCR
Total cellular RNA was extracted using the Trizol reagent (Invitrogen), according to the manufacturer’s instruction. cDNAs were synthesized and real-time PCR was performed using the GoTaq® 2-Step RT-qPCR System (Promega, Madison, WI, USA) in an ABI Prism 7500 Sequence Detection System (Applied Biosystems, Foster City, CA, USA). Gene expression data were normalized to the geometric mean of the β-actin, housekeeping gene, to control for variability in expression levels and calculated as 2−[(Ct of gene)-(Ct of β-actin)], where Ct represents the threshold cycle for each transcript. The used primers in this study were the following: BACE1 (forward: 5′-AGGCAGCTGTCCAGCACATACC-3′, reverse: 3′-TAGCCAGCTGGTGCAGGAGAT-5′), a disintegrin and metalloprotease 17 (ADAM17) (forward: 5′-AGCAGGTGTCGTTGTTCAGA-3′, reverse: 3′-GTGGCGATCACGAGAACAAT-5′), Presenilin 1 (forward: 5′-CAGGTGCTATAAGGTCATCC-3′, reverse: 3′-GTCCACAGCAACGTTATAGG-5′), Presenilin 2 (forward: 5′-CAGCTCATCTACACGCCATT-3′, reverse: 3′-CAGCAGCATCAGTGAAGACA-5′), Nicastrin (forward: 5′-TACGGAACCAGGTGGAGGAT-3′, reverse: 3′-GAAGGCACCAGAGTGGTCAG-5′), APH-1 (forward: 5′-TCCTGACTTCAGCCTTTCTGAC-3′, reverse: 3′-CAAGAGGCTGCGCTGAATAC-5′), Pen-2 (forward: 5′-GCCAAATCAAAGGCTATGTCTG-3′, reverse: 3′-ATGGTGAAGGAGAGGTAGTC-5′), β-actin (forward: 5′-TGGCACCCAGCACAATGAA-3′, reverse: 3′-CTAAGTCATAGTCCGCCTAGAAGCA-5′).
Western blotting
Cells were harvested in sampling buffer (62.5 mmol/L Tris-HCl [pH 6.8], 10% glycerol, 2% SDS) and heated for 5 minutes at 100°C. The concentration of extracted proteins was determined by the Bradford assay using a commercial kit (Bio-Rad, Berkeley, CA, USA). Equal quantities of protein were separated by electrophoresis on 12% SDS/polyacrylamide gels and transferred onto polyvinylidene difluoride membranes (Roche, Indianapolis, IN, USA). The membranes were then probed with rabbit primary antibodies against anti-BACE1, anti-ADAM17, anti-Presenilin 1, anti-Presenilin 2, anti-Nicastrin, anti-APH-1, anti-Pen-2, neprilysin (NEP), insulin-degrading enzyme (IDE), endothelin-converting enzyme 1 (ECE-1), and ECE-2 (1:1,000; Santa Cruz Biotechnology). The protein expression was detected with horseradish peroxidase-conjugated goat anti-rabbit IgG (1:2,000; Amersham Pharmacia Biotech, Piscataway, NJ, USA) and an enhanced chemiluminescence kit (Amersham Pharmacia Biotech) according to the manufacturer’s instructions. Anti-β-actin mouse monoclonal antibody (1:1,000; Cell Signaling, Danvers, MA, USA) acted as a loading control.
Transmission electron microscopy
SH-SY5Y treated with 3-MA and STF-62247 for 24 hours was fixed for 2 hours at room temperature with 2.5% glutaraldehyde in phosphate-buffered saline (pH 7.4) and, subsequently, with 1% OsO4 in 50 mM sodium cacodylate buffer (pH 7.3), dehydrated in an ethanol series and embedded into epon (catalyst). Ultrathin sections of 50 nm were contrasted with uranyl acetate and lead citrate and analyzed in a Tecnai Spirit transmission electron microscope (FEI) with an ORIUS CCD camera (Gatan).
Aβ1–42 measurement
Cells were seed at a constant density to obtain identical experimental conditions in the different tests and to achieve a high accuracy of the measurements. Aβ1–42 levels were determined in the culture supernatant using an ELISA kit (Uscn Life). The assays were performed according to manufacturer’s guidelines. Results were expressed as pg/mL.
Statistical analysis
Student’s t-test was used to evaluate the significant difference between two groups of data in all the pertinent experiments. Data were represented as the mean ± standard error of the mean. P-value <0.05 (using a two-tailed paired t-test) was considered as statistically significant.
Ethics
This study was approved by the Ethics committee of Renmin Hospital, Hubei University of Medicine.
Results
Inhibiting autophagy pathway increased Aβ expression
Mounting hypothesis has shown that extracellular Aβ plays an important role in AD pathogenesis.22,33,34 Consistent with other research results, the level of Aβ1–42 peptide was remarkably increased by the autophagy inhibitor (3-MA), compared with control and the autophagy inducer (STF-62247) (P<0.001) (Figure 1). Different from the other research results, both the autophagy inhibitor (3-MA) and inducer (STF-62247) enhanced Aβ1–42 expression whereas the level of Aβ1–42 stimulated by 3-MA was higher than that by STF-62247 (P<0.005) (Figure 1).
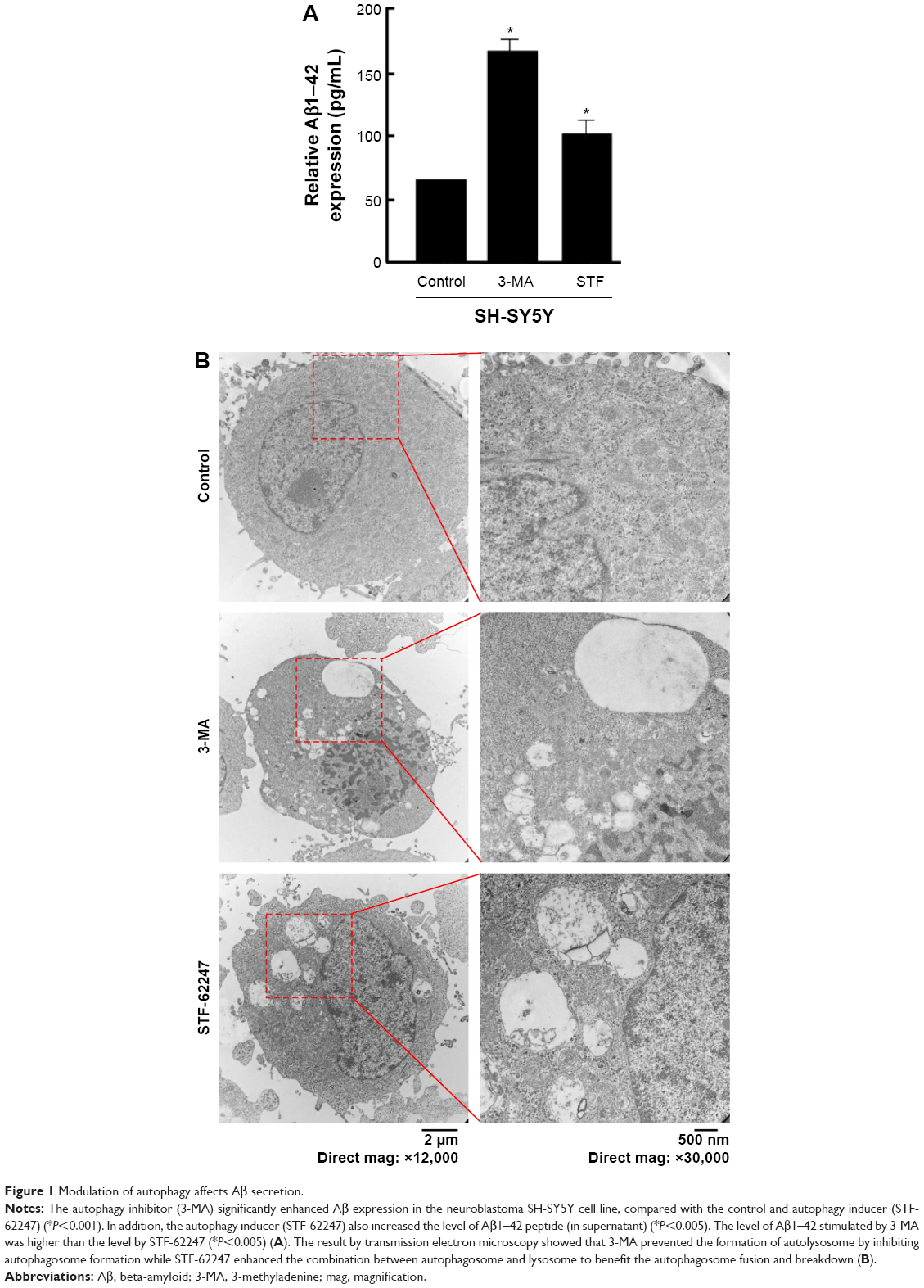 | Figure 1 Modulation of autophagy affects Aβ secretion. |
Autophagy dysfunction augmented the activity of γ-secretase complex
APP is cleaved sequentially at the extracellular site by β-secretase (BACE) and γ-secretase to release Aβ peptides.35–37 APP can be also cleaved by α-and γ-secretases to produce P3. The α-secretase represents two members of the family of ADAM: tumor necrosis factor-converting enzyme (ADAM17) and ADAM10. β-Secretase, a membrane-bound aspartic protease, is also called BACE.36,38 γ-Secretase is a multi-subunit protease complex, consisting of four individual proteins: Presenilin, Nicastrin, APH-1 (anterior pharynx-defective 1), and Pen-2.35,38
In the present study, the glioma cell line SH-SY5Y was treated with autophagy inhibitor (3-MA) and autophagy inducer (STF-62247). Then, the cell harvested to analyze by real-time quantitative PCR and Western blotting. These results demonstrated that α- and β-secretases, respectively exhibited as BACE1 and ADAM17, were not different between 3-MA and STF-62247 treatments. The components of the γ-secretase complex (Presenilin 1, Nicastrin, and Pen-2) were activated by 3-MA, compared with the STF-62247 treatment (P<0.001) (Figure 2). Both autophagy inhibitor and inducer improved the activity of α-, β-, and γ-secretases (P<0.005).
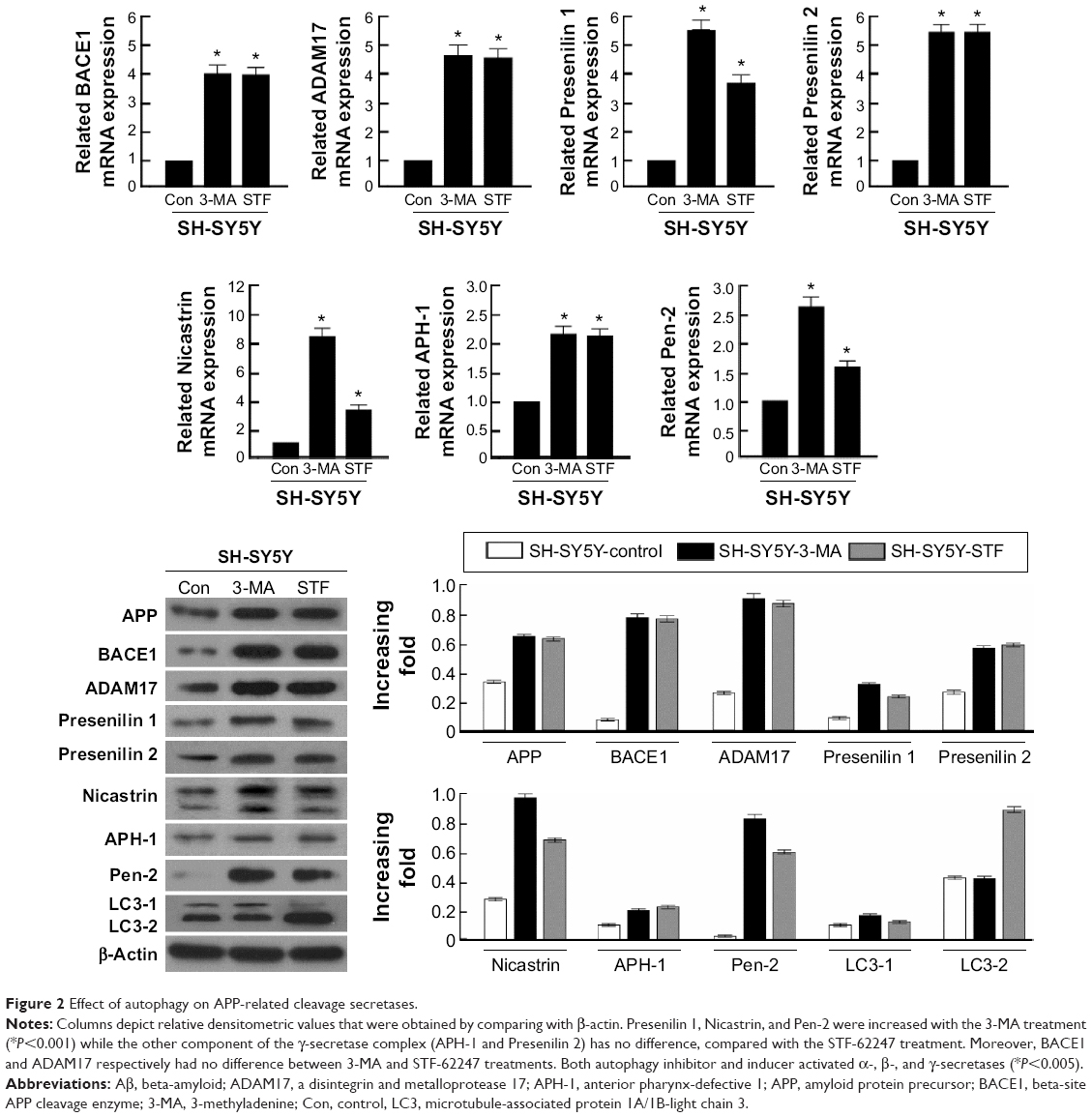 | Figure 2 Effect of autophagy on APP-related cleavage secretases. |
Autophagy had no effect on the Aβ clearance markers
Aβ production and failure of Aβ clearance are key factors in the development of AD. An overall impairment in Aβ clearance has been found where clearance rates for both Aβ1–42 and Aβ1–40 were impaired in AD.35,39 Several Aβ-degrading enzymes such as NEP, IDE, ECE-1, and ECE-2 are critical in Aβ accumulation determined in part by the imbalance between the production of Aβ and its removal from the brain.35,40,41 Therefore, the present study further investigated the influence of autophagy on the Aβ clearance markers (NEP, IDE, ECE-1, and ECE-2) although several research supported that the activation of autophagy benefited Aβ clearance. However, these findings revealed that there was no difference between the treatment of the autophagy inhibitor (3-MA) and autophagy inducer (STF-62247) (P>0.05) (Figure 3).
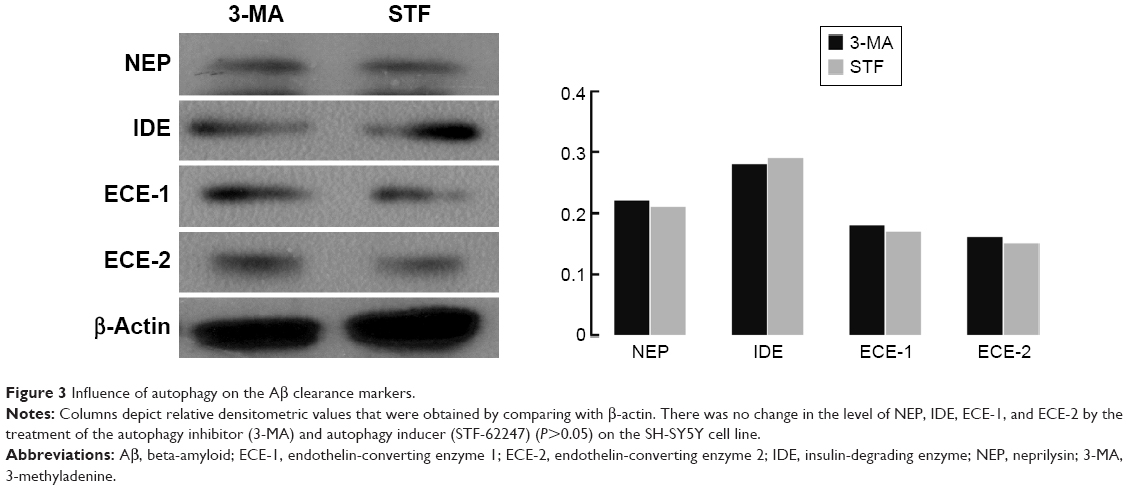 | Figure 3 Influence of autophagy on the Aβ clearance markers. |
Discussion
Autophagy is one major cellular pathway associated with the removal of aggregated proteins. A few studies elucidated that autophagy plays a critical role in multiple pathological lesions of AD, such as dysregulating APP turnover and enhancing the activity of β- and/or γ-secretases.42–44 Autophagy affects an array of molecular pathways that may play a role in both Aβ generation and Aβ clearance.22,45,46 However, the precise mechanism or role of autophagy in Aβ generation and Aβ clearance remained unclear.22 We found that both autophagy inhibitor and inducer are associated with Aβ generation via regulating α-, β-, and/or γ-secretases. The relationship between autophagy inhibitor and the level of Aβ is robust enough for autophagy inducer, which heightened Aβ expression through upregulating α-, β-, and/or γ-secretases. Nevertheless, there was no divergence in Aβ clearance under the treatment of autophagy inhibitor and inducer.
Our study has certain distinctive outcome. Formerly, inducing autophagy pathway keeps a foothold to restrain Aβ generation whereas inhibiting autophagy pathway is a friend for Aβ generation. Yet, the present results were different from the past whereas this study illustrated that both autophagy inhibitor and inducer account for Aβ generation. Consistent with the former findings, inhibiting autophagy pathway was a more strongly enhancer to the event of Aβ generation. Thus, it seems that our results were self-contradictory and paradox. It may be reasonable to take into consideration that Aβ plays a dual role as both physical and pathological substance. Functional Aβ is abundant in most environmental biofilms. Several potential activities have been discovered for Aβ, such as activating kinase enzymes, protection against oxidative stress, regulation of cholesterol transport, functioning as a transcription factor, and anti-microbial activity.47–50 Intracellular Aβ may impel a variety of cellular events such as protein degradation, axonal transport, neuronal firing, and autophagy to apoptosis.47,51–53 Therefore, it is indicated that autophagy plays dual roles in the degradation and secretion of Aβ since inducing and inhibiting autophagy pathway shares a common access to heighten Aβ production under the Aβ physical process (Figure 4).
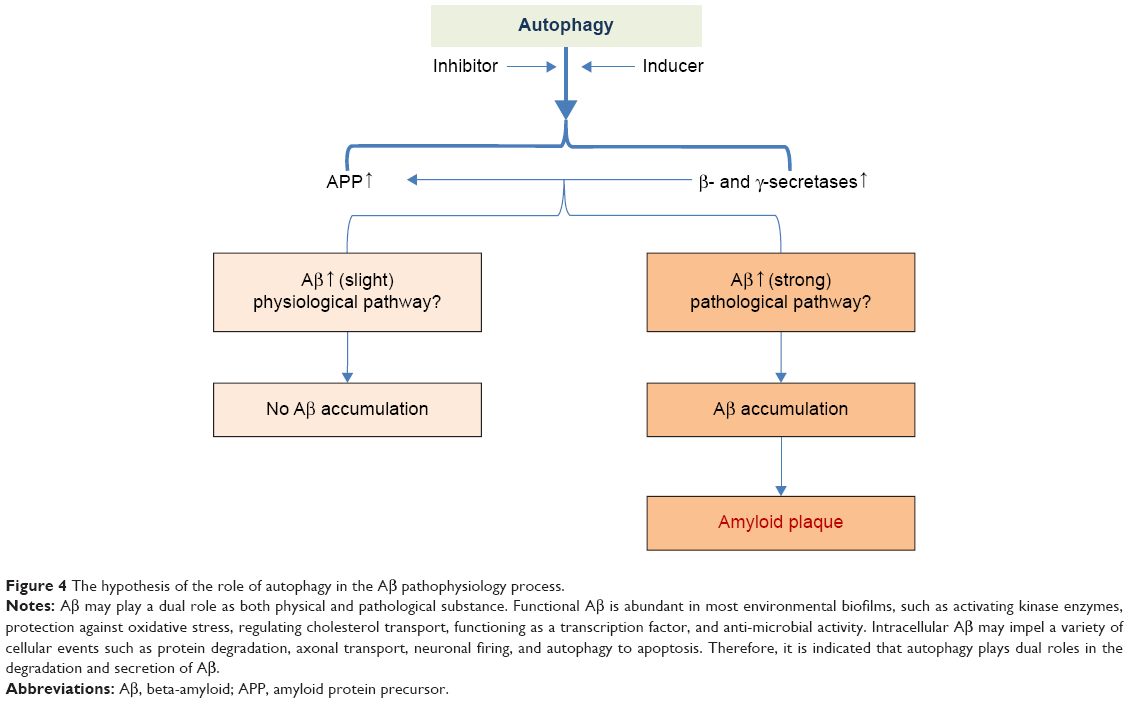 | Figure 4 The hypothesis of the role of autophagy in the Aβ pathophysiology process. |
It is well known that Aβ is generated from APP by the sequential cleavage of two proteolytic enzymes: β-(BACE) and γ-secretase.54,55 A few studies demonstrated that the role of autophagy has been linked to Aβ generation and aberrant processing of APP.56,57 Autophagy plays an important role in Aβ generation via regulating APP turnover and stimulating the activity of β- and γ-secretases.58,59 However, the key role of autophagy in AD development is still under consideration today.60 Whether autophagy regulates Aβ by influencing the expression of these associated secretases (α-, β-, and/or γ-secretases)? In controversy with the other findings which indicate that autophagy inducer can inhibit the expression of β- and γ-secretases, and Aβ generation, it was occurred that both autophagy inhibitor and inducer increased the activity of α-, β-, and γ-secretases in the present study. In consideration to the physiological function of APP, α-, β-, and γ-secretases,49,61 it may be a normal response to the stimulation of autophagy inducer and acute stress.
Importantly, this study demonstrated that autophagy inhibitor augmented the activity of γ-secretase complex by upregulating its components (Presenilin 1, Nicastrin, and Pen-2), compared with autophagy inducer. Accordingly, it was inferred that autophagy failure contributed to Aβ generation via activating γ-secretase complex (Figure 4).
Several studies have supported that failure of Aβ clearance in the brains of patients with AD has been involved in autophagy dysfunction.62,63 The Aβ deposition and formation of Aβ plaques will be accelerated because of defects in its removal, mediated through a combination of diffusion along perivascular extracellular matrix, transport across vessel walls into the blood stream, and enzymatic degradation.44,64–66 Several Aβ-degrading enzymes such as NEP, IDE, ECE-1, and ECE-2 are critical in Aβ clearance, most of which are produced by neurons and glial cells.40,41 Scientific evidence has revealed the role of autophagy in Aβ clearance involved in Aβ-degrading enzymes.40,44 However, the clearance markers of Aβ (NEP, IDE, ECE-1, and ECE-2) were not different between autophagy inducer and inhibitor treatment. These results showed that, autophagy inhibitor may activate γ-secretase and promote Aβ generation and accumulation, but has no influence on Aβ clearance.
In the current study, both autophagy inhibitor and inducer increased the activity of α-, β-, and γ-secretases, and enhanced Aβ production. Moreover, autophagy inhibitor may more activate γ-secretase and promote Aβ production and accumulation, compared with its inducer. Both autophagy inhibitor and inducer had no influence on Aβ clearance. However, other results indicate that inducing autophagy pathway keeps a foothold to restrain Aβ generation whereas inhibiting autophagy pathway is a friend for Aβ generation. Aberrant autophagy induction leads into a concentration of AVs rich in APP, Aβ and the elements crucial for its formation while the dysfunction of autophagic clearance plays an important role in AD pathogenesis.60 Overall, the precise role of in AD pathogenesis is still under contention.11 Therefore, it is indispensable to further elucidate the potential routes for autophagy-mediated Aβ production and clearance in the AD pathophysiology mechanisms.
Acknowledgments
This study was supported by the National Nature Science Foundation of China (81070878/H0902) and Nature Science Foundation of Guangdong (S20120200-10867) to Professor Bin Zhao, the Hubei Province Health and Family Planning Scientific Research Project (WJ2015MB219), the Hubei Provincial Nature Science Foundation (2015CFB260), and the Shiyan Nature Science Foundation (15K70) to Dr Zhiyou Cai.
Disclosure
The authors report no conflicts of interest in this work.
References
Mhatre SD, Michelson SJ, Gomes J, Tabb LP, Saunders AJ, Marenda DR. Development and characterization of an aged onset model of Alzheimer’s disease in Drosophila melanogaster. Exp Neurol. 2014;261C:772–781. | ||
Fargo K, Bleiler L. Alzheimer’s Association report. Alzheimers Dement. 2014;10:e47–e92. | ||
Choi SJ, Jung SS, You YS, et al. Prevalence of Alzheimer’s dementia and its risk factors in community-dwelling elderly Koreans. Psychiatry Investig. 2008;5:78–85. | ||
Hsieh S, Hodges JR, Piguet O. Recognition of positive vocalizations is impaired in behavioral-variant frontotemporal dementia. J Int Neuropsychol Soc. 2013;19:483–487. | ||
Teri L, Logsdon R, Yesavage J. Measuring behavior, mood, and psychiatric symptoms in Alzheimer disease. Alzheimer Dis Assoc Disord. 1997;11(Suppl 6):50–59. | ||
Hartig W, Kacza J, Paulke BR, et al. In vivo labelling of hippocampal beta-amyloid in triple-transgenic mice with a fluorescent acetylcholinesterase inhibitor released from nanoparticles. Eur J Neurosci. 2010;31:99–109. | ||
Heese K, Akatsu H. Alzheimer’s disease – an interactive perspective. Curr Alzheimer Res. 2006;3:109–121. | ||
da Cruz e Silva OA, Henriques AG, Domingues SC, da Cruz e Silva EF. Wnt signalling is a relevant pathway contributing to amyloid beta-peptide-mediated neuropathology in Alzheimer’s disease. CNS Neurol Disord Drug Targets. 2010;9:720–726. | ||
Melendez A, Levine B. Autophagy in C. elegans. Pasadena (CA): Worm Book; 2009:1–26. | ||
Chaumorcel M, Souquere S, Pierron G, Codogno P, Esclatine A. Human cytomegalovirus controls a new autophagy-dependent cellular antiviral defense mechanism. Autophagy. 2008;4:46–53. | ||
Funderburk SF, Marcellino BK, Yue Z. Cell “self-eating” (autophagy) mechanism in Alzheimer’s disease. Mt Sinai J Med. 2010;77:59–68. | ||
Zhou X, Yang C, Liu Y, et al. Lipid rafts participate in aberrant degradative autophagic-lysosomal pathway of amyloid-beta peptide in Alzheimer’s disease. Neural Regen Res. 2014;9:92–100. | ||
Yang Y, Chen S, Zhang J, et al. Stimulation of autophagy prevents amyloid-beta peptide-induced neuritic degeneration in PC12 cells. J Alzheimers Dis. 2014;40:929–939. | ||
Moreira PI, Siedlak SL, Wang X, et al. Increased autophagic degradation of mitochondria in Alzheimer disease. Autophagy. 2007;3:614–615. | ||
Arduino DM, Esteves AR, Cardoso SM. Mitochondria drive autophagy pathology via microtubule disassembly: a new hypothesis for Parkinson disease. Autophagy. 2013;9:112–114. | ||
Yang Q, Mao Z. Parkinson disease: a role for autophagy? Neuroscientist. 2010;16:335–341. | ||
Koga H, Martinez-Vicente M, Arias E, Kaushik S, Sulzer D, Cuervo AM. Constitutive upregulation of chaperone-mediated autophagy in Huntington’s disease. J Neurosci. 2011;31:18492–18505. | ||
Martinez-Vicente M, Talloczy Z, Wong E, et al. Cargo recognition failure is responsible for inefficient autophagy in Huntington’s disease. Nat Neurosci. 2010;13:567–576. | ||
Wong YC, Holzbaur EL. The regulation of autophagosome dynamics by huntingtin and HAP1 is disrupted by expression of mutant huntingtin, leading to defective cargo degradation. J Neurosci. 2014;34:1293–1305. | ||
Rajawat YS, Hilioti Z, Bossis I. Aging: central role for autophagy and the lysosomal degradative system. Ageing Res Rev. 2009;8:199–213. | ||
Tan CC, Yu JT, Tan MS, Jiang T, Zhu XC, Tan L. Autophagy in aging and neurodegenerative diseases: implications for pathogenesis and therapy. Neurobiol Aging. 2014;35:941–957. | ||
Nixon RA. Alzheimer neurodegeneration, autophagy, and Abeta secretion: the ins and outs (comment on DOI:10.1002/bies.201400002). Bioessays. 2014;36:547. | ||
Li L, Zhang S, Zhang X, et al. Autophagy enhancer carbamazepine alleviates memory deficits and cerebral amyloid-beta pathology in a mouse model of Alzheimer’s disease. Curr Alzheimer Res. 2013;10:433–441. | ||
Yu WH, Cuervo AM, Kumar A, et al. Macroautophagy – a novel Beta-amyloid peptide-generating pathway activated in Alzheimer’s disease. J Cell Biol. 2005;171:87–98. | ||
Lee JK, Jin HK, Park MH, et al. Acid sphingomyelinase modulates the autophagic process by controlling lysosomal biogenesis in Alzheimer’s disease. J Exp Med. 2014;211:1551–1570. | ||
Wolfe DM, Lee JH, Kumar A, Lee S, Orenstein SJ, Nixon RA. Autophagy failure in Alzheimer’s disease and the role of defective lysosomal acidification. Eur J Neurosci. 2013;37:1949–1961. | ||
Nixon RA. Autophagy, amyloidogenesis and Alzheimer disease. J Cell Sci. 2007;120:4081–4091. | ||
Allsop D, Yamamoto T, Kametani F, Miyazaki N, Ishii T. Alzheimer amyloid beta/A4 peptide binding sites and a possible ‘APP-secretase’ activity associated with rat brain cortical membranes. Brain Res. 1991;551:1–9. | ||
Hata S, Fujishige S, Araki Y, et al. Alcadein cleavages by amyloid beta-precursor protein (APP) alpha- and gamma-secretases generate small peptides, p3-Alcs, indicating Alzheimer disease-related gamma-secretase dysfunction. J Biol Chem. 2009;284:36024–36033. | ||
Deng Y, Wang Z, Wang R, et al. Amyloid-β protein (Aβ) Glu11 is the major β-secretase site of β-site amyloid-β precursor protein-cleaving enzyme 1 (BACE1), and shifting the cleavage site to Aβ Asp1 contributes to Alzheimer pathogenesis. Eur J Neurosci. 2013;37:1962–1969. | ||
Fahrenholz F. Alpha-secretase as a therapeutic target. Curr Alzheimer Res. 2007;4:412–417. | ||
Cai Z, Li B, Li K, Zhao B. Down-regulation of amyloid-β through AMPK activation by inhibitors of GSK-3β in SH-SY5Y and SH-SY5Y-AβPP695 cells. J Alzheimers Dis. 2012;29:89–98. | ||
Fernandez MA, Klutkowski JA, Freret T, Wolfe MS. Alzheimer presenilin-1 mutations dramatically reduce trimming of long amyloid β-peptides (Aβ) by gamma-secretase to increase 42-to-40-residue Aβ. J Biol Chem. 2014;289(45):31043–31052. | ||
Swomley AM, Forster S, Keeney JT, et al. Abeta, oxidative stress in Alzheimer disease: evidence based on proteomics studies. Biochim Biophys Acta. 2014;1842:1248–1257. | ||
Cai Z, Zhao B, Li K, et al. Mammalian target of rapamycin: a valid therapeutic target through the autophagy pathway for Alzheimer’s disease? J Neurosci Res. 2012;90:1105–1118. | ||
Cai Z, Zhao B, Ratka A. Oxidative stress and beta-amyloid protein in Alzheimer’s disease. Neuromolecular Med. 2011;13:223–250. | ||
Cai Z, Yan LJ, Li K, Quazi SH, Zhao B. Roles of AMP-activated protein kinase in Alzheimer’s disease. Neuromolecular Med. 2012;14:1–14. | ||
Cai Z, Zhao Y, Zhao B. Roles of glycogen synthase kinase 3 in Alzheimer’s disease. Curr Alzheimer Res. 2012;9:864–879. | ||
Mawuenyega KG, Sigurdson W, Ovod V, et al. Decreased clearance of CNS beta-amyloid in Alzheimer’s disease. Science. 2010;330:1774. | ||
Cai Z, Hussain MD, Yan LJ. Microglia, neuroinflammation, and beta-amyloid protein in Alzheimer’s disease. Int J Neurosci. 2014;124:307–321. | ||
Yan LJ, Xiao M, Chen R, Cai Z. Metabolic dysfunction of astrocyte: an initiating factor in beta-amyloid pathology? Aging Neurodegener. 2013;1:7–14. | ||
Shin JY, Park HJ, Kim HN, et al. Mesenchymal stem cells enhance autophagy and increase beta-amyloid clearance in Alzheimer disease models. Autophagy. 2014;10:32–44. | ||
Steele JW, Gandy S. Latrepirdine (Dimebon(R)), a potential Alzheimer therapeutic, regulates autophagy and neuropathology in an Alzheimer mouse model. Autophagy. 2013;9:617–618. | ||
Nixon RA, Wegiel J, Kumar A, et al. Extensive involvement of autophagy in Alzheimer disease: an immuno-electron microscopy study. J Neuropathol Exp Neurol. 2005;64:113–122. | ||
Jaeger PA, Wyss-Coray T. Beclin 1 complex in autophagy and Alzheimer disease. Arch Neurol. 2010;67:1181–1184. | ||
Ling D, Salvaterra PM. A central role for autophagy in Alzheimer-type neurodegeneration. Autophagy. 2009;5:738–740. | ||
Torres M, Jimenez S, Sanchez-Varo R, et al. Defective lysosomal proteolysis and axonal transport are early pathogenic events that worsen with age leading to increased APP metabolism and synaptic Abeta in transgenic APP/PS1 hippocampus. Mol Neurodegener. 2012;7:59. | ||
Ghavami S, Shojaei S, Yeganeh B, et al. Autophagy and apoptosis dysfunction in neurodegenerative disorders. Prog Neurobiol. 2014;112:24–49. | ||
Kuperstein F, Brand A, Yavin E. Amyloid Abeta1-40 preconditions non-apoptotic signals in vivo and protects fetal rat brain from intrauterine ischemic stress. J Neurochem. 2004;91:965–974. | ||
Ansari N, Khodagholi F. Molecular mechanism aspect of ER stress in Alzheimer’s disease: current approaches and future strategies. Curr Drug Targets. 2013;14:114–122. | ||
Vagnoni A, Glennon EB, Perkinton MS, Gray EH, Noble W, Miller CC. Loss of c-Jun N-terminal kinase-interacting protein-1 does not affect axonal transport of the amyloid precursor protein or Aβ production. Hum Mol Genet. 2013;22:4646–4652. | ||
Vagnoni A, Perkinton MS, Gray EH, Francis PT, Noble W, Miller CC. Calsyntenin-1 mediates axonal transport of the amyloid precursor protein and regulates Aβ production. Hum Mol Genet. 2012;21:2845–2854. | ||
Vossel KA, Zhang K, Brodbeck J, et al. Tau reduction prevents Abeta-induced defects in axonal transport. Science. 2010;330:198. | ||
Winton MJ, Lee EB, Sun E, et al. Intraneuronal APP, not free Aβ peptides in 3xTg-AD mice: implications for tau versus Aβ-mediated Alzheimer neurodegeneration. J Neurosci. 2011;31:7691–7699. | ||
George AJ, Holsinger RM, McLean CA, et al. APP intracellular domain is increased and soluble Abeta is reduced with diet-induced hypercholesterolemia in a transgenic mouse model of Alzheimer disease. Neurobiol Dis. 2004;16:124–132. | ||
Son SM, Song H, Byun J, et al. Accumulation of autophagosomes contributes to enhanced amyloidogenic APP processing under insulin-resistant conditions. Autophagy. 2012;8:1842–1844. | ||
Makioka K, Yamazaki T, Kakuda S, Okamoto K. Variations in the effects on synthesis of amyloid beta protein in modulated autophagic conditions. Neurol Res. 2009;31:959–968. | ||
Tung YT, Wang BJ, Hu MK, et al. Autophagy: a double-edged sword in Alzheimer’s disease. J Biosci. 2012;37:157–165. | ||
Tamboli IY, Hampel H, Tien NT, et al. Sphingolipid storage affects autophagic metabolism of the amyloid precursor protein and promotes Abeta generation. J Neurosci. 2011;31:1837–1849. | ||
Ulamek-Koziol M, Furmaga-Jablonska W, Januszewski S, et al. Neuronal autophagy: self-eating or self-cannibalism in Alzheimer’s disease. Neurochem Res. 2013;38:1769–1773. | ||
Chun YS, Park Y, Oh HG, et al. O-GlcNAcylation promotes non-amyloidogenic processing of amyloid-beta protein precursor via inhibition of endocytosis from the plasma membrane. J Alzheimers Dis. 2015;44(1):261–275. | ||
Hung SY, Huang WP, Liou HC, Fu WM. Autophagy protects neuron from Abeta-induced cytotoxicity. Autophagy. 2009;5:502–510. | ||
Rajadas J, Sun W, Li H, et al. Enhanced Aβ(1–40) production in endothelial cells stimulated with fibrillar Aβ(1–42). PLoS One. 2013;8:e58194. | ||
Cecarini V, Bonfili L, Cuccioloni M, et al. Wild type and mutant amyloid precursor proteins influence downstream effects of proteasome and autophagy inhibition. Biochim Biophys Acta. 2014;1842:127–134. | ||
Li W, Poteet E, Xie L, Liu R, Wen Y, Yang SH. Regulation of matrix metalloproteinase 2 by oligomeric amyloid beta protein. Brain Res. 2011;1387:141–148. | ||
Knobloch M, Konietzko U, Krebs DC, Nitsch RM. Intracellular Abeta and cognitive deficits precede beta-amyloid deposition in transgenic arcAbeta mice. Neurobiol Aging. 2007;28:1297–1306. |
 © 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.