Back to Journals » Cancer Management and Research » Volume 8
Atypical teratoid/rhabdoid tumors: challenges and search for solutions
Authors Biswas A, Kashyap L, Kakkar A, Sarkar C, Julka P
Received 27 January 2016
Accepted for publication 27 June 2016
Published 16 September 2016 Volume 2016:8 Pages 115—125
DOI https://doi.org/10.2147/CMAR.S83472
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Kenan Onel
Ahitagni Biswas,1 Lakhan Kashyap,1 Aanchal Kakkar,2 Chitra Sarkar,2 Pramod Kumar Julka1
1Department of Radiotherapy & Oncology, 2Department of Pathology, All India Institute of Medical Sciences, New Delhi, India
Abstract: Atypical teratoid/rhabdoid tumor (AT/RT) is a highly malignant embryonal central nervous system tumor commonly affecting children <3 years of age. It roughly constitutes 1%–2% of all pediatric central nervous system tumors. Recent data show that it is the most common malignant central nervous system tumor in children <6 months of age. Management of this aggressive tumor is associated with a myriad of diagnostic and therapeutic challenges. On the basis of radiology and histopathology alone, distinction of AT/RT from medulloblastoma or primitive neuroectodermal tumor is difficult, and hence this tumor has been commonly misdiagnosed as primitive neuroectodermal tumor for decades. Presence of a bulky heterogeneous solid-cystic mass with readily visible calcification and intratumor hemorrhage, occurring off-midline in children <3 years of age, should alert the radiologist toward the possibility of AT/RT. Presence of rhabdoid cells on histopathology and polyphenotypic immunopositivity for epithelial, mesenchymal, and neuroectodermal markers along with loss of expression of SMARCB1/INI1 or SMARCA4/BRG1 help in establishing a diagnosis of AT/RT. The optimal management comprises maximal safe resection followed by radiation therapy and multiagent intensive systemic chemotherapy. Gross total excision is difficult to achieve in view of the large tumor size and location and young age at presentation. Leptomeningeal spread is noted in 15%–30% of patients, and hence craniospinal irradiation followed by boost to tumor bed is considered standard in children older than 3 years. However, in younger children, craniospinal irradiation may lead to long-term neurocognitive and neuroendocrine sequel, and hence focal radiation therapy may be a pragmatic approach. In this age group, high-dose chemotherapy with autologous stem cell rescue may also be considered to defer radiation therapy, but this approach is also associated with significant treatment-related morbidity and mortality. Novel small molecule inhibitors hold promise in preclinical studies and should be considered in patients with relapsed or refractory tumor.
Keywords: atypical teratoid/rhabdoid tumor, intracranial, medulloblastoma, primitive neuroectodermal tumor
Introduction
Rhabdoid tumor was originally described by Beckwith and Palmer as a variant of Wilms tumor with a rhabdosarcomatous component.1 Apart from kidney,1 rhabdoid tumors have been reported from many organs, including soft tissues2 and central nervous system (CNS).3 Atypical teratoid/rhabdoid tumor (AT/RT) of CNS was first recognized in 1987, when it was simply referred to as “rhabdoid tumor”.4 It was defined as a distinct entity in 19965 and included in World Health Organization classification of CNS tumors in 2000.6 AT/RT currently constitutes one of three major CNS embryonal tumors in the 2007 World Health Organization classification of CNS tumors and is accorded World Health Organization grade IV due to its highly malignant nature.7 The tumor cells show histologic diversity with a combination of rhabdoid cells and neuroectodermal, epithelial, and mesenchymal elements but lack divergent tissue development pathognomonic of teratoma.8,9 Hence, it is designated as AT/RT. Despite progress in treatment modalities, this highly malignant tumor of CNS carries dismal prognosis. So far, there is no consensus regarding optimal management of this tumor. Critical location of the tumor precludes complete resection and young age of patients restricts optimal integration of radiation therapy (RT) in the management of this aggressive tumor.
Epidemiology
The accurate incidence of AT/RT is not yet known. This may be because of the recent recognition of AT/RT as a distinct pathologic entity and lack of prospective studies in patients with this tumor, with most of the series being retrospective case reviews or pathologic reviews. AT/RT is predominantly observed in children younger than 3 years.10 As per hospital-based series, prevalence of AT/RT in pediatric brain tumor ranges from 1% to 2%.11–13 A population-based study by Austrian Brain Tumor Registry showed an age-standardized incidence rate of 1.38 per 100,000 person years in children and peak incidence was found in the first 2 years of life.10 Median age at diagnosis ranges from 1.2 years to 2.3 years in various large series.14–16 AT/RT has a slight male preponderance till the age of 3 years.17,18 As per Central Brain Tumor Registry of the United States, AT/RT accounts for 1.6% of all pediatric CNS tumors and 4.4% of all CNS tumors in children of 0- to 5-year age group, with an average annual incidence of 0.07 per 100,000.19 Recent data show that AT/RT is the most common malignant CNS tumor in children <6 months of age.20
AT/RT has been described in virtually all CNS locations, including the cerebellopontine angle cistern, meninges, cranial nerves, spinal canal, and extradural location.21 There is discrepancy regarding the most common site of involvement of AT/RT, with some series suggesting the supratentorial region,22,23 while other series suggesting the infratentorial region.24 Some series have shown equal involvement of both supratentorial and infratentorial regions.15,17 This discrepancy may be due to the small number of patients in the reported series of AT/RT. Around 14%–21% of patients in different series have been found to have disseminated disease at diagnosis.17,18,22,23 Historically, survival in patients with AT/RT has been dismal, with a reported median survival of ~1 year.14,15 The poor prognostic factors associated with AT/RTs are age <2 years, metastatic disease at diagnosis, and delayed initiation of radiotherapy. Patients with supratentorial tumor location have relatively favorable outcome.14–16
Adult AT/RT is very rare, with isolated case reports existing in literature.25 Majority of adult-onset AT/RTs are located in the supratentorial region, with sellar and suprasellar regions being the preferential site of involvement.26 In contrast to pediatric AT/RT, adult-onset AT/RT has favorable clinical course, and many reported cases have shown long-term survival.27
Diagnosis
Clinical features of AT/RT depend on the location of tumor and age of the patient. Patients usually present with symptoms of raised intracranial tension, including headache, vomiting, lethargy, failure to thrive, regression of developmental milestones, irritability, and macrocephaly in very young children. Involvement of cerebellar hemisphere may lead to symptoms of ataxia, head tilt, and nystagmus. Patients can also present with cranial nerve palsies (cranial nerves VII and VIII) if cerebellopontine angle is involved by tumor.28–30
Computed tomography of brain demonstrates hyperdense lesion attributable to the high cellularity of the tumor and heterogeneous enhancement on postcontrast images. Calcification may be seen in up to 40% of tumors.30 Magnetic resonance imaging (MRI) is the imaging modality of choice in patients with AT/RT (Figure 1).29,30 On MRI, AT/RTs show heterogeneous iso-intense signal on both T1- and T2-weighted MR images. Contrast-enhanced MRI shows variable enhancement (heterogeneous, peripheral nodular, intense, and mild). Peripherally located cystic components are commonly demonstrated on MR images and are a useful distinguishing feature.29–31 Majority of the supratentorial AT/RTs arise from cerebral hemispheres, and frontal lobe is commonly involved.32 Supratentorial AT/RT demonstrates thick irregular heterogeneously enhancing wall encircling central cystic or necrotic lesion. In one series, this feature was shown to have high specificity and negative predictive value for AT/RT.33
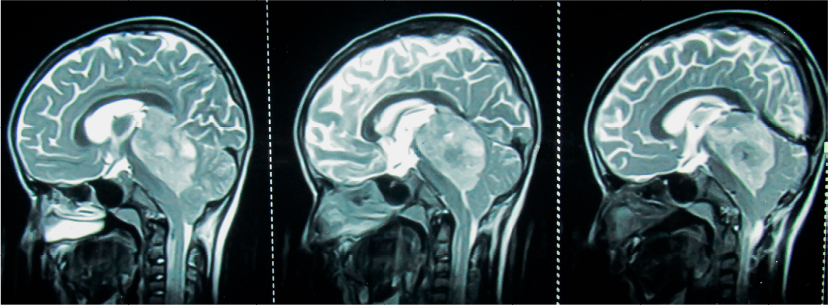 | Figure 1 Midsagittal (left panel) and parasagittal (middle and right panel) T2 weighted MRI of brain in a 8 year old male child with AT/RT showing a bulky (5.3 cm × 3.1 cm), heterogeneous, intracranial mass in the posterior fossa involving the fourth ventricle, cerebellar vermis, midbrain and pineal region, pushing the brainstem anteriorly and cerebellum posteriorly; the tumor is lifting the tentorium cerebelli and causing herniation of the cerebellar tonsils in the middle panel; the tumor is abutting the straight sinus in the right panel; intra-tumor necrosis is better appreciated in middle and right panel. Abbreviation: MRI, magnetic resonance imaging; AT/RT, atypical teratoid/rhabdoid tumor. |
The main radiological differential diagnosis of AT/RT is medulloblastoma (MB). Other differentials include supratentorial primitive neuroectodermal tumor (SPNET), ependymoma, teratoma, choroid plexus tumors, pilocytic astrocytoma, and desmoplastic infantile ganglioglioma.29 There are certain radiologic features that may help in differentiating AT/RT from MB such as off-midline location and presence of eccentric cyst, calcification, and intratumor hemorrhage.29,30 Both AT/RT and MB show diffusion restriction on diffusion-weighted MRI.34
Cerebrospinal fluid involvement via leptomeningeal dissemination can be found in 15%–30% of patients; hence, MRI of entire neuraxis and cerebrospinal fluid cytology form integral part of diagnostic evaluation.29,30 Additional investigation includes renal ultrasound to rule out rhabdoid tumor of kidney,30 as patients with rhabdoid tumor predisposition syndrome can develop multiple rhabdoid tumors.35
Histopathology
AT/RTs are heterogeneous tumors that are often difficult to recognize on histopathology alone (Figure 2). They show complex histologic patterns resulting from an intimate admixture of primitive neuroctodermal elements along with rhabdoid cells.5 The rhabdoid phenotype is characterized by large cells with abundant eosinophilic cytoplasm, paranuclear globular cytoplasmic inclusions that correspond to aggregates of intermediate filaments, eccentrically placed nuclei with vesicular chromatin, and a prominent eosinophilic nucleolus. The primitive neuroectodermal component is characterized by undifferentiated small round blue cells. The mesenchymal component is represented by spindle cells within a basophilic myxoid background. The epithelial component, which is rarely detected, can be observed as squamous, papillary, adenomatous, or ribbon-like structures.28,29 Mitotic figures are frequent (including atypical mitotic figures), as are areas of necrosis and hemorrhage. On the basis of histopathology alone, differentiation of AT/RT from primitive neuroectodermal tumors and MB remains difficult.5,29
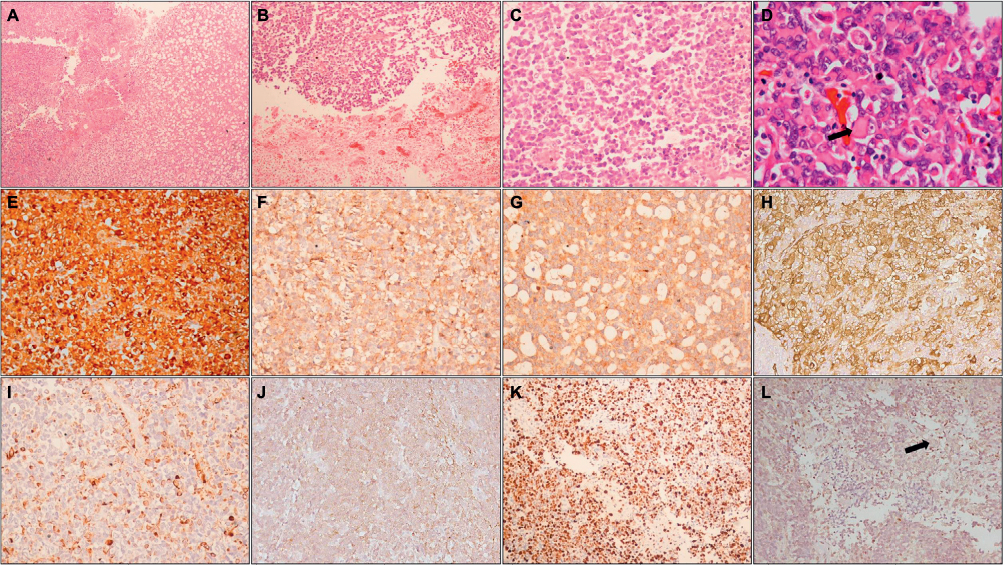 | Figure 2 Histopathology and immunohistochemistry of AT/RT. Notes: Photomicrographs showing a heterogeneous tumor (A; HE, 100×) with areas of necrosis (B; HE, 100×) composed of small round cells admixed with cells with eosinophilic cytoplasm (C; HE, 200×); higher magnification shows typical rhabdoid cells (arrow) (D; HE, 400×). On IHC, tumor cells are positive for vimentin (E; IHC, 200×), EMA (F; IHC, 200×), synaptophysin (G; IHC, 200×), SMA (H; IHC, 200×), and focally for GFAP (I; IHC, 200×) and cytokeratin (J; IHC, 200×); MIB-1-LI is high (K; IHC, 200×) and tumor cells show loss of INI1, while endothelial cells (arrow) show retained expression (L; IHC, 200×). Abbreviations: HE, hematoxylin and eosin; IHC, immunohistochemistry; EMA, epithelial membrane antigen; SMA, smooth muscle actin; GFAP, glial fibrillary acidic protein; MIB-1-LI, MIB-1 labeling index; INI1, integrase interactor 1; AT/RT, atypical teratoid/rhabdoid tumor. |
On immunohistochemistry, AT/RTs show polyphenotypic reactivity, demonstrating immunopositivity for vimentin, epithelial membrane antigen, smooth muscle actin, synaptophysin, and, to a lesser extent, cytokeratin, glial fibrillary acidic protein, and neurofilament protein.5,29,30,36 AT/RTs are highly proliferative tumors and show high MIB-1 labeling indices.5,29,30
Majority of AT/RTs show loss of SMARCB1/INI1, while loss of SMARCA4/BRG1 expression is seen in a small proportion of cases.36,37 Integrase interactor 1 (INI1), the gene product of SMARCB1, is a constitutively expressed protein that is present in all normal tissues, as well as in most neoplasms. In AT/RTs, there is loss of nuclear expression of this protein, due to deletion or mutation of the SMARCB1 locus on 22q11.2, a genetic hallmark of this tumor.29,30 A recently described subset of embryonal tumors designated as cribriform neuroepithelial tumor also shows loss of INI1 expression without evidence of rhabdoid morphology – this group of tumors is now being considered an epithelioid variant of AT/RT.38 Rarely, tumors with histological features typical of AT/RT show retained INI1 expression. These AT/RTs show loss of nuclear expression of BRG1, resulting from mutation and inactivation of the SMARCA4 gene, and are associated with a poorer prognosis.37,39 Thus, loss of either of these markers is essential for establishing a diagnosis of AT/RT. Immunohistochemistry also has potential for predicting prognosis, with one study showing association of FLI-1 and cyclin D1 immunopositivity with improved survival.40
Molecular classification
The prognosis of patients with AT/RT is grave. However, some patients respond favorably to standard treatment, suggesting the existence of molecular intertumor heterogeneity. In a landmark study involving 192 patients with AT/RT, Johann et al41 identified three distinct molecular subtypes by genetic and epigenetic analysis of tumor tissue. Loss of SMARCB1 expression was noted in 188 (98%) patients. Four cases had retained SMARCB1 expression, of which three cases were confirmed to have a SMARCA4 mutation. In AT/RT-TYR subgroup, patients were younger (usually <1 year) and had mostly infratentorial tumor. This subgroup was characterized by broad SMARCB1 deletion and overexpression of melanosomal genes – TYR and MITF. In AT/RT-SHH subgroup, patients had both supratentorial and infratentorial tumors. This subgroup was characterized by focal SMARCB1 aberration and overexpression of Sonic Hedgehog (SHH) pathway genes – MYCN and GLI2. In AT/RT-MYC subgroup, patients were older (usually >1 year) and had mostly supratentorial tumor. This subgroup was characterized by focal SMARCB1 deletion and overexpression of MYC, HOTAIR, and HOX cluster of genes. There was no sex predilection noted in any of the aforementioned molecular subtypes. Genes that were highly expressed in almost all AT/RTs compared with normal brain included components of the Polycomb Repressive Complex 2 (PRC2) – EZH2, SUZ12, and EED – confirming previous reports that suggest an antagonistic relationship between members of the SWI/SNF complex and PRC2 complex in chromatin remodeling and epigenetic silencing of genes.
Surgery
Surgery provides rapid relief of symptoms in patients with AT/RT. Extent of surgical resection is influenced by age of the patient, location, and size of the tumor.29,30 Extension of posterior fossa tumor to cerebellopontine angle may make surgical resection difficult due to involvement of cranial nerves. Impact of extent of resection on survival outcome is not conclusive. In studies by Biswas et al30 and Lafay-Cousin et al,42 overall survival (OS) significantly improved with gross total resection compared with subtotal/near total resection. Other series have reported no significant difference in survival based on the extent of surgical resection.17,22,23 In light of these data, the aim of surgery should be maximal safe resection with adequate preservation of neurological function. Apart from resection, surgical intervention is required for placement of ventriculoperitoneal shunt to relieve raised intracranial pressure and for placement of ventricular catheter and reservoir to deliver intraventricular chemotherapy.
Chemotherapy
Chemotherapy is an integral component of multimodality management of patients with AT/RT. Adjuvant chemotherapy in AT/RTs is intensive, and published series have used two backbones of chemotherapy: Children’s Cancer Group (CCG)-9921 and Intergroup Rhabdomyosarcoma III (IRS III) protocols. In CCG-9921 induction chemotherapy regimen, a four drug combination, consisting of either vincristine, cisplatin, cyclophosphamide, and etoposide, or vincristine, carboplatin, ifosfamide, and etoposide were used.43 This induction regimen was found to be active in pediatric brain tumors (overall response rate 42%). IRS III Regimen 36, originally designed to treat parameningeal rhabdomyosarcoma, has been used in treatment of patients with AT/RT.44 IRS III Regimen 36 consists of vincristine, cisplatin, doxorubicin, cyclophosphamide, dacarbazine, etoposide, actinomycin-D, and triple intrathecal chemotherapy with methotrexate, hydrocortisone, and cytarabine.45
Gardner et al46 evaluated the role of postoperative intensive chemotherapy with stem cell rescue in young children with AT/RT with the intent of avoiding irradiation. Chemotherapy regimen consisted of five cycles of induction chemotherapy followed by consolidation chemotherapy with autologous stem cell rescue. The induction chemotherapy regimen consisted of vincristine, cisplatin, etoposide, and cyclophosphamide in “Head Start I” (HS I) study (n=6). High-dose intravenous methotrexate was added to the same regimen in Head Start II (HS II) study (n=7). Consolidation chemotherapy consisted of carboplatin, thiotepa, and etoposide. RT was administered in patients with partial response or stable disease and omitted in patients with complete response on MRI following induction chemotherapy. The estimated event-free survival (EFS) and OS rates at 3 years among the 13 children were 23%. The authors concluded that long-term survival can be achieved in a subset of young children with CNS AT/RT with the use of postoperative high-dose methotrexate-based multidrug chemotherapy and autologous stem cell rescue without RT.
Chi et al47 evaluated the role of intensive multimodality treatment in a study involving 25 children with AT/RT. They used modified IRS III regimen consisting of vincristine, actinomycin-D, cyclophosphamide, cisplatin, doxorubicin, and temozolomide. Triple intrathecal chemotherapy with methotrexate, hydrocortisone, and cytarabine was also administered by alternate intralumbar and intraventricular routes. Induction chemotherapy was administered in three phases of 6 weeks each: pre-irradiation induction, chemo-irradiation induction (focal RT – eleven patients; craniospinal irradiation [CSI] – four patients), and post-irradiation induction. This was followed by maintenance and continuation phases of chemotherapy up to 51 weeks. The authors reported 2-year progression-free survival and OS rates of 53% and 70%, respectively. Though the clinical outcome with this approach was encouraging, there was one treatment-related death and frequent dose adjustments of chemotherapeutics due to grade 3 and 4 toxicities.
In a retrospective analysis of 22 patients with AT/RT confirmed by immunonegativity for SMARCB1/INI1 from the Medical University of Vienna (MUV), Slavc et al48 compared the clinical outcome of patients treated uniformly with intensive chemotherapy and delayed radiotherapy (cohort A; n=9) and patients treated variably as per the different initial diagnoses (cohort B; n=13). The MUV AT/RT regimen consisted of three 9-week courses of a dose-dense regimen comprising doxorubicin, cyclophosphamide, vincristine, ifosfamide, cisplatin, etoposide, and high-dose methotrexate augmented with intrathecal chemotherapy followed by high-dose chemotherapy (HDCT) with autologous hematopoietic stem cell rescue. Focal radiotherapy (54 Gy in 30 fractions over 6 weeks) was deferred to the very end of treatment (between 38 weeks and 43 weeks). The 5-year OS rates were 100% and 28.8% in patients in cohorts A and B, respectively, underlining the importance of HDCT and intrathecal chemotherapy in the management of patients with AT/RT.
Zaky et al49 evaluated the role of postoperative intensive induction chemotherapy followed by myeloablative chemotherapy with autologous stem cell rescue in 19 patients with AT/RT enrolled on Head Start III (HS III) protocol. The induction regimen consisted of five cycles of vincristine, cisplatin, etoposide, cyclophosphamide, and high-dose methotrexate alternating with vincristine, etoposide, cyclophosphamide, and temozolomide. After induction phase, patients received HDCT with thiotepa, etoposide, and carboplatin followed by autologous stem cell recue. Radiotherapy was administered in patients with pre-consolidation residuum or with age between 6 years and 10 years. However, only four patients finished induction chemotherapy and three patients proceeded to consolidation phase. There was significant hematologic toxicity and five treatment-related deaths reported in the study. The median survival was only 6.1 months, and the 3-year EFS and OS rates were 21% and 26%, respectively, underscoring the necessity of upfront use of RT after surgery.
In the recently closed ACNS0333 multicentre Phase III clinical trial conducted by Children’s Oncology Group, 70 patients with AT/RT received two cycles of induction chemotherapy with vincristine, high-dose methotrexate (with leucovorin rescue), etoposide, cyclophosphamide, and cisplatin after surgery. Patients were reevaluated after the completion of induction chemotherapy, and those with residual disease were considered for second-look surgery and three-dimensional conformal radiotherapy to brain (and spine if needed) 5 days a week for 5–6 weeks. Patients with disease progression on induction chemotherapy were removed from the study. Within 2–6 weeks of induction chemotherapy or radiotherapy, patients received three courses of HDCT with carboplatin and thiotepa followed by autologous peripheral blood stem cell rescue. The primary end points of the study were evaluation of OS, EFS, and toxic death, and the secondary end point was assessment of overall toxicity.
Biswas et al30 evaluated the role of ifosfamide, carboplatin, and etoposide or vincristine, actinomycin-D, and cyclophosphamide as adjuvant chemotherapy in a retrospective series of 15 patients with AT/RT. Chemotherapy was usually administered after completion of RT (CSI). However, patients <3 years of age received upfront chemotherapy with deferred RT. The median OS was 10 months, and the actuarial rate of OS at 2 years was 24.1%, suggesting the effectiveness of this pragmatic approach within the resource constraints of a developing nation.
HDCT with autologous stem cell rescue has been used in younger children in order to defer radiotherapy. However, in view of formidable toxicity profile and limited evidence, the use of HDCT should be carefully considered.49 Despite being a chemosensitive tumor, AT/RT usually recurs within 6 months and progresses relentlessly on recurrence.29,30,50 In patients with recurrent CNS embryonal tumor, use of single-agent temozolomide may lead to prolonged disease stabilization without any severe toxicity.51
Radiotherapy
Radiotherapy is an important component of treatment in patients with AT/RT. As the majority of patients diagnosed with AT/RT are younger than 3 years, radiotherapy has been often deferred in the postoperative setting as this is the critical time for neurodevelopment in a child. Variable treatment volume (craniospinal axis, whole brain, or tumor bed with margin) has been used while planning RT in these patients. CSI has been used in older children (>3 years) due to high propensity of leptomeningeal dissemination (15%–30%) of the tumor. The dose of RT required in patients with AT/RT has not been standardized perhaps due to rarity of the tumor and limited use of RT in many case series. Radiotherapy dose to tumor bed ranges from 50 Gy to 56 Gy, while dose to neuraxis ranges from 23.4 Gy to 36 Gy in conventional fractionation.18,52 In a retrospective series of 15 patients with AT/RT by Biswas et al,30 postoperative RT (CSI followed by boost to tumor bed) was started in six patients and completed in five. Use of CSI was a significant predictor of OS on univariate analysis (P=0.0087). In another retrospective series of 17 patients with CNS AT/RT by Chen et al,52 total radiotherapy dose of >50 Gy was associated with significantly improved failure-free survival but not OS on multivariate analysis. Intensity-modulated radiotherapy helps in delivering higher dose to tumor while sparing critical organs at risk, but there is concern of high integral dose to the developing brain.
A meta-analysis by Athale et al22 showed significant benefit of addition of RT to the treatment protocol in patients with AT/RT. The mean survival of patients who received RT in addition to chemotherapy was 18.4 months as compared with 8.5 months in those who did not receive RT (P=0.097). There was significant beneficial effect of RT on OS in patients <3 years of age. Buscariollo et al17 used Surveillance, Epidemiology and End Results (SEER) database to analyze survival outcome in 144 patients with AT/RT treated from 1973 to 2008. Overall, 33% of patients received postoperative RT. The median OS in the entire cohort was 10 months. On multivariate analysis, use of RT was found to be an independent predictor of OS (hazard ratio 0.1; P=0.02). This landmark analysis showed significant association between use of RT and improved OS, and this benefit of RT was more pronounced in younger patients <3 years of age.
Use of postoperative RT is associated with acute and late treatment-related morbidity, the late effects being of more concern in a growing child. Spiegler et al53 evaluated change in neurocognitive function after cranial irradiation in childhood. Serial clinical neuropsychological assessment was performed 0.79–4.71 years from diagnosis (median time of first assessment to last assessment from diagnosis). The authors reported an early intellectual decline followed by attenuation of the decline. Besides, there was a significant decline in visual motor integration, visual memory, verbal fluency, and executive functioning. Apart from neurocognitive decline, other significant late toxicities include growth suppression, hearing impairment, hypothyroidism, adrenocortical hormone deficiency, encephalopathy, cataracts, fertility issues, second malignancy, and radiation necrosis.54 Development in radiation technology has provided opportunity to minimize radiation sequelae by incorporating intensity-modulated radiotherapy, helical tomotherapy, and proton therapy in the management of these tumors.
Recently, there has been considerable interest in the use of proton beam therapy in the management of patients with AT/RT. Physical advantage of proton beam over photon beam is due to the presence of Bragg’s peak, which, when properly spread out, adequately covers the target volume with practically no exit dose. Thus, proton radiation allows precise delivery of conformal radiation to tumor with simultaneous sparing of surrounding critical structures. Proton pencil beam scanning technology allows delivery of intensity-modulated proton therapy, which further improves target conformity and sparing of adjacent structures.55 Proton beam therapy helps in reducing acute hematological and gastrointestinal toxicities and thus improves tolerance to concurrent and adjuvant chemotherapy. De Amorim Bernstein et al56 evaluated early clinical outcomes using postoperative proton radiation in ten children with CNS AT/RT. The patients were treated with three-dimensional conformal proton therapy to a median dose of 50.4 Gy relative biological effectiveness (RBE) to the tumor bed. Nine out of ten patients were alive and disease free at a median follow-up of 27.3 months, and none of the patients had major radiotherapy-related toxicity. McGovern et al57 evaluated clinical outcome and acute toxicity profile in a larger series of 31 patients with AT/RT undergoing postoperative proton beam radiation. Seventeen patients younger than 3 years received focal RT up to a dose of 50.4 Gy RBE. Fourteen patients older than 3 years received CSI (24–30.6 Gy RBE) followed by boost to tumor bed up to a dose of 54 Gy RBE. The 2-year EFS and OS rates were 47.6% and 68.3%, respectively. Within 4 months of completion of RT, 16% of patients developed clinical and imaging findings of radiation reaction in the brainstem and adjacent structures, which resolved with the use of steroids or bevacizumab. In a series of 60 consecutive patients with pediatric CNS malignant tumors treated with proton beam therapy at Indianapolis, 31% of patients developed radiographic sign of radiation necrosis (multiple small areas of parenchymal enhancement remote from the surgical site).58 The median time to development of radiation necrosis was 5 months. Among patients with imaging findings of radiation necrosis, 25% had severe symptoms needing medical intervention. The associated risk factors were use of multiple chemotherapeutic agents (>3) (P=0.03) and histology of AT/RT (P=0.03). The median time to complete radiological resolution of features of radiation necrosis was 5.3 months. Whether the long-term benefits of proton therapy (eg, decrease in the risk of hypothyroidism, cardiac toxicity, pulmonary fibrosis, and infertility) outweigh the risk of radiation necrosis (particularly brain stem injury) needs to be carefully considered.20
Literature regarding salvage radiotherapy in patients with AT/RT is limited due to dismal prognosis after relapse. In patients with disease relapse, RT should be considered if not used as a part of primary treatment. CD133, which is marker of cancer stem cells, has been found to be expressed by AT/RT cells, and the amount of CD133-positive cells in tumor correlates positively with resistance to RT.59 Radiation tolerance of brain and relative radioresistance of AT/RT further limit the benefit of reirradiation. Radiosurgery can be considered in patients requiring reirradiation.29,30 Large fraction size in radiosurgery has potential radiobiological advantage in the treatment of radioresistant tumor.29,30 There is also a possibility of sparing adjacent normal structure without significantly increasing the integral dose to brain.60
Novel therapeutic targets
Despite the use of intensive multimodality treatment, survival outcome has been dismal in AT/RT. Although loss of SMARCB1 (BAF47/INI1/SNF5) gene is the defining genetic alteration in this tumor, the biology contributing to development and aggressiveness of the tumor is not completely understood.61 SMARCB1 is a component of SWI/SNF chromatin remodeling complex, and loss of function of SMARCB1 can dysregulate thousands of genes across the genome.62 Recently, various molecular biology studies have tried to identify potential therapeutic targets in these dysregulated pathways.36,41
A preclinical study by Jayanthan et al63 has demonstrated the potential role of multikinase inhibitors in AT/RT. The authors detected expression of receptor tyrosine kinases c-Kit, platelet-derived growth factor receptor β, vascular endothelial growth factor receptor 2, and Flt3 in AT/RT cell lines. Multikinase inhibitors sorafenib and sunitinib have been shown to inhibit these target receptors. The authors demonstrated dose-dependent inhibition of AT/RT cell growth by these agents. There was also synergistic activity of irinotecan with sorafenib and sunitinib, probably due to the ability of irinotecan in increasing the activity of vascular endothelial growth factor-directed therapy. Analysis of gene expression profiling by Sredni et al64 revealed overexpression of ErbB2 and ErbB3 in AT/RT, accompanied by ErbB2–ErbB3 downstream activation of the Ras/Raf/MEK/ERK pathway. The authors observed progressive cell death of AT/RT cell line with increasing concentration and exposure time to lapatinib (dual tyrosine kinase inhibitor of ErbB1 and ErbB2). Fouladi et al65 conducted a Phase II trial evaluating the efficacy of lapatinib in children with refractory CNS malignancies (recurrent MB, ependymoma, and high-grade glioma). Lapatinib was well tolerated in children with recurrent CNS malignancies but had little single-agent activity.
Weingart et al66 identified mitogen-activated protein kinase as a potential therapeutic target in patients with AT/RT. The authors found frequent overexpression of the cell programming factors LIN28A and LIN28B in AT/RT primary tumor samples and cell lines. They demonstrated that LIN28 is important for overall growth, proliferation, clonogenicity, and tumorigenicity of AT/RT. LIN28A suppression was associated with downregulation of KRAS expression and phospho-ERK expression (a key readout of mitogen-activated protein kinase pathway activation). The broad expression of p-ERK suggests MEK inhibition a promising approach. The authors demonstrated sensitivity of LIN28A- and LIN28B-expressing AT/RT to MEK inhibitor selumetinib.
Loss of INI expression is associated with increased expression of CCND1 (cyclin D1), AURKA (Aurora A kinase), GLI, and insulin-like growth factor-I receptor (IGF-IR) signaling.20 Cyclin D1 propagates cell through G1–S checkpoint, while Aurora A and IGF-IR signaling also play an important role in the molecular pathogenesis of AT/RT. These pathways provide potential therapeutic targets for retinoids, rexinoids, and ribociclib (against cyclin D1 and CDK4/6), alisertib (against Aurora A kinase), arsenic trioxide As2O3 (against GLI 1), and IGF-IR inhibitors (against IGF-IR).20,67 The chromatin antagonizing effect of EZH2 may be therapeutically targeted by tazemetostat.20 Histone deacetylase (HDAC) inhibitor vorinostat may also be used in combination with conventional chemotherapy in patients with AT/RT as HDAC1 and HDAC2 are overexpressed in primary tumors and cell lines.20 Banderali et al68 in an in vitro study identified chloride-selective volume-sensitive ion channel as a potential therapeutic target and demonstrated the synergistic effect of ion-channel inhibition with antineoplastic agents in AT/RT cell lines.
Survival, patterns of failure, and prognostic factors
In a study of 52 patients with AT/RT, Rorke et al5 reported median time to progression and OS to be 4.5 months and 6 months, respectively. Another study of eight patients with AT/RT by Bambakidis et al69 showed a median OS of 9 months. The median OS in a study of 144 patients by Buscariollo et al17 was 10 months (13 months in patients with localized disease and 3 months in patients with metastatic disease). In the Canadian population-based study of 50 pediatric patients with CNS AT/RT reported by Lafay-Cousin et al,42 median time to progression and median OS were noted to be 5.5 months and 13.5 months, respectively. With the passage of time and refinement of treatment strategies, there has been modest improvement in survival outcome in patients with AT/RT (OS rate 65%–70% at 2 years; 55%–60% at 5 years).47,48,57 Long-term survivors are being increasingly seen. In the Austrian study by Slavc et al,48 all nine MUV-AT/RT regimen-treated patients were alive after a median follow-up of 76 months. A prolonged survival of >6 years has been reported by Hirth et al in a patient with AT/RT of the right temporal lobe initially treated with gross total excision and multiagent chemotherapy and a second resection, triple intrathecal chemotherapy and gamma knife radiosurgery on relapse.70
In spite of intensive multimodality management, AT/RT typically recurs within 6 months,29,30 the patterns of failure being local recurrence, diffuse leptomeningeal spread in brain and spinal cord, and rarely distant metastases. In a retrospective study of 17 patients with CNS AT/RT by Chen et al,52 14 (82.4%) patients relapsed – two patients had local recurrence and 12 patients had disseminated intracranial and spinal seeding. Eleven patients eventually died of progressive disease, the median time from relapse to death being only 6 months. In a meta-analysis of observational studies of childhood CNS AT/RT (1995–2007) by Athale et al,22 overall mortality was noted in 67% of patients with disease progression being the major cause of death. Craniospinal spread was found in 58% of patients at progression. In an interesting study of 133 patients with pediatric embryonal CNS tumor from North America by Perreault et al,71 overall 49 (36.8%) patients relapsed (MB 29.2%, nonpineal SPNET 56%, AT/RT 40%, and pinealoblastoma 55.6%). The majority of failures were local (79%) in nonpineal SPNET, diffuse leptomeningeal (100%) in pinealoblastoma, and diverse (local 27%, distant 35%, diffuse leptomeningeal 38%) in MB. Among ten patients with AT/RT in this series, four patients relapsed – two had local failure and two had diffuse leptomeningeal failure at first relapse. The median time to relapse were 16 months, 11.5 months, 5.5 months, and 20 months, respectively, in patients with MB, nonpineal SPNET, AT/RT, and pinealoblastoma.
There are various putative factors associated with long-term survival in patients with this aggressive tumor. In a population-based study of 19 patients with AT/RT by Austrian Brain Tumor Registry from 1996 to 2006, all eight long-term survivors underwent multimodality therapy with HDCT in seven, intrathecal chemotherapy in six, and focal radiation in all.10 Extent of resection and intensified treatment with MUV AT/RT protocol were significant predictors of OS. In the Canadian population-based study of 50 patients with AT/RT by Lafay-Cousin et al,42 patients undergoing gross total resection and HDCT had significantly improved OS. In the study of 144 patients with AT/RT from National Cancer Institute SEER database (1973–2008), metastatic disease and use of RT were significant predictors of OS.17 In a meta-analysis of 79 patients with AT/RT by Athale et al, use of intrathecal chemotherapy led to significantly improved 2-year OS and lower prevalence of distant CNS metastases. The relative role of each treatment modality, namely, gross total resection, RT, HDCT, and intrathecal chemotherapy, toward therapeutic success in this aggressive tumor is difficult to dissect, but multimodal management appears to be the key.30
Conclusion
AT/RTs are aggressive CNS malignancies that require multimodality approach consisting of maximal safe surgery, chemotherapy, and radiotherapy. Intensive multiagent chemotherapy with alkylating agents, anthracyclines, and platinum should be administered in these patients. HDCT with hematopoietic stem cell rescue can be carefully considered in very young children, where RT has to be deferred. Addition of radiotherapy has conferred an OS benefit in patients with AT/RT, and hence, radiotherapy should be administered as a part of multimodality treatment. CSI followed by boost to tumor bed should be used in children >3 years preferably by innovative techniques such as helical tomotherapy or proton beam therapy for optimal target coverage with simultaneous sparing of surrounding critical structures. In children younger than 3 years, an individualized pragmatic approach to RT with focus on reduction of dose and volume (focal radiotherapy) should be considered. Novel therapeutic target inhibitors, being investigated in various in vitro studies, should be incorporated in Phase I/II clinical trials in patients with relapsed or refractory AT/RT. Once the safety and efficacy of these novel small molecule inhibitors are established, integration of these newer agents in the primary systemic management of this aggressive tumor is imperative. National AT/RT registry should be established to consolidate data regarding epidemiology and treatment of these aggressive tumors and to develop optimal management guidelines for patients diagnosed with this relatively rare tumor.
Disclosure
The authors report no conflicts of interest in this work.
References
Beckwith J, Palmer N. Histopathology and prognosis of Wilms tumor results from the first national Wilms’ tumor study. Cancer. 1978;41(5):1937–1948. | ||
Lynch HT, Shurin SB, Dahms BB, Izant RJ Jr, Lynch J, Danes BS. Paravertebral malignant rhabdoid tumor in infancy. In vitro studies of a familial tumor. Cancer. 1983;52(2):290–296. | ||
Bonnin JM, Rubinstein LJ, Palmer NF, Beckwith JB. The association of embryonal tumors originating in the kidney and in the brain. A report of seven cases. Cancer. 1984;54(10):2137–2146. | ||
Lefkowitz I, Rorke L, Packer R. Atypical teratoid tumor of infancy: definition of an entity (abstract). Ann Neurol. 1987;22:448–449. | ||
Rorke LB, Packer RJ, Biegel JA. Central nervous system atypical teratoid/rhabdoid tumors of infancy and childhood: definition of an entity. J Neurosurg. 1996;85(1):56–65. | ||
Gonzales M. The 2000 World Health Organization classification of tumours of the nervous system. J Clin Neurosci. 2001;8(1):1–3. | ||
Louis DN, Ohgaki H, Wiestler OD, et al. The 2007 WHO classification of tumours of the central nervous system. Acta Neuropathol. 2007;114(2):97–109. | ||
Burger P, Yu I, Tihan T, et al. Atypical teratoid/rhabdoid tumor of the central nervous system: a highly malignant tumor of infancy and childhood frequently mistaken for medulloblastoma. Am J Surg Pathol. 1998;22(9):1083–1092. | ||
Ho DM, Hsu CY, Wong TT, Ting LT, Chiang H. Atypical teratoid/rhabdoid tumor of the central nervous system: a comparative study with primitive neuroectodermal tumor/medulloblastoma. Acta Neuropathol. 2000;99(5):482–488. | ||
Woehrer A, Slavc I, Waldhoer T, et al. Incidence of atypical teratoid/rhabdoid tumors in children. Cancer. 2010;116(24):5725–5732. | ||
Rickert CH, Paulus W. Epidemiology of central nervous system tumors in childhood and adolescence based on the new WHO classification. Childs Nerv Syst. 2001;17(9):503–511. | ||
Wong T, Ho D, Chang K, et al. Primary pediatric brain tumors. Cancer. 2005;104(10):2156–2167. | ||
Kaderali Z, Lamberti-Pasculli M, Rutka JT. The changing epidemiology of paediatric brain tumours: a review from the Hospital for Sick Children. Childs Nerv Syst. 2009;25(7):787–793. | ||
Dufour C, Beaugrand A, Le Deley M, et al. Clinicopathologic prognostic factors in childhood atypical teratoid and rhabdoid tumor of the central nervous system. Cancer. 2011;118(15):3812–3821. | ||
von Hoff K, Hinkes B, Dannenmann-Stern E, et al. Frequency, risk-factors and survival of children with atypical teratoid rhabdoid tumors (AT/RT) of the CNS diagnosed between 1988 and 2004, and registered to the German HIT Database. Pediatr Blood Cancer. 2011;57(6):978–985. | ||
Pai Panandiker A, Merchant T, Beltran C, et al. Sequencing of local therapy affects the pattern of treatment failure and survival in children with atypical teratoid rhabdoid tumors of the central nervous system. Int J Radiat Oncol Biol Phys. 2012;82(5):1756–1763. | ||
Buscariollo DL, Park HS, Roberts KB, Yu JB. Survival outcomes in atypical teratoid rhabdoid tumor for patients undergoing radiotherapy in a surveillance, epidemiology, and end results analysis. Cancer. 2011;118(17):4212–4219. | ||
Tekautz TM, Fuller CE, Blaney S, et al. Atypical teratoid/rhabdoid tumors (ATRT): improved survival in children 3 years of age and older with radiation therapy and high-dose alkylator-based chemotherapy. J Clin Oncol. 2005;23(7):1491–1499. | ||
Ostrom QT, Chen Y, M. de Blank P, et al. The descriptive epidemiology of atypical teratoid/rhabdoid tumors in the United States, 2001–2010. Neuro Oncol. 2014;16(10):1392–1399. | ||
Frühwald MC, Biegel JA, Bourdeaut F, Roberts CW, Chi SN. Atypical teratoid/rhabdoid tumors-current concepts, advances in biology, and potential future therapies. Neuro Oncol. 2016;18(6):764–778. | ||
Oh CC, Orr BA, Bernardi B, et al. Atypical teratoid/rhabdoid tumor (ATRT) arising from the 3rd cranial nerve in infants: a clinical-radiological entity? J Neurooncol. 2015;124(2):175–183. | ||
Athale UH, Duckworth J, Odame I, Barr R. Childhood atypical teratoid rhabdoid tumor of the central nervous system. J Pediatr Hematol Oncol. 2009;31(9):651–663. | ||
Hilden JM, Meerbaum S, Burger P, et al. Central nervous system atypical teratoid/rhabdoid tumor: results of therapy in children enrolled in a registry. J Clin Oncol. 2004;22(14):2877–2884. | ||
Dho YS, Kim SK, Cheon JE, et al. Investigation of the location of atypical teratoid/rhabdoid tumor. Childs Nerv Syst. 2015;31(8):1305–1311. | ||
Shitara S, Akiyama Y. Atypical teratoid/rhabdoid tumor in sellar turcica in an adult: a case report and review of the literature. Surg Neurol Int. 2014;5(1):75. | ||
Park HG, Yoon JH, Kim SH, et al. Adult-onset sellar and suprasellar atypical teratoid rhabdoid tumor treated with a multimodal approach: a case report. Brain Tumor Res Treat. 2014;2(2):108–113. | ||
Takahashi K, Nishihara H, Katoh M, et al. A case of atypical teratoid/rhabdoid tumor in an adult, with long survival. Brain Tumor Pathol. 2010;28(1):71–76. | ||
DiPatri A, Sredni S, Grahovac G, Tomita T. Atypical teratoid rhabdoid tumors of the posterior fossa in children. Childs Nerv Syst. 2015;31(10):1717–1728. | ||
Biswas A, Goyal S, Puri T, et al. Atypical teratoid rhabdoid tumor of the brain: case series and review of literature. Childs Nerv Syst. 2009;25(11):1495–1500. | ||
Biswas A, Julka PK, Bakhshi S, Suri A, Rath GK. Intracranial atypical teratoid rhabdoid tumor: current management and a single institute experience of 15 patients from north India. Acta Neurochir (Wien). 2015;157(4):589–596. | ||
Cheng YC, Lirng JF, Chang FC, et al. Neuroradiological findings in atypical teratoid/rhabdoid tumor of the central nervous system. Acta Radiol. 2005;46(1):89–96. | ||
Warmuth-Metz M, Bison B, Dannemann-Stern E, Kortmann R, Rutkowski S, Pietsch T. CT and MR imaging in atypical teratoid/rhabdoid tumors of the central nervous system. Neuroradiology. 2008;50(5):447–452. | ||
Au Yong KJ, Jaremko JL, Jans L, et al. How specific is the MRI appearance of supratentorial atypical teratoid rhabdoid tumors? Pediatr Radiol. 2012;43(3):347–354. | ||
Jin B, Feng XY. MRI features of atypical teratoid/rhabdoid tumors in children. Pediatr Radiol. 2013;43(8):1001–1008. | ||
Sredni ST, Tomita T. Rhabdoid tumor predisposition syndrome. Pediatr Dev Pathol. 2015;18(1):49–58. | ||
Kakkar A, Biswas A, Goyal N, et al. The expression of Cyclin D1, VEGF, EZH2, and H3K27me3 in atypical teratoid/rhabdoid tumors of the CNS: a possible role in targeted therapy. Appl Immunohistochem Mol Morphol. Epub 2015 Oct 13. | ||
Hasselblatt M, Gesk S, Oyen F, et al. Nonsense mutation and inactivation of SMARCA4 (BRG1) in an atypical Teratoid/Rhabdoid Tumor showing retained SMARCB1 (INI1) expression. Am J Surg Pathol. 2011;35(6):933–935. | ||
Hasselblatt M, Oyen F, Gesk S, et al. Cribriform neuroepithelial tumor (CRINET): a nonrhabdoid ventricular tumor with INI1 loss and relatively favorable prognosis. J Neuropathol Exp Neurol. 2009;68(12):1249–1255. | ||
Hasselblatt M, Nagel I, Oyen F, et al. SMARCA4-mutated atypical teratoid/rhabdoid tumors are associated with inherited germline alterations and poor prognosis. Acta Neuropathol. 2014;128(3):453–456. | ||
Al-Hussaini M, Dissi N, Souki C, Amayiri N. Atypical teratoid/rhabdoid tumor, an immunohistochemical study of potential diagnostic and prognostic markers. Neuropathology. Epub 2015 July 24. | ||
Johann PD, Erkek S, Zapatka M, et al. Atypical teratoid/rhabdoid tumors are comprised of three epigenetic subgroups with distinct enhancer landscapes. Cancer Cell. 2016;29(3):379–393. | ||
Lafay-Cousin L, Hawkins C, Carret AS, et al. Central nervous system atypical teratoid rhabdoid tumours: the Canadian Paediatric Brain Tumour Consortium experience. Eur J Cancer. 2012;48(3):353–359. | ||
Geyer J. Multiagent chemotherapy and deferred radiotherapy in infants with malignant brain tumors: a report from the Children’s Cancer Group. J Clin Oncol. 2005;23(30):7621–7631. | ||
Weinblatt M, Kochen J. Rhabdoid tumor of the central nervous system. Med Pediatr Oncol. 1992;20(3):258–258. | ||
Crist W, Gehan EA, Ragab AH, et al. The third intergroup rhabdomyosarcoma study. J Clin Oncol. 1995;13(3):610–630. | ||
Gardner SL, Asgharzadeh S, Green A, Horn B, McCowage G, Finlay J. Intensive induction chemotherapy followed by high dose chemotherapy with autologous hematopoietic progenitor cell rescue in young children newly diagnosed with central nervous system atypical teratoid rhabdoid tumors. Pediatr Blood Cancer. 2008;51(2):235–240. | ||
Chi S, Zimmerman M, Yao X, et al. Intensive multimodality treatment for children with newly diagnosed CNS atypical teratoid rhabdoid tumor. J Clin Oncol. 2008;27(3):385–389. | ||
Slavc I, Chocholous M, Leiss U, et al. Atypical teratoid rhabdoid tumor: improved long-term survival with an intensive multimodal therapy and delayed radiotherapy. The Medical University of Vienna Experience 1992–2012. Cancer Med. 2013;3(1):91–100. | ||
Zaky W, Dhall G, Ji L, et al. Intensive induction chemotherapy followed by myeloablative chemotherapy with autologous hematopoietic progenitor cell rescue for young children newly-diagnosed with central nervous system atypical teratoid/rhabdoid tumors: the head start III experience. Pediatr Blood Cancer. 2014;61(1):95–101. | ||
Kufe D, Bast R, Hait W, et al. Cancer Medicine. 7th ed. London: BC Decker; 2006:1955. | ||
Wang CH, Hsu TR, Wong TT, Chang KP. Efficacy of temozolomide for recurrent embryonal brain tumors in children. Childs Nerv Syst. 2009;25(5):535–541. | ||
Chen YW, Wong TT, Ho DM, et al. Impact of radiotherapy for pediatric CNS atypical teratoid/rhabdoid tumor (single institute experience). Int J Radiat Oncol Biol Phys. 2006;64(4):1038–1043. | ||
Spiegler BJ, Bouffet E, Greenberg ML, Rutka JT, Mabbott DJ. Change in neurocognitive functioning after treatment with cranial radiation in childhood. J Clin Oncol. 2004;22(4):706–713. | ||
Christopherson K, Rotondo R, Bradley J, et al. Late toxicity following craniospinal radiation for early-stage medulloblastoma. Acta Oncol. 2014;53(4):471–480. | ||
Clasie B, Depauw N, Fransen M, et al. Golden beam data for proton pencil-beam scanning. Phys Med Biol. 2012;57(5):1147–1158. | ||
De Amorim Bernstein K, Sethi R, Trofimov A, et al. Early clinical outcomes using proton radiation for children with central nervous system atypical teratoid rhabdoid tumors. Int J Radiat Oncol Biol Phys. 2013;86(1):114–120. | ||
McGovern SL, Okcu MF, Munsell MF, et al. Outcomes and acute toxicities of proton therapy for pediatric atypical teratoid/rhabdoid tumor of the central nervous system. Int J Radiat Oncol Biol Phys. 2014;90(5):1143–1152. | ||
Kralik SF, Ho CY, Finke W, Buchsbaum JC, Haskins CP, Shih CS. Radiation necrosis in pediatric patients with brain tumors treated with proton radiotherapy. AJNR Am J Neuroradiol. 2015;36(8):1572–1578. | ||
Chiou SH, Kao CL, Chen YW, et al. Identification of CD133-positive radioresistant cells in atypical teratoid/rhabdoid tumor. PLoS One. 2008;3(5):e2090. | ||
Squire SE, Chan MD, Marcus KJ. Atypical teratoid/rhabdoid tumor: the controversy behind radiation therapy. J Neurooncol. 2006;81(1):97–111. | ||
Roberts C, Biegel J. The role of SMARCB1/INI1 in the development of rhabdoid tumors. Cancer Biol Ther. 2009;8(5):412–416. | ||
Wei D, Goldfarb D, Song S, et al. SNF5/INI1 deficiency redefines chromatin remodeling complex composition during tumor development. Mol Cancer Res. 2014;12(11):1574–1585. | ||
Jayanthan A, Bernoux D, Bose P, Riabowol K, Narendran A. Multi-tyrosine kinase inhibitors in preclinical studies for pediatric CNS AT/RT: evidence for synergy with Topoisomerase-I inhibition. Cancer Cell Int. 2011;11(1):44. | ||
Sredni S, Patel K, Costa F, de Fátima Bonaldo M, Tomita T. Activation of ErbB2- ErbB3 signaling pathway supports potential therapeutic activity of ErbB inhibitors in AT/RT. J Neurooncol. 2014;118(1):201–203. | ||
Fouladi M, Stewart CF, Blaney SM, et al. A molecular biology and phase II trial of lapatinib in children with refractory CNS malignancies: a pediatric brain tumor consortium study. J Neurooncol. 2013;114(2):173–179. | ||
Weingart M, Roth J, Hutt-Cabezas M, et al. Disrupting LIN28 in atypical teratoid rhabdoid tumors reveals the importance of the mitogen activated protein kinase pathway as a therapeutic target. Oncotarget. 2015;6(5):3165–3177. | ||
Ginn K, Gajjar A. Atypical teratoid rhabdoid tumor: current therapy and future directions. Front Oncol. 2012;2:114. | ||
Banderali U, Jayanthan A, Hoeksema KA, Narendran A, Giles WR. Ion channels in pediatric CNS Atypical Teratoid/Rhabdoid Tumor (AT/RT) cells: potential targets for novel therapeutic agents. J Neurooncol. 2011;107(1):111–119. | ||
Bambakidis NC, Robinson S, Cohen M, Cohen AR. Atypical teratoid/rhabdoid tumors of the central nervous system: clinical, radiographic and pathologic features. Pediatr Neurosurg. 2002;37:64–70. | ||
Hirth A, Pedersen PH, Wester K, Mörk S, Helgestad J. Cerebral atypical teratoid/rhabdoid tumour of infancy: long-term survival after multimodal treatment, also including triple intrathecal chemotherapy and gamma knife radiosurgery-case report. Pediatr Hematol Oncol. 2003;20:327–332. | ||
Perreault LRM, Carret AS, Zhang G, et al. Relapse pattern in pediatric embryonal central nervous system tumors. J Neurooncol. 2013;115:209–215. |
 © 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.