Back to Journals » Journal of Inflammation Research » Volume 18
Atorvastatin Alleviates Sepsis-Induced Cardiomyopathy by Targeting the METTL3/IGF2BP1/CXCL2 Pathway
Authors Zhang L, Yin Y, Zheng L, Wang Y, Zhao J, Mu F ![]() , Jin F, Gong R, Wang J
, Jin F, Gong R, Wang J ![]()
Received 7 April 2025
Accepted for publication 8 September 2025
Published 21 October 2025 Volume 2025:18 Pages 14609—14628
DOI https://doi.org/10.2147/JIR.S532987
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr Rongxue Wu
Lulu Zhang,1,2,* Yanping Yin,1,3,* Lingling Zheng,1 Yiwen Wang,1,2 Jinyi Zhao,1 Fei Mu,1 Fuxing Jin,1,2 Rui Gong,1 Jingwen Wang1
1Department of Pharmacy, Xijing Hospital, The Fourth Military Medical University, Xi’an, People’s Republic of China; 2College of Pharmacy, Shaanxi University of Chinese Medicine, Xianyang, People’s Republic of China; 3College of Life Sciences, Northwestern University, Xi’an, People’s Republic of China
*These authors contributed equally to this work
Correspondence: Jingwen Wang, Department of Pharmacy, Xijing Hospital, Fourth Military Medical University, Xi’an, People’s Republic of China, Email [email protected] Rui Gong, Department of Pharmacy, Xijing Hospital, Fourth Military Medical University, Xi’an, People’s Republic of China, Email [email protected]
Background: Sepsis-induced cardiomyopathy (SICM) is a common and serious complication in patients with sepsis and septic shock. Its pathological and physiological mechanisms are highly complex and often lead to dysfunction of multiple organ systems, and the current clinical intervention strategies have limited efficacy. Statins have been shown to reduce mortality of sepsis patients and alleviate multi-organ damage caused by sepsis, including SICM. However, the molecular mechanisms by which atorvastatin (ATO) exerts cardioprotective effects have not been fully elucidated. This study aims to systematically explore the cardioprotective effect of ATO on SICM through in vivo and in vitro experiments, and to elucidate the molecular mechanism underlying ATO’s cardioprotective effects.
Methods: Mice were exposed to multi-bacterial sepsis through cecal ligation and puncture (CLP) surgery, and were continuously pretreated with ATO for 6 days before surgery. Cardiac function was evaluated by echocardiography. Immunohistochemistry and Western blotting were used to investigate the protective effect of ATO on heart injury in septic mice. AutoDock software, CETSA and DARTS experiments were used to verify the specific binding of ATO to METTL3. Furthermore, the regulatory role of METTL3-mediated m6A modification in septic myocardial inflammatory injury and its molecular mechanisms were elucidated by m6A-MeRIP-seq, RNA immunoprecipitation and Western blotting.
Results: ATO could obviously improve the cardiac function of septic mice, alleviate the heart tissue damage, and inhibit the release of inflammatory factors and myocardial injury factors. Mechanically, ATO binds to METTL3 with high affinity and inhibits its expression, thereby suppressing the overall m6A modification level in heart tissue of septic mice and in lipopolysaccharide (LPS)-stimulated cardiomyocytes. Importantly, METTL3 promoted CXCL2 expression by mediating m6A modification of CXCL2 mRNA and enhancing the stability of CXCL2 mRNA in an IGF2BP1 dependent manner. The high expression of CXCL2 eventually triggers the inflammatory response and ferroptosis of cardiomyocytes, resulting in septic myocardial injury.
Conclusion: This study elucidates that the METTL3 (m6A)/IGF2BP1/CXCL2 axis promotes the pathological progression of SICM, and ATO exerts cardioprotective effects by targeting this pathway, providing new insights into the significance of RNA m6A modification in SICM.
Keywords: atorvastatin, sepsis-induced cardiomyopathy, m6A modification, METTL3, CXCL2
Introduction
Sepsis is a rapidly progressing, life-threatening condition characterized by multi-organ dysfunction due to a dysregulated characterized by multiple organ dysfunction that ensues from a dysregulated host response to infection,1 and is recognized as a global health priority by the World Health Organization.2 Sepsis-induced myocardial dysfunction, also called SICM, is a common and serious complication in patients with sepsis.3 In China, within the ICU patients, sepsis has an occurrence rate of 20.6%, and its 90-day fatality rate reaches 35.5%, imposing substantial economic and social burdens.4 Among them, about 40% of patients experience myocardial injury, and the associated mortality rate ranges from 70–90%.5 The development of SICM is a complex process, which is caused by disrupted calcium homeostasis, inflammation, apoptosis, oxidative stress, and mitochondrial dysfunction, inflammation, cell apoptosis, oxidative stress, and mitochondrial dysfunction.6 The current treatment strategies for SICM are similar to conventional therapy for sepsis, mainly using supportive therapy to control the source of infection, including fluid resuscitation, oxygen therapy, and vasoactive drugs that improve cardiac function (such as norepinephrine and dobutamine).7 However, these therapies have limited effectiveness in improving the survival rate of SICM patients.8,9 So, it is urgent to develop novel targeted therapy strategies targeting the pathological mechanisms of SICM.
Statins not only have lipid-lowering effects, but also exert multiple pharmacological effects by regulating inflammatory responses, immune homeostasis, and coagulation pathways.10 Study has shown that statins can protect endothelial function, alleviate sepsis-related inflammatory injury, and significantly inhibit the release of key inflammatory factors such as TLR4, IL-6, TNF-α and IL-1β.11 Epidemiological evidence further confirmed that statin treatment can reduce the incidence rate and mortality of patients with sepsis-related diseases,12 and also alleviate the complications of sepsis, such as myocardial injury,13 lung injury,14 cognitive dysfunction,15 and other related diseases. However, it is worth noting that although statins exert cardioprotective effects in SICM, their specific molecular regulatory mechanisms have not been fully elucidated. Therefore, in-depth analysis of the pathogenesis of SICM and the targets of stains will lay a key theoretical foundation for the development of targeted clinical prevention and treatment strategies.
N6-methyladenosine (m6A), as the most common post-transcriptional modification in eukaryotic RNAs,16 widely affects gene expression by dynamically regulating core metabolic processes, such as RNA splicing, processing, translation, degradation, and mRNA stability.17 The dynamics of this modification are precisely coordinated by three types of functional proteins: the methyltransferase complex (METTL3 as the catalytic core, methyltransferase like 14 (METTL14) /Wilms’tumor 1-associatingprotein (WTAP) as the structural cofactor) acts as “writers” to catalyze methylation; Demethylases (such as FTO, ALKBH5) act as “erasers” to remove modifications; Reading proteins (including YTHDF1/2/3, YTHDC1/2, IGF2BP1/2/3, etc.) serve as “readers” to recognize modification signals. The imbalance of this regulatory network has been proven to drive various pathological processes such as cancer,18 the development of the nervous system,19 immune disorders,20 and cardiovascular diseases.21 As a key RNA modification, m6A regulates gene expression and may influence inflammation and cell survival in cardiac tissue. The role of the core catalytic subunit METTL3 in autoimmune diseases, inflammatory diseases and sepsis has received widespread attention.22–24 For example, METTL3 promoted non-small-cell lung carcinoma progression by inhibiting the RIG-I-MAVS innate immune pathway.25 In sepsis-induced myocardial injury, METTL3/YTHDF1-mediated m6A modification stabilized USP12 mRNA, thereby promoting FOXO3 deubiquitination and exacerbating myocardial cell apoptosis in sepsis-induced myocardial dysfunction.26 Moreover, METTL3 alleviated lipopolysaccharide (LPS) induced inflammation in macrophages by inhibiting nuclear factor kappa-B (NF-κB) pathway. Importantly, the inflammatory regulatory function of METTL3 is closely related to the pathological mechanism of SICM.27 When sepsis occurs, a large number of pro-inflammatory cytokines are released, which can directly damage myocardial cells, inhibit myocardial contractile function, and lead to myocardial dysfunction.28,29 Therefore, in-depth analysis of the mechanism of m6A modification (especially the METTL3-mediated regulatory axis) in sepsis and SICM will provide a key theoretical basis for finding inhibitors of myocardial inflammatory signaling with m6A modification as the entry point, and further promote the innovation of clinical prevention and treatment strategies.
Interleukin 17 (IL-17), as a core mediator of immune inflammation, drives the pathological process of inflammation in infections and autoimmune diseases.30 Meta analysis showed that IL-17 concentration in non-surviving sepsis patients was 5.96 times higher than that in survivors,31 confirming its potential as an independent predictor of sepsis mortality risk. Excessive IL-17A can disrupt immune homeostasis, potentially leading to organ dysfunction including acute lung injury, acute respiratory distress syndrome, and cardiac dysfunction,32 indicating that it may play crucial roles in sepsis and its induced multiple organ dysfunction. In addition, studies have shown that IL-17 can activate the specific transcription factor NF-κB and promote its nuclear translocation, specifically binding to the CXCL2 gene promoter to enhance its transcription, ultimately leading to a significant increase in CXCL2 protein expression.33 The up-regulated CXCL2 in sepsis further mediates the infiltration of neutrophils into the inflammatory site, amplifying tissue injury.34
Since ATO may modulate inflammation, and IL-17–CXCL2 signaling drives inflammatory injury, understanding this link could reveal part of ATO’s cardioprotective mechanism. Although statins show benefit in sepsis, the exact molecular mechanisms by which atorvastatin protects the heart in SICM remain unclear, particularly with respect to RNA methylation pathways. The aim of this study is to elucidate the regulatory mechanism of m6A modification mediated by METTL3 in myocardial inflammatory injury in SICM, and to reveal the mechanism by which ATO, as an inhibitor of METTL3, alleviates myocardial injury by targeting the inhibition of the METTL3/CXCL2 axis. These results will provide innovative molecular targets and theoretical basis for the clinical application of ATO in the prevention and treatment of SICM.
Materials and Methods
Reagents and Antibodies
ATO (10059) was purchased from the China Food and Drug Administration. LPS (O55:B5) was purchased from 100 Puxitang Biotechnology Co., LTD (Beijing, China). The primary antibody used in this study were as follows: Anti-COX2 (1:800, 27308-1-AP, Proteintech), Anti-METTL3 (1:1000, 15073-1-AP, Proteintech), Anti-CXCL2 (1:500, 26791-1-AP, Proteintech), Anti-TLR4 (1:1000, 19811-1-AP, Proteintech), and Anti-IL-17 (1:500, 66148-1-Ig, Proteintech). Anti-IL-6 (1:1000, ab290735, Abcam), Anti-GPX4 (1:1000, ab125066, Abcam), Anti-SLC7A11 (1:1000, ab307601, Abcam), and Anti-GAPDH (1:10,000, 40493–1, Signalway).
Animal Studies
C57BL/6J male mice (6–8 weeks) were purchased from animal center of the Fourth Military Medical University (Xi’an, China) and randomly divided into Sham, CLP, CLP+ATO (10 mg/kg), CLP+ATO (20 mg/kg) and CLP+ATO (40 mg/kg) groups. All animals were weighed before the experiment and stratified according to their weight range. Within each layer, animals were assigned to different groups by a random number generator. Before constructing the SICM mouse model through cecal ligation and puncture (CLP), mice were pretreated with ATO by gavage for 6 consecutive days. 24 hours after CLP, the mice were anesthetized with isoflurane (3% for induction and 2% for maintenance) and 1 cm incision was made along the midline of the mouse abdomen to expose the cecum. Subsequently, 75% of the cecum was ligated with nonabsorbable surgical suture. The ligation area was punctured with a 21-gauge needle and a small number of intestinal contents were extruded. Finally, the cecum was placed back into the abdominal cavity and the peritoneum and skin were sutured separately. Fluid resuscitation was performed by subcutaneous injection of sterile saline (1 mL per animal), and the mice slowly woke up after 1 hour. Ensure animal stability by continuously monitoring vital signs (heart rate, body temperature).
Echocardiography
The cardiac function of mice was evaluated by using an ultra-high resolution small animal ultrasound imaging system (Fujifilm Visualsonics VEVO 3100 LT). Image acquisition and analysis were carried out under the blind condition that the operator was unaware of the experimental group. The ejection fraction (EF%), fractional shortening (FS%), LVPWs, LVPWd, LVAWs and LVAWd of the left ventricle were measured by using Vevo LAB 5.7.1 software. The standardized echocardiographic procedure for all mice was random and objective.
Histology
All operation procedures were carried out as described in the previously.35 All histological images were analyzed without knowing the experimental groups. Briefly, the heart was collected and fixed with 4% paraformaldehyde for 24 h. The next day, the heart was dehydrated with gradient ethanol and embedded in paraffin. The embedded tissues were cut into 5 μm sections. After dewaxing and hydration, the hematoxylin and eosin (H&E) staining was used to evaluate the pathological damage of the heart. All surgical procedures strictly followed the above protocol.
Immunohistochemistry
The mouse hearts fixed with 4% paraformaldehyde were cut into 5 μm sections and dewaxed with xylene. After that, the endogenous peroxidase activity was inactivated with 3% hydrogen peroxide to reduce background, and then sodium citrate buffer was used for antigen repair. Subsequently, the sections were incubated with anti-TLR4 antibody (1:1000, 19811-1-AP) overnight at 4°C. The next day, the sections were washed twice with PBS, and then incubated with the corresponding horseradish peroxidase (HRP) -labeled secondary antibody at 37°C for 1 h. Finally, the sections were stained with 3, 3’-diaminobenzidine (DAB) solution. The microscope (Olympus BX53, Japan) was used to capture images and the CaseViewer 2.4.0 software was used for quantitative analysis.
ELISA Assays
The expression levels of TNF-α, IL-6 and IL-1β in the serum and cell supernatant were measured using the corresponding ELISA kits (TNF-α: EMC102a.96, IL-6: EMC004.96, IL-1β: EMC001b.96; Neobioscience Biotechnology, Shenzhen, China). The levels of CK-MB, LDH, cTnI, AST, MDA and GSH in the serum were detected using corresponding assay kits (JianCheng, Nanjing, China). The level of Fe2+ in the cell supernatant was detected using a specific assay kit (Solarbio, China). All assays were performed according to the respective manufacturer’s instructions.
Quantification of m6A RNA Modification Level
The quantification of m6A RNA methylation levels in heart tissue and HL-1 cardiomyocytes were detected using an m6A RNA Methylation Quantification Kit (EpiQuik, P-9005-96) according to the manufacturer’s instructions. In brief, RNA was extracted from the samples using TRIzol reagent (Invitrogen, USA), and then added into the strip well provided by the kit and incubated at 37°C for 90 min. After washing with washing buffer three times, the capture antibody, detection antibody and enhancer solution were added successively. Finally, the color development solution was added and the absorbance at 450 nm was measured using a microplate reader.
The Culture and Cell Transfection of HL-1 Cardiomyocytes
HL-1 cardiomyocytes were purchased from Haixing Biology Technology Co., Ltd, and cultured in DMEM/HIGH GLUCOSE medium containing 10% fetal bovine serum (FBS), streptomycin, and penicillin, and in a humidified incubator at 37°C and 5% CO2.
The small interfering RNA (siRNA) targeting METTL3 (Si-METTL3), IGF2BP1 (Si-IGF2BP1) and CXCL2 (Si-CXCL2), as well as METTL3 overexpression plasmids (OV-METTL3), were constructed by General Biology Co., Ltd (Anhui, China). SiRNA was transfected into HL-1 cardiomyocytes using Lipofectamine RNAiMAX (Invitrogen, USA), and overexpression plasmid was transfected into HL-1 cardiomyocytes using Lipofectamine 2000 (Invitrogen, USA). After transfection for 48 h, the cells were collected for subsequent experimental analysis. The corresponding sequences were shown in Table S1.
Cell Viability
HL-1 cardiomyocytes were seeded into 96-well plates (1×104 cells/well) and incubated overnight in a 37°C, 5% CO2 incubator to achieve a confluence of 70–80%. Subsequently, the cells were pretreated with ATO for 24 h and then treated with LPS for another 24 h. Finally, the Cell Counting Kit-8 (CCK-8) reagent (MedChemExpress, USA) was added and the absorbance was detected at a wavelength of 450 nm to assess cell viability.
Quantitative Real-Time PCR (qRT-PCR)
Total RNAs were extracted from the samples using TRIzol reagent (Invitrogen, USA). The extracted total RNA was dissolved in RNase-free water and the concentration and purity were determined using Nanodrop 2000 (Thermo ScientificTM). Subsequently, the quantified RNA samples were reverse transcribed into cDNA using the SweScript All-in-One RT SuperMix (ServiceBio, Wuhan, China) according to the manufacturer’s instructions. qRT-PCR was conducted using SYBR Green Master Mix (Applied Biosystems, Forster city, CA) according to the manufacturer’s instructions. The expression level of target genes was normalized to GAPDH. The primer sequences used were shown in Table S1.
Western Blot
Total protein was extracted from mouse heart tissue and HL-1 cardiomyocytes using RIPA lysis buffer (Ncnbio, WB3100), and quantified using the BCA protein quantification kit (Ncmbio, WB6501). Subsequently, the quantified protein samples were separated by SDS-PAGE gel, and then transferred to the methanol-activated PVDF membrane. The membranes were blocked with 5% milk at room temperature (RT) for 1–2 h and then incubated with the primary antibodies overnight at 4°C. The next day, the membranes were thoroughly washed three times with PBST buffer and then incubated at RT with the corresponding HRP-labeled secondary antibody for 1 h. Finally, the protein bands were visualized using an extremely sensitive ECL Chemiluminescence kit (Ncmbio, P10060).
Molecular Docking Study
The tertiary structures of METTL3 (PDB ID: 8bn8), METTL14 (PDB ID: 702i), ALKBH5 (PDB ID: 5fto), and FTO (PDB ID: 7ckk) proteins were obtained from the PDB database (https://www.rcsb.org/). The 3D structure of ATO was then obtained from the PubChem databases (https://pubchem.ncbi.nli.nih.gov/), and energy optimization was performed in Chem3D software. PymoL was used to remove water molecules and ligands etc. of target proteins, and the ligands and proteins were saved as pdb files, and AutoDock Tools was used to convert proteins, ligands and drug molecules into pdbgt files. AutoDock Vina was used to run the docking program to find the active pocket of target protein centered on the ligand and set the Grid Box coordinates and box size to obtain the docking score. Finally, PyMOL was used to visualize the results of molecular docking.
Cellular Thermal Shift Assay (CETSA)
After treatment with ATO (8 nM) or DMSO for 24 h, HL-1 cardiomyocytes were collected and lysed with RIPA lysis buffer. The soluble proteins in the supernatant were separated by centrifugation at 12,000 rpm for 15 min at 4°C. The protein samples were divided into seven parts and heated at 40, 43, 46, 49, 51, 54, and 57°C for 3 min, respectively. After the protein samples were mixed with the loading buffer and denatured, the protein expression level of METTL3 was detected by Western blotting.
Drug Affinity Responsive Target Stability (DARTS) Assay
HL-1 cardiomyocytes were washed twice with PBS, and then lysed with M-Per lysate buffer and the supernatant was collected. The 10×TNC buffer (Tris-HCl, NaCl, and CaCl2) was added to the supernatant in a ratio of 1:10. After the protein concentration was determined by BCA protein quantification kit, the protein sample was divided into control and ATO group. Among them, in the ATO group, ATO was added to the sample at a final concentration of 8 nM, and an equal volume of DMSO was added to the control group. The samples were then incubated on vertical shaker at RT for 1 h and added with pronase solution (dissolved pronase: protein =0; 1:1000; 1:2000; 1:3000; 1:10,000; 1:20000) and let stand at RT for 15 min. Finally, the loading buffer was added to the samples and the mixtures were denatured by heating, and the protein expression level of METTL3 was detected by Western blotting.
M6A-Methylated RNA Immunoprecipitation Sequencing (M6A-MeRIP-Seq)
Total RNA was extracted from cardiomyocytes transfected with Si-METTL3 or negative control (Si-NC). Total RNA (1–3 μg) was added to 300 μL of 1×IP buffer (50 mM Tris-HCl, pH7.4, 150 mM NaCl, 0.1% NP40, and 40 U/μL RNase inhibitor), which has been pre-mixed with Anti-m6A antibody (2 μg). The reaction mixture was incubated on a vertical rotator at 4°C for 2 h for m6A-RNA immunoprecipitation. Subsequently, the Dynabeads™ beads pre-washed with IP buffer were added to the mixture and incubated overnight at 4°C. The next day, the immunoprecipitation complex was thoroughly washed four times with IP buffer to remove non-specific binding. Next, the enriched m6A modified RNAs were eluted using elution buffer and immediately extracted with acid phenol-chloroform and precipitated with ethanol. After labeling and hybridization, the hybridization arrays were washed and fixed, and scanned using an Agilent Scanner G2505C (Agilent Technologies, Santa Clara). Finally, the libraries were sequenced using the Illumina NovaSeq platform. After removing low-quality reads, the remaining clean reads were then aligned to the reference genome by STAR and Hisat2 software (v2.0.4). GO and KEGG analyses were performed for functional analysis.
RNA Immunoprecipitation (RIP)
HL-1 cardiomyocytes were collected after transfection with si-METTL3 for 48 h and lysed with RIP lysis buffer. The Dynabeads™ beads were thoroughly washed with RIP washing buffer and incubated with Anti-m6A antibody (1:500, Synaptic Systems) on a 4°C vertical shaker for 3 h to allow the antibody to bind to the beads. Then, the antibody-beads complex was mixed with cell lysate and incubated overnight at 4°C to allow the antibody to capture m6A modified RNAs. The next day, the immunoprecipitation complex were washed six times with RIP washing buffer to remove non-specific binding, and then purified with proteinase K. Finally, the enriched RNAs were analyzed by qRT-PCR. Normal IgG antibody was used as negative control.
RNA Stability Analysis
HL-1 cardiomyocytes were transfected with Si-METTL3 or Si-IGF2BP1 and treated with 5 μg/mL actinomycin D (Act D) for 0, 6 and 12 h, respectively. Subsequently, the total RNAs were extracted using TRIzol reagent. The mRNA expression of CXCL2 was detected by qRT-PCR, and GAPDH was used as an internal reference gene. The relative quantification was calculated by the 2−ΔΔCt method normalized to GAPDH.
Statistical Analysis
Statistical analysis was performed using Prism 8.0 software. Data were presented as mean ± SD, and the unpaired (two-tailed) Student’s test and one-way analysis of variance (ANOVA) were used to analyze differences. * P < 0.05, ** P < 0.01, *** P < 0.001. P < 0.05 was considered statistically significant. Statistical analysis of data in at least three biological replicates or independent experiments.
Results
ATO Improves Cardiac Dysfunction in Septic Mice
Studies have shown that statins can significantly improve cardiac dysfunction in septic mice.36 Therefore, CLP was used to establish a mouse model of sepsis, and to further explore the protective effect of ATO on heart injury in septic mice. To evaluate the effect of ATO on cardiac function in sepsis mice, we used echocardiography to detect indicators of cardiac function in each group of mice. The results showed that compared with the Sham group, the cardiac function of mice in the CLP group was obviously impaired, manifested by a decrease in EF%, FS%, LVAWs, LVPWs, LVPWd and the increase of LVAWd. Pretreatment with ATO (20 or 40 mg/kg) can markedly improve the impaired cardiac function of septic mice (Figure 1A–G). In addition, no significant differences were observed in heart rate (HR) and heart weight/body weight ratio (HW/BW) among the groups of mice (Figure 1H–I). In terms of histological analysis, H&E staining showed that the CLP group mice had disordered arrangement of myocardial fibers and increased infiltration of inflammatory cells, while ATO pretreatment could effectively alleviate these pathological changes (Figure 1J). Moreover, compared with the CLP group, pretreatment with ATO significantly reduced the expression levels of CK-MB, LDH, cTnI, and AST (Figure 1K-N). The above results indicated that ATO pretreatment can effectively improve cardiac dysfunction in septic mice. Among them, the most significant effect was observed when the concentration of ATO appeared to be 20 mg/kg.
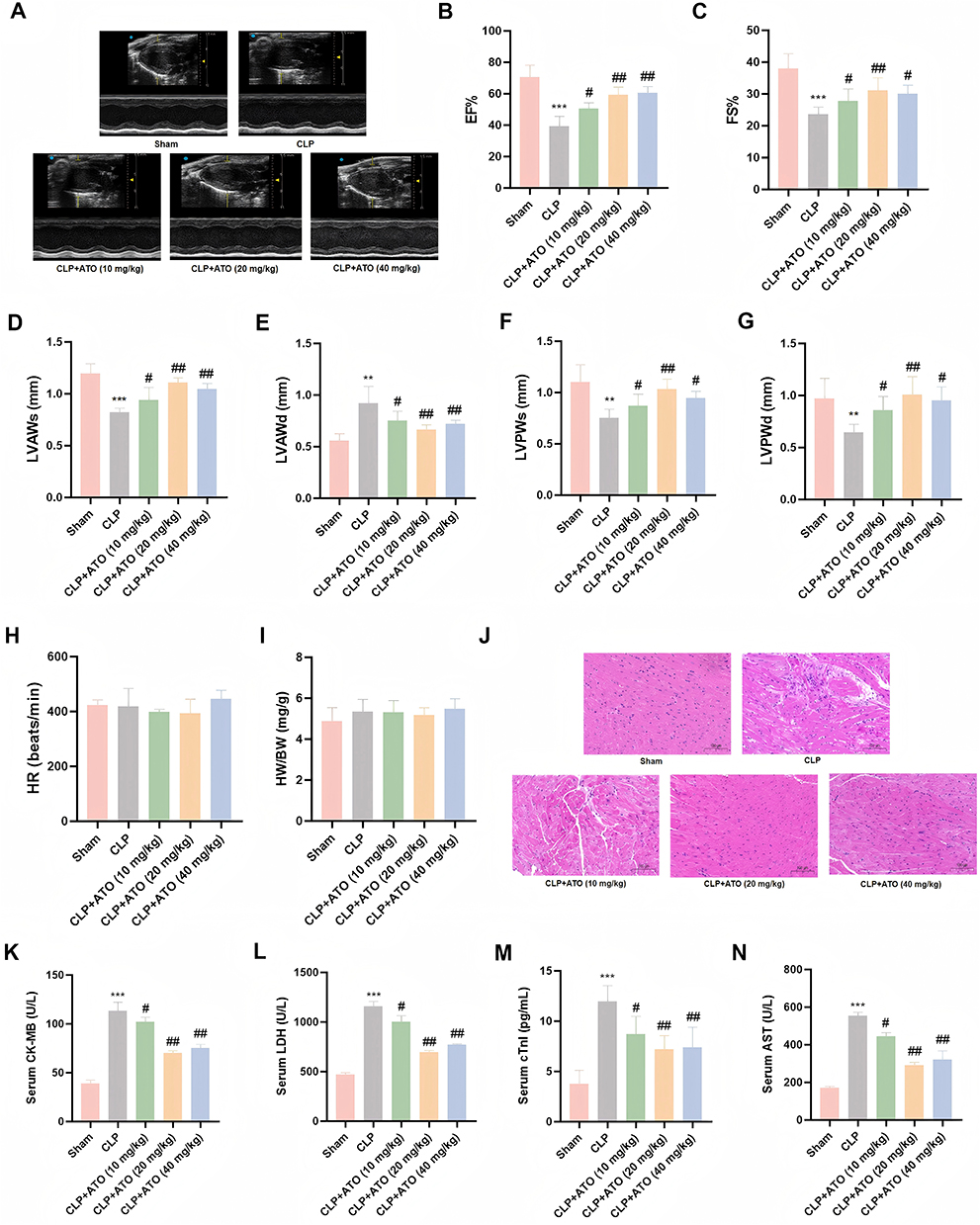 |
Figure 1 ATO can improve cardiac dysfunction, reduce heart tissue damage and inflammatory infiltration in SICM mice. (A) Representative images of M-mode echocardiography. (B–H) Quantitative analysis of EF%, FS%, LVAWs, LVAWd, LVPWs, LVPWd, HR in each group of mice (n = 6). (I) The center weight ratio of mice in each group (n = 6). (J) Representative images of hematoxylin–eosin staining; scale bar =100 μm (n = 3). (K–N) Expression levels of CK-MB, LDH, cTnI and AST in serum of mice in each group (n = 6). Data are represented as mean±SEM; ** P < 0.01, *** p < 0.001 vs Sham; # P < 0.05, ## P < 0.01 vs CLP. |
ATO Inhibits the Release of Inflammatory Factors in Septic Mice and the Levels of m6A Modification and the Expression of METTL3 in Heart Tissue
Due to the crucial role of inflammatory response in sepsis and SICM, we further measured the release levels of inflammatory factors in the serum of each group of mice and found that compared with the Sham group, the expression levels of TNF-α, IL-6 and IL-1β were increased in the sepsis group. Pretreatment with ATO, especially at doses of 20 and 40 mg/kg, markedly inhibited the high expression of TNF-α, IL-6, and IL-1β in septic mice (Figure 2A–C). To further investigate the regulation of inflammatory signaling pathway by ATO, we used immunohistochemistry to detect the expression of TLR4 in heart tissue and found that pretreatment with ATO significantly reduced the upregulation of TLR4 induced by sepsis (Figure 2D). In addition, Western blotting analysis showed that the protein expression of COX2 and IL-6 in the heart tissue of septic mice were significantly increased, and pretreatment with ATO could effectively inhibit the expression of these two proteins (Figure 2E), suggesting that ATO may exert cardioprotective effects by regulating the inflammatory signaling pathway.
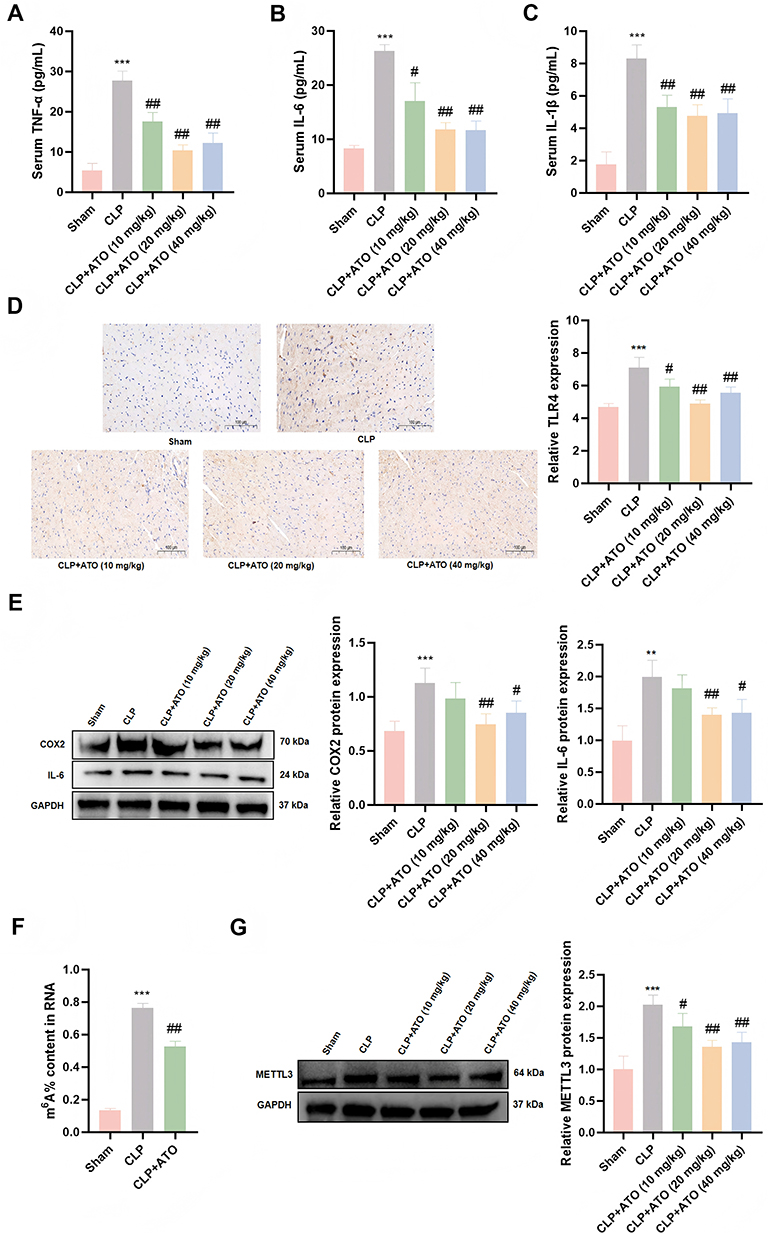 |
Figure 2 ATO inhibits myocardial inflammation and the expression of m6A modification and METTL3 protein in heart tissue of septic mice. (A–C) ATO reduces the levels of TNF-α, IL-6 and IL-1β in SICM mice (n = 6). (D) The effect of ATO on the expression of TLR4 protein in the heart of SICM mice was detected by immunohistochemistry; scale bar =100 μm (n = 3). (E) Western Blot was used to detect the protein expression levels of COX2 and IL-6 (n = 3). (F) Elisa was used to detect the level of m6A modification. (G) Western Blot was used to detect the protein expression levels of METTL3. **P < 0.01, ***p < 0.001 vs Sham; #p < 0.05, ##p < 0.01 vs CLP. |
As the most common post-transcriptional modification in eukaryotic mRNA,16 m6A has been shown to be involved in the diagnosis, treatment and prognosis of sepsis.37 Previous studies have shown that m6A and its related regulatory factors can affect the occurrence and development of SICM by regulating inflammatory responses, apoptosis, pyroptosis and other pathways.38,39 In view of this, we first detected the changes in m6A modification levels in heart tissue. ELISA results showed that there was a dramatic increase in m6A modification level in the hearts of mice in the sepsis group, while ATO pretreatment could effectively reduce the increase of this modification level (Figure 2F). Given that METTL3 is a core component of the m6A methyltransferase complex,40 we further detected its protein expression level by Western blotting and found that the expression of METTL3 was obviously upregulated in the sepsis group, while ATO pretreatment could significantly suppress the high expression of METTL3 induced by sepsis (Figure 2G). The above results indicated that pretreated with ATO can alleviate myocardial inflammatory infiltration and injury in septic mice by inhibiting the release of inflammatory factors. It is worth noting that our results suggested that METTL3-mediated m6A modification may be involved in the protective process of ATO against myocardial inflammatory injury in sepsis.
ATO Alleviates LPS Induced Inflammatory Damage in Cardiomyocytes
Sepsis is an excessive inflammatory response triggered by infection, and its pathological progression is mainly driven by inflammatory cascade reaction. To investigate the protective mechanism of ATO against SICM, we constructed an in vitro sepsis induced myocardial injury model by stimulating HL-1 cardiomyocytes with LPS. The results showed that LPS treatment suppressed the cell viability of HL-1 cardiomyocytes in a dose-dependent manner (0, 1, 2, 5, 10 μg/mL) (Figure 3A), accompanied by increased release of LDH, and significantly upregulated the expression levels of inflammatory proteins COX2 and IL-6 (Figure 3B and C). However, LPS-induced cell damage was alleviated by pretreatment with ATO, manifested as increased cell viability (Figure 3D), a reduction in LDH release (Figure 3E), and a decrease in the level of pro-inflammatory factor IL-1β (Figure 3F). Furthermore, Western blotting analysis further demonstrated that ATO pretreatment could significantly inhibit the high protein expression levels of COX2 and IL-6 induced by LPS (Figure 3G). Thus, these findings concluded that ATO pretreatment can alleviate LPS-induced cardiomyocyte injury by inhibiting the expression of inflammatory factors.
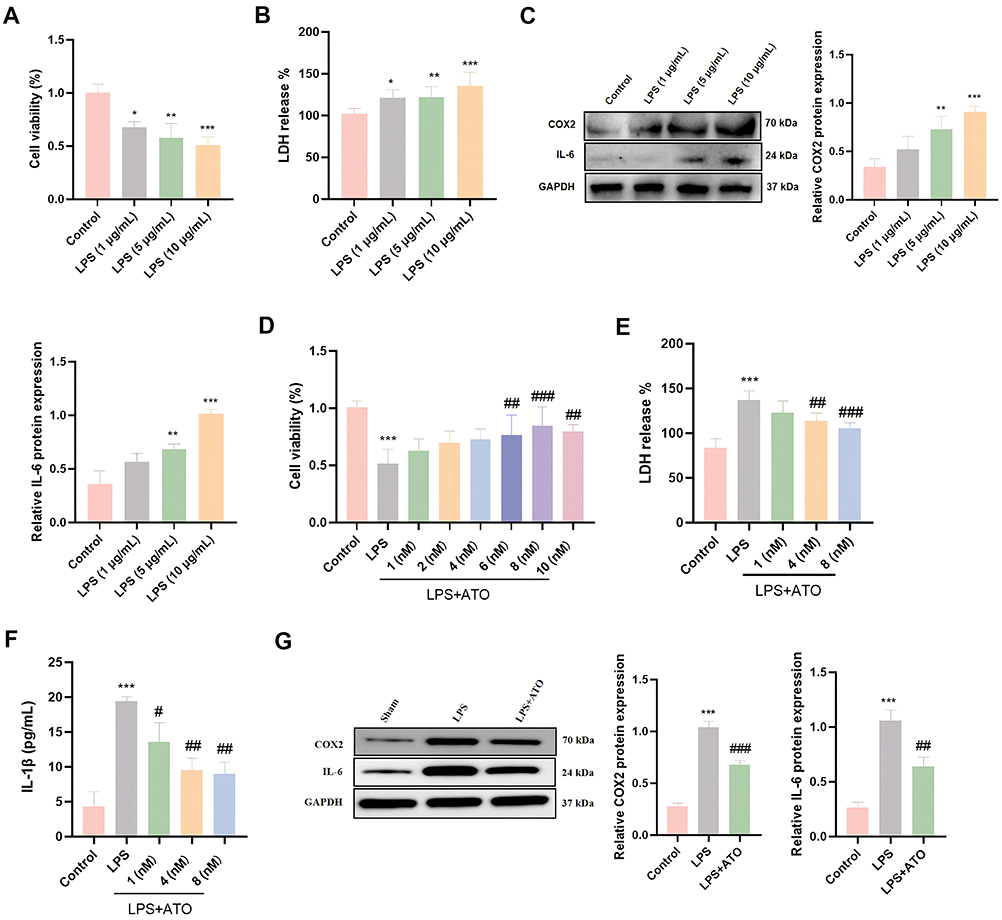 |
Figure 3 ATO alleviates LPS-induced myocardial cell injury in vitro. (A) CCK-8 was used to detect the viability of HL-1 cardiomyocytes treated with different doses of LPS; n = 6. (B) LDH content after LPS treatment was measured by Elisa (n = 6). (C) Western Blot was used to detect the protein expression levels of COX2 and IL-6 after LPS treatment (n = 3). (D) CCK-8 was used to detect the viability of HL-1 cardiomyocytes after ATO treatment (n = 6). (E and F) The contents of LDH and IL-1β after ATO treatment were detected by Elisa (n = 6). (G) Western Blot was used to detect the protein expression levels of COX2 and IL-6 after ATO treatment (n = 3). *p < 0.05, **p < 0.01, ***p < 0.001 vs Control; #p < 0.05, ##p < 0.01, ###p < 0.001 vs LPS. |
ATO Directly Interacts with METTL3
To further explore whether ATO exerts cardioprotective effects by regulating m6A modification, we detected the level of m6A modification and the expression of METTL3 in HL-1 cardiomyocytes. The results showed that LPS stimulation can significantly increased the m6A modification level in cardiomyocytes, and significantly upregulated the expression of METTL3, while ATO pretreatment inhibited the increase of m6A modification level and METTL3 expression induced by LPS (Figure 4A and B). These in vitro experimental results are highly consistent with the findings in animal models, further confirming that ATO may exert cardioprotective effects by regulating METTL3-mediated m6A modification. To elucidate the specific mechanism by which ATO regulates METTL3 expression, we performed molecular docking simulations using Autodock software. To elucidate the specific mechanism by which ATO regulates METTL3 expression, we performed molecular docking simulations using Autodock software. The results indicated that ATO has a relatively high binding affinity with METTL3 protein (−8.48 kcal/mol) (Figures 4C and S1). Further DARTS and CETSA experiments confirmed that ATO can bind to METTL3 to form a stable complex, which is manifested as enhanced anti-protease degradation ability (Figure 4D), and improved thermal stability (Figure 4E). The above results suggested that ATO may alleviate sepsis-related myocardial injury by directly targeting METTL3 and inhibiting m6A modification.
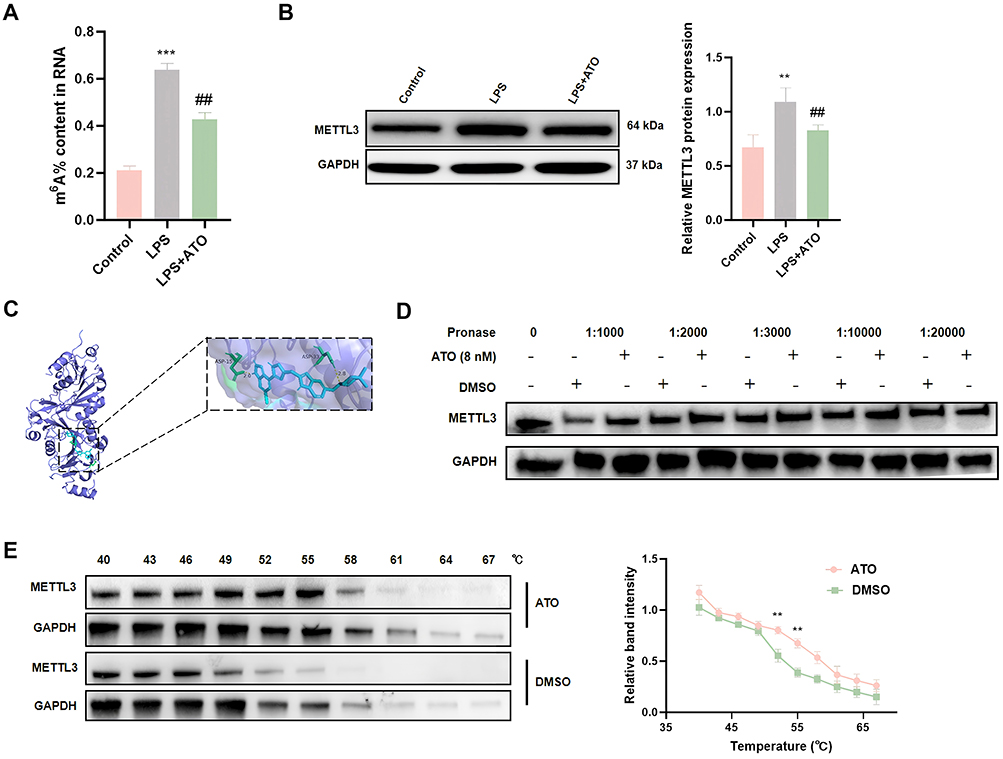 |
Figure 4 ATO binds to METTL3 with high affinity and inhibits its expression. (A) Elisa was used to detect the level of m6A modification. (B) Western Blot was used to detect the protein expression levels of METTL3. (C) Molecular docking was used to detect the binding potential of ATO with m6A regulators METTL3. (D) Degradation stability of METTL3 incubated with ATO in cell lysates as determined by drug affinity responsive target stability (n = 3). (E) The thermal stability of METTL3 incubated with ATO in HL-1 cardiomyocytes was measured by temperature-dependent cell thermal shift assay (n = 3). **p < 0.01, ***p < 0.001 vs Sham; ##p < 0.01 vs LPS; n = 3. |
ATO Inhibits LPS Induced Myocardial Cell Inflammatory Injury by Regulating the METTL3/m6A Signaling Pathway
To elucidate the role and mechanism of METTL3 in ATO-mediated cardioprotective effects, we knocked down the expression of METTL3 using siRNA. And it was found that the knockdown of METTL3 can significantly inhibit the increased expression levels of myocardial injury markers (CK-MB and LDH), inflammatory factors (TNF-α and IL-1β) and inflammation-related protein COX2 in HL-1 cardiomyocytes treated with LPS (Figure 5A–E). To further verify whether the cardioprotective effect of ATO depends on the regulation of METTL3, we overexpressed or knocked down METTL3 in HL-1 cardiomyocytes stimulated with LPS and pretreated with ATO. The results showed that under the co-treatment condition of LPS+ATO, METTL3 overexpression can reversed the protective effect of ATO, leading to a further increase in the levels of myocardial injury markers and inflammatory factors. Conversely, knockdown of METTL3 can further enhanced the inhibitory effect of ATO on the expression of the above indicators (Figure 5F–I). The above results demonstrated that ATO can alleviate LPS-induced myocardial cell inflammatory injury by regulating METTL3-mediated m6A modification.
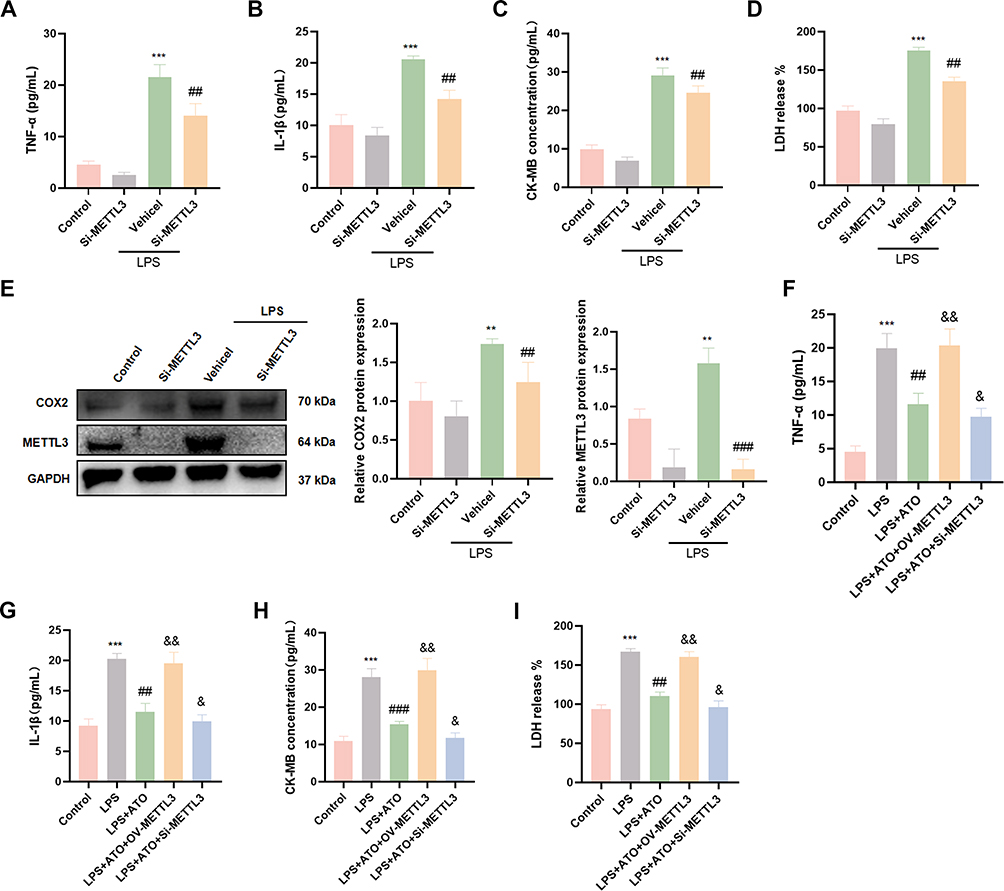 |
Figure 5 ATO inhibits LPS-induced myocardial injury by down-regulating METTL3. (A–D) Elisa was used to detect the expression levels of CK-MB, LDH, TNF-α and IL-1β in HL-1 cardiomyocytes after Si-METTL3 treatment (n = 6). (E) Western Blot was used to detect the protein expression levels of METTL3, COX2 and IL-6 in HL-1 cardiomyocytes after Si-METTL3 treatment (n = 3). (F–I) Elisa was used to detect the expression levels of CK-MB, LDH, TNF-α and IL-1β in HL-1 cardiomyocytes after OV-METTL3 treatment (n = 6). **p < 0.01, ***p < 0.001 vs Control; ##p < 0.01, ###p < 0.001 vs LPS; &p < 0.05, &&p < 0.01 vs LPS+ATO. |
METTL3 Enhances CXCL2 mRNA Stability Through an m6A-IGF2BP1-Dependent Mechanism to Promote Septic Myocardial Cell Inflammatory Injury
To further reveal the molecular mechanism by which METTL3 regulates SICM, we performed m6A-MeRIP-seq after METTL3 knockdown in HL-1 cardiomyocytes. The result showed that the knockdown of METTL3 led to a significant alteration in the genome-wide m6A modification levels (Figure 6A). GO analysis showed that the down-regulated genes of m6A modification were significantly enriched in the following biological processes: phosphorus metabolism process, macromolecular biosynthesis regulation, etc. Cellular components such as cytoplasm and lysosomes were significantly affected by differentially expressed genes. The main molecular functions include protein binding and catalytic activity (Figure S2). Further analysis revealed that differentially expressed m6A modified genes were mainly enriched in signaling pathways related to inflammation and immune regulation, such as TNF signaling pathway, MAPK signaling pathway, and IL-17 signaling pathway (Figure 6B). Among them, it has been confirmed that the IL-17 signaling pathway was related to the severity of sepsis.31 And the expression of CXC chemokine ligand 2 (CXCL2), a key protein in the IL-17 signaling pathway, was increased in patients with sepsis,41 suggesting that CXCL2 may play a critical regulatory role in the pathological progression of sepsis. Therefore, we focused on the key effector molecule CXCL2, whose m6A modification level on mRNA was significantly reduced after METTL3 knockdown (Figure 6C). Western blotting analysis further confirmed that the knockdown of METTL3 downregulated the protein expression of IL-17 and CXCL2, while overexpression showed the opposite effect (Figure 6D).
Figure 6 Continued. Figure 6 METTL3/IGF2BP1 axis mediates CXCL2 mRNA stability through m6A modification in the pathological progression of SICM. Analysis of m6A-MeRIP-seq results after METTL3 knockdown. (A) Cluster Venn map, (B) KEGG pathway analysis and (C) heat map of differentially expressed genes. (D) Western Blot was used to detect the protein expression levels of IL-17 and CXCL2 after Si-METTL3 and OV-METTL3 treatment. (E) The prediction software SRAMP predicted the potential m6A modification sites on the CXCL2 genome. (F) RIP-qRT-PCR was used to detect the expression level of CXCL2 mRNA after METTL3 knockdown. (G) qRT-PCR detected the expression of candidate m6A readers (YTHDF1, YTHDF2, YTHDF3, IGF2BP1, IGF2BP2, IGF2BP3) in HL-1 cardiomyocytes. (H) The silencing efficiency of Si-IGF2BP1 and (I) the level of CXCL2 mRNA by qRT-PCR. (J) Western blot analysis detected the level of CXCL2 protein. (K) The stability of CXCL2 mRNA was determined by qRT-PCR. * p < 0.05, ** p < 0.01, *** p < 0.001 vs Si-NC; ## p < 0.01 vs OV-NC; n = 3.
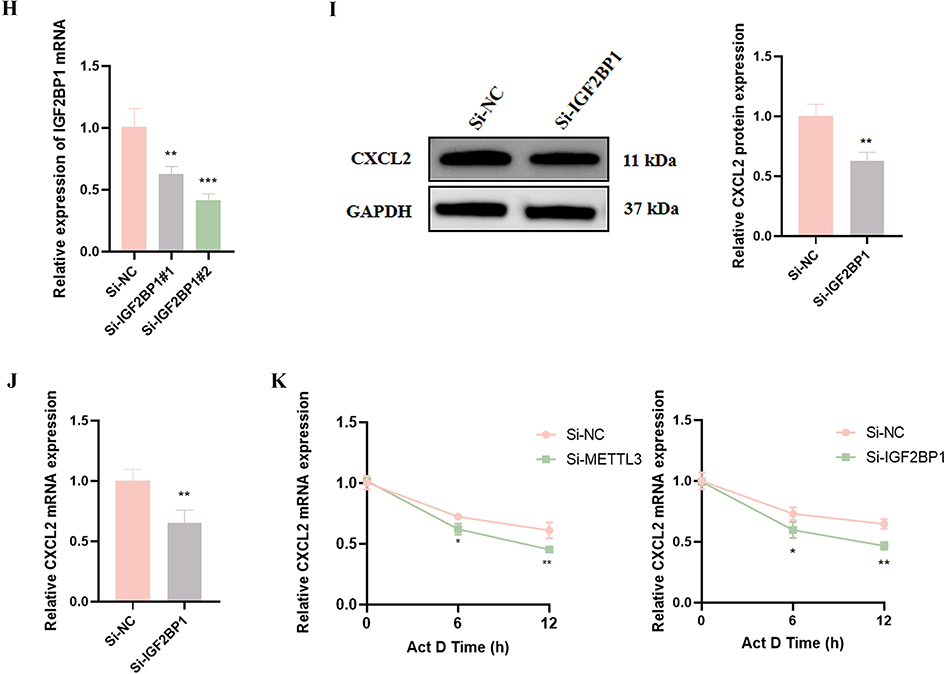
Using the sequence-based m6A modification site predictor (SRAMP, http://www.cuilab.cn/sramp), we identified multiple potential m6A modification sites on the CXCL2 genome (Figure 6E). Subsequently, RIP-qRT-PCR further confirmed the existence of m6A modification in CXCL2 mRNA, and the modification level was significantly reduced after METTL3 knockdown (Figure 6F). To further explore the downstream regulatory mechanism, we detected the expression profile of m6A “readers” in LPS-treated HL-1 cardiomyocytes and found that the expression of IGF2BP1 was significantly upregulated after LPS stimulation (Figure 6G). The knockdown of IGF2BP1 reduced the mRNA and protein expression of CXCL2 (Figure 6H–J). Further analysis of RNA stability revealed that knockdown METTL3 or IGF2BP1 accelerates the degradation of CXCL2 mRNA (Figure 6K). In summary, these findings revealed a molecular mechanism by which METTL3-mediated m6A modification enhances CXCL2 mRNA stability and promotes its expression through the IGF2BP1 dependent pathway. This regulatory axis plays an important role in LPS induced myocardial cell injury.
METTL3 Exacerbates Sepsis-Induced Myocardial Injury Through CXCL2-Mediated Inflammatory Responses and Ferroptosis
Next, we elucidated the key role of CXCL2 as a downstream effector molecule of METTL3 in myocardial cell inflammatory injury through a series of functional recovery experiments. CXCL2, as an important pro-inflammatory chemokine, can exacerbate inflammatory responses by recruiting immune cells such as neutrophils.42 We simultaneously transfected the OV-METTL3 and CXCL2 siRNA into HL-1 cardiomyocytes and detected the expression levels of myocardial injury factors (CK-MB and LDH) and inflammatory factors (TNF-α and IL-1β). The results showed that OV-METTL3 significantly promoted the expression of LPS induced myocardial injury markers (CK-MB, LDH) and inflammatory factors (TNF-α, IL-1 β), while CXCL2 silencing effectively reversed this effect (Figure 7A–D). It was reported that overexpression of CXCL2 can induce ferroptosis.43 So, we also measured the levels of Fe2+, lipid peroxidation product MDA and antioxidant GSH in cardiomyocytes and the results indicated that OV-METTL3 promoted the LPS-induced accumulation of Fe2+ and lipid peroxidation product MDA generation, while reducing antioxidant GSH levels. These changes were all reversed by CXCL2 silencing (Figure 7E–G). Similarly, CXCL2 silencing upregulated the expression of ferroptosis-inhibiting proteins SLC7A11 and GPX4, while METTL3 overexpression reversed this protective effect (Figure 7H). These results revealed a new mechanism of METTL3 regulating LPS-induced myocardial cell inflammatory injury through CXCL2, providing a new theoretical perspective for understanding the molecular mechanism of sepsis induced myocardial injury.
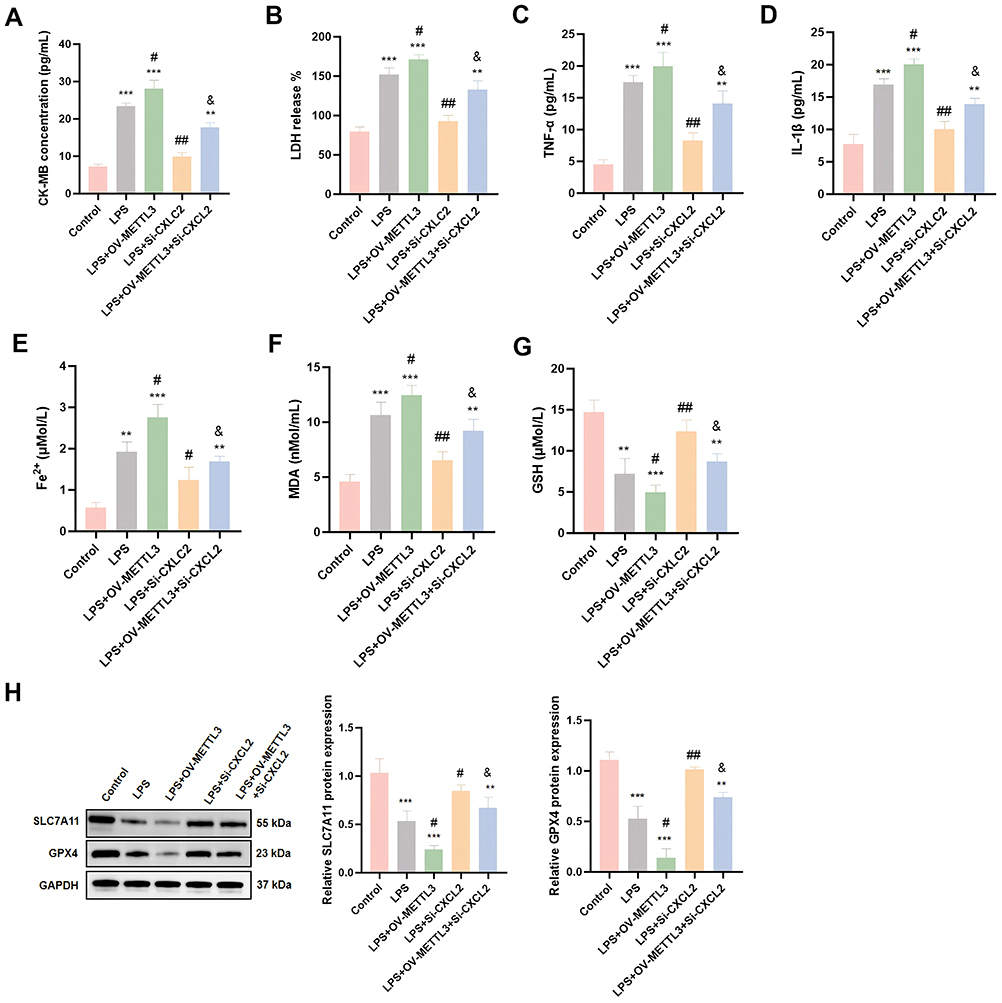 |
Figure 7 METTL3/IGF2BP1/CXCL2 axis regulates LPS-induced inflammatory injury of cardiomyocytes. (A–G) Elisa was used to detect the expression levels of CK-MB, LDH, TNF-α, IL-1β, Fe2+, MDA and GSH in HL-1 cardiomyocytes after OV-METTL3 and Si-CXCL2 treatment (n = 6). (H) Western Blot was used to detect the protein expression levels of SLC7A11 and GPX4 in HL-1 cardiomyocytes after OV-METTL3 and Si-CXCL2 treatment (n = 3). **p < 0.01, ***p < 0.001 vs Control; #p < 0.05, ##p < 0.01 vs LPS; &p < 0.05 vs LPS+OV-METTL3. |
Discussion
Statins, as inhibitors of 3-hydroxymethylglutaryl-coenzyme A reductase, not only have lipid-lowering effects, but also exhibit various cholesterol independent effects, including antioxidant, immune regulatory, and anti-inflammatory activities. Recently, they have become a research hotspot for host-directed therapy of infectious diseases.44 For example, rosuvastatin could reduce the release of pro-inflammatory cytokines in RAW 264.7 macrophages induced by LPS by inhibiting NF-κB activation.45 In sepsis models, ATO has shown positive therapeutic potential, manifested by improving mean arterial pressure and reducing microvascular inflammation. Its anti-inflammatory effect is closely associated with the downregulation of pro-inflammatory nitric oxide synthase II.46 In addition, ATO can also induce heme oxygenase-1 by activating extracellular regulated protein kinase and p38 mitogen-activated protein kinase pathway, thereby attenuating LPS induced TNF-α expression.47 These findings collectively confirm the extensive non cholesterol dependent anti-inflammatory effects of statins and their value in the treatment of sepsis. However, these prior studies predominantly attribute the protective effects of statins to the canonical NF-κB or MAPK mediated anti-inflammatory cascades, leaving the upstream epigenetic or transcriptomic determinants largely unexplored. In contrast, this study further confirmed that ATO can effectively alleviate sepsis-induced myocardial dysfunction, manifested by improving cardiac function, reducing pathological damage to myocardial tissue, and suppressing the release of inflammatory cytokines. Importantly, we found that the protective effect of ATO on SICM is achieved by inhibiting the METTL3 (m6A)/IGF2BP1/CXCL2 signaling pathway (Figure 8). This finding not only provides a deeper theoretical basis for the treatment of sepsis-related heart injury with ATO, but also provides directions for exploring new treatment strategies.
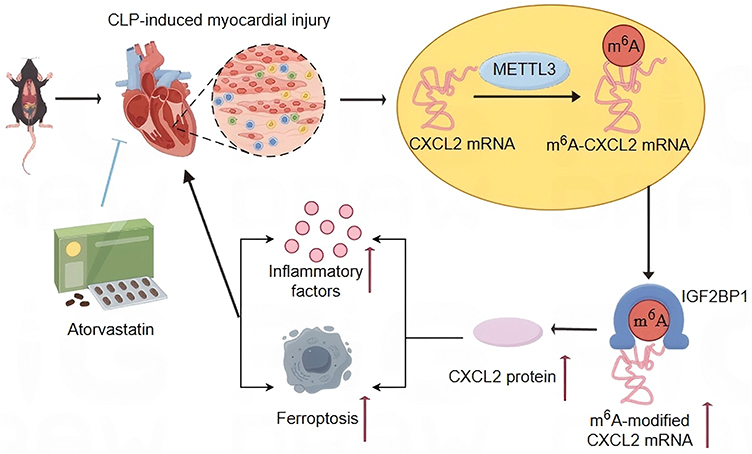 |
Figure 8 ATO inhibits the occurrence of inflammation and ferroptosis through METTL3/IGF2BP1/CXCL2 axis inhibition, thereby exerting cardiac protection. “↑” indicates elevated mRNA or inflammatory cytokine expression levels, or promotion of cardiomyocyte ferroptosis. |
This study focuses on the role of RNA m6A modification. As one of the most common modifications on RNAs, m6A modification is crucial in various physiological and pathological processes, and its levels were elevated in sepsis patients and animal models.48,49 This study observed a significant increase in m6A modification levels in the heart tissue of septic mice, while ATO treatment could reduce the m6A modification levels. In vitro, the results also showed that ATO pretreatment can reduce LPS induced high m6A modification level in HL-I cardiomyocytes. To elucidate the target of ATO regulating m6A modification levels, we conducted molecular docking (AutoDock software), CETSA, DARTS experiments and Western blotting analysis, and found that ATO could bind to METTL3 with high affinity and inhibited its expression. This inhibition of METTL3 ultimately alleviated myocardial cells inflammatory injury, manifested by significant downregulation of inflammatory factors (TNF-α, IL-6, and IL-1β) and myocardial injury markers (CK-MB and LDH). In summary, this study reveals that METTL3-mediated m6A modification is an important mechanism for ATO to alleviate SICM.
To further reveal the molecular mechanism of METTL3 in regulating myocardial inflammatory injury, we knocked down the expression of METTL3 in cardiomyocytes and performed m6A-MeRIP-seq analysis. The results showed that the genes with significantly downregulated m6A modification levels were mainly enriched in signaling pathways closely related to inflammation and immunity, such as TNF, MAPK and IL-17. Among them, the IL-17 signaling pathway plays a critical role in the occurrence and development of sepsis related organ damage. The activation of this pathway triggers a large number of neutrophils to infiltrate tissues and organs, releasing reactive oxygen species and proteases at the site of inflammation, leading to inflammatory damage and death of tissue cells, thereby triggering or exacerbating organ dysfunction. To further investigate the downstream impact of METTL3, we focused on the IL-17 signaling pathway, known for its role in sepsis-related organ damage. It is worth noting that CXCL2 is a key effector molecule downstream of the IL-17 signaling pathway. Study has found that IL-17 can promote the expression of chemokines such as CXCL2, thereby recruiting neutrophils to heart tissue and exacerbating myocardial inflammation and injury.50 In sepsis, it has been confirmed that CXCL2 expression is increased and its level is positively correlated with myocardial injury markers.34 The main finding of this study is that the METTL3/IGF2BP1 enhances the mRNA stability and expression level of CXCL2 by mediating m6A modification, thereby exacerbating SICM.
Studies have revealed that CXCL2 is not only a pro-inflammatory chemokine, but may also serve as a bridge between inflammation and ferroptosis.51 CXCL2 induced the migration of inflammatory cells such as neutrophils and monocytes to the inflammatory or infected areas by binding to the chemokine receptor CXCR2, stimulating them to release large amounts of inflammatory cytokines and amplifying the inflammatory cascade reaction. The inflammatory responses are often accompanied by significant oxidative stress, which is an important factor inducing ferroptosis.52 It has been reported that CXCL2 overexpression can make cells more susceptible to ferroptosis by exacerbating oxidative stress or interfering with intracellular iron metabolism.43 In our study, we found that knockdown of CXCL2 can effectively reverse the effects of METTL3 overexpression on ferroptosis-related indicators, manifested as a decrease in intracellular Fe2+ and lipid peroxidation product MDA levels, an increase in antioxidant GSH levels, and upregulation of key ferroptosis-inhibiting proteins SLC7A11 and GPX4 expression. In summary, METTL3 enhances the expression and stability of CXCL2, not only triggering inflammatory responses in cardiomyocytes, but also promoting ferroptosis process, collectively exacerbating SICM. This finding lays an important foundation for a deeper understanding of the pathogenesis of SICM and exploring new therapeutic targets.
There are several limitations to this study that need to be fully considered in future work. Firstly, the study design adopted a prophylactic dosing strategy rather than a therapeutic intervention, which may limit the direct evaluation of the natural progression of myocardial injury and its relationship with treatment dynamics. Secondly, due to the lack of positive control drugs for the treatment of SICM, it is difficult to accurately compare the efficacy differences of different treatment strategies. Thirdly, the potential toxic side effects of ATO in the context of SICM intervention, especially in long-term or high-dose applications, have not been systematically evaluated. Fourthly, the analysis of the mechanism of ferroptosis is not comprehensive enough, and the complex regulatory network of lipid peroxidation process (initiation, propagation and termination) under the intervention of ATO needs to be further analyzed. Finally, in this study, the 24-hour observation period focused on acute-phase responses and failed to assess long-term survival and organ function recovery. In the future, the monitoring period needs to be extended to investigate potential delayed effects.
Conclusion
In summary, we demonstrated that ATO significantly alleviates SICM by targeting and inhibiting METTL3-mediated m6A modification. The core mechanism is that ATO binds to METTL3 with high affinity and inhibits its expression, significantly reducing m6A modification level in heart tissue, thereby blocking the m6A modification of CXCL2 mRNA mediated by METTL3, and inhibiting the stability of CXCL2 mRNA in an IGF2BP1 dependent manner, ultimately suppressing the expression of CXCL2. The inhibition of this pathway effectively alleviates inflammation and ferroptosis of cardiomyocytes, thereby improving cardiac function and tissue structure damage. This mechanism elucidates the key pathogenic pathway of SICM from the perspective of epigenetics, providing innovative targets for clinical intervention.
Ethics Approval and Consent to Participate
The study was approved by the ethics committees at Laboratory Animal Center, The Fourth Military Medical University, Xi’an, China, which were carried out in compliance with the ARRIVE guidelines ARRIVE.53 The animal experiments were conducted in accordance with the Guiding Opinions on the Humane Treatment of Laboratory Animals issued by the Ministry of Science and Technology of the People’s Republic of China, as well as the national standard GB/T 35892-2018 entitled Laboratory Animal—Guidelines for Ethical Review of Animal Welfare.
Author Contributions
All authors made a significant contribution to the work reported, whether that is in the conception, study design, execution, acquisition of data, analysis and interpretation, or in all these areas; took part in drafting, revising or critically reviewing the article; gave final approval of the version to be published; have agreed on the journal to which the article has been submitted; and agree to be accountable for all aspects of the work.
Funding
This work was supported by the National Natural Science Foundation of China (No. 72074218, 81903837, 82400418).
Disclosure
The authors report no conflicts of interest in this work.
References
1. Singer M, Deutschman CS, Seymour CW, et al. The third international consensus definitions for sepsis and septic shock (Sepsis-3). JAMA. 2016;315(8):801–810. doi:10.1001/jama.2016.0287
2. Reinhart K, Daniels R, Kissoon N, Machado FR, Schachter RD, Finfer S. Recognizing sepsis as a global health priority - A WHO resolution. N Engl J Med. 2017;377(5):414–417. doi:10.1056/NEJMp1707170
3. Pei XB, Liu B. Research progress on the mechanism and management of septic cardiomyopathy: a comprehensive review. Emerg Med Int. 2023;2023:8107336. doi:10.1155/2023/8107336
4. Xie J, Wang H, Kang Y, et al. The epidemiology of sepsis in Chinese ICUs: a national cross-sectional survey. Crit Care Med. 2020;48(3):e209–e218. doi:10.1097/ccm.0000000000004155
5. Wang Z, Wang Y, Dong C, et al. Po-Ge-Jiu-Xin decoction alleviate sepsis-induced cardiomyopathy via regulating phosphatase and tensin homolog-induced putative kinase 1 /parkin-mediated mitophagy. J Ethnopharmacol. 2025;337(Pt 3):118952. doi:10.1016/j.jep.2024.118952
6. Hollenberg SM, Singer M. Pathophysiology of sepsis-induced cardiomyopathy. Nat Rev Cardiol. 2021;18(6):424–434. doi:10.1038/s41569-020-00492-2
7. Hiraiwa H, Kasugai D, Okumura T, Murohara T. Clinical implications of septic cardiomyopathy: a narrative review. Medicine. 2024;103(17):e37940. doi:10.1097/md.0000000000037940
8. Liu DH, Ning YL, Lei YY, et al. Levosimendan versus dobutamine for sepsis-induced cardiac dysfunction: a systematic review and meta-analysis. Sci Rep. 2021;11(1):20333. doi:10.1038/s41598-021-99716-9
9. Liu H, Xu C, Hu Q, Wang Y. Sepsis-induced cardiomyopathy: understanding pathophysiology and clinical implications. Arch Toxicol. 2024. doi:10.1007/s00204-024-03916-x
10. Rothberg MB, Bigelow C, Pekow PS, Lindenauer PK. Association between statins given in hospital and mortality in pneumonia patients. J Gen Intern Med. 2012;27(3):280–286. doi:10.1007/s11606-011-1826-2
11. Deng LJ, Deng WQ, Fan SR, et al. m6A modification: recent advances, anticancer targeted drug discovery and beyond. Mol Cancer. 2022;21(1):52. doi:10.1186/s12943-022-01510-2
12. Li M, Noordam R, Trompet S, et al. The impact of statin use on sepsis mortality. J Clin Lipidol. 2024;18(6):e915–e925. doi:10.1016/j.jacl.2024.07.006
13. Ren G, Zhou Q, Lu M, Wang H. Rosuvastatin corrects oxidative stress and inflammation induced by LPS to attenuate cardiac injury by inhibiting the NLRP3/TLR4 pathway. Can J Physiol Pharmacol. 2021;99(9):964–973. doi:10.1139/cjpp-2020-0321
14. Nežić L, Amidžić L, Škrbić R, et al. Amelioration of endotoxin-induced acute lung injury and alveolar epithelial cells apoptosis by simvastatin is associated with up-regulation of survivin/NF-kB/p65 pathway. Int J Mol Sci. 2022;23(5):2596. doi:10.3390/ijms23052596
15. Catalão CHR, Santos-Junior NN, da Costa LHA, et al. Simvastatin prevents long-term cognitive deficits in sepsis survivor rats by reducing neuroinflammation and neurodegeneration. Neurotox Res. 2020;38(4):871–886. doi:10.1007/s12640-020-00222-z
16. Edupuganti RR, Geiger S, Lindeboom RGH, et al. N(6)-methyladenosine (m(6)A) recruits and repels proteins to regulate mRNA homeostasis. Nat Struct Mol Biol. 2017;24(10):870–878. doi:10.1038/nsmb.3462
17. Zhang Y, Geng X, Li Q, et al. m6A modification in RNA: biogenesis, functions and roles in gliomas. J Exp Clin Cancer Res. 2020;39(1):192. doi:10.1186/s13046-020-01706-8
18. Koch J, Lyko F. Refining the role of N(6)-methyladenosine in cancer. Curr Opin Genet Dev. 2024;88:102242. doi:10.1016/j.gde.2024.102242
19. Yen YP, Chen JA. The m(6)A epitranscriptome on neural development and degeneration. J Biomed Sci. 2021;28(1):40. doi:10.1186/s12929-021-00734-6
20. Mu S, Zhao K, Zhong S, Wang Y. The role of m6A methylation in tumor immunity and immune-associated disorder. Biomolecules. 2024;14(8):1042. doi:10.3390/biom14081042
21. Liu ZY, You QY, Liu ZY, Lin LC, Yang JJ, Tao H. m6A control programmed cell death in cardiac fibrosis. Life Sci. 2024;353:122922. doi:10.1016/j.lfs.2024.122922
22. Palmieri V, Innocenti F, Guzzo A, Guerrini E, Vignaroli D, Pini R. Left ventricular systolic longitudinal function as predictor of outcome in patients with sepsis. Circ Cardiovasc Imaging. 2015;8(11):e003865. discussion e003865. doi:10.1161/circimaging.115.003865
23. Wang H, Hu X, Huang M, et al. Mettl3-mediated mRNA m(6)A methylation promotes dendritic cell activation. Nat Commun. 2019;10(1):1898. doi:10.1038/s41467-019-09903-6
24. Shen H, Xie K, Li M, Yang Q, Wang X. N(6)-methyladenosine (m(6)A) methyltransferase METTL3 regulates sepsis-induced myocardial injury through IGF2BP1/HDAC4 dependent manner. Cell Death Discov. 2022;8(1):322. doi:10.1038/s41420-022-01099-x
25. Huang T, Ao X, Liu J, et al. m6A methyltransferase METTL3 promotes non-small-cell lung carcinoma progression by inhibiting the RIG-I-MAVS innate immune pathway. Transl Oncol. 2025;51:102230. doi:10.1016/j.tranon.2024.102230
26. Wang Z, Sun S, Huang L, et al. METTL3/YTHDF1-mediated m(6)A modification stabilizes USP12 to deubiquitinate FOXO3 and promote apoptosis in sepsis-induced myocardial dysfunction. Mol Immunol. 2024;177:17–31. doi:10.1016/j.molimm.2024.12.001
27. Wang J, Yan S, Lu H, Wang S, Xu D. METTL3 attenuates LPS-induced inflammatory response in macrophages via NF-κB signaling pathway. Mediators Inflamm. 2019;2019:3120391. doi:10.1155/2019/3120391
28. Liu YC, Yu MM, Shou ST, Chai YF. Sepsis-induced cardiomyopathy: mechanisms and treatments. Front Immunol. 2017;8:1021. doi:10.3389/fimmu.2017.01021
29. Li W, Hua S, Yang J, et al. Investigating immune dysregulation and hub genes in septic cardiomyopathy development. Sci Rep. 2024;14(1):21608. doi:10.1038/s41598-024-72724-1
30. Zenobia C, Hajishengallis G. Basic biology and role of interleukin-17 in immunity and inflammation. Periodontol 2000. 2015;69(1):142–159. doi:10.1111/prd.12083
31. Retnoningrum D, Mulyono B, Intansari US, Jaludamascena A. Interleukin-17 as predictor mortality of septic patients: a systematic review and meta-analysis. Wiad Lek. 2024;77(6):1134–1140. doi:10.36740/WLek202406104
32. Ge Y, Huang M, Yao YM. Biology of interleukin-17 and its pathophysiological significance in sepsis. Front Immunol. 2020;11:1558. doi:10.3389/fimmu.2020.01558
33. Burke SJ, Lu D, Sparer TE, et al. NF-κB and STAT1 control CXCL1 and CXCL2 gene transcription. Am J Physiol Endocrinol Metab. 2014;306(2):E131–149. doi:10.1152/ajpendo.00347.2013
34. Guo LY, Yang F, Peng LJ, Li YB, Wang AP. CXCL2, a new critical factor and therapeutic target for cardiovascular diseases. Clin Exp Hypertens. 2020;42(5):428–437. doi:10.1080/10641963.2019.1693585
35. Cai B, Ma W, Ding F, et al. The long noncoding RNA CAREL controls cardiac regeneration. J Am Coll Cardiol. 2018;72(5):534–550. doi:10.1016/j.jacc.2018.04.085
36. Wang Y, Zhang L, Zhao X, Yang W, Zhang R. An experimental study of the protective effect of simvastatin on sepsis-induced myocardial depression in rats. Biomed Pharmacother. 2017;94:705–711. doi:10.1016/j.biopha.2017.07.105
37. Zhu L, Zhang H, Zhang X, Xia L. RNA m6A methylation regulators in sepsis. Mol Cell Biochem. 2024;479(9):2165–2180. doi:10.1007/s11010-023-04841-w
38. Liang L, Liu S, Wu Q, Chen R, Jiang S, Yang Z. m6A-mediated upregulation of miRNA-193a aggravates cardiomyocyte apoptosis and inflammatory response in sepsis-induced cardiomyopathy via the METTL3/ miRNA-193a/BCL2L2 pathway. Exp Cell Res. 2023;430(1):113712. doi:10.1016/j.yexcr.2023.113712
39. Zhao D, Zhuang J, Wang L, et al. Unveiling key biomarkers and mechanisms in septic cardiomyopathy: a comprehensive transcriptome analysis. J Inflamm Res. 2024;17:11451–11467. doi:10.2147/jir.S486763
40. Li H, Zhang Q, Feng Q, You Q, Guo X. The development of small molecules targeting methyltransferase-like 3. Drug Discov Today. 2023;28(4):103513. doi:10.1016/j.drudis.2023.103513
41. Chen M, Pan L, Chen D, et al. PAK1 promotes inflammation induced by sepsis through the Snail/CXCL2 signaling pathway. ACS Infect Dis. 2024;10(4):1370–1378. doi:10.1021/acsinfecdis.4c00052
42. Zhou C, Gao Y, Ding P, Wu T, Ji G. The role of CXCL family members in different diseases. Cell Death Discov. 2023;9(1):212. doi:10.1038/s41420-023-01524-9
43. Yi Q, Liang Q, Liu Y, Gong Z, Yan Y. Application of genomic selection and experimental techniques to predict cell death and immunotherapeutic efficacy of ferroptosis-related CXCL2 in hepatocellular carcinoma. Front Oncol. 2022;12:998736. doi:10.3389/fonc.2022.998736
44. Parihar SP, Guler R, Brombacher F. Statins: a viable candidate for host-directed therapy against infectious diseases. Nat Rev Immunol. 2019;19(2):104–117. doi:10.1038/s41577-018-0094-3
45. Tang Z, Ning Z, Li Z. The beneficial effects of Rosuvastatin in inhibiting inflammation in sepsis. Aging. 2024;16(12):10424–10434. doi:10.18632/aging.205937
46. McGown CC, Brookes ZL, Hellewell PG, Ross JJ, Brown NJ. Atorvastatin reduces endotoxin-induced microvascular inflammation via NOSII. Naunyn Schmiedebergs Arch Pharmacol. 2015;388(5):557–564. doi:10.1007/s00210-015-1100-y
47. Wang XQ, Luo NS, Salah ZQ, Lin YQ, Gu MN, Chen YX. Atorvastatin attenuates TNF-alpha production via heme oxygenase-1 pathway in LPS-stimulated RAW264.7 macrophages. Biomed Environ Sci. 2014;27(10):786–793. doi:10.3967/bes2014.114
48. Li F, Zhang Y, Peng Z, Wang Y, Zeng Z, Tang Z. Diagnostic, clustering, and immune cell infiltration analysis of m6A regulators in patients with sepsis. Sci Rep. 2023;13(1):2532. doi:10.1038/s41598-022-27039-4
49. Liang X, Hu X, Li J, et al. m6A methylation in myocardial tissue of septic mice analyzed using MeRIP/m6A-sequencing and RNA-sequencing. Funct Integr Genomics. 2024;24(5):173. doi:10.1007/s10142-024-01452-6
50. Jiang K, Xu Y, Wang Y, Yin N, Huang F, Chen M. Unveiling the role of IL-17: therapeutic insights and cardiovascular implications. Cytokine Growth Factor Rev. 2024;77:91–103. doi:10.1016/j.cytogfr.2024.05.001
51. Pervaiz S, Bellot GL, Lemoine A, Brenner C. Redox signaling in the pathogenesis of human disease and the regulatory role of autophagy. Int Rev Cell Mol Biol. 2020;352:189–214. doi:10.1016/bs.ircmb.2020.03.002
52. Yan HF, Zou T, Tuo QZ, et al. Ferroptosis: mechanisms and links with diseases. Signal Transduct Target Ther. 2021;6(1):49. doi:10.1038/s41392-020-00428-9
53. Kilkenny C, Browne W, Cuthill IC, Emerson M, Altman DG. Animal research: reporting in vivo experiments: the ARRIVE guidelines. Br J Pharmacol. 2010;160(7):1577–1579. doi:10.1111/j.1476-5381.2010.00872.x
 © 2025 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2025 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.