Back to Journals » Journal of Inflammation Research » Volume 14
Astragaloside IV Protects from PM2.5-Induced Lung Injury by Regulating Autophagy via Inhibition of PI3K/Akt/mTOR Signaling in vivo and in vitro
Authors Pei C, Wang F, Huang D ![]() , Shi S
, Shi S ![]() , Wang X, Wang Y
, Wang X, Wang Y ![]() , Li S, Wu Y
, Li S, Wu Y ![]() , Wang Z
, Wang Z ![]()
Received 25 March 2021
Accepted for publication 31 August 2021
Published 16 September 2021 Volume 2021:14 Pages 4707—4721
DOI https://doi.org/10.2147/JIR.S312167
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Professor Ning Quan
Caixia Pei,* Fei Wang,* Demei Huang, Shihua Shi, Xiaomin Wang, Yilan Wang, Shuiqin Li, Yongcan Wu, Zhenxing Wang
Hospital of Chengdu University of Traditional Chinese Medicine, Chengdu, Sichuan Province, People’s Republic of China
*These authors contributed equally to this work
Correspondence: Zhenxing Wang; Yongcan Wu
Hospital of Chengdu University of Traditional Chinese Medicine, Chengdu, Sichuan Province, People’s Republic of China
Email [email protected]; [email protected]
Introduction: Prolonged exposure to air polluted with airborne fine particulate matter (PM2.5) can increase respiratory disease risk. Astragaloside IV (AS-IV) is one of the main bioactive substances in the traditional Chinese medicinal herb, Astragalus membranaceus Bunge. AS-IV has numerous pharmacological properties; whereas there are few reports on the prevention of PM2.5-induced lung injury by AS-IV through modulation of the autophagic pathway. This study aimed to investigate the protective effects and the underlying mechanisms of AS-IV in PM2.5-induced lung injury rats and rat alveolar macrophages (NR8383 cells).
Methods: The pneumotoxicity model was established by intratracheal injection of PM2.5 in rats, and PM2.5 challenge in NR8383 cells. The severity of lung injury was evaluated by wet weight to dry weight ratio and McGuigan pathology scoring. Inflammatory factors and oxidative stress were detected through ELISA. The expressions of p-PI3K, p-Akt, and p-mTOR proteins were analyzed by immunohistochemistry. Immunofluorescence and transmission electron microscopy were used to detect autophagosomes. The expressions of autophagy marker protein (LC3B and p62), PI3K/Akt/mTOR signaling and NF-κB translocation were detected by Western blot in lung tissue and NR8383 cells.
Results: After PM2.5 stimulation, rats showed severe inflammation and oxidative stress, along with inhibition of autophagy in lung tissue. AS-IV not only decreased pulmonary inflammation and oxidative stress by inhibiting nuclear factor kappa B translocation, but also regulated autophagy by inhibiting PI3K/Akt/mTOR signaling. After treatment with 3-methyladenine (a classic PI3K inhibitor, blocking the formation of autophagosomes), the protective effect of AS-IV on PM2.5-induced lung injury was further strengthened. In parallel, using Western blot, immunohistochemistry, and transmission electron microscopy, we demonstrated that AS-IV restore autophagic flux mainly through regulating the degradation of autophagosomes rather than suppressing the formation in vivo and in vitro.
Conclusion: Our data indicated that AS-IV protects from PM2.5-induced lung injury in vivo and in vitro by inhibiting the PI3K/Akt/mTOR pathway to regulate autophagy and inflammation.
Keywords: PM2.5, lung injury, astragaloside IV, autophagy, inflammation
Introduction
Environmental pollution by fine particulate matter (PM2.5), which has an aerodynamic diameter of ≤2.5 μm, has become a significant public health threat in China and other developing countries.1,2 Several epidemiological studies have shown that long-term exposure to high levels of PM2.5 increases the risk of many respiratory diseases, such as influenza,3,4 asthma,5 bronchitis,6 chronic obstructive pulmonary disease (COPD),7,8 pulmonary fibrosis,9 and lung cancer.10,11 As a result, PM2.5-induced lung injury poses an enormous social and economic burden.12,13 Currently, respiratory diseases caused by PM2.5 have attracted widespread attention from scholars.14,15 However, most studies present epidemiological and environmental toxicological data. At present, there are no effective therapies for controlling the progression of PM2.5-induced lung injury. Therefore, there is a need to seek preventive and therapeutic approaches to prevent PM2.5-induced lung injury.
Several PM2.5 toxicology studies have shown that PM2.5-induced lung injury is closely associated with autophagy and inflammation.16,17 Autophagy, a type II programmed cell death modality, is usually considered a protective mechanism for cells, while excessive autophagy induces autophagic cell death.18 Normal autophagy helps balanced inflammatory, which in turn has a protective effect against disease. Altered autophagy can lead to maladaptive inflammatory responses, increased inflammatory diseases.19,20 Several recent studies have shown that autophagy is an anti-inflammatory mechanism that prevents endothelial damage triggered by various endogenous or infectious agents and prevents unnecessary or excessive inflammation.21,22 In contrast, some studies have suggested that over-activated autophagy exacerbates inflammatory damage, which in turn promotes lung disease.23,24 Alveolar macrophages are the most common immune cells residing in the alveoli, which plays pivotal regulatory roles in immune and inflammatory systems. Previous studies have shown that abnormal autophagy in PM2.5-induced alveolar macrophages are closely related to the pathophysiological progression of acute and chronic lung inflammation. Therefore, regulating the balance between autophagy and inflammation may be a potential mechanism to combat PM2.5-induced lung injury.25,26 Despite some interventions showing great potential, no therapies are available that specifically modulate autophagy in humans.
Astragaloside IV (AS-IV) is one of the main bioactive substances in the traditional Chinese medicinal herb, Astragalus membranaceusBunge. Previous studies have shown that AS-IV has pharmacological effects as an anti-oxidant, anti-inflammatory, immunomodulatory, antiviral, and antitumor agent.27 In recent years, AS-IV has received attention in the prevention of respiratory diseases, such as asthma,28 pulmonary fibrosis29 and lung cancer.30 However, there are few reports on the prevention of PM2.5-induced lung injury by AS-IV through modulation of the autophagic pathway.31 Therefore, in this study, we investigated the protective effect of AS-IV pre-treatment on PM2.5-induced lung injury and explored the potential mechanisms by which AS-IV protects against PM2.5-induced lung injury via the autophagy-inflammation pathway in vivo and in vitro.
Materials and Methods
Materials
AS-IV (HPLC ≥ 98.81%) and fine particulate matter standards were respectively purchased from Chengdu Munster Company (China) and National Institute of Standards and Technology (USA). 3-Methyladenine (3-MA) was purchased from Selleck, and polyvinylidene fluoride membranes were purchased from Bio-Rad (USA). Antibodies against the following targets were obtained from Abcam (Cambridge, UK): LC3B (ab48394), p62 (ab56416), PI3K (ab182651), Akt (ab185633), p-Akt (ab38449), mTOR (ab2732), p-mTOR (ab137133), Lamin B (ab16048), and GAPDH (ab181602). An antibody against p-PI3K (AF3242) was obtained from Affinity Biosciences (USA). An antibody against p65 (8242S) was obtained from Cell Signaling Technology (Danvers, MA, USA). Horseradish peroxidase (HRP)-labeled goat anti-rabbit IgG secondary antibody (GB23303) and 4°C tissue radioimmunoprecipitation assay lysates were obtained from Servicebio (Wuhan, China).
Ethics Statement
All animal procedures were approved by the Animal Studies Ethics Committee of Chengdu University of Traditional Chinese Medicine. Animal handling followed the dictates of the National Animal Welfare Law of China. Animal welfare conformed to the National Institutes of Health animal welfare guidelines.
Animals and Experimental Design
Male Sprague-Dawley rats (120–150g) were obtained from Chengdu Dashuo Experimental Animal Co., Ltd. All rats were raised in a standard environment (temperature, 25°C ± 1°C; humidity, 55% ± 5%; photoperiod, 12:12 hours). After adapting for 1 week, rats were randomly assigned into seven groups (n = 7 per group): (1) control; (2) AS-IV (100 mg/kg); (3) PM2.5 (7.5 mg/kg); (4) AS-IV 50 (AS-IV 50 mg/kg + PM2.5 7.5 mg/kg); (5) AS-IV 100 (AS-IV 100 mg/kg + PM2.5 7.5 mg/kg); (6) 3-MA (3-MA 15 mg/kg + PM2.5 7.5 mg/kg); (7) AS-IV 100 + 3-MA (AS-IV 100 mg/kg + 3-MA 15 mg/kg + PM2.5 7.5 mg/kg). Before PM2.5 administration, all groups were treated by intraperitoneal injection with AS-IV (dissolved in normal saline containing 0.1% dimethyl sulfoxide [DMSO]) once a day for 3 days consecutively, except for the control and PM2.5 groups. The AS-IV 100 + 3-MA group was first intraperitoneally injected with AS-IV (100 mg/kg), and 3-MA (15 mg/kg) was intraperitoneally injected 1 hour later once a day for 3 days consecutively.32 The control and AS-IV groups were administered an equivalent volume of sterile saline containing 1% DMSO by the same method. The rat model of lung injury induced by PM2.5 was established in accordance with previous literature.23,31,33 All groups except the control and AS-IV groups received an intratracheal injection of PM2.5 dust suspension (7.5 mg/kg dissolved in a certain volume of saline according to the animal’s weight). Intratracheal instillation of PM2.5 suspension was repeated after 24 hours. The control and AS-IV groups received the same volume of saline. All rats were anesthetized and sacrificed with 0.4% pentobarbital after PM2.5 instillation for 12 hours.
Bronchoalveolar Lavage Fluid (BALF) Acquisition
The whole left lung was for BALF collection. BALF was gathered via three intratracheal injections of 2 mL sterile saline. BALF was centrifuged at 5000 r/minute for 10 minutes at 4°C. The cell-free supernatant was maintained and stored at −80°C for cytokine detection.
Lung Wet Weight to Dry Weight (W/D) Ratio
After rats were sacrificed, the inferior lobe of the right lung was excised and weighed for wet weight. Then, lung tissue was placed in an incubator at 60°C for 48 hours, and dry weight was obtained. The lung W/D ratio was calculated.
Histological Analysis
The superior lobe of the right lung was extracted and immediately fixed in 4% paraformaldehyde for 24 hours, embedded in paraffin, sectioned to 4 μm, and dewaxed. Slices were stained with hematoxylin and eosin (H&E) and observed under an inverted light microscope. The extent of lung tissue damage was assessed using the McGuigan pathology scoring method and was independently scored by two specialized pathologists.34
Cytokines and Oxidative Stress
Tumor necrosis factor-α (TNF-α), interleukin (IL)-6, and C-reactive protein (CRP) in BALF were measured using enzyme-linked immunosorbent assay (ELISA) kits (MultiSciences Biotech, Hangzhou, China). The contents of malondialdehyde (MDA), catalase (CAT), and superoxide dismutase (SOD) in lung tissue homogenates were determined using available assay kits (Jiancheng Bioengineering Institute, Nanjing, China).
Immunofluorescence
Immunofluorescence sections were prepared in the same way as H&E-stained sections. Then, sections were incubated with anti-LC3B monoclonal antibodies prepared according to the manufacturer’s instructions. Secondary antibodies were added to cover the tissue and incubated at room temperature for 50 minutes. Slices were observed using a confocal microscope (C2; Nikon, Tokyo, Japan), and images were acquired. The relative fluorescence intensity of LC3B was analyzed by Image J software.35
Immunohistochemistry
p-PI3K, p-Akt, and p-mTOR were analyzed by immunohistochemistry. Paraffin sections were routinely dewaxed and antigenically repaired with 3% hydrogen peroxide to block peroxidase. Sections were incubated with primary antibodies against p-PI3K (1:100), p-Akt (1:250), and p-mTOR (1:100) at 4°C overnight. Subsequently, sections were incubated with HRP-labeled secondary antibodies at room temperature for 1 hour. Sections were stained with 3,3′ -diaminobenzidine (DAB) and counterstained with hematoxylin. Under the microscope, each section was observed, and brow particles were considered to indicate positive results.
Cells Cultures and Treatments
Rat alveolar macrophages (NR8383 cells) were purchased from National Collection of Authenticated Cell Cultures (Shanghai, China) and cultured in Ham’s F12K medium (Thermo) containing 20% heat-inactivated fetal bovine serum (FBS, PAA, Australia) and 1% penicillin-streptomycin (100 Units/mL) with a sub-confluent density at 37 °C in a 5% CO2 atmosphere. Then, the NR8383s were divided into eight groups (control, DMSO, AS-IV 50, PM2.5, AS-IV 10+PM2.5, AS-IV 50+PM2.5, 3-MA+PM2.5, and 3-MA+AS-IV+PM2.5). The control group cultured with normal culture medium, the DMSO group was treated with 0.1% DMSO and the AS-IV group was treated with AS-IV (50 µM). AS-IV 10+PM2.5 group and AS-IV 50+PM2.5 group were pre-treated with AS-IV (10 and 50 µM) for 12 h, then the cells were exposure to PM2.5 (50 ug/mL) for 24 h. In the last two groups, cells were pretreated with 3-MA (5mM) for 12 h followed by treatment with PM2.5 (50 ug/mL),23,36 and treated with different drugs with or without AS-IV (50 µM) for 12 h.
Cell Viability Assay
CCK-8 kit (Biosharp, Hefei, China) was used to analyzed the total number of live cells. Specific detailed CCK-8 testing steps were according to the guidelines provided by the supplier. After cell grouping and treatment, cells were incubated with 100 μL of CCK8 reagent for 2 h at 37°C. The absorbance was measured at 450nm using a microplate spectrophotometer.
Western Blot
The left lung was homogenized in lysis buffer (containing phenylmethylsulfonyl fluoride (PMSF), protease, and phosphatase inhibitor). The homogenate was centrifuged at 12,000 ×g for 10 minutes at 4°C, and then the supernatant was collected as total protein. Similarly, the different groups of NR8383 cells were lysed in lysis buffer. The protein concentration was quantified using a bicinchoninic acid protein assay kit (Thermo Fisher Scientific, USA). Equal protein concentrations were separated using sodium dodecyl sulfate-polyacrylamide gel electrophoresis at 8–12% and then transferred onto polyvinylidene fluoride membranes. Membranes were blocked using 5% milk or 5% bovine serum albumin for 1 hour at room temperature. Subsequently, membranes were incubated with primary antibodies at 4°C overnight and incubated with HRP-labeled goat anti-rabbit secondary antibody for 2 hours at room temperature. Finally, protein bands were visualized by enhanced chemiluminescence and detected with a Bio-Rad image analysis system. The gray value of protein bands was analyzed with Image J software. Nuclear and Cytoplasmic Protein Extraction Kit (Beyotime Biotechnology, Shanghai, China) was used to extract the cytoplasmic/nuclear proteins of p65 according to the manufacturer’s protocol.
Quantitative GFP-LC3 Analysis
Cells were transfected with either GFP-LC3 (Hanbio, Shanghai, China) constructs 24 h prior to treatment. After drug treatment, the cellular localization of GFP-LC3 was visualized using a confocal microscope (C2; Nikon, Tokyo, Japan). GFP-LC3-positive or endogenous LC3-positive punctate structures were obtained by counting at least 100 positive cells in each working condition of three independent experiments.
Transmission Electron Microscopy (TEM)
Lung tissue from the upper lobe of the right lung was removed, quickly placed on a glass slide containing chloral fixative, and shaken to completely immerse the tissue in fixative for 24 hours. After dehydration with different concentrations of ethanol, tissues were immersed in acetone and 812 embedding agent, and samples were inserted into the embedding plate pre-placed with pure 812 embedding agent and placed in a 37°C oven overnight. Similarly, Cells were fixed in 0.1 M PBS with 2.5% glutaraldehyde, and then treated in 1% osmium tetroxide buffer. Fixed cells were post-fixed in 2% OsO4, dehydrated in graded alcohol and flat embedded in Epon 812. Ultra-thin sections (50–60 nm) were imaged using TEM (HT7700, Hitachi, Tokyo, Japan), focusing on the autophagic vacuoles (autophagosomes, Avi; autolysosomes, Avd) of alveolar macrophages.37
Statistical Analysis
Data are presented as mean ± standard error. Data were analyzed by one-way analysis of variance (ANOVA) followed by Tukey's post hoc test using GraphPad Prism 8.0, and a P value of <0.05 was considered statistically significant.
Results
Effects of as-IV on PM2.5-Induced Lung Injury in Rats
As shown in Figure 1A, H & E staining of lung tissues showed that the lung tissues of the Con and AS-IV groups were intact without obvious inflammatory infiltration or damage. After PM2.5 stimulation, lung tissue showed alveolar congestion, alveolar wall thickening, and pulmonary interstitial neutrophil and lymphocyte infiltration. With AS-IV pre-treatment, there was a dose-dependent reduction in lung tissue damage. As shown in Figure 1B, AS-IV distinctly suppressed the lung injury score of PM2.5-induced lung injury in rats. As depicted in Figure 1C, compared with the control group, the W/D ratio was significantly higher after PM2.5 stimulation (P < 0.05), while the W/D ratio declined after pre-administration of AS-IV. No significant difference was observed between the AS-IV group and the control group in terms of W/D ratio (P > 0.05). To analyze the effects of AS-IV on the PM2.5-induced inflammatory response, secretion of TNF-α, IL-6, and CRP in BALF was measured by ELISA. As shown in Figure 1D, PM2.5 markedly increased the production of CRP, TNF-α, and IL-6 in BALF compared with the control and AS-IV groups, whereas pre-administration of AS-IV considerably inhibited the expression of inflammatory cytokines in BALF. As shown in Figure 1E, PM2.5 stimulation significantly upregulated MDA activity in lung tissue. In contrast, SOD and CAT were markedly reduced in lung tissues after PM2.5 stimulation. AS-IV reversed the PM2.5-induced changes in SOD, CAT, and MDA. These results indicate that AS-IV decreases pulmonary inflammation and oxidative stress in the lung tissues of rats exposed to PM2.5. Interestingly, compared with AS-IV pre-treatment groups, 3-MA (a classic PI3K inhibitor, blocking the formation of autophagosomes) has a similar protective effect in vivo. Furthermore, AS-IV combined with 3-MA could further improved PM2.5-induced lung injury in rats, suggesting that AS-IV might act by inhibiting PI3K pathway.
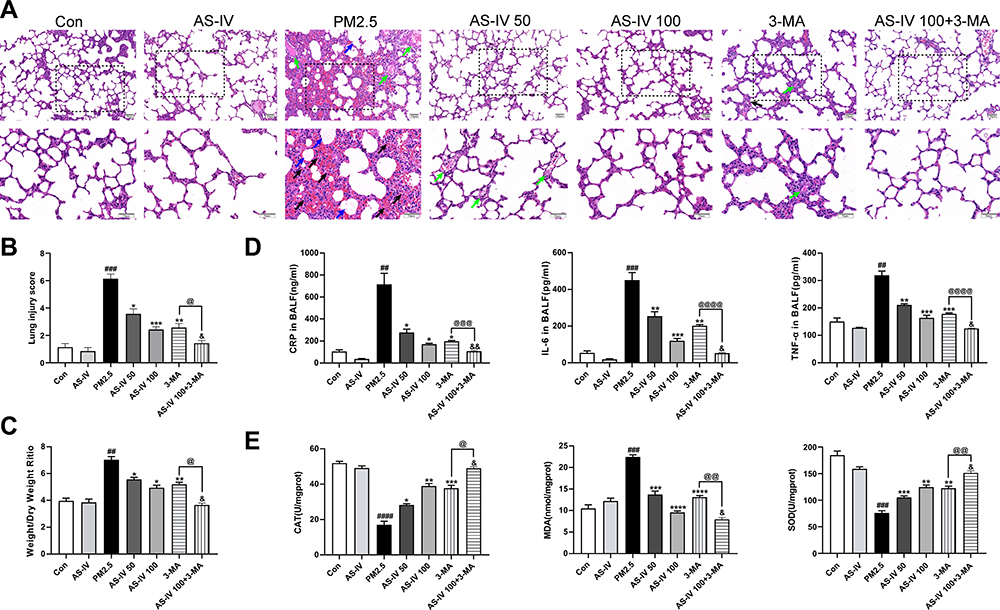 |
Figure 1 Effects of AS-IV on PM2.5-induced lung injury in rats. (A) HE staining of lung tissues from Con, AS-IV, PM2.5, AS-IV 50, AS-IV 100, 3-MA, and AS-IV 100+3-MA groups. (Black arrows represent hemorrhage, green arrows represent clear inflammatory cells, and blue arrows represent inflammatory exudate). (Magnification 200× for the upper row and 400× for the lower row, Scale bar = 50 μm). (B) Semiquantitative histopathological score of lung injury. (C) Effect of AS-IV on lung wet/dry ratio of PM2.5-induced lung injury. (D) Effect of AS-IV on CRP, IL-6, and TNF-α in the BALF in PM2.5-induced lung injury. (E) Effect of AS-IV on CAT, MDA, and SOD in the lung tissues in PM2.5-induced lung injury. Data are mean±SEM (n=6-7). ##P<0.01, ###P<0.001, and ####P<0.0001 compared with control group; *P<0.05, **P<0.01, ***P<0.001, and ****P<0.0001 compared with PM2.5 group; &P<0.05, and &&P<0.01 compared with AS-IV 100 group; @P<0.05, @@P<0.01, @@@P<0.001, and @@@@P<0.0001 compared with 3-MA group. |
AS-IV Restores Impaired Autophagy in Lung Tissues from Rats Exposed to PM2.5
To clarify the effect of AS-IV on autophagy in PM2.5-induced lung injury, we measured the levels of key autophagy molecules, including LC3B-II/I and p62, by Western blot. As shown in Figure 2A and B, the expression of LC3B-II/I in the PM2.5 group was significantly increased, meanwhile the expression of p62 was significantly enhanced (P<0.01), suggestive of compromised autophagic degradation. AS-IV pre-treatment decreased the levels of LC3B-II/I and p62 in the lung tissues of rats exposed to PM2.5. To demonstrate that activation of autophagy is the primary mechanism by which AS-IV exert its protective effects outlined above, we administered 3-MA (an inhibitor of autophagosome biogenesis) alone and co-administered 3MA with AS-IV, then evaluated outcomes. Compared with the AS-IV 100 group, AS-IV combined with 3-MA effectively suppressed LC3B-II/I (P<0.001) (Figure 2C and D). Immunofluorescence showed that AS-IV significantly decreased the relative fluorescence intensity of LC3B (Figure 2E and F); meanwhile, the frequency of LC3II-positive cells in the lung tissues treated with AS-IV and 3-MA is decreased compared with AS-IV alone. Moreover, AS-IV decreased the levels of p62. Interestingly, there was not a statistically significant difference between AS-IV 100 group and AS 100+3-MA group, indicating that AS-IV restored autophagic flux mainly through regulating the degradation of autophagosomes rather than suppressing the formation. It was challenging to detect autophagic vacuoles (autophagosomes, Avi; autolysosomes, Avd) in lung tissue in the control and AS-IV groups using TEM. Avi were increased in the lung exposed to PM2.5 without a corresponding increase in Avd. However, AS-IV pre-administration decreased the number of Avi with increasing the number of Avd. Compared with the 3-MA group, AS-IV balanced the number of Avi and Avd, suggesting that AS-IV activates autophagic flux by increasing autophagosome–lysosome fusion (Figure 2G and H). Together, these observations indicate that AS-IV pre-treatment might restore impaired autophagy in PM2.5-induced lung injury.
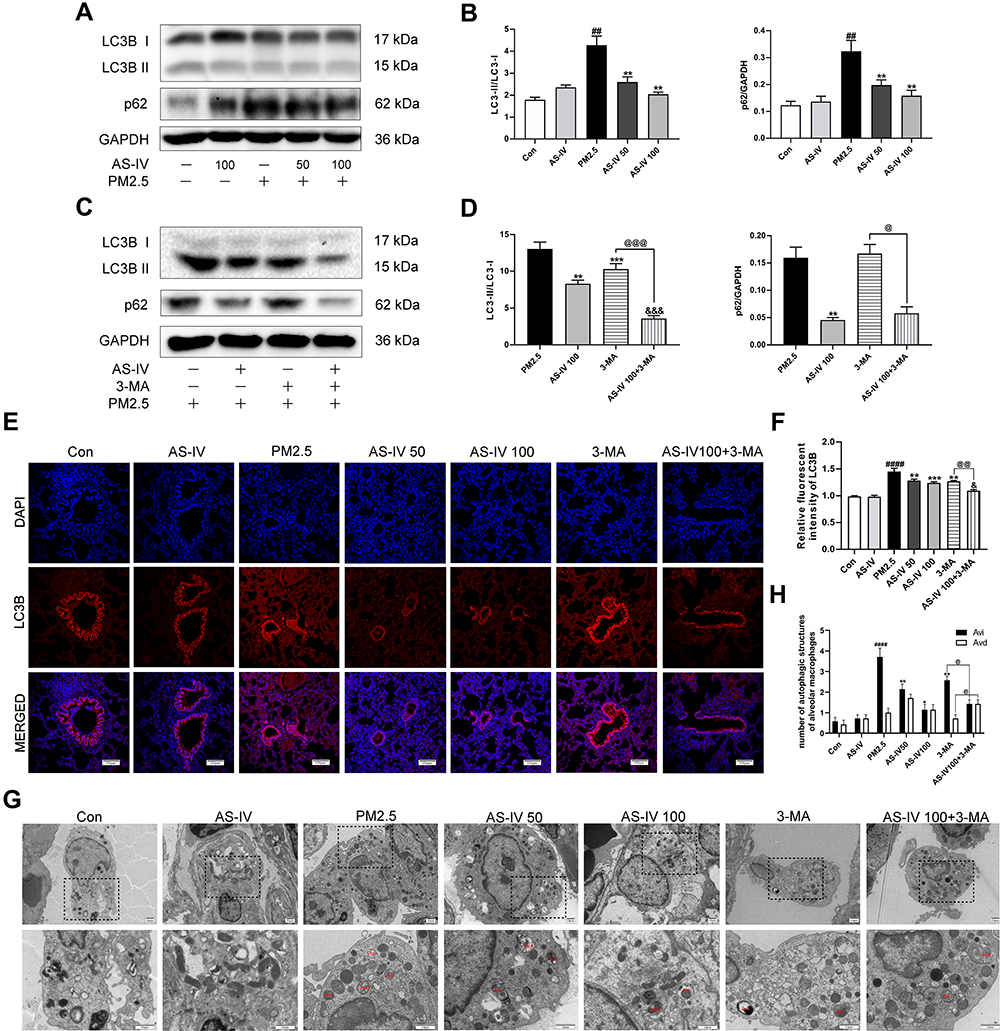 |
Figure 2 AS-IV restores impaired autophagy in lung tissues from rats exposed to PM2.5. (A–D) The levels of LC3B and p62 were determined by Western blot analysis (n=6-7). (E) Immunofluorescence staining of LC3B in lung tissues (Magnification 200×, Scale bar = 100 μm). (F) The relative fluorescent intensity of LC3B in lung tissues (n=6-7). (G) TEM was utilized to visualize autophagic vacuoles (autophagosomes, Avi; autolysosomes, Avd) in alveolar macrophages. (n =3, Magnification 2000× for the upper row and 5000× for the lower row, Scale bar = 10 μm). (H) Qualitative analysis of the number of autophagic vacuoles in alveolar macrophages under TEM. Data are mean±SEM. ##P<0.01, and ####P<0.0001 compared with control group; *P<0.05, **P<0.01, and ***P<0.001 compared with PM2.5 group; &P<0.05, and &&&P<0.001 compared with AS-IV 100 group; @P<0.05, @@P<0.01, and @@@P<0.001 compared with 3-MA group. |
AS-IV Inhibits the PI3K/Akt/mTOR Pathway in Lung Tissues from Rats Exposed to PM2.5
To further elucidate the mechanism of the in-vivo findings, the PI3K/Akt/mTOR pathway was examined (Figure 3A–D). Western blot revealed that AS-IV significantly inhibited the relative contents of p-PI3K, p-Akt, and p-mTOR protein (P<0.01), which were further decreased after adding 3-MA (P<0.05). In addition, we examined the expression of p-PI3K, p-Akt, and p-mTOR in lung tissue using immunohistochemistry. As shown in Figure 3E and F, PM2.5 stimulation elevated the positive expression of p-PI3K, p-Akt, and p-mTOR in lung tissues, while AS-IV suppressed expression in a dose-dependent manner. As a PI3K inhibitor, compared with AS-IV pre-treatment, 3-MA further inhibited the expression of p-PI3K, p-Akt, and p-mTOR to some extent in Western blot analysis and immunohistochemistry. These results suggest that AS-IV inhibits the PI3K/Akt/mTOR pathway in lung tissues of rats with PM2.5-induced lung injury.
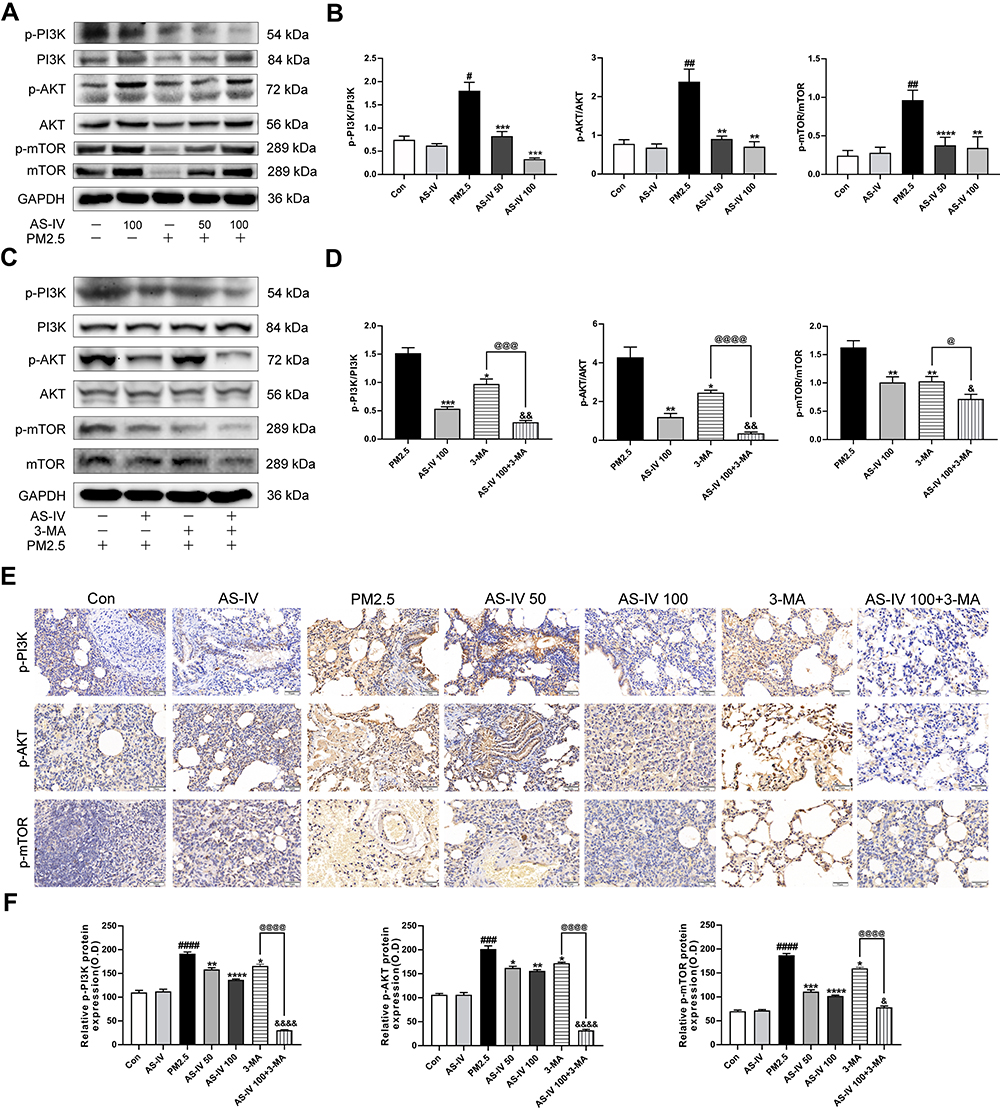 |
Figure 3 AS-IV inhibits the PI3K/Akt/mTOR pathway in lung tissues from rats exposed to PM2.5. (A–D) The levels of p-PI3K, PI3K, p-AKT, AKT, p-mTOR, and mTOR were determined by Western blot analysis. (E) Effects of p-PI3K, p-AKT, and p-mTOR expression in lung tissues by immunohistochemistry staining. (Magnification 200×, Scale bar = 50 μm). (F) Statistical chart of positive expression of p-PI3K, p-AKT, and p-mTOR proteins. Graphs represent mean±SEM (n=6-7). ##P<0.01, ###P<0.001, and ####P<0.0001 compared with control group; *P<0.05, **P<0.01, and ***P<0.001 compared with PM2.5 group; &P<0.05, &&P<0.01, and &&&P<0.001 compared with AS-IV 100 group; @P<0.05, @@@P<0.001, and @@@@P<0.0001 compared with 3-MA group. |
AS-IV Inhibits the NF-κB Translocation in Lung Tissues from Rats Exposed to PM2.5
In order to study whether NF-κB is activated in lung tissues induced by PM2.5, we measured the protein the expression level of NF-κB (Figure 4A–D). Compared with the control group, the nuclear expression of p65 levels was significantly promoted by PM2.5 (P<0.01), and cytoplasmic p65 significantly decreased (P<0.01). However, AS-IV dose-dependently reduced p65 protein translocating into the nuclear. Meanwhile, the above trend was further suppressed by 3-MA (P<0.01). Collectively, these findings suggest that AS-IV inhibits the NF-κB translocation through induction of PI3K-dependent autophagy.
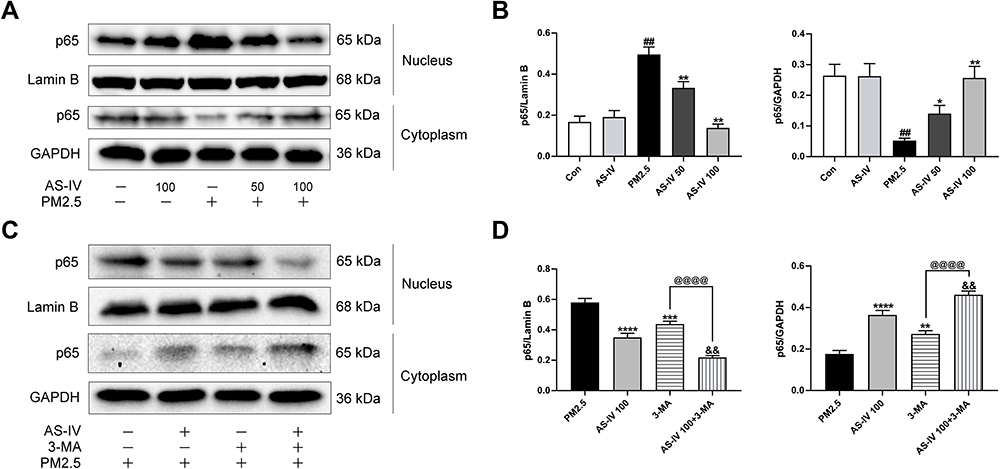 |
Figure 4 AS-IV inhibits the NF-κB translocation in lung tissues from rats exposed to PM2.5. (A–D) The levels of cytoplasmic and nuclear p65 were determined by Western blot analysis. Graphs represent mean±SEM (n=6-7). ##P<0.01 compared with control group; *P<0.05, **P<0.01, ***P<0.001, and ****P<0.0001 compared with PM2.5 group; &&P<0.01 compared with AS-IV 100 group; @@@@P<0.0001 compared with 3-MA group. |
AS-IV Restores Impaired Autophagy in PM2.5-Induced NR8383 Cells
To verify AS-IV can regulate autophagy, we conducted experiments with NR8383 rat alveolar macrophages to AS-IV in vitro. As shown in Figure 5A, compared with non-treated controls, the viability of NR8383 cells was reduced by PM2.5, which indicated that 50 ug/mL PM2.5 induced apparent cellular damage. Concurrently, cell viability increased significantly after AS-IV treatment at concentrations between 1 and 100 μM. According to the above results, 10 μM and 50 μM of AS-IV were used for further research. As illustrated in Figure 5B and C, the protein expression of LC3B-II/I and p62 was elevated by PM2.5, suggesting that PM2.5 leaded to defective autophagy. In parallel, AS-IV decreased the levels of LC3B-II/I and p62, while 3-MA only effectively suppressed LC3B-II/I. Interestingly, there was no significant difference in the level of p62 between AS-IV 50 group and AS 50+3-MA group, which was consistent with that observed in vivo. Consistent with the protein levels, PM2.5 significantly increased the accumulation of autophagosomes in these cells transfected with GFP-LC3 (Figure 5D and E). AS-IV significantly suppressed the amount of GFP-LC3-labeled puncta formation, suggesting that AS-IV decreased the accumulation of autophagosomes. Additionally, AS-IV and 3-MA cotreatment dramatically reduced the accumulation of GFP-LC3-labeled puncta compared with a single treatment. Similar to the in vivo studies, TEM showed that PM2.5 promoted the accumulation of Avi, but AS-IV balanced the number of Avi and Avd, further emphasizing the ability of AS-IV to restore impaired autophagic flux. (Figure 5F). Collectively, these data suggested that AS-IV restored autophagic flux mainly through regulating the degradation of autophagosomes rather than suppressing the formation.
 |
Figure 5 AS-IV restores impaired autophagy in PM2.5-induced NR8383 cells. (A) Cell viability was measured in AS-IV-treated (0.01, 0,1, 1, 10, 25, 50, 75, or 100 μM; 12 h) NR8383 cells by CCK-8 assays (n=3). (B and C) The levels of LC3B and p62 were determined by Western blot analysis (n=3). (D and E) NR8383 cells were treated with AS-IV (50 μM), 3-Ma (5 mM), or PM2.5 (50 μg/mL), or in combination for 24 h after transfection with GFP-LC3 plasmid (Scale bar = 20 μm). LC3 punctate-positive cells were observed by fluorescence microscopy and quantitation of the percentage of cells with punctate GFP-LC3 fluorescence per total GFP-LC3-positive cells. Data represent mean SEM calculated from three experiments of 100 transfected cells each. (F) TEM was utilized to visualize autophagic vacuoles (autophagosomes, Avi; autolysosomes, Avd) in NR8383 cells (n =3, Magnification 12,000× for the upper row and 20,000× for the lower row, Scale bar=1 μm). Graphs represent mean±SEM. ####P<0.0001 compared with DMSO group; *P<0.05, **P<0.01, ***P<0.001, and ****P<0.0001 compared with PM2.5 group; &P<0.05 and &&P<0.01 compared with AS-IV 50 group; @@@@P<0.0001 compared with 3-MA group. |
AS-IV Inhibits the PI3K/Akt/mTOR Pathway and NF-κB Translocation in PM2.5-Induced NR8383 Cells
As demonstrated in Figure 6A and B, similar to the results in vivo, PM2.5 significantly increased the levels of p-PI3K, p-Akt and p-mTOR in PM2.5-induced NR8383 cells. AS-IV treatment decreased the phosphorylation proteins expression of PI3K, Akt and mTOR. In addition, PM2.5 stimulated NF-ĸB p65 translocation significantly, while AS-IV treatment markedly inhibited p65 protein translocating into the nuclear in a dose-dependent manner (Figure 6C and D). Furthermore, 3-MA promoted the effects caused by AS-IV. These results suggest that PI3K/Akt/mTOR pathway and NF-κB translocation might deeply involve in the process of autophagy of AS-IV in PM2.5-induced rat alveolar macrophages.
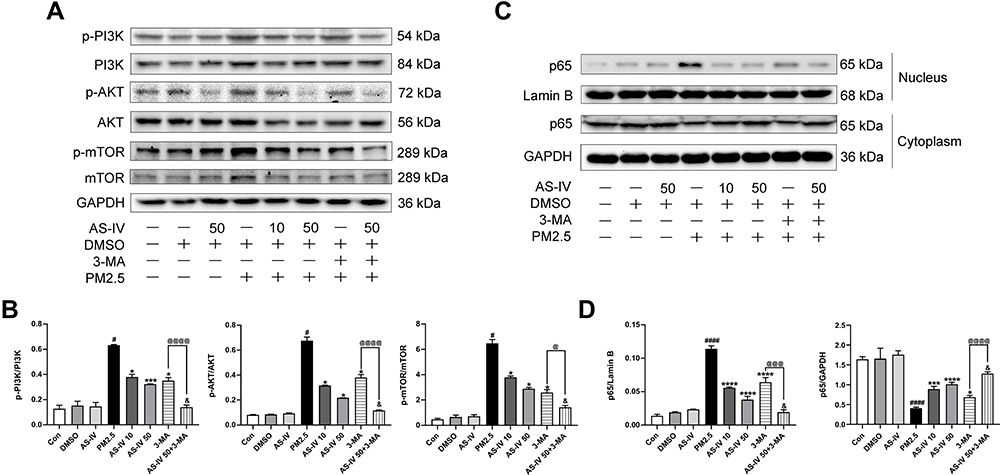 |
Figure 6 AS-IV inhibits the PI3K/Akt/mTOR pathway and NF-κB translocation in PM2.5-induced NR8383 cells. (A and B) The levels of p-PI3K, PI3K, p-AKT, AKT, p-mTOR, and mTOR were determined by Western blot analysis. (C and D) The levels of cytoplasmic and nuclear p65 were determined by Western blot analysis. Graphs represent mean±SEM (n=3). #P<0.05, and ####P<0.0001 compared with DMSO group; *P<0.05, ***P<0.001, and ****P<0.0001 compared with PM2.5 group; &P<0.05 compared with AS-IV 50 group; @P<0.05, @@@P<0.001, and @@@@P<0.0001 compared with 3-MA group. |
Discussion
Currently, there is no effective strategy to prevent and treat PM2.5-induced lung injury. Inflammatory damage and oxidative stress induced by an imbalance in autophagy are closely related to PM2.5-induced lung injury. Therefore, regulating the relationship between autophagy and inflammation may be a potential prevention and control strategy.19,26 This study revealed that AS-IV protects from PM2.5-induced lung injury by inhibiting the PI3K/Akt/mTOR signaling pathway to restore impaired autophagy in vivo and in vitro. To explore this possibility, 3-MA was used to block autophagy through inhibiting PI3K. In addition, this study provides a new understanding of the preventive effect of AS-IV on PM2.5-induced lung injury.
PM2.5 damages lung tissue, triggers the release of inflammatory mediators that participate in airway inflammation and lung tissue damage, and ultimately increases morbidity and mortality from respiratory disease.38 However, the pathogenesis of PM2.5-induced lung injury is complicated.39 AS-IV is widely used to prevent respiratory diseases, but its protective effect on PM2.5-induced lung injury is rarely reported. Although many advances have been made in understanding the molecular mechanisms of PM2.5-induced lung injury,40–42 there is still a lack of preventive and therapeutic strategies. In our study, we found that AS-IV significantly ameliorated pulmonary edema in rats with PM2.5-induced lung injury. Compared with the PM2.5 group, the AS-IV 50 and AS-IV 100 groups showed clear and intact lung tissue structures with no bleeding, neutrophil infiltration, alveolar wall thickening, or other classic pathological changes associated with lung injury.
The inflammatory response is accompanied by degranulation and activation of topical mast cells and macrophages, as well as release of various proinflammatory mediators.43 Previous studies have shown that a variety of proinflammatory cytokines, such as IL-6, IL-8, and IL-10, are elevated in PM2.5-induced lung injury.44,45 Among them, TNF-α and IL-6, which are key components of the cytokine network, are essential inflammatory mediators for lung injury initiation.46,47 CRP is a protein that rises sharply in plasma during tissue damage and plays a vital role in the innate immune process.48,49 After AS-IV pre-treatment, proinflammatory cytokines (IL-6 and TNF-α) and CRP were significantly reduced in BALF. These results preliminarily indicate that AS-IV may attenuate PM2.5-induced lung injury by inhibiting inflammation.
Disruption of the homeostasis between oxidation or antioxidants in cells may be a critical mechanism in PM2.5-induced lung injury.50–52 PM2.5 mediates cellular oxidative stress through different pathways, resulting in lung injury under the combined action of proinflammatory cytokines.53–55 Previous studies have shown that reactive oxygen species (ROS) play an indispensable role in the AS-IV-induced anti-inflammatory effect, including in endothelial,56 epithelial,57 and mesenchymal stem cell lines.58 Our results show that excessive ROS caused by PM2.5 induced nuclear transcription factor activation factor NF-κB and further released proinflammatory factors and cytokines, including TNF-α and IL-6, leading to lung inflammation. AS-IV increased CAT and SOD in lung tissue, inhibited MDA activity, and reduced ROS. However, further studies are needed to evaluate the role of AS-IV in regulating inflammation and oxidative stress to protect from PM2.5-induced lung injury.
Recently, some studies have shown that autophagy, as an important mechanism that maintains the dynamic balance of the cell microenvironment and cell self-renewal, is involved in the progression of respiratory diseases caused by PM2.5.17,59,60 During acute pulmonary inflammation, activation of autophagy appears to be a protective mechanism for the host to respond to bacterial and viral infections.61 Meanwhile, regulating autophagy has also become a potential mechanism to alleviate PM2.5-induced lung injury.62 The results showed that autophagosomes were increased in the lung exposed to PM2.5, but AS-IV pre-administration balanced the number of autophagosomes and autolysosomes in vivo and in vitro. The increase in the number of autophagosomes reflects either increased autophagosome formation due to enhanced autophagic activity or an accumulation of autophagosomes due to impaired autophagic degradation.63 We then tested whether autophagy was induced in vivo and in vitro by analyzing commonly used autophagy markers including LC3 and p62. LC3 conversion (LC3-I to LC3-II) reflects the development of autophagic flux;64 p62 directly binds to LC3 and is degraded by autophagy activation in autolysosome,65 therefore, its accumulation always suggests a block in autophagosome degradation. The simultaneous increase of LC3 conversion and p62 expression level could indicate that the late degradation stage of autophagy is blocked.66,67 Our findings reveal that the levels of LC3B-II/LC3B-I and p62 in lung tissue increased simultaneously after modeling, which implies that PM2.5 induces autophagy impairment via inhibiting autophagy degradation resulting in autophagosome accumulation in vivo and in vitro. As a previously reported,68 dramatic increase in p62 and LC3-II expression, together with accumulation of autophagosomes, suggested that autophagic flux was impaired in PM2.5-induced blood-testis barrier. Furthermore, AS-IV restored impaired autophagy by decreasing LC3B-II/LC3B-I ratio, and inhibiting p62 protein expression in vivo and in vitro. Interestingly, 3-MA (a classic PI3K inhibitor, blocking the formation of autophagosomes) further reduced the levels of LC3B-II/LC3B-I, but not p62. The above results indicated that AS-IV restored autophagic flux mainly through regulating the degradation of autophagosomes rather than suppressing the formation.69,70 It has been reported that AS-IV can inhibit autophagy in LPS-induced pulmonary epithelial cell injury.71 In fact, for the regulation of autophagy, our results showed exactly the opposite outcome in vivo and in vitro. Basing on the paradoxical role of AS-IV in regulating autophagy, we tentatively suggest that different injured models, injury degrees, and administration times may differentially affect autophagy regulation, leading to varied outcomes.72 Therefore, further research would be needed to explore the regulation of autophagy by AS-IV in different disease models.
Further, we explored the possible mechanism of activation of autophagy by AS-IV. Autophagy is an important mechanism to maintain cellular homeostasis, and multiple signaling pathways are involved in this process.73 Since the PI3K/Akt/mTOR pathway is one of the most common factors related to the progression of inflammation and oxidative stress, the PI3K/Akt/mTOR pathway and its related molecules are regarded as prevention and treatment targets for lung injury.74,75 Therefore, we hypothesized that AS-IV enhances autophagy through the PI3K/Akt/mTOR signaling pathway in PM2.5-induced lung injury. We used 3-MA to illustrate whether AS-IV protects from PM2.5-induced NR8383 cells and rats through PI3K targets.76 The results demonstrate that AS-IV suppresses the PI3K/Akt/mTOR signaling pathway in vivo and in vitro, and 3-MA significantly promotes this trend. More importantly, there was no significant differences in protective effect between AS-IV 100 group and 3-MA group. AS-IV and 3-MA cotreatment further reduced histopathological damage, decreased the lung injury score, reduced the lung dry/wet weight ratio, and decreased pulmonary inflammation and oxidative stress in the lung tissues. Also, under the action of 3-MA, AS-IV was effective at reducing NF-κB translocation in vivo and in vitro. The above results suggest that AS-IV protects from PM2.5-induced lung injury by regulating autophagy via inhibition of PI3K/Akt/mTOR pathway. As an important downstream target of PI3K, mTOR is a convergent target of autophagy modulation and has a negative regulatory effect on autophagy.77 Similar to our results, a previous study showed that rapamycin, an inhibitor of mTOR, protects from PM2.5-induced pulmonary inflammation in rat by activating autophagy.78 These findings indicate that activation of autophagy may be a potential strategy to prevent PM2.5-induced lung injury. However, the mechanism of AS-IV-activated autophagy to attenuate PM2.5-induced lung injury requires further investigation.
Conclusion
The present study demonstrated that AS-IV protects from PM2.5-induced lung injury by inhibiting the PI3K/Akt/mTOR signaling pathway to restore impaired autophagy in vivo and in vitro. Meanwhile, our results show that AS-IV may be useful clinically to prevent and treat pulmonary diseases caused by PM2.5.
Acknowledgments
We thank Zherui Shen for her great contribution to the experiments on the cell fraction.
Author Contributions
All authors contributed to data analysis, drafting and revising the article, have agreed on the journal to which the article will be submitted, gave final approval of the version to be published, and agree to be accountable for all aspects of the work.
Funding
This project was funded by National Natural Science Foundation of China (No. 82104824), China Postdoctoral Science Foundation (No. 2020M683645XB), and the Key R&D Program of Sichuan Provincial Department of Science and Technology (No. 2020YFS0312).
Disclosure
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. Cohen AJ, Brauer M, Burnett R, et al. Estimates and 25-year trends of the global burden of disease attributable to ambient air pollution: an analysis of data from the Global Burden of Diseases Study 2015. Lancet. 2017;389(10082):1907–1918. doi:10.1016/S0140-6736(17)30505-6
2. Liu C, Chen R, Sera F, et al. Ambient particulate air pollution and daily mortality in 652 cities. N Engl J Med. 2019;381(8):705–715. doi:10.1056/NEJMoa1817364
3. Horne BD, Joy EA, Hofmann MG, et al. Short-term elevation of fine particulate matter air pollution and acute lower respiratory infection. Am J Respir Crit Care Med. 2018;198:759–766. doi:10.1164/rccm.201709-1883OC
4. Lau SY, Cheng W, Yu Z, et al. Independent association between meteorological factors, PM2.5, and seasonal influenza activity in Hangzhou, Zhejiang province, China. Influenza Other Respir Viruses. 2020;15:513–520. doi:10.1111/irv.12829
5. Mirabelli MC, Vaidyanathan A, Flanders WD, Qin X, Garbe P. Outdoor PM2.5, ambient air temperature, and asthma symptoms in the past 14 days among adults with active asthma. Environ Health Perspect. 2016;124:1882–1890. doi:10.1289/EHP92
6. Ghosh R, Rossner P, Honkova K, Dostal M, Sram RJ, Hertz-Picciotto I. Air pollution and childhood bronchitis: interaction with xenobiotic, immune regulatory and DNA repair genes. Environ Int. 2016;87:94–100. doi:10.1016/j.envint.2015.10.002
7. Hendryx M, Luo J, Chojenta C, Byles JE. Air pollution exposures from multiple point sources and risk of incident chronic obstructive pulmonary disease (COPD) and asthma. Environ Res. 2019;179:108783. doi:10.1016/j.envres.2019.108783
8. Huang HC, Lin FC, Wu MF, et al. Association between chronic obstructive pulmonary disease and PM2.5 in Taiwanese nonsmokers. Int J Hyg Environ Health. 2019;222:884–888. doi:10.1016/j.ijheh.2019.03.009
9. Shull JG, Pay MT, Lara Compte C, et al. Mapping IPF helps identify geographic regions at higher risk for disease development and potential triggers. Respirology. 2020;26:352–359. doi:10.1111/resp.13973
10. Hamra GB, Guha N, Cohen A, et al. Outdoor particulate matter exposure and lung cancer: a systematic review and meta-analysis. Environ Health Perspect. 2014;122:906–911. doi:10.1289/ehp/1408092
11. Qin S, Li B, Wang X, et al. Metal element detection and carcinogenicity risk assessment of PM2.5 samples. Environ Toxicol Chem. 2020;39:1273–1276. doi:10.1002/etc.4730
12. Atkinson RW, Kang S, Anderson HR, Mills IC, Walton HA. Epidemiological time series studies of PM2.5 and daily mortality and hospital admissions: a systematic review and meta-analysis. Thorax. 2014;69:660–665. doi:10.1136/thoraxjnl-2013-204492
13. Falcon-Rodriguez CI, Osornio-Vargas AR, Sada-Ovalle I, Segura-Medina P. Aeroparticles, composition, and lung diseases. Front Immunol. 2016;7:3. doi:10.3389/fimmu.2016.00003
14. Fann N, Kim SY, Olives C, Sheppard L. Estimated changes in life expectancy and adult mortality resulting from declining PM2.5 exposures in the contiguous United States: 1980–2010. Environ Health Perspect. 2017;125:097003. doi:10.1289/EHP507
15. Bowe B, Xie Y, Yan Y, Al-Aly Z. Burden of cause-specific mortality associated with PM2.5 air pollution in the United States. JAMA Netw Open. 2019;2:e1915834. doi:10.1001/jamanetworkopen.2019.15834
16. Xu X, Wang H, Liu S, et al. TP53-dependent autophagy links the ATR-CHEK1 axis activation to proinflammatory VEGFA production in human bronchial epithelial cells exposed to fine particulate matter (PM2.5). Autophagy. 2016;12:1832–1848. doi:10.1080/15548627.2016.1204496
17. Zhu XM, Wang Q, Xing WW, et al. PM2.5 induces autophagy-mediated cell death via NOS2 signaling in human bronchial epithelium cells. Int J Biol Sci. 2018;14(5):557–564. doi:10.7150/ijbs.24546
18. Antonioli M, Di Rienzo M, Piacentini M, Fimia GM. Emerging mechanisms in initiating and terminating autophagy. Trends Biochem Sci. 2017;42:28–41. doi:10.1016/j.tibs.2016.09.008
19. Deretic V, Levine B. Autophagy balances inflammation in innate immunity. Autophagy. 2018;14:243–251. doi:10.1080/15548627.2017.1402992
20. Levine B, Mizushima N, Virgin HW. Autophagy in immunity and inflammation. Nature. 2011;469:323–335. doi:10.1038/nature09782
21. Guo L, Stripay JL, Zhang X, et al. CaMKIα regulates AMP kinase-dependent, TORC-1-independent autophagy during lipopolysaccharide-induced acute lung neutrophilic inflammation. J Immunol. 2013;190:3620–3628. doi:10.4049/jimmunol.1102975
22. Zhao H, Chen H, Xiaoyin M, et al. Autophagy activation improves lung injury and inflammation in sepsis. Inflammation. 2019;42:426–439. doi:10.1007/s10753-018-00952-5
23. Xu XC, Wu YF, Zhou JS, et al. Autophagy inhibitors suppress environmental particulate matter-induced airway inflammation. Toxicol Lett. 2017;280:206–212. doi:10.1016/j.toxlet.2017.08.081
24. Zeng M, Sang W, Chen S, et al. 4-PBA inhibits LPS-induced inflammation through regulating ER stress and autophagy in acute lung injury models. Toxicol Lett. 2017;271:26–37. doi:10.1016/j.toxlet.2017.02.023
25. Matsuzawa-Ishimoto Y, Hwang S, Cadwell K. Autophagy and inflammation. Annu Rev Immunol. 2018;36:73–101. doi:10.1146/annurev-immunol-042617-053253
26. Racanelli AC, Kikkers SA, Choi A, Cloonan SM. Autophagy and inflammation in chronic respiratory disease. Autophagy. 2018;14:221–232. doi:10.1080/15548627.2017.1389823
27. Zhang J, Wu C, Gao L, Du G, Qin X. Astragaloside IV derived from Astragalus membranaceus: a research review on the pharmacological effects. Adv Pharmacol. 2020;87:89–112. doi:10.1016/bs.apha.2019.08.002
28. Yang X, Wang F. The effect of astragaloside IV on JAK2-STAT6 signalling pathway in mouse model of ovalbumin-induced asthma. J Anim Physiol Anim Nutr (Berl). 2019;103:1578–1584. doi:10.1111/jpn.13114
29. Li LC, Xu L, Hu Y, et al. Astragaloside IV improves bleomycin-induced pulmonary fibrosis in rats by attenuating extracellular matrix deposition. Front Pharmacol. 2017;8:513. doi:10.3389/fphar.2017.00513
30. Lai ST, Wang Y, Peng F. Astragaloside IV sensitizes non-small cell lung cancer cells to cisplatin by suppressing endoplasmic reticulum stress and autophagy. J Thorac Dis. 2020;12:3715–3724. doi:10.21037/jtd-20-2098
31. Wu Y, Xiao W, Pei C, et al. Astragaloside IV alleviates PM2.5-induced lung injury in rats by modulating TLR4/MyD88/NF-κB signalling pathway. Int Immunopharmacol. 2020;91:107290. doi:10.1016/j.intimp.2020.107290
32. Zhan L, Zhang Y, Su W, et al. The roles of autophagy in acute lung injury induced by myocardial ischemia reperfusion in diabetic rats. J Diabetes Res. 2018;2018:5047526. doi:10.1155/2018/5047526
33. Chen ZH, Wu YF, Wang PL, et al. Autophagy is essential for ultrafine particle-induced inflammation and mucus hyperproduction in airway epithelium. Autophagy. 2016;12:297–311. doi:10.1080/15548627.2015.1124224
34. McGuigan RM, Mullenix P, Norlund LL, Ward D, Walts M, Azarow K. Acute lung injury using oleic acid in the laboratory rat: establishment of a working model and evidence against free radicals in the acute phase. Curr Surg. 2003;60(4):412–417. doi:10.1016/S0149-7944(02)00775-4
35. Chen S, Tian R, Luo D, Xiao Z, Li H, Lin D. Time-course changes and role of autophagy in primary spinal motor neurons subjected to oxygen-glucose deprivation: insights into autophagy changes in a cellular model of spinal cord ischemia. Front Cell Neurosci. 2020;14:38. doi:10.3389/fncel.2020.00038
36. Yang P, Song R, Li N, et al. Silica dust exposure induces autophagy in alveolar macrophages through switching Beclin1 affinity from Bcl-2 to PIK3C3. Environ Toxicol. 2020;35(7):758–767. doi:10.1002/tox.22910
37. Hua F, Li K, Yu JJ, et al. TRB3 links insulin/IGF to tumour promotion by interacting with p62 and impeding autophagic/proteasomal degradations. Nat Commun. 2015;6:7951. doi:10.1038/ncomms8951
38. Wyatt LH, Devlin RB, Rappold AG, Case MW, Diaz-Sanchez D. Low levels of fine particulate matter increase vascular damage and reduce pulmonary function in young healthy adults. Part Fibre Toxicol. 2020;17:58. doi:10.1186/s12989-020-00389-5
39. Watts N, Amann M, Arnell N, et al. The 2020 report of the Lancet countdown on health and climate change: responding to converging crises. Lancet. 2021;397:129–170. doi:10.1016/S0140-6736(20)32290-X
40. Zheng Y, Fan J, Chen HW, Liu EQ. Trametes orientalis polysaccharide alleviates PM2.5-induced lung injury in mice through its antioxidant and anti-inflammatory activities. Food Funct. 2019;10:8005–8015. doi:10.1039/c9fo01777a
41. Yang L, Liu G, Fu L, Zhong W, Li X, Pan Q. DNA repair enzyme OGG1 promotes alveolar progenitor cell renewal and relieves PM2.5-induced lung injury and fibrosis. Ecotoxicol Environ Saf. 2020;205:111283. doi:10.1016/j.ecoenv.2020.111283
42. Yang L, Liu G, Li X, et al. Small GTPase RAB6 deficiency promotes alveolar progenitor cell renewal and attenuates PM2.5-induced lung injury and fibrosis. Cell Death Dis. 2020;11:827. doi:10.1038/s41419-020-03027-2
43. Lamkanfi M, Dixit VM. Mechanisms and functions of inflammasomes. Cell. 2014;157:1013–1022. doi:10.1016/j.cell.2014.04.007
44. Fei YX, Zhao B, Yin QY, et al. Ma Xing Shi Gan decoction attenuates PM2.5 induced lung injury via inhibiting HMGB1/TLR4/NFκB signal pathway in rat. Front Pharmacol. 2019;10:1361. doi:10.3389/fphar.2019.01361
45. Feng S, Duan E, Shi X, et al. Hydrogen ameliorates lung injury in a rat model of subacute exposure to concentrated ambient PM2.5 via Aryl hydrocarbon receptor. Int Immunopharmacol. 2019;77:105939. doi:10.1016/j.intimp.2019.105939
46. Ward PA. Role of complement, chemokines, and regulatory cytokines in acute lung injury. Ann NY Acad Sci. 1996;796:104–112. doi:10.1111/j.1749-6632.1996.tb32572.x
47. Deng JC, Standiford TJ. Growth factors and cytokines in acute lung injury. Compr Physiol. 2011;1:81–104. doi:10.1002/cphy.c090011
48. Black S, Kushner I, Samols D. C-reactive Protein. J Biol Chem. 2004;279:48487–48490. doi:10.1074/jbc.R400025200
49. Toubi E, Vadasz Z. Innate immune-responses and their role in driving autoimmunity. Autoimmun Rev. 2019;18:306–311. doi:10.1016/j.autrev.2018.10.005
50. Weichenthal SA, Godri-Pollitt K, Villeneuve PJ. PM2.5, oxidant defence and cardiorespiratory health: a review. Environ Health. 2013;12:40. doi:10.1186/1476-069X-12-40
51. Bao ZJ, Fan YM, Cui YF, Sheng YF, Zhu M. Effect of PM2.5 mediated oxidative stress on the innate immune cellular response of Der p1 treated human bronchial epithelial cells. Eur Rev Med Pharmacol Sci. 2017;21:2907–2912.
52. Yue W, Tong L, Liu X, et al. Short term Pm2.5 exposure caused a robust lung inflammation, vascular remodeling, and exacerbated transition from left ventricular failure to right ventricular hypertrophy. Redox Biol. 2019;22:101161. doi:10.1016/j.redox.2019.101161
53. Li R, Zhou R, Zhang J. Function of PM2.5 in the pathogenesis of lung cancer and chronic airway inflammatory diseases. Oncol Lett. 2018;15:7506–7514. doi:10.3892/ol.2018.8355
54. Yang B, Guo J, Xiao C. Effect of PM2.5 environmental pollution on rat lung. Environ Sci Pollut Res Int. 2018;25:36136–36146. doi:10.1007/s11356-018-3492-y
55. Chen X, Chen Y, Chen Y, Wang X, He M. Protective effect of lutein on oxidative stress damage caused by acute PM2.5 exposure in rats. Ann Palliat Med. 2020;9:2028–2036. doi:10.21037/apm-20-1138
56. Zhu Z, Li J, Zhang X. Astragaloside IV protects against oxidized low-density lipoprotein (ox-LDL)-induced endothelial cell injury by reducing oxidative stress and inflammation. Med Sci Monit. 2019;25:2132–2140. doi:10.12659/MSM.912894
57. Hsieh HL, Liu SH, Chen YL, Huang CY, Wu SJ. Astragaloside IV suppresses inflammatory response via suppression of NF-κB, and MAPK signalling in human bronchial epithelial cells. Arch Physiol Biochem. 2020;1–10. doi:10.1080/13813455.2020.1727525
58. Li Z, Zhang S, Cao L, et al. Tanshinone IIA and Astragaloside IV promote the angiogenesis of mesenchymal stem cell-derived endothelial cell-like cells via upregulation of Cx37, Cx40 and Cx43. Exp Ther Med. 2018;15:1847–1854. doi:10.3892/etm.2017.5636
59. Xu H, Xu X, Wang H, et al. LKB1/p53/TIGAR/autophagy-dependent VEGF expression contributes to PM2.5-induced pulmonary inflammatory responses. Sci Rep. 2019;9:16600. doi:10.1038/s41598-019-53247-6
60. Lu X, Li R, Yan X. Airway hyperresponsiveness development and the toxicity of PM2.5. Environ Sci Pollut Res Int. 2021;1–8. doi:10.1007/s11356-020-12051-w
61. Painter JD, Galle-Treger L, Akbari O. Role of autophagy in lung inflammation. Front Immunol. 2020;11:1337. doi:10.3389/fimmu.2020.01337
62. Wu YF, Li ZY, Dong LL, et al. Inactivation of MTOR promotes autophagy-mediated epithelial injury in particulate matter-induced airway inflammation. Autophagy. 2020;16:435–450. doi:10.1080/15548627.2019.1628536
63. D’Agostino M, Scerra G, Cannata Serio M, Caporaso MG, Bonatti S, Renna M. Unconventional secretion of α-Crystallin B requires the Autophagic pathway and is controlled by phosphorylation of its serine 59 residue. Sci Rep. 2019;9(1):16892. doi:10.1038/s41598-019-53226-x
64. Yin N, Wu C, Qiu J, et al. Protective properties of heme oxygenase-1 expressed in umbilical cord mesenchymal stem cells help restore the ovarian function of premature ovarian failure mice through activating the JNK/Bcl-2 signal pathway-regulated autophagy and upregulating the circulating of CD8+CD28- T cells. Stem Cell Res Ther. 2020;11(1):49. doi:10.1186/s13287-019-1537-x
65. Bao L, Jaramillo MC, Zhang Z, et al. Induction of autophagy contributes to cisplatin resistance in human ovarian cancer cells. Mol Med Rep. 2015;11(1):91–98. doi:10.3892/mmr.2014.2671
66. Oh SH, Yoon MH, Lim KJ, Yu BS, Jee IG, Jung KT. Nefopam downregulates autophagy and c-Jun N-terminal kinase activity in the regulation of neuropathic pain development following spinal nerve ligation. BMC Anesthesiol. 2018;18(1):97. doi:10.1186/s12871-018-0559-8
67. Lai Z, Gu L, Yu L, et al. Delta opioid peptide [d-Ala2, d-Leu5] enkephalin confers neuroprotection by activating delta opioid receptor-AMPK-autophagy axis against global ischemia. Cell Biosci. 2020;10:79. doi:10.1186/s13578-020-00441-z
68. Wei Y, Cao XN, Tang XL, et al. Urban fine particulate matter (PM2.5) exposure destroys blood-testis barrier (BTB) integrity through excessive ROS-mediated autophagy. Toxicol Mech Methods. 2018;28(4):302–319. doi:10.1080/15376516.2017.1410743
69. Gretzmeier C, Eiselein S, Johnson GR, et al. Degradation of protein translation machinery by amino acid starvation-induced macroautophagy. Autophagy. 2017;13:1064–1075. doi:10.1080/15548627.2016.1274485
70. Li D, Wang C, Li Z. Nano-sized Al2O3 particle-induced autophagy reduces osteolysis in aseptic loosening of total hip arthroplasty by negative feedback regulation of RANKL expression in fibroblasts. Cell Death Dis. 2018;9:840. doi:10.1038/s41419-018-0862-9
71. Liu B, Zhao H, Wang Y, Zhang H, Ma Y. Astragaloside IV attenuates lipopolysaccharides-induced pulmonary epithelial cell injury through inhibiting autophagy. Pharmacology. 2020;105(1–2):90–101. doi:10.1159/000502865
72. Bai L, Mei X, Shen Z, et al. Netrin-1 improves functional recovery through autophagy regulation by activating the AMPK/mTOR signaling pathway in rats with spinal cord injury. Sci Rep. 2017;7:42288. doi:10.1038/srep42288
73. Shahnazari S, Namolovan A, Mogridge J, Kim PK, Brumell JH. Bacterial toxins can inhibit host cell autophagy through cAMP generation. Autophagy. 2011;7:957–965. doi:10.4161/auto.7.9.16435
74. Meng L, Li L, Lu S, et al. The protective effect of dexmedetomidine on LPS-induced acute lung injury through the HMGB1-mediated TLR4/NF-κB and PI3K/Akt/mTOR pathways. Mol Immunol. 2018;94:7–17. doi:10.1016/j.molimm.2017.12.008
75. Huang CY, Deng JS, Huang WC, Jiang WP, Huang GJ. Attenuation of lipopolysaccharide-induced acute lung injury by hispolon in mice, through regulating the TLR4/PI3K/Akt/mTOR and Keap1/Nrf2/HO-1 pathways, and suppressing oxidative stress-mediated ER stress-induced apoptosis and autophagy. Nutrients. 2020;12:1742. doi:10.3390/nu12061742
76. Qu L, Chen C, He W, et al. Glycyrrhizic acid ameliorates LPS-induced acute lung injury by regulating autophagy through the PI3K/AKT/mTOR pathway. Am J Transl Res. 2019;11:2042–2055.
77. Dibble CC, Cantley LC. Regulation of mTORC1 by PI3K signaling. Trends Cell Biol. 2015;25:545–555. doi:10.1016/j.tcb.2015.06.002
78. Shumin Z, Luying Z, Senlin L, et al. Ambient particulate matter-associated autophagy alleviates pulmonary inflammation induced by Platanus pollen protein 3 (Pla3). Sci Total Environ. 2021;758:143696. doi:10.1016/j.scitotenv.2020.143696
 © 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.