Back to Journals » Diabetes, Metabolic Syndrome and Obesity » Volume 14
Astragaloside IV Alleviates Liver Inflammation, Oxidative Stress and Apoptosis to Protect Against Experimental Non-Alcoholic Fatty Liver Disease
Authors Liang XY, Hong Ff, Yang SL ![]()
Received 3 February 2021
Accepted for publication 25 March 2021
Published 28 April 2021 Volume 2021:14 Pages 1871—1883
DOI https://doi.org/10.2147/DMSO.S304817
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Ming-Hui Zou
Xiao-yu Liang,1 Fen-Fang Hong,2 Shu-Long Yang1
1Department of Physiology, College of Medicine, Nanchang University, Nanchang, 330006, People’s Republic of China; 2Experimental Teaching Center, Nanchang University, Nanchang, 330031, People’s Republic of China
Correspondence: Shu-Long Yang
Department of Physiology, College of Medicine, Nanchang University, Nanchang, 330006, People’s Republic of China
Tel +86 13576291532
Email [email protected]
Fen-Fang Hong
Experimental Teaching Center, Nanchang University, Nanchang, 330031, People’s Republic of China
Tel +86 18970965319
Email [email protected]
Purpose: Non-alcoholic fatty liver disease (NAFLD) is the main form of chronic liver disease in the world. Astragaloside IV (ASIV) has been tested in experimental models of different diseases. The purpose of this study was to evaluate the effect and protective mechanism of ASIV on NAFLD.
Methods: Lipopolysaccharide (LPS)- and palmitate acid (PA)-induced RAW264.7 cells and LO2 cells were used as a NAFLD model. The mice NAFLD model was evaluated by hematoxylin-eosin staining (HE staining), and aspartate aminotransferase (AST) and alanine aminotransferase (ALT) levels. Liver lipid metabolism was evaluated by triglyceride (TG) and total cholesterol (TC) kits and oil red O staining. Oxidative stress indicators were examined through biochemical methods. Inflammatory factors were explored through enzyme-linked immuno sorbent assay (ELISA), real-time quantitative PCR and oxidative stress indicator kits. The expression levels of 5-LO (5-lipoxygenase) and leukotriene A4 hydrolase (LTA4H) were checked by real-time quantitative PCR and Western blotting. Apoptosis was detected by Annexin V-FITC/PI cell apoptosis detection kit.
Results: Our results showed that in vivo ASIV significantly reduced liver tissue damage, and serum AST, ALT and serum TG levels in NAFLD mice. In vitro, ASIV reduced cell supernatant TG and TC content increased by PA treatment, and significantly decreased the accumulation of intracellular lipid droplets induced by PA treatment. Additionally, ASIV reduced reactive oxygen species (ROS) and malondialdehyde (MDA) levels, and restored glutathione peroxidase (GSH-Px) levels in PA-treated LO2 cell supernatant. Furthermore, ASIV inhibited the production of proinflammatory cytokines (IL-6 and TNF-α) in RAW264.7 cells induced by LPS. We also found that ASIV downregulated the expression of 5-LO and LTB4 (leukotriene B4) in NAFLD mice. Moreover, ASIV restored apoptotic protein (Bax and Bcl-2) expression in PA-treated LO2 cells.
Conclusion: ASIV may reduce liver steatosis, hepatocyte oxidative stress and apoptosis, and decrease liver inflammation, thereby attenuating the progression of NAFLD and thus might be of therapeutic interest.
Keywords: astragaloside IV, non-alcoholic fatty liver disease, lipid accumulation, oxidative stress, apoptosis
Introduction
Non-alcoholic fatty liver disease (NAFLD) is an emerging health problem worldwide, with significantly increasing morbidity in the past two decades.1,2 NAFLD covers the disease spectrum from isolated hepatic steatosis (simple steatosis) to non-alcoholic steatohepatitis (NASH), which is characterized by hepatocellular damage, persistent liver inflammation and fibrosis, and eventually develops into cirrhosis and liver cell carcinoma (HCC).3,4 Simple steatosis is a relatively benign disease, and the pathological and biological processes of its evolution into NASH are complex and multiphase.5,6 Development of NAFLD involves multiple pathogenic factors such as fat accumulation, lipotoxicity, inflammation, oxidative stress and mitochondrial dysfunction.7 Feeding high fat diet (HFD) has been recognized to cause liver steatosis and inflammation in experimental mice, and there is obvious oxidative damage in the liver.8 It has been reported that mice fed a HFD for 12–16 weeks can replicate the histopathological and pathogenic characteristics of NAFLD.9 Fat accumulation in the liver results from the increased uptake of circulating lipids, increased nascent lipid production, and impaired fatty acid oxidation.10 Many studies have shown that saturated fatty acids, especially palmitic acid (PA), are the most abundant free fatty acids (FFA) in the diet and serum which can increase liver fat and insulin resistance. Additionally, these FFA can activate endoplasmic reticulum stress and accelerate mitochondrial metabolism.11,12 Moreover, the accumulation of reactive oxygen species (ROS) promotes liver cell dysfunction and even cell apoptosis.13 Studies have shown that mitochondrial disorders and oxidative stress are the core of the pathogenesis of NAFLD.14 ROS may be formed by FFA oxidation in peroxisomes and microsomes and activation of macrophages.15 ROS can lead to cell damage by destroying several bioenergy reactions involved in oxidative phosphorylation and trigger a vicious cycle of oxidative damage.16 In liver cells, mitochondrial dysfunction can amplify apoptosis signals, mitochondrial membrane depolarization, oxidative phosphorylation, and ATP (adenosine triphosphate) depletion.17 Therefore, liver cells cannot maintain structural and functional integrity and initiate proapoptotic signals, and mitochondrial events are affected by Bcl-2 family proteins, the regulation of antiapoptotic protein Bcl-2 (B-cell lymphoma-2) and proapoptotic protein Bax (Bcl-2 Associated X Protein).18
The innate immune mechanism is the main driving factor of inflammation and other pathological manifestations in NAFLD.19 Recruited bone marrow-derived macrophages are a key factor in acute and chronic hepatitis, and the inflammatory microenvironment containing lipopolysaccharide (LPS) and palmitate induces the polarization of macrophages to the proinflammatory M1 type-, which produces inflammatory cytokines to maintain chronic inflammation, and can promote fibrosis by releasing fibrotic cytokines.20,21 In addition, 5-lipoxygenase (5-LO) is mainly expressed in inflammatory cells, including monocytes, mast cells and B lymphocytes.22 In the liver, macrophages are the only place where the 5-LO pathway is metabolized23. A variety of studies have confirmed the relationship between the 5-LO metabolic pathway and NASH.24 The study by Martinez-Clemente et al showed that in the hyperlipidemia model of non-alcoholic fatty liver in mice, ApoE -/- mice lacking 5-LO had reduced liver inflammation; and macrophage infiltration; and reduced caspase-3 and NF-κB activities, and the expression of proinflammatory cytokines was reduced.25 Taken together, these findings indicate that the 5-LO pathway has a key role in the progression of NAFLD.
Traditional Chinese medicine has been used to treat hepatic disease with few side effects for thousands of years.26 ASIV is the main active ingredient of Astragalus, a well-known Chinese medicine, and has been proven to have various pharmacological effects such as antioxidative stress, antiinflammatory, antiapoptosis, and antifibrotic effects.27 It has been reported that ASIV reduces PA-induced lipid accumulation and endoplasmic reticulum stress in hepatocytes.28 Emerging evidence also shows that ASIV reduces liver damage by reducing oxidative stress and inflammation.29 However, the effects of ASIV on NAFLD and its underlying mechanism are not completely understood.
In the present study, using an in vivo and in vitro NAFLD model, we explored whether the mechanisms underlying the protective effects of ASIV on NAFLD involve oxidative stress, lipid accumulation, inflammation and apoptosis in the liver.
Materials and Methods
Reagents
Penicillin-Streptomycin Liquid, Roswell Park Memorial Institute (RPMI) 1640 medium and fetal bovine serum (FBS) were purchased from Solarbio Co., Ltd (Beijing, China), BAX (1:1,000,#Q07812,cell signaling),BCL-2 (1:2,000,bs-0032R,Bioss) and GAPDH (1:1,000,ZS-25778,ZSGB-BIO), ASIV was purchased from shzeye biomart (Shanghai, China). Enzyme-linked immunosorbent assay (ELISA) kits for TNF-α and ELISA kits for IL-6 and ELISA kits for LTB4 were purchased from Multisciences CO., Ltd. (hangzhou, china). 5-LO (1:1,000,C49G1, Cell signaling) and LTA4H (1:1,000,SC-367021, SANTA) were purchased from Cell Signaling, SANTA CRUZ or Abcam. LPS and PA were purchased from American Sigma. Carboxymethyl Cellulose (CMC) was purchased from sigma (USA), Serum plasma alanine aminotransferase (ALT) and aspartate aminotransferase (AST) Activity Assay Kit were purchased from Nanjing JianCheng Bioengineering Institute (Jiangsu, China), total cholesterol (TC) and triglyceride (TG) kits and reactive oxygen species (ROS) and malondialdehyde (MDA) and glutathione peroxidase (GSH-Px) were purchased from Jian Cheng Biotechnology Company (Nanjing, China),Annexin V apoptosis detection kit were purchased from eBioscience (USA), BCA Protein Assay Kit were purchased from Beyotime Biotechnology (Shanghai, China).
Animals and Experiments
The animal protocol of this study were approved by the Medical Experimental Animal administrative Committee of Jiangxi Province, China. All animal experiments are followed the Guide for the Care and Use of Laboratory Animals published the US National Institutes of Health Publication. Make animal models according to literature methods,29,30 male 8-week-old Kunming mice with 18±2g body weight were purchased from the experimental animal center of the Jiangxi university of traditional Chinese medicine (Jiangxi, China). Animals were kept in specific pathogen free(SPF) condition and fed a standard diet with free access to water in temperature-controlled room (24 ± 1°C) with humidity of 40%–80%. For our experiments, the mice were randomly divided into five groups (n=8 in each group): control group, HFD group, ASIV-L (20 mg/kg) group, ASIV-M (40 mg/kg) group, ASIV-H (80 mg/kg) group. Mice fed with HFD for 16 weeks, while the control animals were fed with standard rodent diet. Three weeks later, the HFD-fed mice were intragastric administrated different doses of ASIV (dissolved in 0.5% CMC), while the control mice received the same volume of 0.5%CMC. The body weight was measured weekly. At the end of the intervention, all the animals fasted overnight and were anesthetized with Ketamine (240 mg/kg) and Xylazine (30 mg/kg) intraperitoneally, and then the blood and liver of all groups were collected, which was stored individually on −80 °C, until assay.
Histological Examination
Liver tissues were fixed in 4% paraformaldehyde, embedded in paraffin and cut into 4-µm sections. Histological assessment of the liver tissue sections was performed following hematoxylin and eosin (HE) staining. Each slide was photographed in randomly selected five fields under the same magnification (×100).
Biochemical Measurements
Serum plasma alanine aminotransferase (ALT) and aspartate aminotransferase (AST) activities as liver injury markers, were measured using an ALT and AST Activity Assay Kit (Nanjing JianCheng Bioengineering Institute, Jiangsu, China). The OD value was measured with a microplate reader (Leica Microsystems) at 510 nm. The contents of liver TG were analyzed with a commercially available enzyme detection kit (Nanjing JianCheng Bioengineering Institute, Jiangsu, China).
Cell Culture
LO2 cells and RAW264.7 cells were propagated in RPMI 1640 supplemented with 10% FBS and containing 100 U/mL penicillin and 100 U/mL streptomycin. Cells were grown in vitro at 37°C and 5% CO2. When the cell fusion reached 80%, LO2 cells were stimulated with 0.25 mM PA for 24h, then with different concentrations of ASIV (low L, 20 µg/mL; medium M, 60 µg/mL; high H, 100 µg/mL) for 24 h; 1 ug/mL LPS-treated RAW264.7 cells for 8 h, then with different concentrations of ASIV (low L, 20 µg/mL; medium M, 60 µg/mL; high H, 100 µg/mL) for 24 h. LO2 cells and RAW264.7 cells were obtained from the National Infrastructure of Cell line Resource (Wuhan, China).
Cell Treatment and Viability Test
Cell viability was quantified by the Thiazole Blue (MTT) assay. Cells were seeded in a 96-well plate (5×104 cells/well) and treated with ASIV for 24h, and then incubated with 20ul/well MTT (0.5mg/mL) for 4 hours. After incubating, the culture medium was removed, and 150ul/well DMSO was added. Absorbance was measured at 450nm with a microplate spectrophotometer.
Test for the Contents of Total Cholesterol (TC) and Triglyceride (TG)
The cell supernatant was obtained as previously described, the TC and TG concentrations were detected by using the TG and TC kits according to the manufacturer’s instructions (Jian Cheng Biotechnology Company, Nanjing, China).
Measurement of Intracellular ROS, MDA, GSH-Px
The LO2 cells (4×105cells/well) were seeded in a 6-well plate, after treatment with different concentrations of ASIV (low L, 20µg/mL; medium M, 60µg/mL; high H, 100µg/mL) and 0.25 mM PA, the ROS and MDA and GSH-Px (Jian Cheng Biotechnology Company, Nanjing, China) was performed to determine the levels of reactive oxygen species (ROS) and malondialdehyde (MDA) and glutathione peroxidase (GSH-Px).
Enzyme-Linked Immunosorbent Assay
Cells at a confluence of approximately 80%–90% were plated in 6-well plates at the density of 1×105 cells/well. Then, the RAW264.7 cells were treated with LPS and different concentrations of ASIV (low L, 20µg/mL; medium M, 60µg/mL; high H, 100µg/mL). After incubation, collect the cell supernatant and determine the concentration of LTB4, IL-6 and TNF-α with an ELISA kit according to the manufacturer’s instructions.
Oil Red O Staining
Oil red o staining was used to assess the change of intracellular lipid droplets. After treated with corresponding drugs, the cells were washed twice with PBS and fixed with 4% paraformaldehyde for 25 min, and the cells were stained with oil red o working fluid for 15 min. Then, the cells were counterstained with hematoxylin for 5 minutes.
Western Blotting
RIPA lysis buffer was used for total protein extraction and all of the proteins were quantified using the BCA protein assay reagent. The same amount of protein was separated by SDS-PAGE and transferred to polyvinylidene fluoride membrane. Then, the membrane was blocked with skim milk for 1 hour, and combined with primary antibody Bax (1:1,000,#Q07812,cell signaling),Bcl-2 (1:2,000,bs-0032R,Bioss), 5-LO (1:1,000,C49G1, Cell signaling), LTA4H (1:1,000,SC-367021, SANTA) and GAPDH (1:1,000,ZS-25778,ZSGB-BIO) at 4°C overnight. After incubating with HRP-conjugated secondary antibody (1:2000,ZB-2301,ZSGB-BIO) for 1 hour, the membrane was washed with TBST buffer and subjected to chemiluminescence treatment. The signals were analyzed and quantified by Image J software (NIH, Bethesda, MD, USA).
RNA Extraction and Quantitative Real-Time Polymerase Chain Reaction
Total RNA was extracted using TRIzol reagent. CDNA was synthesized using a reverse transcription kit. QPCR was performed in duplicate using SYBR Green I (Takara, Japan) and an ABI 7500 system (Applied Biosystems, Foster, CA). The specific primers were designed by RiboBio (Guangzhou, China). Target gene expression was standardized for GAPDH levels. The relative fold change in target gene expression was calculated using the 2-ΔΔCt method. The primer sequences are listed in Table 1 (Sangon Biotech Co., Ltd., Shanghai, China).
 |
Table 1 Primer Sequences Used in This Study |
Apoptosis Assays
Flow cytometer was used to evaluate the apoptosis of LO2 cells. After treated with corresponding drugs, the LO2 cells were washed with PBS, and stained with fluorescent dye-conjugated Annexin V (5μL) and propidium iodide (5 μL) at 4°C and in the dark for 2 h. According to the manufacturer’s instructions, the detection was performed using Annexin V apoptosis detection kit (eBioscience, USA). Finally, the data was analyzed with FlowJo software (Becton-Dickinson-San Jose, USA).
Statistical Analysis
All data are expressed as mean ± standard deviation (SD). Statistical analysis was performed by SPSS v18.0 statistical software (IBM Corp., Armonk, NY, USA). The t-test was used to analyze the statistical comparison between the two groups, and the analysis of variance (ANOVA) was used for the post hoc test, and then the comparison between the multiple groups was performed. P<0.05,<0.01 was considered statistically significant, denoted by *and #, respectively.
Results
Effects of ASIV on Body Weight and Liver Weight of NAFLD Model Mice
Changes in the body weight of mice in each group were measured after 6 weeks of intervention and at the end of the experiment after 12 weeks (Figure 1A). A high-fat emulsion diet increased body weight in the HFD group compared to the control group after 6 weeks (P<0.01). Treatment with ASIV significantly attenuated the body weight increase compared to the HFD group after 12 weeks (P<0.01). As shown in Figure 1B, the liver weights of the mice were significantly reduced in the ASIV group in response to 6 weeks ASIV treatment compared to the HFD group (P<0.01).
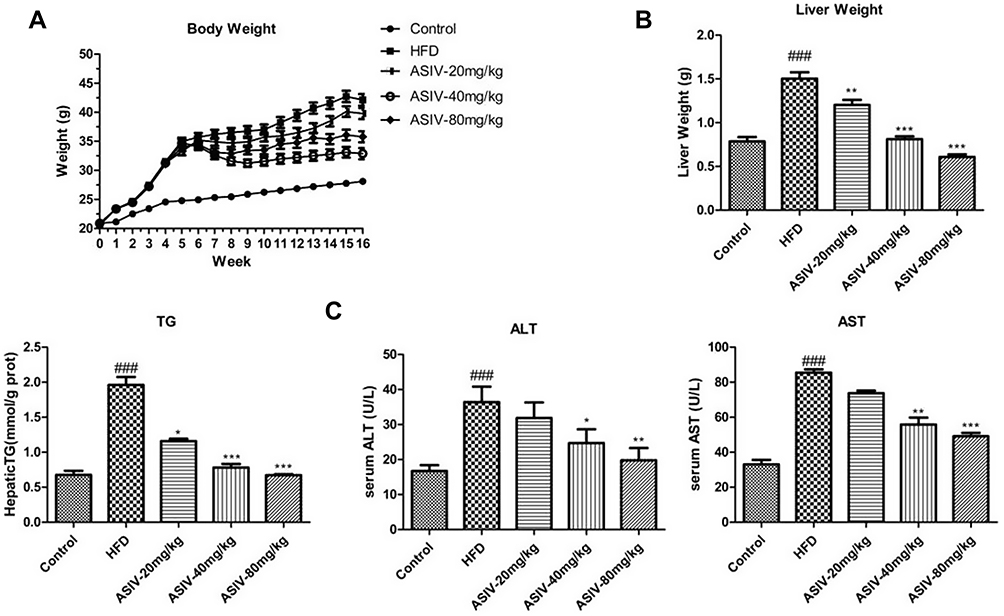 |
Figure 1 ASIV alleviated lipid accumulation in mice fed with HFD. (A) Body weight, (B) liver weight of mice, (C) TG, serum ALT and AST levels; Data expressed as mean ± S.D. (n =8). ###P< 0.01, compared to the Control. *,**P < 0.05, ***P < 0.01, compared to the HFD. |
ASIV Alleviated Liver Injury in HFD-Induced Fatty Liver
Serum TG, ALT and AST levels increased markedly in the HFD group compared to the control group (Figure 1C, P<0.01) which confirmed the successful establishment of the NAFLD model in our study. These findings were in parallel with histopathological alterations observed in HE staining of liver sections. HE staining indicated that ASIV treatment of mice significantly attenuated liver pathological injury (Figure 2) and liver inflammation. These results indicated that ASIV treatment can significantly attenuate liver injury.
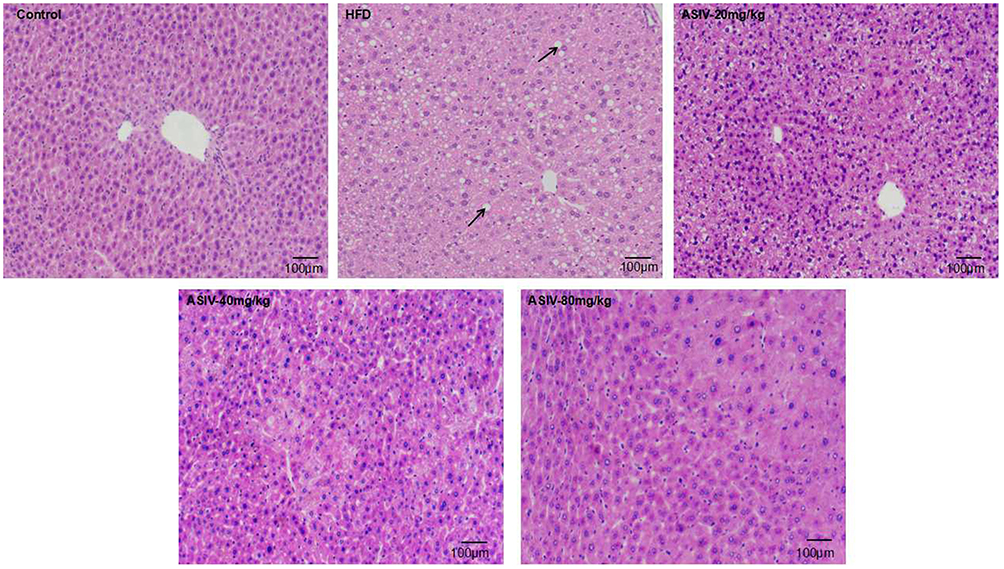 |
Figure 2 ASIV alleviated liver histopathological damage in mice fed with HFD. E staining of liver section of mice. Black arrow indicated lipid accumulation. Lipid was accumulated within hepatocytes (white circle) (magnification 100X). |
Effect of ASIV on the Viability of PA-Induced LO2 Cells
To explore the pathogenesis of NAFLD, 1 μg/mL LPS and 0.25 mM PA were used in RAW264.7 cells and LO2 cells to establish NAFLD models in vitro. We tested the effect of ASIV at a concentration range of 0–200 μg/mL on cell viability. Cell viability decreased significantly at 200 μg/mL ASIV concentrations. Therefore, we selected 20 μg/mL, 60 μg/mL and 100 μg/mL of ASIV for interference with RAW264.7 cell and LO2 cell induction by LPS and PA respectively.
Effects of ASIV on Lipid Accumulation in PA-Induced LO2 Cells
To confirm whether ASIV affects PA-induced LO2 cell lipid accumulation, LO2 cells were treated with 0.25 mM PA for 24 h, and then cells were treated with ASIV for 24 h. AS shown in Figure 3, it was observed that lipid droplets were significant in PA-induced LO2 cells. Furthermore, the addition of ASIV to PA-induced cells significantly decreased the lipid accumulation. Consistently, compared with the control group, the intracellular TG and TC contents were dramatically increased by PA treatment. However, TC and TG levels in the ASIV+PA group were significantly lower than those in the PA group. Thus, ASIV can inhibit PA-induced lipid accumulation in LO2 cells.
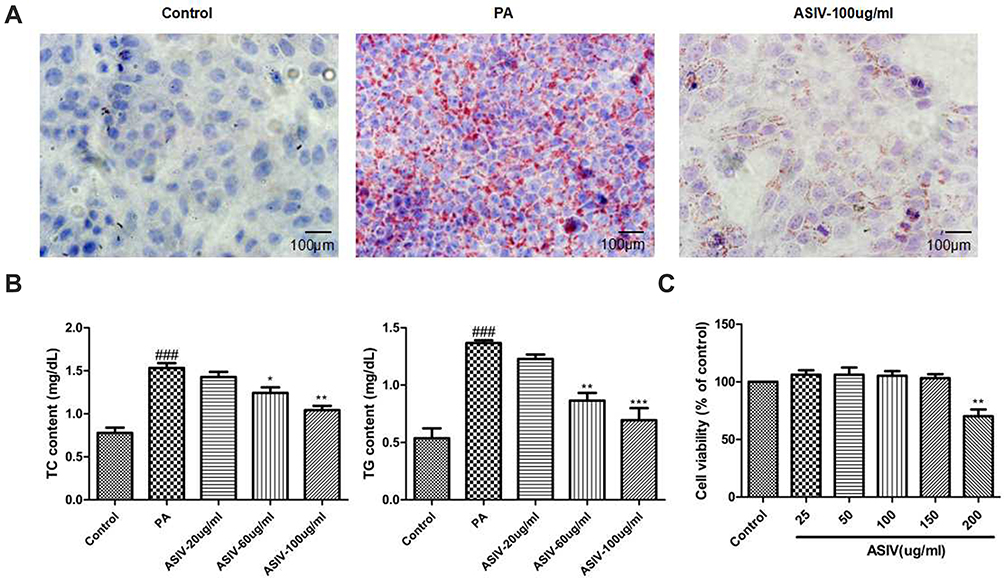 |
Figure 3 ASIV attenuates lipid accumulation in LO2 cells induced by PA. (A) Oil Red O staining were evaluated to examine the level of lipid accumulation (magnification 100X). (B) The concentration of TG and TC in LO2 cells. (C) Cell viability was tested using MTT assay. ###P< 0.01 compared to the Control, while *,**P< 0.05 and ***P < 0.01 compared to the PA-treated group. |
Effects of ASIV on Oxidative Stress in PA-Induced LO2 Cells
We explored the effect of ASIV on PA-induced ROS, MDA and GSH-Px generation in LO2 cells. As depicted in Figure 4, PA exposure markedly enhanced the content of ROS and MDA, while GSH-Px activity significantly decreased, however, incubation with ASIV dramatically abolished these trends. Our results suggested that ASIV suppressed PA-induced oxidative stress in LO2 cells.
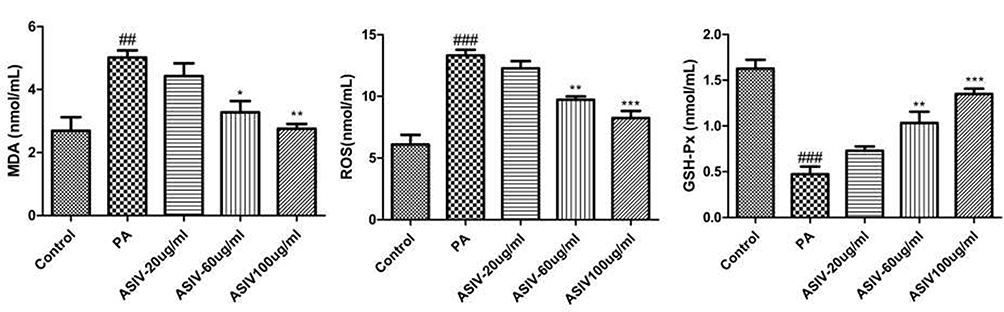 |
Figure 4 ASIV inhibits PA-induced oxidative stress in LO2 cells. The levels of ROS, MDA, and the activity of f GSH-Px were measured by commercial kits. ##P < 0.05 and ###P< 0.01 compared to the Control, while *,**P < 0.05 and ***P < 0.01 compared to the PA-treated group. |
Effects of ASIV on Apoptosis in PA-Induced LO2 Cells
To validate the contribution of ASIV to PA-induced LO2 cell apoptosis, we revealed that PA was mitigated by an Annexin V-FITC/PI double staining assay. Consistently, as shown in Figure 5, incubation with PA upregulated the mRNA levels of Bax and Bax protein expression, whereas down-regulated the mRNA level of Bcl-2 and Bcl-2 protein expression. Together, these data demonstrate that ASIV inhibits PA-induced apoptosis in LO2 cells.
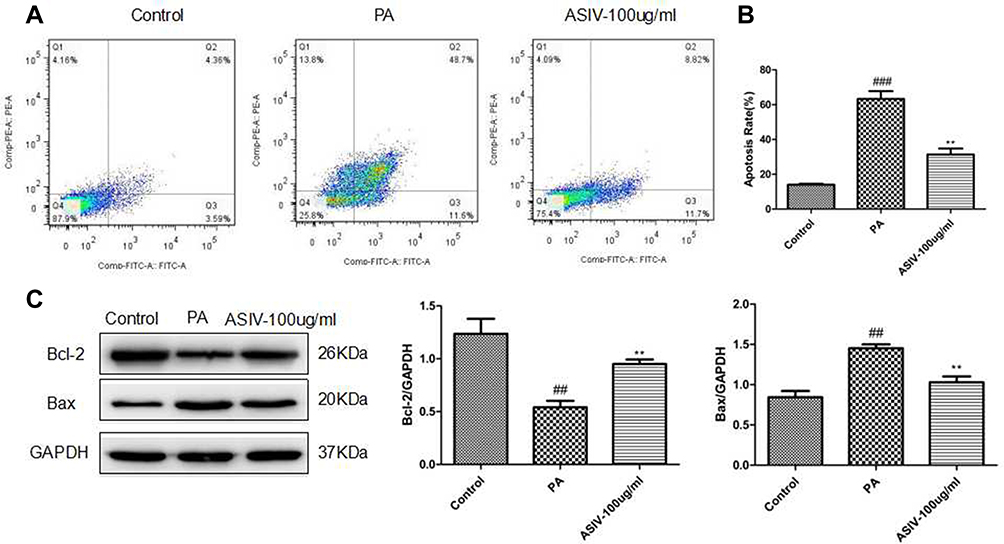 |
Figure 5 ASIV suppressed apoptosis in PA-induced LO2 cells. (A) Apoptosis of LO2 cells was examined by flow cytometry. (B) Apoptosis rate was qualified. (C) The expression of apoptosis-related proteins including Bcl-2 and Bax was tested using Western blot. ##P< 0.05 and ###P< 0.01 compared to the control, while **P < 0.05 compared to the PA-treated group. |
ASIV Can Effectively Improve the Production of Inflammatory Factors in RAW264.7 Cells Induced by LPS
To assess whether ASIV inhibits inflammation, we examined the levels of the inflammatory factors TNF-α and IL-6 in the cell culture supernatants. As shown in Figure 6, we examined the secretion of IL-6 and TNF-α in RAW264.7 cells, which were markedly decreased in response to ASIV treatment. Consistent with the ELISA results, we used qRT-PCR to examine IL-6 and TNF-α levels. The LPS treated group significantly improved the mRNA levels of IL-6 and TNF-α, while ASIV inhibited such increases. Collectively, these data support that ASIV effectively inhibits inflammation in RAW264.7 cells.
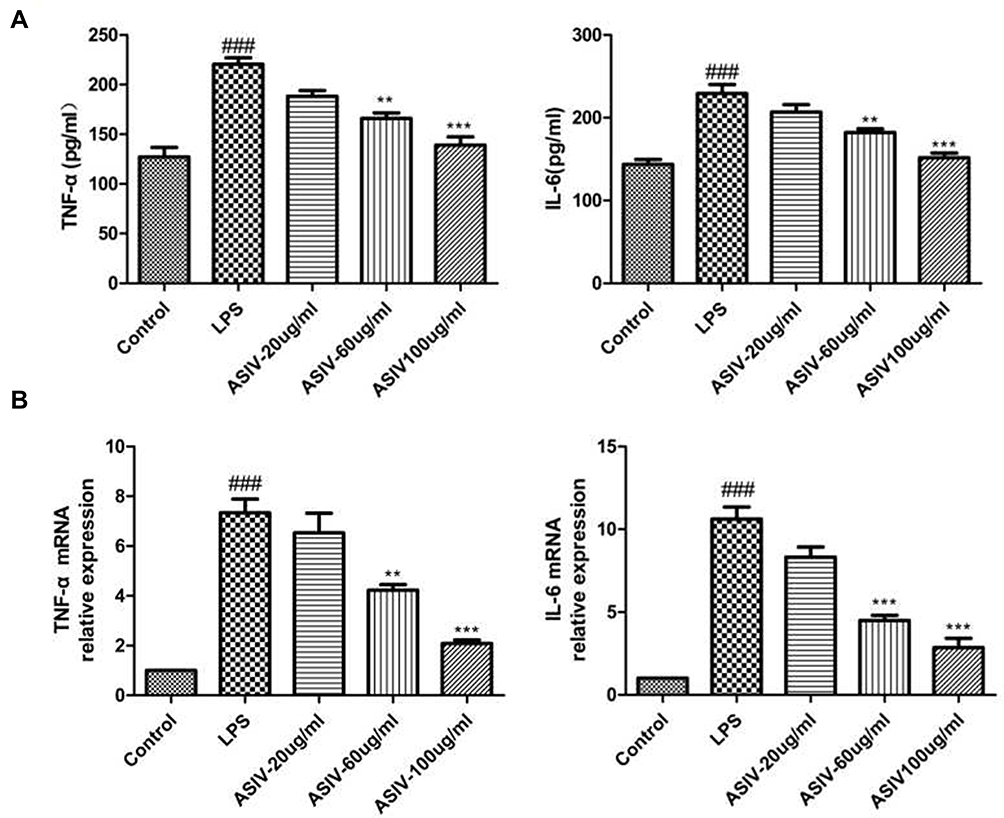 |
Figure 6 LPS-induced the expression of inflammatory cytokines in RAW264.7 cells. (A) The contents of TNF-α and IL-6 were measured using ELISA kits. (B) IL-6 and TNF-α mRNA expression in RAW264.7 cells was analysed by real-time PCR. ###P< 0.01 compared to the control, while **P < 0.05 and ***P < 0.01 compared to the LPS-treated group. |
ASIV Protects NAFLD Mice from HFD-Induced Inflammation by Inhibiting the 5-LO/LTB4 Pathway
To confirm further the possible regulatory mechanisms by which ASIV protects NAFLD mice, the levels of inflammation related proteins (5-LO and LTA4H) were examined by Western blotting. AS shown in Figure 7, we found that ASIV reduced the levels of 5-LO and LTA4H in NAFLD mice. The mRNA levels of 5-LO and LTA4H showed a similar tendency. ELISA revealed that ASIV dramatically upregulated the concentrations of LTB4 in NAFLD mice. Finally, we found that the 5-LO metabolite LTB 4 levels markedly improved in NAFLD mice. Furthermore, ASIV treatment reversed the elevation of LTB4 levels, indicating that 5-LO/LTB4 signaling was enhanced in NAFLD mice. However, ASIV could reverse this tendency.
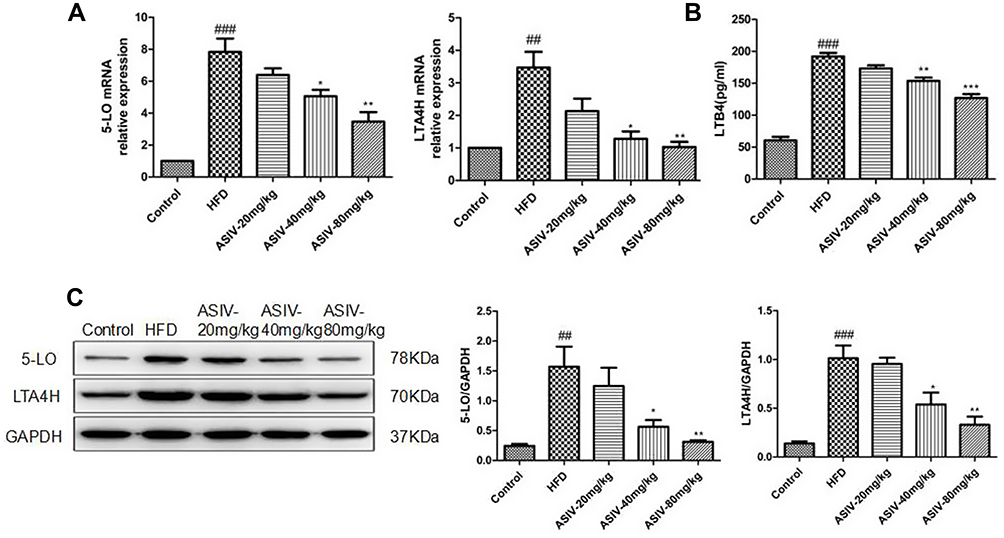 |
Figure 7 ASIV regulated 5-LOX/LTB4 pathing in NAFLD mice. (A) 5-LOX and LTA4H mRNA expression in cells was analyzed by real-time PCR. (B)The contents of LTB4 were measured using ELISA kits. (C) 5-LO and LTA4H protein levels in cells were analysed by Western blotting. ##P < 0.05 and ###P< 0.01 compared to the control, while *,**P < 0.05 and ***P < 0.01 compared to the HFD-treated group. |
Discussion
The progression of non-alcoholic fatty liver to non-alcoholic steatohepatitis is mainly due to the accumulation of liver lipids that triggers lipotoxicity, promotes macrophage activation, and has multiple effects with inflammation and oxidative stress, resulting in fibrosis and other adverse outcomes.31 Since the pathogenesis of NASH is a complex molecular network of reprogramming, effective treatment strategies should target a variety of pathological pathways and have a protective effect on all aspects of liver function.32 In this study, we reported that astragaloside IV is involved in multiple beneficial effects in the disease.
Under physiological conditions, free fatty acids, triglycerides and total cholesterol in the body are in a dynamic balance.33 Hepatic steatosis is caused by the imbalance between the increase and loss of lipid accumulation in the liver, increased lipolysis of visceral adipose tissue, activation of hepatic new fat formation, and diet rich in high calories and fat, Liver intramitochondrial beta-oxidation is reduced, leading to increased production and accumulation of plasma free fatty acids, oxidized low-density lipoproteins and triglycerides.34 A large number of studies have revealed that mice fed a high-fat diet have increased body weight and liver index, and obvious steatosis, hepatitis, dyslipidemia and liver lipid peroxidation. Here, we further evaluated liver lipid metabolism changes by applying in vivo mice and in vitro LO2 cell NAFLD models. Consistent with previous studies,30 our data demonstration that the body weights and liver weight in the mice disease model were significantly increased, the levels of liver injury markers (ALT and AST) and triglycerides were significantly increased, and the liver tissue pathology showed obvious inflammatory infiltration and steatosis. ASIV treatment significantly reduced the above indicators, representing that ASIV effectively improved lipid metabolism disorders and liver function in NAFLD mice. In vivo studies first clarified the close relationship between ASIV and NAFLD. Previous studies have exposed that ASIV reduces lipid accumulation in HepG2 cells and mice primary liver cells.28 Compared with his research, our results in LO2 cells display that compared with the in vitro NAFLD model group, ASIV administration significantly reduced TC and TG levels. Oil red O staining also indicated that ASIV treatment significantly reduced the formation of intracellular lipid droplets. The above in vitro and in vivo experiments consistently prove that ASIV can reduce lipid accumulation in NAFLD.
Lipotoxicity refers to the accumulation of ectopic lipids in nonfat tissues and entry into harmful pathways, leading to inflammation, and oxidative stress and cell apoptosis.35 Mitochondrial dysfunction can impair fat homeostasis in the liver and cause excessive production of ROS.36 These ROS are believed to play an important role in inducing the fatal liver cell damage associated with NAFLD.36 Mitochondria adapt to increased liver lipid content by increasing Beta-oxidation, however, this leads to an increase in the production of ROS, this increase in mitochondrial ROS production may play an important role in the occurrence of insulin resistance in the later stages of NAFLD.37 Oxidative stress occurs through the release of ROS, the combination of ROS and polyunsaturated fatty acids (PUFA) leads to lipid peroxidation and produces aldehyde byproducts (MDA), which cause oxidative damage to cells.38 Changes in lipid peroxidation markers (GSH-Px and MDA) and other substances have been observed in NAFLD/NASH patients, and their concentration is related to the degree of disease.39 In this experiment, we tested the markers of oxidative stress, the content of ROS, MDA and GSH-Px. The results exhibited that after PA induction, the content of ROS and MDA increased significantly, and the content of GSH-Px decreased sharply, representative that the cells experienced severe oxidative stress, dysfunction of the antioxidant system; however in the PA-induced cells treated with ASIV, the decrease in ROS and MDA levels and the increase in GSH-Px levels designate that the antioxidant capacity of the cells was improved, and relieved cell stress damage. This finding suggests that ASIV may protect against NAFLD by inhibiting hepatic oxidative stress in the liver.
Hepatocyte apoptosis is an important mechanism for the development of NAFLD disease.40 Apoptotic hepatocytes stimulate immune cells and hepatic stellate cells to develop liver fibrosis by producing inflammasomes and cytokines.41 Apoptosis is mediated by the death receptor external pathway or internal mitochondrial organelle pathway.42 In steatosis, with the accumulation of free saturated fatty acids, mitochondrial transition pores (mPTP) and mitochondrial DNA are damaged, ATP synthesis efficiency is reduced, ROS is overproduced, proapoptotic Bax protein is activated, Bcl-2 protein is inhibited, and outer mitochondrial membrane permeability (MOMP) and Bax channels are opened during apoptosis.43 Our results illustration that ASIV can significantly ameliorate PA-induced hepatocyte apoptosis, reduce Bax protein expression, and restore Bcl-2 protein expression. In general, consistent with previous reports, the expression changes of Bax and Bcl-2 mediated by mitochondrial damage play an important role in NAFLD cell apoptosis.15 These results demonstrate that ASIV may produce its protective effects by inhibiting hepatic cell apoptosis in NAFLD.
Inflammation is the basis of many pathophysiological processes, and macrophages play a key role in the initiation, maintenance and regression of inflammation.44 NASH is an inflammatory disease, and continuous inflammation and immune cell infiltration are important driving forces of disease progression.45 Most of the current research on NASH disease focuses on the study of liver lipid accumulation and metabolism. If simple steatosis cannot be controlled, then inflammation in the liver will begin, during which macrophages can be activated and released.46 Inflammation-related cytokines can recruit a large number of bone marrow-derived macrophages into the liver, and differentiate into M1 type macrophages that promote disease progression.47 M1 type macrophages play a role in the later stage of disease development, and the effect is greater than that of Kupffer cells, which continuously accumulate in the liver and release a steady stream of inflammatory cytokines.19,20 RAW264.7 cells are mice macrophages and are often used to construct in vitro models of NASH.48 Our research used LPS-induced RAW264.7 cells as a model to study the role and mechanism of ASIV in NAFLD.
The main source of proinflammatory cytokines (TNF-α and IL-6) is released in the pathophysiological process of NASH is macrophages.49 TNF-α is the central link in the cascade of inflammation, which can increase the release of other proinflammatory factors and accelerate the process of inflammation.50 IL-6 is both a proinflammatory factor and an anti-inflammatory factor, involved in a variety of biological processes. TNF-α and IL-6 can directly reflect the severity of inflammation in the body.51 Therefore, we examined the effect of ASIV on TNF-α and IL-6 in LPS-treated RAW264.7 cells. Our results displayed that ASIV downregulated the levels of TNF-α and IL-6 in the supernatant of RAW264.7 cells. At the same time, ASIV also inhibited the upregulation of TNF-α and IL-6 mRNA in RAW264.7 cells induced by LPS. These data imply that ASIV can effectively inhibit the overproduction of inflammatory mediators in macrophages.
Leukotriene is a classic pro-inflammatory lipid metabolism intermediate product. Arachidonic acid (AA), which is converted from essential fatty acid n-6 polyunsaturated fatty acid (PUFA), is formed under catalysis by 5-lipoxygenase, and then expressed by the high-affinity receptor BLT-1 and low-affinity receptor BLT-2.52 Broad-spectrum and high-efficiency inflammatory effects include the release, among others, of chemokines and lymphocytes, with 5-LO and LTB4 as the core signaling molecules mediating the process of inflammation triggering and amplification.53 The 5-LO pathway is the main proinflammatory pathway in macrophages.54 Previous studies have shown that the 5-LO pathway generates the inflammatory mediator LTB4 which is an important factor leading to immune liver injury.11 In our study, ASIV significantly reduced the expression of 5-LO and LTB4 in NAFLD mice. These findings denote that ASIV may attenuate NAFLD mice inflammation by inhibiting the 5-LO/LTB4 pathway.
Polyphenols are known to be the most common antioxidants in the human diet, which are mainly found in fruits, vegetables and herbs, including compounds such as chlorogenic acid, catechins, curcumin and quercetin.55 Studies have revealed that polyphenol compounds can prevent oxidative stress and regulate insulin resistance, and play an important role in the treatment of non-alcoholic fatty liver disease.56 Cardiovascular complications are the main cause of death in NAFLD, recently studies have exposed that resveratrol can reduce BMI and waist circumference in patients with NAFLD, but blood lipids, serum atherosclerosis index, liver enzymes and blood pressure have not changed significantly.57 Our research illustrations that ASIV can reduce blood lipids and body weight in mice. In follow-up studies, we can further pay attention to the role of ASIV in the complications of NAFLD.
In summary, our in vivo experiments have proven that ASIV can inhibit lipid accumulation, inflammation and liver damage in NAFLD mice, and our in vitro experiments further illustration that ASIV reduces lipid metabolism, oxidative stress and apoptosis in LO2 cells, thereby preventing NAFLD. These findings provide new ideas and new ways to treat of NAFLD using ASIV.
Acknowledgments
This work was supported by National Natural Science Foundation of China (№ 81660151, 81660751, 82060661) and Jiangxi Provincial Natural Science Foundation of China (No 20171BAB205085).
Disclosure
The authors have no conflicts of interest in this work.
References
1. Younossi Z, Tacke F, Arrese M, et al. Global perspectives on nonalcoholic fatty liver disease and nonalcoholic steatohepatitis. Hepatology. 2019;69(6):2672–2682. doi:10.1002/hep.30251
2. Golabi P, Stepanova M, Pham HT, et al. Non-alcoholic steatofibrosis (NASF) can independently predict mortality in patients with non-alcoholic fatty liver disease (NAFLD). BMJ Open Gastroenterol. 2018;5(1):e000198. doi:10.1136/bmjgast-2018-000198
3. Younossi ZM, Otgonsuren M, Henry L, et al. Association of nonalcoholic fatty liver disease (NAFLD) with hepatocellular carcinoma (HCC) in the United States from 2004 to 2009. Hepatology. 2015;62(6):1723–1730. doi:10.1002/hep.28123
4. Estes C, Anstee QM, Arias-Loste MT, et al. Modeling NAFLD disease burden in China, France, Germany, Italy, Japan, Spain, United Kingdom, and United States for the period 2016–2030. J Hepatol. 2018;69(4):896–904. doi:10.1016/j.jhep.2018.05.036
5. Manne V, Handa P, Kowdley KV. Pathophysiology of nonalcoholic fatty liver disease/nonalcoholic steatohepatitis. Clin Liver Dis. 2018;22(1):23–37. doi:10.1016/j.cld.2017.08.007
6. Bessone F, Razori MV, Roma MG. Molecular pathways of nonalcoholic fatty liver disease development and progression. Cell Mol Life Sci. 2019;76(1):99–128.
7. Neuschwander-Tetri BA. Non-alcoholic fatty liver disease. BMC Med. 2017;15(1):45. doi:10.1186/s12916-017-0806-8
8. Wardani HA, Rahmadi M, Ardianto C, Balan SS, Kamaruddin NS, Khotib J. Development of nonalcoholic fatty liver disease model by high-fat diet in rats. J Basic Clin Physiol Pharmacol. 2019;30(6). doi:10.1515/jbcpp-2019-0258
9. Van Herck MA, Vonghia L, Francque SM. Animal models of nonalcoholic fatty liver disease-a starter’s guide. Nutrients. 2017;9(10):1072. doi:10.3390/nu9101072
10. Nevzorova YA, Boyer-Diaz Z, Cubero FJ, Gracia-Sancho J. Animal models for liver disease - A practical approach for translational research. J Hepatol. 2020;73(2):423–440. doi:10.1016/j.jhep.2020.04.011
11. Pei K, Gui T, Kan D, et al. An overview of lipid metabolism and nonalcoholic fatty liver disease. Biomed Res Int. 2020;2020:4020249. doi:10.1155/2020/4020249
12. Puri P, Wiest MM, Cheung O, et al. The plasma lipidomic signature of nonalcoholic steatohepatitis. Hepatology. 2009;50(6):1827–1838. doi:10.1002/hep.23229
13. Spahis S, Delvin E, Borys JM, Levy E. Oxidative stress as a critical factor in nonalcoholic fatty liver disease pathogenesis. Antioxid Redox Signal. 2017;26(10):519–541. doi:10.1089/ars.2016.6776
14. Masarone M, Rosato V, Dallio M, et al. Role of oxidative stress in pathophysiology of nonalcoholic fatty liver disease. Oxid Med Cell Longev. 2018;2018:9547613. doi:10.1155/2018/9547613
15. Lee J, Park JS, Roh YS. Molecular insights into the role of mitochondria in non-alcoholic fatty liver disease. Arch Pharm Res. 2019;42(11):935–946. doi:10.1007/s12272-019-01178-1
16. Paradies G, Paradies V, Ruggiero FM, Petrosillo G. Oxidative stress, cardiolipin and mitochondrial dysfunction in nonalcoholic fatty liver disease. World J Gastroenterol. 2014;20(39):14205–14218. doi:10.3748/wjg.v20.i39.14205
17. Nassir F, Ibdah JA. Role of mitochondria in nonalcoholic fatty liver disease. Int J Mol Sci. 2014;15(5):8713–8742. doi:10.3390/ijms15058713
18. Fontes A, Alemany-Pages M, Oliveira PJ, Ramalho-Santos J, Zischka H, Azul AM. Antioxidant versus pro-apoptotic effects of mushroom-enriched diets on mitochondria in liver disease. Int J Mol Sci. 2019;20(16):3987. doi:10.3390/ijms20163987
19. Cha JY, Kim DH, Chun KH. The role of hepatic macrophages in nonalcoholic fatty liver disease and nonalcoholic steatohepatitis. Lab Anim Res. 2018;34(4):133–139. doi:10.5625/lar.2018.34.4.133
20. Kazankov K, Jorgensen SMD, Thomsen KL, et al. The role of macrophages in nonalcoholic fatty liver disease and nonalcoholic steatohepatitis. Nat Rev Gastroenterol Hepatol. 2019;16(3):145–159. doi:10.1038/s41575-018-0082-x
21. Luo ZL, Ren JD, Huang Z, et al. The role of exogenous hydrogen sulfide in free fatty acids induced inflammation in macrophages. Cell Physiol Biochem. 2017;42(4):1635–1644. doi:10.1159/000479405
22. Xu XM, Deng JJ, Yuan GJ, et al. 5-Lipoxygenase contributes to the progression of hepatocellular carcinoma. Mol Med Rep. 2011;4(6):1195–1200. doi:10.3892/mmr.2011.547
23. Lee SJ, Seo KW, Kim CD. LPS increases 5-lo expression on monocytes via an activation of Akt-Sp1/NF-kappaB pathways. Korean J Physiol Pharmacol. 2015;19(3):263–268. doi:10.4196/kjpp.2015.19.3.263
24. Nakamura Y, Kozuka M, Naniwa K, et al. Arachidonic acid cascade inhibitors modulate phorbol ester-induced oxidative stress in female ICR mice skin: differential roles of 5-lipoxygenase and cyclooxygenase-2 in leukocyte infiltration and activation. Free Radic Biol Med. 2003;35(9):997–1007. doi:10.1016/S0891-5849(03)00440-4
25. Martinez-Clemente M, Claria J, Titos E. The 5-lipoxygenase/leukotriene pathway in obesity, insulin resistance, and fatty liver disease. Curr Opin Clin Nutr Metab Care. 2011;14(4):347–353. doi:10.1097/MCO.0b013e32834777fa
26. Shi T, Wu L, Ma W, et al. Nonalcoholic fatty liver disease: pathogenesis and treatment in traditional Chinese medicine and western medicine. Evid Based Complement Alternat Med. 2020;2020:8749564. doi:10.1155/2020/8749564
27. Li L, Hou X, Xu R, Liu C, Tu M. Research review on the pharmacological effects of astragaloside IV. Fundam Clin Pharmacol. 2017;31(1):17–36. doi:10.1111/fcp.12232
28. Zhou B, Zhou DL, Wei XH, Zhong RY, Xu J, Sun L. Astragaloside IV attenuates free fatty acid-induced ER stress and lipid accumulation in hepatocytes via AMPK activation. Acta Pharmacol Sin. 2017;38(7):998–1008. doi:10.1038/aps.2016.175
29. Li L, Huang W, Wang S, et al. Astragaloside IV attenuates acetaminophen-induced liver injuries in mice by activating the Nrf2 signaling pathway. Molecules. 2018;23(8).
30. Huang F, Wang J, Yu F, et al. Protective effect of meretrix meretrix oligopeptides on high-fat-diet-induced non-alcoholic fatty liver disease in mice. Mar Drugs. 2018;16(2):39. doi:10.3390/md16020039
31. Dludla PV, Nkambule BB, Mazibuko-Mbeje SE, et al. N-acetyl cysteine targets hepatic lipid accumulation to curb oxidative stress and inflammation in NAFLD: a comprehensive analysis of the literature. Antioxidants (Basel). 2020;9(12).
32. Wong VWS. Nonalcoholic fatty liver disease in Asia: a story of growth. J Gastroenterol Hepatol. 2013;28(1):18–23. doi:10.1111/jgh.12011
33. Friedman SL, Neuschwander-Tetri BA, Rinella M, Sanyal AJ. Mechanisms of NAFLD development and therapeutic strategies. Nat Med. 2018;24(7):908–922. doi:10.1038/s41591-018-0104-9
34. Byrne CD, Targher G. NAFLD: a multisystem disease. J Hepatol. 2015;62(1 Suppl):S47–S64. doi:10.1016/j.jhep.2014.12.012
35. Mendez-Sanchez N, Cruz-Ramon VC, Ramirez-Perez OL, Hwang JP, Barranco-Fragoso B, Cordova-Gallardo J. New aspects of lipotoxicity in nonalcoholic steatohepatitis. Int J Mol Sci. 2018;19(7):2034. doi:10.3390/ijms19072034
36. Yang J, Fernandez-Galilea M, Martinez-Fernandez L, et al. Oxidative stress and non-alcoholic fatty liver disease: effects of omega-3 fatty acid supplementation. Nutrients. 2019;11(4):872. doi:10.3390/nu11040872
37. Ore A, Akinloye OA. Oxidative stress and antioxidant biomarkers in clinical and experimental models of non-alcoholic fatty liver disease. Medicina. 2019;55(2).
38. Xu L, Nagata N, Ota T. Impact of glucoraphanin-mediated activation of Nrf2 on non-alcoholic fatty liver disease with a focus on mitochondrial dysfunction. Int J Mol Sci. 2019;20(23):5920. doi:10.3390/ijms20235920
39. Egnatchik RA, Leamy AK, Noguchi Y, Shiota M, Young JD. Palmitate-induced activation of mitochondrial metabolism promotes oxidative stress and apoptosis in H4IIEC3 rat hepatocytes. Metabolism. 2014;63(2):283–295. doi:10.1016/j.metabol.2013.10.009
40. Kanda T, Matsuoka S, Yamazaki M, et al. Apoptosis and non-alcoholic fatty liver diseases. World J Gastroenterol. 2018;24(25):2661–2672. doi:10.3748/wjg.v24.i25.2661
41. Afonso MB, Castro RE, Rodrigues CMP. Processes exacerbating apoptosis in non-alcoholic steatohepatitis. Clin Sci. 2019;133(22):2245–2264. doi:10.1042/CS20190068
42. Beier JI, Banales JM. Pyroptosis: an inflammatory link between NAFLD and NASH with potential therapeutic implications. J Hepatol. 2018;68(4):643–645. doi:10.1016/j.jhep.2018.01.017
43. Schuster S, Johnson CD, Hennebelle M, et al. Oxidized linoleic acid metabolites induce liver mitochondrial dysfunction, apoptosis, and NLRP3 activation in mice. J Lipid Res. 2018;59(9):1597–1609. doi:10.1194/jlr.M083741
44. Martinez FO, Helming L, Gordon S. Alternative activation of macrophages: an immunologic functional perspective. Annu Rev Immunol. 2009;27:451–483. doi:10.1146/annurev.immunol.021908.132532
45. Mosser DM, Edwards JP. Exploring the full spectrum of macrophage activation. Nat Rev Immunol. 2008;8(12):958–969. doi:10.1038/nri2448
46. Ju C, Tacke F. Hepatic macrophages in homeostasis and liver diseases: from pathogenesis to novel therapeutic strategies. Cell Mol Immunol. 2016;13(3):316–327. doi:10.1038/cmi.2015.104
47. Ministrini S, Montecucco F, Sahebkar A, Carbone F. Macrophages in the pathophysiology of NAFLD: the role of sex differences. Eur J Clin Invest. 2020:e13236.
48. Kanuri G, Bergheim I. In vitro and in vivo models of non-alcoholic fatty liver disease (NAFLD). Int J Mol Sci. 2013;14(6):11963–11980. doi:10.3390/ijms140611963
49. Cobbina E, Akhlaghi F. Non-alcoholic fatty liver disease (NAFLD) - pathogenesis, classification, and effect on drug metabolizing enzymes and transporters. Drug Metab Rev. 2017;49(2):197–211. doi:10.1080/03602532.2017.1293683
50. Ceccarelli S, Panera N, Mina M, et al. LPS-induced TNF-α factor mediates pro-inflammatory and pro-fibrogenic pattern in non-alcoholic fatty liver disease. Oncotarget. 2015;6(39):41434–41452. doi:10.18632/oncotarget.5163
51. Schmidt-Arras D, Rose-John S. IL-6 pathway in the liver: from physiopathology to therapy. J Hepatol. 2016;64(6):1403–1415. doi:10.1016/j.jhep.2016.02.004
52. Sonnweber T, Pizzini A, Nairz M, Weiss G, Tancevski I. Arachidonic acid metabolites in cardiovascular and metabolic diseases. Int J Mol Sci. 2018;19(11):3285. doi:10.3390/ijms19113285
53. Di Gennaro A, Haeggstrom JZ. The leukotrienes: immune-modulating lipid mediators of disease. Adv Immunol. 2012;116:51–92.
54. Sun QY, Zhou HH, Mao XY. Emerging roles of 5-lipoxygenase phosphorylation in inflammation and cell death. Oxid Med Cell Longev. 2019;2019:2749173. doi:10.1155/2019/2749173
55. de Araujo FF, de Paulo Farias D, Neri-Numa IA, Pastore GM. Polyphenols and their applications: an approach in food chemistry and innovation potential. Food Chem. 2021;338:127535.
56. Shabalala SC, Dludla PV, Mabasa L, et al. The effect of adiponectin in the pathogenesis of non-alcoholic fatty liver disease (NAFLD) and the potential role of polyphenols in the modulation of adiponectin signaling. Biomed Pharmacother. 2020;131:110785. doi:10.1016/j.biopha.2020.110785
57. Farzin L, Asghari S, Rafraf M, Asghari-Jafarabadi M, Shirmohammadi M. No beneficial effects of resveratrol supplementation on atherogenic risk factors in patients with nonalcoholic fatty liver disease. International Journal for Vitamin and Nutrition Research. 2020;90(3–4):279–289. doi:10.1024/0300-9831/a000528
 © 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.