")
Back to Journals » Drug Design, Development and Therapy » Volume 12
Asthma and poly(ADP-ribose) polymerase inhibition: a new therapeutic approach
Authors Zaffini R, Gotte G, Menegazzi M
Received 5 September 2017
Accepted for publication 11 December 2017
Published 12 February 2018 Volume 2018:12 Pages 281—293
DOI https://doi.org/10.2147/DDDT.S150846
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Sukesh Voruganti
Raffaela Zaffini, Giovanni Gotte, Marta Menegazzi
Department of Neuroscience, Biomedicine and Movement Science, Biochemistry Section, University of Verona, Verona, Italy
Abstract: Asthma is a chronic lung disease affecting people of all ages worldwide, and it frequently begins in childhood. Because of its chronic nature, it is characterized by pathological manifestations, including airway inflammation, remodeling, and goblet cell hyperplasia. Current therapies for asthma, including corticosteroids and beta-2 adrenergic agonists, are directed toward relieving the symptoms of the asthmatic response, with poor effectiveness against the underlying causes of the disease. Asthma initiation and progression depends on the T helper (Th) 2 type immune response carried out by a complex interplay of cytokines, such as interleukin (IL) 4, IL5, and IL13, and the signal transducer and activator of transcription 6. Much of the data resulting from different laboratories support the role of poly(ADP-ribose) polymerase (PARP) 1 and PARP14 activation in asthma. Indeed, PARP enzymes play key roles in the regulation and progression of the inflammatory asthma process because they affect the expression of genes and chemokines involved in the immune response. Consistently, PARP inhibition achievable either upon genetic ablation or by using pharmacological agents has shown a range of therapeutic effects against the disease. Indeed, in the last two decades, several preclinical studies highlighted the protective effects of PARP inhibition in various animal models of asthma. PARP inhibitors showed the ability to reduce the overall lung inflammation acting with a specific effect on immune cell recruitment and through the modulation of asthma-associated cytokines production. PARP inhibition has been shown to affect the Th1–Th2 balance and, at least in some aspects, the airway remodeling. In this review, we summarize and discuss the steps that led PARP inhibition to become a possible future therapeutic strategy against allergic asthma.
Keywords: allergic airway disease, PARP1, PARP14, remodeling, STAT6, Th1–Th2 balance, Th2 response
Introduction
More than five decades after the discovery of the poly(ADP-ribosyl)ation pathway,1 the potential benefits of treating some human life-threatening diseases, such as cancer and serious cardiovascular conditions, with inhibitors of that enzymatic system are a promising reality. In fact, many poly(ADP-ribose) polymerase (PARP) inhibitors have been enrolled in clinical trials as a combination therapy for several types of cancer, and many of these were licensed by the Food and Drug Administration (FDA) and/or the European Medicines Agency (EMA). To name a few, olaparib and rucaparib are licensed by FDA against recurrent ovarian cancer. Olaparib is also licensed by EMA as a maintenance therapy agent in patients displaying germline or somatic BRCA mutations.2 Finally, more than 10 years of clinical PARP research, and even more of preclinical phase, have been translated into promising therapies for patients.3,4 Starting from these exciting results, research in the PARP field is evolving to uncover the roles of this family of enzymes in multiple biological and pathological pathways related to many diseases, with the aim to discover new therapeutic approaches in combination or as single therapies.5
Chronic airway inflammatory diseases, such as asthma, are life-threatening conditions that impact on the quality of life and health care systems. Globally, over 300 million people are affected by asthma, and the incidence of the disease is increasing, affecting 1 in 10 children and 1 in 12 adults.6–8 Individuals suffering from this condition widely range from teenage children to elderly people. According to World Health Organization, asthma is annually responsible for over 180,000 deaths.9 Allergic asthma is a complex syndrome resulting from the interplay of genetic and environmental factors. It is characterized by three essential modifications of the airway features: inflammation, hyperresponsiveness, and remodeling.7,8,10 These manifestations lead to repeated breathlessness attacks, wheezing, coughing, and chest tightness often occurring after exposure to allergens, pollutants, infections, or drugs.7,11 Airway inflammation is a protective response to cellular injuries and represents also the main feature of asthma. However, when this beneficial response occurs in an uncontrolled way, it causes excessive cellular and tissue damage through the release of reactive oxygen species (ROS). These events result in chronic inflammation and destruction of normal tissue, enhancing DNA damage.12,13 Considering this mechanism, PARP enzymes (PARPs) have been shown to mediate inflammation in a wide range of animal models of diseases.13,14
Allergic asthma can be controlled by a combination of a corticosteroid (an anti-inflammatory agent) and a short- or long-acting beta-2 adrenergic agonist. Unfortunately, 10% of patients do not respond to these therapies, and even if those drugs are effective in controlling symptoms, they do not impact the progressive airway remodeling.15 Hence, new therapeutic approaches are needed.
In the last two decades, several studies highlighted the protective effects of PARPs inhibition in various animal models of asthma. The focus of this review is centered on the current understanding of the PARP inhibition as a possible new therapeutic approach to counteract human asthma.
PARPs involvement in allergic airway inflammation: evidences from studies on humans
Previous studies have shown a genetic correlation of PARPs with asthma and allergic rhinitis in humans. Concerning one of the PARP isoenzymes (PARP1), Tezcan et al16 analyzed 122 Turkish subjects affected by stable asthma in comparison with 180 normal controls coming from the same geographic region and also identified PARP1 polymorphism by performing polymerase chain reaction-based restriction analyses. The results of this study showed a positive correlation between wild-type PARP1 762 V allele and risk of developing asthma, which was indeed five times higher than in subjects lacking the allele. In contrast, PARP1 762 AA genotype conferred a 3.4-fold risk reduction.16 The object of another study is to investigate whether a single-nucleotide polymorphism of the PARP1 gene promoter was associated with a genetic susceptibility to allergic rhinitis in 110 subjects affected by this disease and 130 used as controls. In this study, the heterozygous genotype of the polymorphism promoter (−1,672) was found to be significantly associated with susceptibility to allergic rhinitis.17
Recently, the relevance and significance of these studies have been supported by the evidence described by Ghonim et al18 that the enzyme is activated in cells or tissues of asthmatic humans. To do so, they compared lung specimens and peripheral blood mononuclear cells of six asthmatic patients with those of four normal subjects. However, the validity of these results is reduced by the few number of human samples screened.
Anyway, these data point out, on the one hand, a close link between PARPs and allergic airway inflammation diseases in humans and, on the other hand, suggest that PARP1 gene polymorphism may represent one of the factors conferring protection or susceptibility to asthma and allergic rhinitis.
PARPs cellular functions: focus on inflammation
PARPs is a family of 18 enzymes capable of cleaving the ADP-ribose moiety of NAD+ and carrying it onto several nuclear substrates in a monomeric or, most likely, an oligomeric form.19 This posttranslational protein modification is named ADP-ribosylation reaction. A new nomenclature, based on the type of the enzymatic reaction catalyzed and the PARP structural features, has been proposed for this family of enzymes. Hence, the PARP family members are now referred to as ADP-ribosyltransferase diphtheria toxin-like (ARTD) family of proteins.20 However, in this review, the old, more common, “PARP” nomenclature has been used.
The first PARP enzyme discovered was PARP1 (ARTD1) enzyme, whose enzymatic action is strongly activated by single DNA strand breaks.1 At the beginning, PARP1 function was mainly associated with the repair mechanism of damaged DNA.1 Afterward, PARP1 and other PARP enzymes were recognized to be involved also in many other important cellular functions such as chromatin remodeling, cell differentiation, cell death or proliferation, and gene expression.14 The pleiotropic effects of these enzymes are mediated through the modification of the physicochemical properties of the recipient molecules. This modification is caused by the long anionic chains attached to the glutamate residues of different substrates and consisting of a maximum of 250 ADP-ribose monomers (PAR).21 PARP1 itself, which is responsible for about 90% of the total cellular poly ADP-ribosylation activities, is poly ADP-ribosylated together with histones, nuclear repair enzymes, and transcription factors.22 PARylation is a transient protein modification, as the polymer is rapidly split into ADP-ribose monomers and polymers by a poly(ADP-ribose) glycohydrolase.23 Free polymers also function as signaling molecules by attaching themselves to PAR binding motives present in various proteins.24 Indeed, PARylation is a multifunctional regulatory mechanism that is implicated in various cellular functions.25,26 In the last decade, several data highlighted the complex role of PARP1 in the modulation of gene expression. PARP1-dependent transcriptional regulation may be ascribable to histone modification and change in chromatin composition, which in turn could facilitate gene transcription.27–29 For a long time, several studies using PARP inhibitors demonstrated the pivotal role of PARP1 in inflammation.21,25
Inflammation processes are associated with high levels of oxidative stress. ROS are produced by a mitochondrial respiratory chain dysfunction, by activation of NADPH oxidase, and mostly by inducible nitric oxide synthase (iNOS) expression.30 It is worth mentioning that several cytokines and chemokines, secreted by the immunocompetent cells infiltrated at the inflammation site, elicit iNOS induction.31 In the tissue, iNOS generates also superoxide besides a high amount of nitric oxide; these two free radicals interact with each other to form the potent oxidant molecule peroxynitrite.32 Among the ROS and nitrogen species, peroxynitrite is the most harmful agent inducing deep injuries on lipids, proteins, and DNA.33 When peroxynitrite attacks the sugar phosphate backbone, it results in DNA strand breaks33,34 that elicit tissue cytotoxicity by triggering PARP activation.35 While apoptosis is a typical consequence of low to moderate peroxynitrite concentrations, the exposure of cells to increasing concentrations of the oxidant has been associated with broad necrosis.36,37 Concerning cell death, it has been known for a long time that, after intense inflammation stimuli, the overactivation of PARP1 causes an intracellular depletion of its NAD+ substrate. This effect is accompanied by a decrease of the cellular energy level and a consequent induction of necrosis.38 Recently, an interesting study by Fouquerel et al39 demonstrated that PARP1 can induce cell necrosis in response to high level of DNA strand breaks through a different mechanism. The first glycolytic enzyme hexokinase 1, which is primarily associated with the outer mitochondrial membrane, is ADP-ribosylated after DNA damage and translocated to the cytosol with a concomitant loss of its enzymatic activity. Indeed, the block of the glycolytic pathway observed after induction of DNA strand breaks can be ascribable to hexokinase 1 inactivation. Thus, the direct glycolysis inhibition, more than NAD+ depletion, could be responsible for the cell necrosis occurring in DNA-damaged cells.39 Moreover, some sirtuin members have been shown to play an important role in cell survival/death. NAD+ depletion, mediated by PARP1 activation in response to oxidative DNA damage, inhibits sirtuin1 deacetylase activity. In this context, the increase of PARP1 activity triggers the release of the apoptosis-inducing factor from mitochondria and causes apoptosis through a caspase-independent pathway.40,41 In addition, the PAR polymer itself was shown to work as a signal, eliciting an unusual form of cell death named parthanatos.42
Accordingly, PARP1 enzyme, when fully activated, is involved in several types of cell death: necrosis, parthanatos, and apoptosis caused by both caspase-dependent and caspase-independent pathways.41,43
The nuclear factor-kB (NF-kB) is the master transcriptional promoter of inflammatory mediators, such as cytokines, chemokines, adhesion molecules, and iNOS.44 Moreover, NF-kB is a molecule signaling the cell redox state because it is activated by ROS.45 Thus, the link between free radical damage, PARP1 and NF-kB activity, in the inflammation context is demonstrated.45–47 Because PARP1 is a sensor of genotoxic stress, a PARP1-deficient mouse cell model has been used to investigate the mechanism whereby DNA damage induces NF-kB activation.48 Stilmann et al48 reported that the PARylation of PARP1 triggers a cascade of events resulting in NF-kB nuclear translocation and activation. These events are accompanied by the assembly of PARP1 in a multiprotein complex where protein–protein interactions between PARP1, inhibitor kB kinase, and other proteins, but not NF-kB subunits themselves, are needed. On the contrary, Hassa et al49 showed that a direct interaction between PARP1 and NF-kB subunits is required to achieve the full NF-kB activation. This result demonstrates that PARP1 acts as an NF-kB coactivator through protein–protein interaction, independently of its enzyme activity.49 In a different way, by using a macrophage murine model, Liu et al50 showed a direct PARylation of the p65 NF-kB subunit and a consequent NF-kB activation. Accordingly, Zerfaoui et al51 demonstrated that PARylation of p65 NF-kB subunit elicits the nuclear retention of this transcription factor by decreasing its nuclear protein export. In conclusion, all data support the evidence that PARP1 elicits the transcription of several inflammatory genes by triggering NF-kB activation, although through different mechanisms.
In addition, PARP1 also modulates the activity of other transcription factors: activator protein 1, regulating cytokines production, Th differentiation,52 and the nuclear factor of activated T cells, playing a pivotal role in the regulation of T cell functions.53,54 These, besides NF-kB, are important inflammatory mediators.
For all these reasons, PARP1 inhibition could overcome inflammation because of multiple molecular events. For an overview of the effects of PARP inhibition or deletion occurring in different types of inflammatory diseases, see the reviews by Berger et al14 and Cuzzocrea.47
As concerning, other PARP family members beyond PARP1, PARP2 (ARTD2), and PARP3 (ARTD3) are also activated by DNA strand breaks. They are the only members of this family being DNA damage-dependent enzymes.55 Interestingly, both caspase 3 and caspase 8 cleave PARP2, suggesting that PARP2 may also be involved in modulating cellular necrosis/apoptosis.56 PARP2 knockdown reduces inflammation in interleukin (IL)10-deficient mouse models of chronic colitis,57 whereas no data connecting PARP2 or PARP3 to asthma have been found so far.
Many studies showed that PARP14 (ARTD8) is involved in transcription, signal transduction pathways, and cytoskeleton regulation.58 PARP14 is enzymatically active by catalyzing mono-ADP ribosylation and belongs to a subfamily that contains repeats of a domain found in the nonhistone-like region of a histone macro-H2A variant.59 Although the macrodomains of the macro-H2A histone participate in gene silencing,60 the PARP14 macrodomains interact with the signal transducer and activator of transcription (STAT)6 by increasing the IL4-elicited gene expression.59 By the way, the PARP14-elicited transcriptional activation mechanism will be analyzed in the following section.
Mechanisms involving PARP family members in immune system and allergic asthma
Many years ago, we demonstrated that PARP1 gene expression, low in human resting T lymphocytes, rapidly increases after activation and 12-O-tetradecanoylphorbol-13-acetate (TPA)-induced differentiation of human T cells.61,62 In another study, we measured gene expression and the activity levels of PARP1 in 17 rat tissues. Our data demonstrated the presence of higher levels of PARP1 in rat thymus compared to the other tissues but testis.63 Indeed, the induction of PARP1 gene transcription, together with the increase in PARP1 protein level and activity, suggested a functional role of this enzyme in the immune system.61–64
At least two members of this family, PARP1 and PARP14, have already been demonstrated to play an important role in allergic inflammatory diseases, as it is asthma.
Saenz et al65 demonstrated that PARP1 regulates the expression of a large number of genes, including those coding for chemokines and cytokines in T cells. T cells deriving from PARP deficiency (PARP1−/− mice) showed an increased expression of Th1 cytokines, interferon γ, and chemokine (C–C motif) ligands (CCLs) 4 and 9 and decreased production of IL4.65 According to the study by Rosado et al,66 naive cluster of differentiation (CD)4+ cells from PARP1 knockout mice generates less IL4 and expresses GATA3 mRNA at a lower level than wild-type mice cells, even under Th2 stimulation. Nasta et al67 reported that PARP1−/− mice show an increased number of forkhead box P3+ (FoxP3+) regulatory T (Treg) cells in thymus and in peripheral lymphatic organ displaying a normal phenotype. The molecular mechanism underlying the link between Treg cells and PARP1 was investigated by Zhang et al.68 They demonstrated that PARP1 downregulates the expression of transforming growth factor (TGF)βI/II receptors either as a function of its enzymatic activity or independently by aging as a transcription corepressor. During an inflammatory reaction, the differentiation of naive CD4+ cells toward inducible Treg cells is inhibited by PARP1 activation. This effect depends on the decreasing sensitivity of CD4+ cells to TFGβ that blocks the Treg development. PARP1 inhibition or depletion reverts this inhibitory effect by increasing TGFβ receptors and, consequently, FoxP3+ gene expression, which is an essential regulator of Treg differentiation.68
Th17 cells play a pivotal role in induction and propagation of autoimmunity because they are defined by their ability to produce some cytokines, such as IL17A, IL17F, and IL22.69 They were also shown to play a role in neutrophil recruitment and airway resistance in patients affected by asthma.70 Conflicting data have been reported concerning the effects of PARP1 inhibition/deletion in IL17 production.71 Zhang et al68 reported increased levels of IL17 in PARP1−/− mice, in comparison with the study by Nasta et al67 that showed no change. Ghonim et al18 showed that the level of IL17 does not increase in bronchoalveolar lavage fluid (BALF) of PARP1−/− mice but found an increase of this cytokine in PARP1-deficient CD4+ cells in vitro stimulated. In an inflammatory arthritis mouse model, Ahmad et al72 showed that IL17 levels decrease upon PARP pharmacological inhibition with 5-aminoisoquinolinone (5-AIQ). Because, as it will be discussed later, IL17 levels were affected also by PARP14, it is possible that the data obtained upon pharmacological inhibition do not reflect their selective dependence on the PARP1 enzyme. Actually, no conclusive data are available so far.
Several studies showed that PARP1 is involved in B cell differentiation and function, even though PARP1−/− mice display a normal number of B cell in the peripheral lymphatic organs.66,73 Somatic hypermutation and gene conversion, typical of B cell immunoglobulin (Ig) generation, require PARP1, which promotes mutagenic repair mechanisms through its breast cancer 1 C-terminal domain.74 The result obtained upon PARP1 lack or inhibition is an increased fidelity repair of Ig chains.74 PARP1 is also involved in Ig class switch recombination. Depending on the stimuli, B cells from PARP1 knockout or pharmacological inhibition increase the frequency of Ig class recombination to IgA and IgG1, whereas they decrease the switch to IgG2a.66
Underlying the anti-inflammatory effects of PARP1 inhibition or deficiency in allergic inflammatory diseases, such as asthma, Datta et al75 reported a decrease in the amount of IL5 mRNA with a consequent reduction of the number of eosinophils infiltrating the lungs in an allergen-induced mouse model. The molecular mechanism that occurs downstream the binding of IL4 to its receptor has been demonstrated: after allergen stimulation, the inhibition or the complete absence of PARP1 enzymatic activity causes a remarkable increase of STAT6 protein degradation carried out by calpain rather than the proteasome system. Thus, it leads to a reduction of the transcription factor GATA-binding protein 3 (GATA3) mRNA and of protein levels culminating in IL5 transcriptional repression.75 Decreased levels of STAT6 were found in another study based on the pharmacological inhibition of PARP1 after a well-established inflammation, and in remodeling in a mouse model of airway disease, using house dust mite (HDM) as an allergen.76 These results have shown that PARP1 directly regulates STAT6 activation in an allergen stimulation-dependent fashion and that its inhibition can improve all signs of allergic inflammation.
Another PARP family protein, PARP14, was shown to be correlated with allergic airway diseases. PARP14 regulates STAT6-dependent gene transcription acting as a transcriptional switch. Under nonstimulating conditions, PARP14 binds to the promoters and recruits histone deacetylase (HDAC) 2 and 3 repressing gene transcription. Upon IL4 stimulation and STAT6 activation, the catalytic activity of PARP14 increases, resulting in ADP-ribosylation of HDACs that leave the GATA3 promoter, making this transcription factor active.77 Consequently, in allergic inflammation, pharmacologic inhibition or ablation of PARP14 expression results in a reduced STAT6-dependent gene transcription. PARP14 promotes Th2 differentiation, as naive T cells from PARP14−/− mice cultured under Th2 conditions showed a reduced production of Th2 cytokines (IL4, IL5, and IL13).78 By using ChIP-Seq analysis, Riley et al79 identified 2,744 genes whose expression in Th2 cells was dependent on the expression of PARP14. Therefore, this enzyme enhances the expression of Th2 genes as it represses the expression of Th1-associated genes.79 The ability of PARP14 to promote Th2 differentiation is ascribed to its capacity to regulate the activation of STAT6 and its binding to the GATA3 promoter.78,80 It is noteworthy that PARP14-deficient animals showed reduced levels of IgE, which were correlated with the intrinsic role of PARP14 in Ig class switching to IgE.78 PARP14−/− mice displayed a selective impairment in IgA production associated with a reduced marginal zone and increased follicular B cell numbers.81 Recently, the role of PARP14 in other T cell types was elucidated. T follicular helper (Tfh) cells secrete IL4, IL10, and IL21 and play an essential role in allergic responses for the germinal center formation of T cell-dependent antigens, as well as for the IgE production.82 PARP14 is highly expressed in Th17 cells, and PARP14 deficiency or the pharmacological inhibition of its activity result in a decreased Th17 differentiation in vitro and in a model of allergic airway inflammation.83 As a matter of fact, reduced IL-17A levels were detected in the BALF of PARP14-deficient mice in comparison with control mice.83 According to the study by Riley et al,79 PARP14 regulates the expression of IL21, which in turn promotes Th17 and Tfh development and function. It is noteworthy that a number of genes were found to be dependent on the PARP14 activity but not regulated by STAT6, this latter feature suggesting that they play STAT6-independent functions.79 PARP14 also promotes Tfh development through the activation of STAT3, which plays a pivotal role in the differentiation of both Tfh and Th17 cells. PARP14 ablation in mice showed a reduced number and frequency of Tfh in allergic airway inflammation model. Similarly, PARP14−/− T cells showed a decreased differentiation of Tfh and Th17 due to a decreased phosphorylation of STAT3.83
Although the lack of PARP1 and PARP14 causes similar effects in terms of protection against airway inflammation, GATA3 gene expression, and Th2 differentiation, the two enzymes operate in different ways. When PARP1 is inhibited or absent, calpain mediates the degradation of STAT6 in the cell,75 whereas if PARP14 is absent, HDACs remain bound to the GATA3 promoter inhibiting its gene expression.77 Furthermore, PARP14 affects the expression of IL17A, IL17F, and IL21,79,83 whereas the effects of PARP1 on IL17 are unclear71 and probably not relevant. All these findings suggest that PARP1 and PARP14 are at a crossroad in the allergic airway disease pathway, working with distinct but overlapping mechanisms (Figure 1). Moreover, they also suggest that PARP enzymes play a significant role in allergic airway diseases.
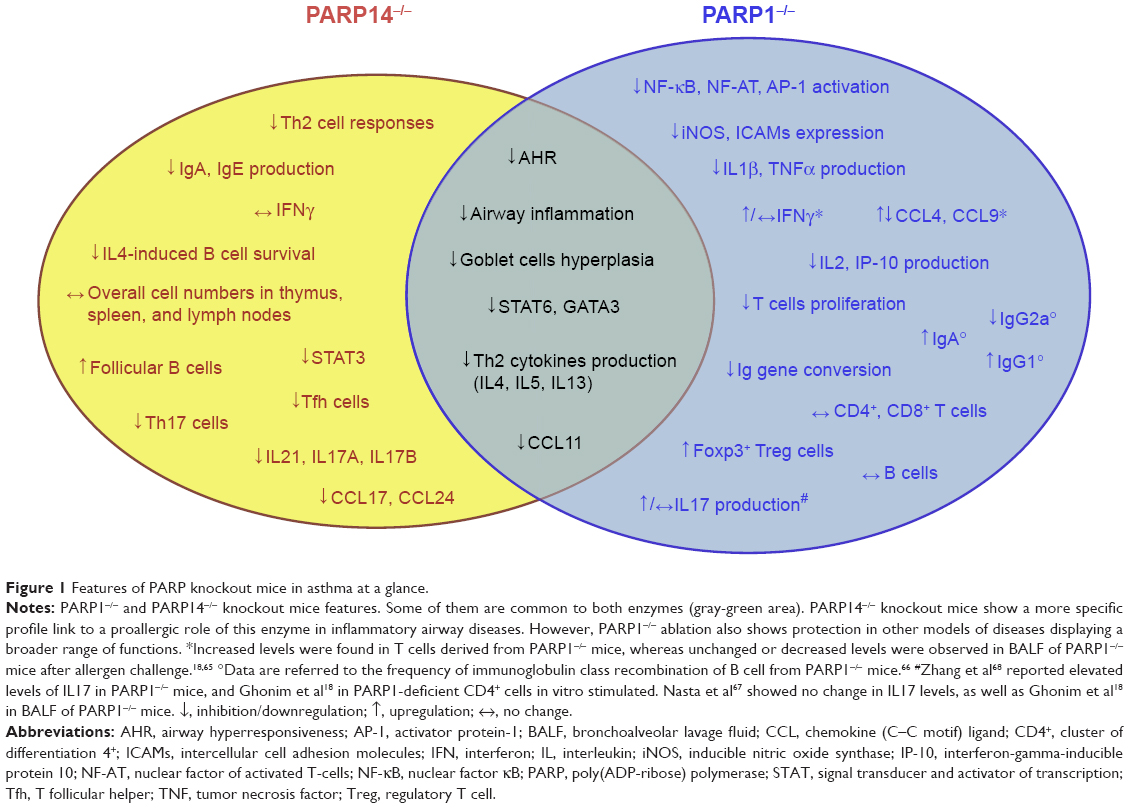 | Figure 1 Features of PARP knockout mice in asthma at a glance. |
PARP inhibition in asthma: evidences from preclinical studies
Experiments performed with PARPs inhibitors and knockout mice have shown that PARP enzymes are involved in several inflammatory diseases by triggering cell death in injured tissues.14,84 Havranek et al85 demonstrated an increased PARP1 expression and activation in an allergen-induced airway inflammation model, which suggests a direct involvement of the enzyme in allergic inflammation. In this study, two murine models of allergic airway inflammation, sensitization, and challenge to ovalbumin (OVA) and intratracheal exposure to IL13 were used to evaluate the PARP1 protein expression, localization, and activity, as well as inflammation and goblet cell hyperplasia. The authors concluded that PARP1 expression and activity are increased by allergen-activated inflammatory mediators. In contrast, PARP1 protein, or its activity, did not change after a single exposure to IL13, a condition under which goblet cells hyperplasia occurred without inflammation.85
From an immunological point of view, asthma is characterized by a deregulated Th2 immune response,86 and the IL4-activated STAT6 pathway is involved in the progression of the disease.87,88 The role of PARP14 and of its enzyme activity in asthma and Th2 differentiation were investigated by Mehrotra et al89 by using a murine model of asthma with OVA as allergen in in vitro Th cell differentiation. Interestingly, PARP14 was shown to act, as previously described, as a transcriptional switch for IL4-dependent STAT6-mediated transcription.89
Historically, nicotinamide, a pleiotropic biological agent, can be considered to be the first PARPs inhibitor, and various studies of Bekier et al90,91 in the early 1970s provided the hints that PARPs could be involved in the pathogenesis of asthma. More recently, Boulares et al92 published a study focused on the role of PARPs activation in asthma. They showed the activation of PARPs through the immunohistochemical detection of PAR (the product of the PARP-catalyzed reaction) in a conventional murine model of asthma using OVA as allergen. The PARPs inhibitor used in this study was 3-aminobenzamide (3-ABA), and the authors found a reduced inflammatory cell migration and a suppressed expression of iNOS. Moreover, they confirmed these results in a PARP1-deficient mouse model in which protection from the OVA-induced lung inflammation was obtained.92 Virag et al93 investigated the role of PARPs activation in asthma using a similar animal model, where the PARPs inhibitor was N-(6-Oxo-5,6-dihydrophenanthridin-2-yl)-(N, N-dimethylamino)acetamide hydrochloride (PJ-34). A reduced number of neutrophils, but not of eosinophils in the BALF, was observed in the single exposure model. However, the authors did not obtain a reduction in the severe inflammation protocol. Cytokines levels in BALF were detected, and PJ-34 suppressed the production of tumor necrosis factor (TNF) α, IL12, and the CCL3 but did not affect the expression of chemokine (C–X–C motif) ligand 2 (CXCL2) or the production of IL5 and IL13.93 Later, a group from our laboratory investigated the role of PARPs activation in a guinea pig asthma model using OVA for sensitization.94 The PARPs inhibitors used were 3-ABA and 5-AIQ. Both inhibitors reduced the severity of cough and the occurrence of dyspnea. Moreover, they delayed the respiratory abnormalities. In this study, it was also shown that PARPs inhibitors are able to prevent the biochemical changes in lungs or BALF measured as myeloperoxidase activity, malondialdehyde formation, tyrosine nitration, serum nitrite levels, and TNFα production.94 Another study, carried out by the Boulares et al, showed that PARPs play a role in eosinophils recruitment into lungs. Administration of the PARPs inhibitor thieno[2,3-c]isoquinolin-5-one (TIQ-A) before allergen challenge resulted in the suppression of Th2 cytokines, including IL4, IL5, and IL13. A suppression of the eosinophils infiltration with associated mucus production in lung airways was also observed, which suggests a specific effect on IL5.95
The studies described above were performed upon administering PARPs inhibitors only before the allergic sensitization or rechallenge. Naura et al96 performed a single injection of TIQ-A in mice, 1 or 6 hours after OVA challenge. Postallergen challenge administration of the drug resulted in the suppression of IL4, IL5, and IgE production and prevented airway hyperresponsiveness to methacholine. IL13 was reported to decrease significantly, causing the interruption of airway mucus secretion and goblet cell hyperplasia.96 Nevertheless, the authors did not investigate the remodeling events typical of chronic asthma because they evaluated the effect of TIQ-A in an asthma acute model by focusing their attention on airway inflammation and hyperresponsiveness. This study documented that PARP inhibitors were active also after sensitization and challenge. The results suggested that PARP inhibitors could be administered within a reasonable time, ie, commensurate with the therapeutic approach against human asthma. More recently, Ghonim et al97 strengthened the validity of the postchallenge drug administration protocol using the OVA-based mouse model of asthma and primary CD4+ cells. In fact, they demonstrated that olaparib, administered 30 minutes after challenge, blocks asthma-like manifestations (inflammation and hyperresponsiveness) by modulating CD4+ cells without prominent effects on B cells function. The protective effects of olaparib were ascribed to the suppression of Th2 cytokines, such as CCL11, IL4, IL5, IL6, IL13, and macrophage colony-stimulating factor.97
The hallmark features of asthma include airway eosinophilic inflammation and structural remodeling characterized by increased thickness of the subepithelial reticular basement membrane. This latter event goes along with peribronchial collagen deposition, increased airway smooth muscle mass, goblet cell hyperplasia, and angiogenesis, which are associated with a progressive and irreversible loss of lung function.97 These structural changes are present even in mild asthma and are permanent, albeit symptoms may remit for some time.98,99 Recent data showed that remodeling could be independent of inflammation. For example, it was reported that both eosinophilic and noneosinophilic patients appear equally responsive to salbutamol, although asthmatic noneosinophilic subjects do not benefit from corticosteroid treatment. This suggests that remodeling could be present in both groups.100
The first evidence that PARP inhibition affects the airway remodeling in asthma was given by Lucarini et al101 who showed that PARP inhibition reduced airway damage and remodeling in guinea pigs using hydroxyl-dimethylaminomethyl-thieno[2,3-c]isoquinolin-5(4H)-one (HYDAMTIQ), a potent and selective PARP1 and PARP2 inhibitor, that was previously tested in stroke-induced inflammation and brain damage. A limitation of this study is represented by the asthma model, OVA in this case, which is not specifically designed to evaluate all the aspects of airway remodeling. As a matter of fact, animal models, with the use of HDM instead of OVA as allergen, better reproduce the remodeling process, in addition to inflammation, occurring in humans affected by asthma.102 Recently, our group and others used a nonconventional mouse model of chronic asthma with HDM as an allergen to investigate the PARP inhibition on inflammation and remodeling.18,76 This protocol led to sustained airway eosinophilic inflammation associated with immune activation in the lung, allergen-specific memory response by splenocytes, and humoral immunity to HDM.103 Differently from OVA, which is an innocuous protein, the HDM extract belongs to environmental antigens and causes cytotoxicity measured as increased lung levels of oxidative damage to proteins, lipids, and nucleic acids.104 Recently, Ghonim et al18 demonstrated that the inhibition of PARP1 and PARP2 obtained by olaparib is effective in blocking established HDM-induced diseases in mice, including airway eosinophilia and hyperresponsiveness. These effects are linked to a marked reduction in Th2 cytokines production and are related to a modulation of CD4+ T cells function as well as to an increase in Treg cells.18 In our work,76 the postallergen challenge PARP inhibition and the 5-week long protocol of HDM administration reflect more closely the pathological modification occurring in human asthma and its therapeutic approach. As expected, this experimental model elicits a strong chronic inflammatory response in mice, characterized by systemic Th2 immunity and airway remodeling. Our results are in line with previous studies concerning the protective effect carried out on inflammation by PARP inhibition: we obtained a robust decrease in airway inflammation, although we did not find a decrease of the IgE level, as well as of IgG1 and IgG2a titers. In our study, we found a small difference between acute and chronic models (10 days versus 5 weeks) in terms of cells number in the BALF. Therefore, PARP inhibition was a little more effective in the acute model possibly because of the induction of a small inflammation level. The results registered can be ascribed to a downregulation of STAT6 DNA binding, and, partly, to a decrease in the mRNA level of CCL11. It is worth mentioning that these inhibitory treatments did not affect the NF-kB DNA-binding activity, although we cannot rule out the possibility that they inhibit the NF-kB transcriptional capacity by removing the PARP1-NF-kB protein–protein interaction.76 Moreover, PARP inhibition by 5-AIQ and 3-ABA clearly induced a reduction of goblet cell hyperplasia. Instead, subepithelial collagen deposition was reduced only by 5-AIQ, while both inhibitors were ineffective against α-smooth actin lung deposition. Thus, the mentioned late events, typical of the remodeling process, were affected by 5-AIQ better than by 3-ABA.76
In in vivo studies of asthma, several PARPs inhibitors have been used, and it should be noticed that their activity includes inhibiting more than one member of the PARP family (Table 1). Consequently, the beneficial effects observed can be ascribed to the synergistic activity, because 5-AIQ targets PARP1, 2, and 3; PJ-34 targets PARP1, 2, and 14; HYDAMTIQ targets PARP1 and 2; olaparib targets PARP1, 2, 3, and 4; and TIQ-A targets PARP1 and 3 (Table 1).105 Thus, the different findings emerging from many studies can be ascribable to the affection of different PARP isoforms in addition to the particular animal model used.
 | Table 1 PARPs inhibitors and their selectivity for the catalytic domain of different members of PARP family |
In summary, the attenuation of lung injury, the improvement of respiratory abnormalities, and the provocation of the anti-inflammatory effect have been obtained in several animal models using different OVA or HDM allergens, upon PARP1/14 deletion (Figure 1) or pharmacological inhibition (Table 2). These effects occurred even when drugs were administered after the antigen challenge. To identify novel therapeutic approach(es) to target human asthma, especially airway remodeling, the use of preclinical models that truly reflect the features of the human disease is essential.
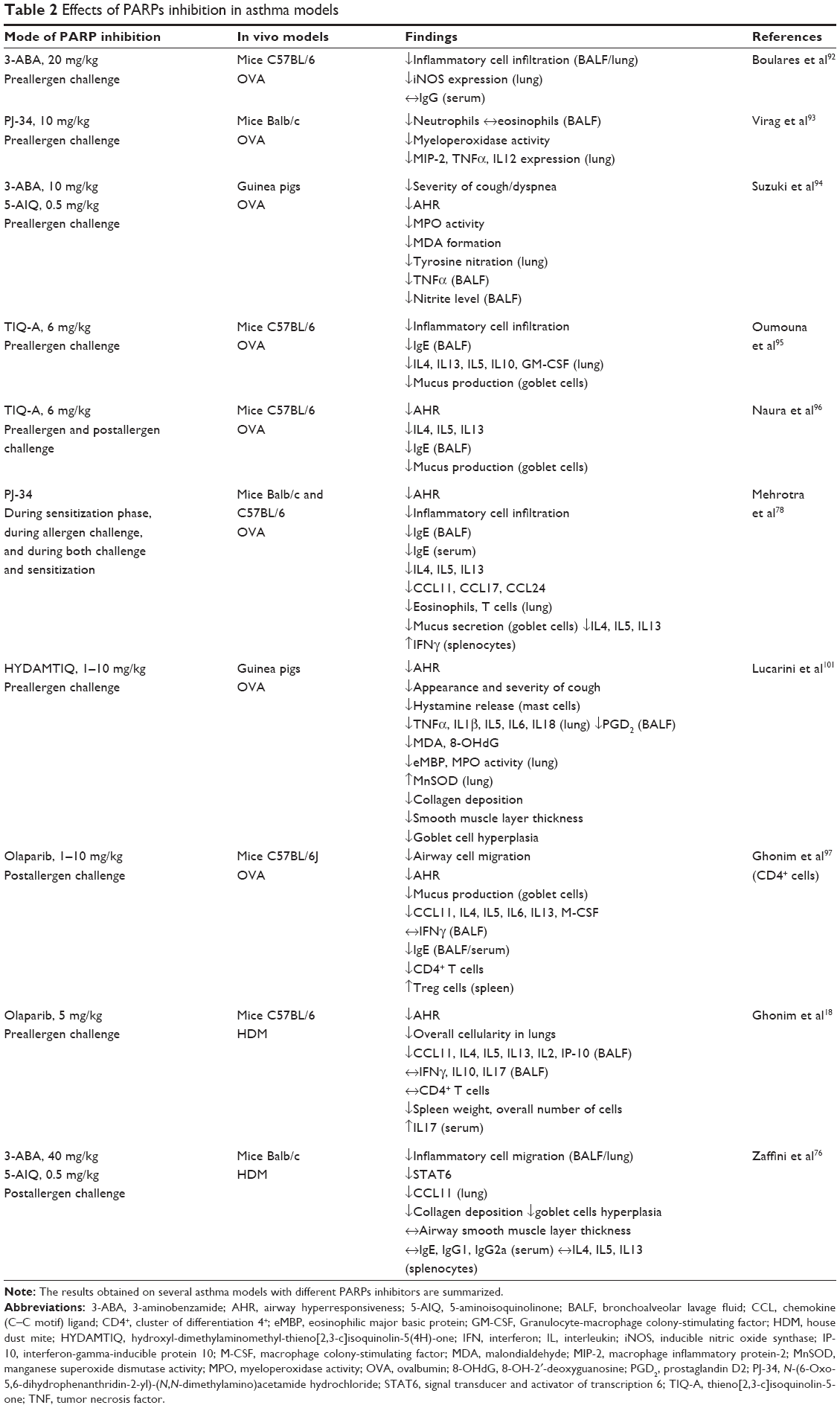 | Table 2 Effects of PARPs inhibition in asthma models |
Conclusion and perspectives
PARP enzymes play a pivotal role in both inflammation and remodeling and in the consecutive airway function, such as airway hyperresponsiveness, as it has been observed in the preclinical studies reported.
Until recently, it was thought that chronic inflammation might drive remodeling. However, it is now definitely clear that inflammation and remodeling are two distinct processes, requiring different biochemical pathways and occurring in a parallel manner and not in a sequential manner.102 The current main approaches to asthma management are essentially based on the inflammatory parameters and not on remodeling and consequent airway hyperresponsiveness. Although novel therapeutic approaches (mainly the use of monoclonal antibody to IgE and Th2 cytokines, such as IL5 and IL13) have improved the control of the disease, they do not result in disease modification, or it seems that they can be applied only to a subgroup of patients.106–108 In fact, we are able to reduce exacerbations by targeting eosinophilic inflammation, but novel approaches and targets are required to impact on lung function.109 On the other hand, PARPs sustain the expression of cytokines, chemokines, and inflammatory mediators, such as TNFα, IL1, IL6, and iNOS. These mediators are also involved in the expression of several adhesion molecules, such as P-selectin, E-selectin, mucosal address in cell adhesion molecule 1, IL8, CCL2, CCL3, CXCL2, metalloproteinase 9. They usually participate in a plethora of stress signaling pathways.110 As many experimental results have shown, the inhibition or deletion of PARPs causes protective effects in acute and chronic airway diseases, which are mainly due to the inhibition of cell migration. Anyway, the link among PARPs, airway remodeling, and key factors related to the disease persistence require further investigations, even if the first evidences are promising (Table 2). Although in our study we registered variable effects of two PARP inhibitors used, the decrease in hallmark of airway remodeling observed in experiments performed by our and another group76,101 could lead to a step change in asthma therapy. Indeed, the weak point of preclinical studies reported here is the use of PARP inhibitors displaying a promiscuous ligand activity. In fact, it is possible that the effects observed are the result of a synergistic inhibition of many PARP enzymes (Table 1). Further investigations should be addressed to the role of each PARP enzyme in the airway remodeling process to elucidate which are the most active PARP isoform(s) in this irreversible process. Particularly, the connections existing among airway remodeling in asthma, TGFβ, NF-kB, and PARPs should be explored in detail.
Discrimination between the different effects of PARPs inhibitors in acute and chronic asthma could be an interesting purpose. However, in this review, an exhaustive comparison among the studies reported is impossible because they follow too different experimental designs.
PARPs inhibitors are known to be active as anti-inflammatory agents, and they are already used in several pathologies: neurodegeneration, myocardial infarction, arthritis, colitis, and other nononcological diseases.14 The main concern on the therapeutic use of PARPs inhibitors in nononcological diseases is due to the wide expression and function of PARPs in physiological key processes, such as metabolism regulation, aging, circadian rhythms, viral infections, and mitochondrial homeostasis to name a few,110 with unpredictable side effects especially for a long-time therapy. Therefore, it would be extremely important in this case to target, finding more selective inhibitors, only the PARP isoforms that are involved in the disease. Nonetheless, it is noteworthy that asthma allows an inhaled therapy, which can limit the side effects of drugs especially if they are administrated at low doses.
Therefore, a future multimechanistic approach to asthma, including PARPs inhibitors, may lead to a more efficacious management of this disease.
Acknowledgment
We are deeply grateful to prof Massimo Libonati for his skillful help in the revision of this work.
Author contributions
All authors contributed toward data analysis, drafting and critically revising the paper and agree to be accountable for all aspects of the work.
Disclosure
The authors report no conflicts of interest in this work.
References
Chambon P, Weil JD, Mandel P. Nicotinamide mononucleotide activation of a new DNA-dependent polyadenylic acid synthesizing nuclear enzyme. Biochem Biophys Res Commun. 1963;11:39–43. | ||
Chao A, Chang TC, Lapke N, et al. Prevalence and clinical significance of BRCA1/2 germline and somatic mutations in Taiwanese patients with ovarian cancer. Oncotarget. 2016;7(51):85529–85541. | ||
D’Arcangelo M, Drew Y, Plummer R. The role of PARP in DNA repair and its therapeutic exploitation. In: Kelley MR, Fishel ML, editors. DNA Repair in Cancer Therapy; Molecular Targets and Clinical Applications. 2nd ed. London: Academic Press Elsevier; 2016:115–134. | ||
Peralta-Leal A, Rodriguez-Vargas JM, Aguilar-Quesada R, et al. PARP inhibitors: new partners in the therapy of cancer and inflammatory diseases. Free Rad Biol Med. 2009;47(1):13–26. | ||
Curtin NJ, Szabo C. Therapeutic applications of PARP inhibitors: anticancer therapy and beyond. Mol Aspects Med. 2013;34(6):1217–1256. | ||
Barnes PJ. Immunology of asthma and chronic obstructive pulmonary disease. Nat Rev Immunol. 2008;8(3):183–192. | ||
Locksley RM. Asthma and allergic inflammation. Cell. 2010;140(6):777–783. | ||
Lambrecht BN, Hammad H. The immunology of asthma. Nat Immunol. 2015;16(1):45–56. | ||
WHO Media Centre [webpage on the Internet]. Bronchial Asthma Fact Sheet No. 206. Available from: http://www.who.int/mediacentre/factsheets/fs206/en/. Accessed January 4, 2017. | ||
Holgate ST. Airway inflammation and remodeling in asthma: current concepts. Mol Biotechnol. 2002;22(2):179–189. | ||
Mukherjee AB, Zhang Z. Allergic asthma: influence of genetic and environmental factors. J Biol Chem. 2011;286(38):32883–32889. | ||
Lee IT, Yang CM. Inflammatory signalings involved in airway and pulmonary diseases. Mediators Inflamm. 2013;2013:791231. | ||
Bao Z, Xiong J, Li W, Chen Z, Shen H, Ying S. Genomic instability in chronic airway inflammatory diseases. Biomed J. 2015;38(2):117–124. | ||
Berger NA, Besson VC, Boulares AH, et al. Opportunities for the repurposing of PARP inhibitors for the therapy of non-oncological diseases. Br J Pharmacol. Epub 2017 Feb 18:doi: 10.1111/bph.13748. | ||
Trevor JL, Deshane JS. Refractory asthma: mechanisms, targets, and therapy. Allergy. 2014;69(7):817–827. | ||
Tezcan G, Gurel CB, Tutluoglu B, Onaran I, Kanigur-Sultuybek G. The Ala allele at Val762Ala polymorphism in poly(ADP-ribose) polymerase-1 (PARP-1) gene is associated with a decreased risk of asthma in a Turkish population. J Asthma. 2009;46(4):371–374. | ||
Ozaydin A, Akbas F, Aksoy F, et al. Investigation of poly(ADP-ribose) polymerase-1 genetic variants as a possible risk for allergic rhinitis. Genet Test Mol Biomarkers. 2014;18(1):57–61. | ||
Ghonim MA, Pyakurel K, Ibba SV, et al. PARP is activated in human asthma and its inhibition by olaparib blocks house dust mite-induced disease in mice. Clin Sci. 2015;129(11):951–962. | ||
Amé JC, Spenlehauer C, De Murcia G. The PARP superfamily. Bioassay. 2004;26(8):882–893. | ||
Hottiger MO, Hassa PO, Luscher B, Schuler H, Koch-Nolte F. Toward a unified nomenclature for mammalian ADP-ribosyltransferases. Trends Biochem Sci. 2010;35(4):208–219. | ||
Virag L. Poly(ADP-ribosyl)ation in asthma and other lung diseases. Pharmacol Res. 2005;52(1):83–92. | ||
Gibson BA, Kraus WL. New insights into the molecular and cellular functions of poly(ADP-ribose) and PARPs. Nat Rev Mol Cell Biol. 2012;13(7):411–424. | ||
Tanuma S, Sato A, Oyama T, Yoshimori A, Abe H, Uchiumi F. New insights into the roles of NAD+-poly(ADP-ribose) metabolism and poly(ADP-ribose) glycohydrolase. Curr Protein Pept Sci. 2016;17(7):668–682. | ||
Miwa M, Ida C, Yamashita S, Tanaka M, Fujisawa J. Poly(ADP-ribose): structure, physicochemical properties and quantification in vivo, with special reference to poly(ADP-ribose) binding protein modules. Curr Protein Pept Sci. 2016;17(7):683–692. | ||
Hassa PO, Hottiger MO. The diverse biological roles of mammalian PARPS, a small but powerful family of poly(ADP-ribose) polymerase. Front Biosci. 2008;13:3046–3082. | ||
Virag L, Szabo C. The therapeutic potential of poly(ADP-ribose) polymerase inhibitors. Pharmacol Rev. 2002;54(3):375–429. | ||
Kraus WL, Lis JT. PARP goes transcription. Cell. 2003;113(6):677–683. | ||
Kraus WL. Transcriptional control by PARP1: chromatin modulation, enhancer-binding, coregulation, and insulation. Curr Opin Cell Biol. 2008;20(3):294–302. | ||
Martinez-Zamudio R, Ha HC. Histone ADP-ribosylation facilitates gene transcription by directly remodeling nucleosomes. Mol Cell Biol. 2012;32(13):2490–2502. | ||
Bai P, Virag L. Role of poly(ADP-ribose) polymerases in the regulation of inflammatory processes. FEBS Let. 2012;586(21):3771–3777. | ||
Kleinert H, Schwarz PM, Förstermann U. Regulation of the expression of inducible nitric oxide synthase. Biol Chem. 2003;384(10–11):1343–1364. | ||
Xia P, Zweier JL. Superoxide and peroxynitrite generation from inducible nitric oxide synthase in macrophages. Proc Nat Acad Sci U S A. 1997;94(13):6954–6958. | ||
Packer P, Beckman JS, Liaudet L. Nitric oxide and peroxynitrite in health and disease. Physiol Rev. 2007;87(1):315–424. | ||
Burney S, Caulfield JL, Niles JC, Wishnok JS, Tannenbaum SR. The chemistry of DNA damage from nitric oxide and peroxynitrite. Mutat Res. 1999;424(1–2):37–49. | ||
Szabo C, Zingarelli B, O’Connor M, Salzman AL. DNA strand breakage, activation of poly(ADP-ribose) synthetase, and cellular energy depletion are involved in the cytotoxicity of macrophages and smooth muscle cells exposed to peroxynitrite. Proc Natl Acad Sci U S A. 1996;93(5):1753–1758. | ||
Bonfoco E, Krainc D, Ankarcrona M, Nicotera P, Lipton SA. Apoptosis and necrosis: two distinct events induced, respectively, by mild and intense insults with N-methyl-D-aspartate or nitric oxide/superoxide in cortical cell cultures. Proc Natl Acad Sci U S A. 1995;92(16):7162–7166. | ||
Virag L, Szabo E, Gergely P, Szabo C. Peroxynitrite-induced cytotoxicity: mechanism and opportunities for intervention. Toxicol Lett. 2003;140–141:113–124. | ||
Ha HC, Snyder SH. Poly(ADP-ribose) polymerase is a mediator of necrotic cell death by ATP depletion. Proc Natl Acad Sci U S A. 1999;96(24):13978–13982. | ||
Fouquerel E, Goellner EM, Yu Z, et al. ARTD1/PARP1 negatively regulates glycolysis by inhibiting hexokinase 1 independent of NAD+ depletion. Cell Rep. 2014;8(6):1819–1831. | ||
Kolthur-Seetharam U, Dantzer F, McBurney MW, de Murcia G, Sassone-Corsi P. Control of AIF-mediated cell death by the functional interplay of SIRT1 and PARP-1 in response to DNA damage. Cell Cycle. 2006;5(8):873–877. | ||
Fouquerel E, Sobol RW. ARTD1 (PARP1) activation and NAD+ in DNA repair and cell death. DNA Repair. 2014;23:27–32. | ||
David KK, Andrabi SA, Dawson TM, Dawson VL. Parthanatos, a messenger of death. Front Biosci. 2009;14:1116–1128. | ||
Yu SW, Wang H, Poitras MF, et al. Mediation of poly(ADP-ribose) polymerase-1-dependent cell death by apoptosis-inducing factor. Science. 2002;297(5579):259–263. | ||
Schuliga M. NF-kappaB signaling in chronic inflammatory airway disease. Biomolecules. 2015;5(3):1266–1283. | ||
Morgan MJ, Liu ZG. Crosstalk of reactive oxygen species and NF-kB signaling. Cell Res. 2011;21(1):103–115. | ||
Oliver FJ, Ménissier-De Murcia J, Nacci C, et al. Resistance to endotoxic shock as a consequence of defective NF-kappaB activation in poly(ADP-ribose) polymerase-1 deficient mice. EMBO J. 1999;18(16):4446–4454. | ||
Cuzzocrea S. Shock, inflammation and PARP. Pharmacol Res. 2005;52(1):72–82. | ||
Stilmann M, Hinz M, Aslan SC, Zimmer A, Schreiber V, Scheidereit C. A nuclear poly(ADP-ribose)-dependent signalosome confers DNA damage-induced IkappaB kinase activation. Mol Cell. 2009;36(3):365–378. | ||
Hassa PO, Covic M, Hasan S, Imhof R, Hottiger MO. The enzymatic and DNA-binding activity of PARP1 are not required for NF-kB coactivator function. J Biol Chem. 2001;276(49):45588–45597. | ||
Liu L, Ke Y, Jiang X, et al. Lipopolysaccharide activates ERK-PARP-1-RelA pathway and promotes nuclear factor-κB transcription in murine macrophages. Hum Immunol. 2012;73(5):439–447. | ||
Zerfaoui M, Errami Y, Naura AS, et al. Poly(ADP-ribose) polymerase-1 is a determining factor in Crm1-mediated nuclear export and retention of p65 NF-kappa B upon TLR4 stimulation. J Immunol. 2010;185(3):1894–1902. | ||
Andreone TL, O’Connor M, Denenberg A, Hake PW, Zingarelli B. Poly(ADP-ribose) polymerase-1 regulates activation of activator protein-1 in murine fibroblasts. J Immunol. 2003;170(4):2113–2120. | ||
Olabisi OA, Soto-Nieves N, Nieves E, et al. Regulation of transcription factor NFAT by ADP-ribosylation. Mol Cell Biol. 2008;28(9):2860–2871. | ||
Valdor R, Schreiber V, Saenz L, et al. Regulation of NFAT by poly(ADP-ribose) polymerase activity in T cells. Mol Immunol. 2008;45(7):1863–1871. | ||
Ali SO, Khan FA, Galindo-Campos MA, Yélamos J. Understanding specific functions of PARP-2: new lessons for cancer therapy. Am J Cancer Res. 2016;6(9):1842–1863. | ||
Benchoua A, Couriaud C, Guegan C, et al. Active caspase-8 translocates into the nucleus of apoptotic cells to inactivate poly(ADP-ribose) polymerase-2. J Biol Chem. 2002;277(37):34217–34222. | ||
Popoff I, Jijon H, Monia B, et al. Antisense oligonucleotides to poly(ADP-ribose) polymerase-2 ameliorate colitis in interleukin-10-deficient mice. J Pharmacol Exp Ther. 2002;303(3):1145–1154. | ||
Vyas S, Matic I, Uchima L, et al. Family-wide analysis of poly(ADP-ribose) polymerase activity. Nat Commun. 2014;5:4426. | ||
Goenka S, Boothby M. Selective potentiation of Stat-dependent gene expression by collaborator of Stat6 (CoaSt6), a transcriptional cofactor. Proc Natl Acad Sci U S A. 2006;103(11):4210–4215. | ||
Ladurner AG. Inactivating chromosomes: a macro domain that minimizes transcription. Mol Cell. 2003;12(1):1–4. | ||
Menegazzi M, Gerosa F, Tommasi M, et al. Induction of poly(ADP-ribose) polymerase in lectin stimulated human T lymphocytes is dependent on protein synthesis. Biochem Biophis Res Commun. 1988;156(2):995–999. | ||
Menegazzi M, Suzuki H, Carcereri de Prati A, et al. Increase of poly(ADP-ribose) polymerase mRNA levels during TPA-induced differentiation of human lymphocytes. FEBS Lett. 1992;297(1–2):59–62. | ||
Menegazzi M, Grassi-Zucconi G, Carcereri de Prati A, et al. Differential expression of poly(ADP-ribose) polymerase and DNA polymerase β in rat tissues. Exp Cell Res. 1991;197(1):66–74. | ||
Rochette-Egly C, Ittel ME, Bilen J, Mandel P. Effect of nicotinamide on RNA and DNA synthesis and on poly(ADP-ribose) polymerase activity in normal and phytohemagglutinin stimulated human lymphocytes. FEBS Lett. 1980;120(1):7–11. | ||
Saenz L, Lozano JJ, Valdor R, et al. Transcriptional regulation by poly(ADP-ribose) polymerase-1 during T cell activation. BMC Genomics. 2008;9:171. | ||
Rosado MM, Bennici E, Novelli F, Pioli C. Beyond DNA repair, the immunological role of PARP-1 and its siblings. Immunology. 2013;139(4):428–437. | ||
Nasta F, Laudisi F, Sambucci M, Rosado MM, Pioli C. Increased Foxp3+ regulatory T cells in poly(ADP-Ribose)polymerase-1 deficiency. J Immunol. 2010;184(7):3470–3477. | ||
Zhang P, Nakatsukasa H, Tu E, et al. PARP-1 regulates expression of TGFβ receptors in T cells. Blood. 2013;12200(13):2224–2232. | ||
Bettelli E, Kom T, Kuchroo VK. Th17: the third member of the effector T cell trilogy. Curr Opin Immunol. 2007;19(6):652–657. | ||
Alcorn JF, Crowe CR, Kolls JK. Th17 cells in asthma and COPD. Annu Rev Physiol. 2010;72:495–516. | ||
Sethi GS, Dharwal V, Naura AS. Poly(ADP-ribose) polymerase-1 in lung inflammatory disorders: a review. Front Immunol. 2017;8:1172–1788. | ||
Ahmad SF, Zoheir KM, Bakheet SA, Ashour AE, Attia SM. Poly(ADP-ribose) polymerase-1 inhibitor modulates T regulatory and IL17 cells in the prevention of adjuvant induced arthritis in mice model. Cytokine. 2014;68(2):76–85. | ||
Ambrose HE, Willimott S, Beswick RW, et al. Poly(ADP-ribose) polymerase-1 (PARP-1) deficient mice demonstrate abnormal antibody responses. Immunology. 2009;127(2):178–186. | ||
Paddock MN, Buelow BD, Takeda S, Scharenberg AM. The BRCT domain of PARP-1 is required for immunoglobulin gene conversion. PLoS Biol. 2010;8(7):e31000428. | ||
Datta R, Naura AS, Zerfaoui M, et al. PARP-1 deficiency blocks IL-5 expression through calpain-dependent degradation of STAT-6 in a murine asthma model. Allergy. 2011;66(7):853–861. | ||
Zaffini R, Di Paola R, Cuzzocrea S, Menegazzi M. PARP inhibition treatment in a nonconventional experimental mouse model of chronic asthma. Naunyn Schmiedebergs Arch Pharmacol. 2016;389(12):1301–1313. | ||
Krishnamurthy P, Kaplan MH. STAT6 and PARP family members in the development of T cell-dependent allergic inflammation. Immune Netw. 2016;16(4):201–210. | ||
Mehrotra P, Hollenbeck A, Riley JP, et al. Poly(ADP-ribose) polymerase 14 and its anzyme activity regulates T(H)2 differentiation and allergic airway disease. J Allergy Clin Immunol. 2013;131(2):521–531. | ||
Riley J, Kulkarni A, Mehrotra P, et al. PARP-14 binds specific DNA sequences to promote Th2 cell gene expression. PLoS One. 2013;8(12):e83127. | ||
Goenka S, Cho SH, Boothby M. Collaborator of STAT6 (CoSt6)-associated poly(ADP-ribose) polymerase activity modulates STAT6-dependent gene transcription. J Biol Chem. 2007;282(26):18732–18739. | ||
Cho SH, Goenka S, Henttinen T, et al. PARP-14, a member of the B aggressive lymphoma family, transduces survival signals in primary B cells. Blood. 2009;113(11):2416–2425. | ||
Crotty S. T follicular helper cell differentiation, function, and roles in disease. Immunity. 2014;41(4):529–542. | ||
Mehrotra P, Krishnamurthy P, Sun J, Goenka S, Kaplan MH. Poly-ADP-ribosyl polymerase-14 promotes T helper 17 and follicular T helper development. Immunology. 2015;146(4):537–546. | ||
Schreiber V, Dantzer F, Ame J-C, de Murcia G. Poly(ADP-ribose): novel functions for an old molecule. Nat Rev Mol Cell Biol. 2006;7(7):517–528. | ||
Havranek T, Aujla PK, Nickola TJ, Rose MC, Scavo LM. Increased poly(ADP-ribose) polymerase (PARP)-1 expression and activity are associated with inflammation but not goblet cell metaplasia in murine models of allergen-induced airway inflammation. Exp Lung Res. 2010;36(7):381–389. | ||
Broide DH, Finkelman F, Bochner BS, Rothenberg ME. Advances in mechanisms of asthma, allergy, and immunology in 2010. J Allergy Clin Immunol. 2011;127(3):689–695. | ||
Kuperman DA, Schleimer RP. Interleukin-4, interleukin-13, signal transducer and activator of transcription factor 6, and allergic asthma. Curr Mol Med. 2008;8(5):384–392. | ||
Sehra S, Bruns HA, Ahyi AN, et al. IL-4 is a critical determinant in the generation of allergic inflammation initiated by a constitutively active STAT-6. J Immunol. 2008;180(5):3551–3559. | ||
Mehrotra P, Riley JP, Patel L, Li F, Voss L, Goenka S. PARP-14 functions as a transcriptional switch for STAT-6-dependent gene activation. J Biol Chem. 2011;286(3):1767–1776. | ||
Bekier E, Czerwinska U. The effect of nicotinamide on the experimental asthma in guinea pigs. Acta Physiol Pol. 1973;24(6):887–889. | ||
Bekier E, Wyczolkowska J, Szyc H, Maslinski C. The inhibitory effect of nicotinamide on asthma-like symptoms and eosinophilia in guinea pigs, anaphylactic mast cell degranulation in mice, and histamine release from rat isolated peritoneal mast cells by compound 48–80. Int Arch Allergy Appl Immunol. 1974;47(5):737–748. | ||
Boulares AH, Zoltoski AJ, Sherif ZA, Jolly P, Massaro D, Smulson ME. Gene knockout or pharmacological inhibition of poly(ADP-ribose) polymerase-1 prevents lung inflammation in a murine model of asthma. Am J Respir Cell Mol Biol. 2003;28(3):322–329. | ||
Virag L, Bai P, Bak I, et al. Effects of poly(ADP-ribose) polymerase inhibition on inflammatory cell migration in a murine model of asthma. Med Sci Monit. 2004;10(3):BR77–BR83. | ||
Suzuki Y, Masini E, Mazzocca C, et al. Inhibition of poly(ADP-ribose) polymerase prevents allergen-induced asthma-like reaction in sensitized guinea pigs. J Pharmacol Exp Ther. 2004;311(3):1241–1248. | ||
Oumouna M, Datta R, Oumouna-Benachour K, et al. PARP-1 inhibition prevents eosinophil recruitment by modulating Th2 cytokines in a murine model of allergic airway inflammation: a potential specific effect on IL-5. J Immunol. 2006;177(9):6489–6496. | ||
Naura AS, Hans CP, Zerfaoui M, et al. Post-allergen challenge inhibition of poly(ADP-ribose) polymerase harbors therapeutic potential for treatment of allergic airway inflammation. Clin Exp Allergy. 2008;38(5):839–846. | ||
Ghonim MA, Pyakurel K, Ibba SV, et al. PARP inhibition by olaparib or gene knockout blocks asthma like manifestation in mice by modulating CD4+ T cell function. J Transl Med. 2015;13:225–236. | ||
Fixman ED, Stewart A, Martin JG. Basic mechanisms of development of airway structural changes in asthma. Eur Respir J. 2007;29(2):379–389. | ||
Pascual RM, Peters SP. The irreversible component of persistent asthma. J Allergy Clin Immunol. 2009;124(5):883–890. | ||
McGrath KW, Icitovic N, Boushey HA, et al; Asthma Clinical Research Network of the National Heart, Lung, and Blood Institute. A large subgroup of mild-to-moderate asthma is persistently noneosinophilic. Am J Resp Crit Care Med. 2012;185(6):612–619. | ||
Lucarini L, Pini A, Gerace E, Pellicciari R, Masini E, Moroni F. Poly(ADP-ribose) polymerase inhibition with HYDAMTIQ reduces allergen-induced asthma-like reaction, bronchial hyper-reactivity and airway remodeling. J Cell Mol Med. 2013;18(3):468–479. | ||
Saglani S, Mathie SA, Gregory LG, Bell MJ, Bush A, Lloyd CM. Pathophysiological features of asthma develop in parallel in house dust mite-exposed neonatal mice. Am J Respir Cell Mol Biol. 2009;41(3):281–289. | ||
Johnson RJ, Wiley ER, Fattouh R, et al. Continuous exposure to house dust mite elicits chronic airway inflammation and structural remodeling Am J Respir Crit Care Med. 2004;169(3):378–385. | ||
Chan TK, Loh XY, Peh HY, et al. House dust mite-induced asthma causes oxidative damage and DNA double-strand breaks in the lungs. J Allergy Clin Immunol. 2016;138(1):84–96. | ||
Wahlberg E, Karlberg T, Kouznetsova E, et al. Family-wide chemical profiling and structural analysis of PARP and tankyrase inhibitors. Nat Biotechnol. 2012;30(3):283–288. | ||
Pelaia G, Walter G, Matucci A, Paolini R, Triggiani M, Paggiaro P. Target therapy in severe asthma today: focus on immunoglobulin E. Drug Des Develop Ther. 2017;11:1979–1998. | ||
Corren J, Lemanske RF, Hanania NA, et al. Lebrikizumab treatment in adults with asthma. N Engl J Med. 2011;365(12):1088–1098. | ||
Pavord ID, Korn S, Howarth P, et al. Mepolizumab for severe eosinophilic asthma (DREAM): a multicentre, double-blind, placebo-controlled trial. Lancet. 2012;380(9842):651–659. | ||
Green RH, Brightling CE, McKenna S, et al. Asthma exacerbations and sputum eosinophil counts: a randomised controlled trial. Lancet. 2002;360(9347):1715–1721. | ||
Bai P. Biology of poly(ADP-ribose) polymerases: the factotums of cell maintenance. Mol Cell. 2015;58(6):947–958. |
 © 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.