Back to Journals » Journal of Inflammation Research » Volume 14
AST-120 Improves Cardiac Dysfunction in Acute Kidney Injury Mice via Suppression of Apoptosis and Proinflammatory NF-κB/ICAM-1 Signaling
Authors Shen WC ![]() , Chou YH, Shi LS, Chen ZW, Tu HJ, Lin XY, Wang GJ
, Chou YH, Shi LS, Chen ZW, Tu HJ, Lin XY, Wang GJ
Received 8 October 2020
Accepted for publication 9 January 2021
Published 24 February 2021 Volume 2021:14 Pages 505—518
DOI https://doi.org/10.2147/JIR.S283378
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Ning Quan
Wen-Ching Shen,1 Yu-Hsiang Chou,2 Li-Shian Shi,3 Zhi-wei Chen,4 Hai-Jian Tu,4 Xin-yi Lin,1 Guei-Jane Wang5– 8
1Department of Basic Medicine, Putian University, Putian City, Fujian Province, People’s Republic of China; 2Department of Internal Medicine, National Taiwan University Hospital Jin-Shan Branch, New Taipei City, Taiwan; 3Department of Biotechnology, National Formosa University, Yun-Lin, Taiwan; 4The Affiliated Hospital of Putian University, Putian City, Fujian Province, People’s Republic of China; 5School of Medicine, Graduate Institute of Clinical Medical Science, China Medical University, Taichung, 40402, Taiwan; 6School of Medicine, Graduate Institute of Biomedical Sciences, China Medical University, Taichung, 40402, Taiwan; 7Department of Medical Research, China Medical University Hospital, Taichung, 40447, Taiwan; 8Department of Health and Nutrition Biotechnology, Asia University, Taichung, 41354, Taiwan
Correspondence: Guei-Jane Wang
School of Medicine, Graduate Institute of Clinical Medical Science, China Medical University, Taichung, 40402, Taiwan
Tel +886-422052121 ext. 4727
Fax +886-422251483
Email [email protected]
Wen-Ching Shen
Department of Basic Medicine, Putian University, 1133 Xueyuan Road, Chengxiang District, Putian City, Fujian Province, 351100, People’s Republic of China
Tel +86-13959593413
Fax +86-2768812
Email [email protected]
Purpose: Acute kidney injury (AKI) is a devastating disorder associated with considerably high morbidity and mortality. Reports have shown that AST-120, an oral charcoal adsorbent, can reduce oxidative stress by lowering serum indoxyl sulfate levels. The effects of AST-120 and indoxyl sulfate on kidney injury and cardiac dysfunction were investigated in vivo and in vitro.
Patients and Methods: Patients were tracked for enrollment upon receiving a diagnosis of AKI. Plasma was collected to determine the renal and inflammatory parameters. Renal ischemia/reperfusion (I/R) induced AKI or sham operation was performed in C57BL/6J mice. Animals were divided into sham, AKI+vehicle, and AKI+AST-120 groups. Plasma and tissues were assembled after 48 h to assess apoptotic and inflammatory responses. We also conducted human umbilical vein endothelial cell (HUVECs) and HL-1 cardiomyocyte culture studies to determine the underlying mechanisms of indoxyl sulfate’s effects. Echocardiography, histopathology, biochemical indexes, ELISA, terminal dUTP nick-end labeling (TUNEL) and Western blot analysis were performed.
Results: The cohort included 25 consecutive patients with AKI and 25 non-AKI. Plasma levels of creatinine, indoxyl sulfate, IL-1β and ICAM-1 were significantly higher in patients with AKI than in non-AKI controls. Plasma levels of blood urea nitrogen, creatinine, indoxyl sulfate, IL-1β and renal tubular injury were increased in mice after renal I/R and were decreased by AST-120 treatment. In addition, AST-120 therapy not only improved the parameters assessed by echocardiography but also substantially attenuated the elevation of plasma BNP. Oral administration of AST-120 significantly downregulated NF-κB/ICAM-1 expression and reduced cell apoptosis in both kidney and heart after renal I/R injury.
Conclusion: Our investigations demonstrated that AST-120 administration improves cardiac dysfunction in AKI mice via the suppression of apoptosis and proinflammatory NF-κB/ICAM-1 signaling.
Keywords: acute kidney injury, cardiac dysfunction, AST-120, inflammation, apoptosis, NF-κB
Introduction
Ischemia/reperfusion (I/R)-induced acute kidney injury (AKI) is an important factor in chronic allograft dysfunction and is characterized by progressive renal dysfunction, afferent arteriolopathy, and inflammatory cell influx.1,2 When the kidneys lose their filtering function due to AKI, dangerous levels of metabolites accumulate in the body, including urea, nitrogenous waste products, and uremic toxins.1 Indoxyl sulfate is an anionic uremic toxin that accumulates in the serum of patients with uremia. Indoxyl sulfate is generated enzymatically in the liver from indole, which is produced from tryptophan by the intestinal flora.2 Indoxyl sulfate treatment is known to damage the proximal tubules of the kidney by various mechanisms, including inflammation, oxidative stress, DNA damage, and apoptosis.3 The adsorption of uremic toxin by carbonic adsorbents in the intestines has been studied extensively as a therapy. AST-120, a type of oral carbonic adsorbent, may prolong the time to initiation of hemodialysis and improve uremic symptoms in patients with kidney disease.4
Much of the mortality risk from AKI is thought to stem from extrarenal complications or the distant organ effects of AKI.5 Reduced kidney function is associated with a risk of cardiovascular events.6 Recent experimental models have elucidated some potential mechanisms of deleterious organ crosstalk, including proinflammatory cascades, oxidative stress, activation of proapoptotic pathways, differential molecular expression, and leukocyte trafficking.7 Leukocyte–endothelial interactions play an important role in the development of cardiovascular diseases.8 Proinflammatory cytokines, including interleukin (IL)-1β, found commonly in vascular lesions, may induce the activation of chemotactic factors and cell adhesion molecules, all of which contribute to the inflammatory process and the pathology of several human diseases, including AKI.6 Cell adhesion molecules belonging to the immunoglobulin superfamily, such as those in the selectin family, including intercellular cell adhesion molecule-1 (ICAM-1), are upregulated to mediate monocyte/macrophage infiltration into vascular lesions.9 The expression of ICAM-1, induced by various stimuli, may initiate networks that augment cell inflammation by activating multiple signaling pathways.10 IL-1β-induced ICAM-1 expression has been shown to be mediated through the activation of nuclear factor kappa-light-chain-enhancer of activated B (NF-κB).11 Indoxyl sulfate levels are increased significantly in patients with decreased renal function, and several studies have found that indoxyl sulfate levels are closely correlated with cytokines and transcription factors.12,15 Transcription factors play an important role in inflammation and apoptosis in both kidney and heart diseases.7 Several studies have indicated that indoxyl sulfate stimulates the phosphorylation of NF-κB in various cell types, including endothelial cells.15 Although previous studies have shown that indoxyl sulfate has inflammatory and apoptosis effects on tubular cells, it is unclear whether it has these effects on vascular endothelial cells.12–16
The present study was undertaken to investigate whether the administration of AST-120 is effective in decreasing I/R-induced renal injury and cardiac dysfunction. We evaluated the effect of AST-120 on the removal of uremic toxins, inflammation, and apoptosis in an experimental model of AKI. The results of our study demonstrate clearly that the removal of uremic toxins by AST-120 treatment is effective in decreasing the progression of injury in both the kidney and the heart, and that this protective effect of AST-120 is related to decreased inflammation and apoptosis.
Patients and Methods
Human Study
A total of 50 persons, including 25 AKI patients and 25 non-AKI patients, were enrolled in National Taiwan University Hospital (Taipei, Taiwan). The definition of AKI was based on the criteria established by the Acute Kidney Injury Network (AKIN).17 To avoid the effects of other causes of chronic or severe heart dysfunction, the study excluded patients with chronic heart disease, with an abnormal echocardiogram on admission, acute coronary syndrome, acute myocardial infarction, acute pulmonary embolism, status post cardiopulmonary resuscitation, cor pulmonale, and respiratory failure with high positive end-expiratory pressure. Plasma was collected on the second day after diagnosis and kept frozen in aliquots until analysis. This study was performed in compliance with the Declaration of Helsinki and was approved by the Institutional Review Board of National Taiwan University Hospital, Taipei, Taiwan (201111012RIB). All participants or their legal representatives provided written informed consent.
AKI Mouse Model
All experimental procedures and protocols involving animals were conducted in accordance with local institutional guidelines for animal care, approved by the Institutional Animal Care Committee of National Taiwan University (Taipei, Taiwan), and complied with the Guide for the Care and Use of Laboratory Animals (NIH publication No. 86–23, revised 1985). C57BL/6 mice (20 ± 2 g body weight, aged 8–9 weeks) were anesthetized briefly with an anesthesia cocktail (80 mg/kg ketamine plus 100 mg/kg xylazine) via intraperitoneal injection, and unilateral AKI was induced by ligature (tied for complete occlusion) for 24 min with a microaneurysm clamp on the left renal artery after the removal of the right kidney for 2 weeks. The body temperature of the mice was maintained at 37 °C by a heating pad. Mice were assigned randomly to one of the following groups: (1) sham group (n = 10): sham-operated mice were subjected to the same surgical procedure, except that the left renal pedicles were not clamped; (2) AKI-V group (n = 10): after unilateral AKI surgery, mice received the same volume of the vehicle for AST-120 as AKI-AST mice; and (3) AKI-AST group (n = 10): after unilateral AKI surgery, mice received oral administration of AST-120 (125 µg/kg/day, b.i.d.) for 2 days.
Specimen Collection and Histopathological Examination
The mice were sacrificed by an intraperitoneal injection of an overdose of sodium pentobarbital 2 days after AKI. Plasma and part of kidney and heart tissues were collected and stored at −80°C until use. The other part of kidney and heart tissues were then post-fixed overnight at 4 °C in 4% paraformaldehyde (Bionovas, Ontario, Canada) and embedded in paraffin. Morphometric changes were examined in sections of renal tissue stained with hematoxylin and eosin (Sigma-Aldrich, MO, USA). The four morphologic indices used were tubule flattening, necrosis, apoptosis, or the presence of casts, resulting in a positive score in a double-blind fashion.
For immunohistochemical analysis of NF-κB expression, paraffin-embedded tissues (5 µm) were cut and dried at 37 °C for 2 h on slides coated with 3-aminopropyltriethoxysilane and then dried for 30 min at 56 °C. The tissue sections were de-paraffinized in 100% xylene and partially rehydrated in graded alcohol solution. Endogenous peroxidase activity was blocked by incubation for 10 min with Novolink Peroxidase Block (Novocastra, Newcastle, UK). Sections were subjected to antigen retrieval for 5 min in 10 mM Tris buffer (pH 8.0, 1 mg/mL trypsin). Following protein blocking for 15 min at room temperature with Novolink Protein Block, the sections were incubated for 16 h at 4 °C with an anti-NF-κB antibody (Epitomics) with 10% normal goat serum (NGS) in phosphate-buffered saline (PBS), then washed three times with PBST (PBS, Tween 0.05%, pH 7.6), and incubated for 20 min at room temperature with Novolink post-primary blocking solution and Novolink polymer. Finally, the sections were visualized using the Novolink DAB chromogen, and the slides were counterstained with hematoxylin.
For the immunofluorescence assay, HUVECs were fixed for 15 min at room temperature in 4% paraformaldehyde and non-specific binding was blocked by incubation with 10% NGS in PBS for 90 min. The cells were then incubated overnight at 4 °C in a 1:50 dilution of ICAM-1 antibodies with 10% NGS, then washed with PBS (for 5 min, three times), and incubated for 1 h at 37 °C with fluorescein isothiocyanate (FITC)-conjugated goat anti-rabbit IgG antibodies and PBS–NGS. To localize NF-κB expression in situ, confluent HUVECs (controls or cells treated for 24 h with indoxyl sulfate or a different treatment) on slides were incubated in the absence or presence of 0.2 ng/mL IL-1β for 30 min, fixed in 4% paraformaldehyde in PBS, at pH 7.4, for 15 min at room temperature, then were blocked with 10% NGS in PBS for 1 h at room temperature, and incubated overnight at 4 °C in a 1:50 dilution of rabbit anti-human NF-κB p65 antibodies (Epitomics). After washing, the slides were incubated for 1 h at 37 °C with FITC-conjugated goat anti-rabbit IgG antibodies (1:200 dilution in PBS–NGS; Epitomics). Images were taken using a fluorescence microscope.
Evaluation of Renal Function
To provide evidence of I/R-induced changes in renal function, plasma creatinine and blood urea nitrogen (BUN) levels in human and mouse specimens were measured in the Department of Laboratory Medicine at Putian University Hospital.
Measurement of Plasma IL-1β and Brain Natriuretic Peptide
Human and mouse IL-1β, and mouse brain natriuretic peptide (BNP), were determined using commercially available enzyme-linked immunosorbent assay (ELISA) kits (RayBiotech, GA, USA). Human ICAM-1 was measured by ELISA (Thermo Fisher Scientific) with a sensitivity of 2.2 ng/mL.
Indoxyl Sulphate Quantification by Liquid Chromatography
The mouse plasma sample was thawed in a water bath at room temperature. Twenty microliters of the plasma sample was mixed with 80 μL of acetonitrile (J. T. Baker, NJ, USA) in a 1.5-mL microcentrifuge tube and vortex-mixed for 60 s. The tube was then centrifuged at 1860 x g for 10 min, and the supernatant was transferred to a sample vial. A 10-μL aliquot of the supernatant was injected into the high-performance liquid chromatography (HPLC) system. HPLC was performed with a Waters HPLC System Series 600 (Waters, MA, USA) equipped with a fluorescence detector (Surveyor FL Plus Detector, Thermo Fisher Scientific, MA, USA). A reverse-phase C18 column (SHISHEDO CAPCELL PAK type SG C18, 5 μm, 150 mm × 4.6 mm) was used and the mobile phase was composed of acetonitrile/water (8:92 v/v) containing 0.2% trifluoroacetic acid (Alfa Aesar, MA, USA). An isocratic elution was applied, and the mobile phase was delivered at a flow rate of 1.0 mL/min. The excitation and emission wavelengths of the fluorescence detector were set at 280 and 375 nm, respectively. Calibration samples of indoxyl sulfate were prepared at 1.7, 3.9, 8.8, 9.8, 44.4, and 100 μM in plasma. The calibration curve was calculated with the peak area against the spiked concentrations. The indoxyl sulfate concentration in subject samples was then determined according to the measured peak area.
Preparation of Cell Lysates and Western Blot Analysis
To prepare cell lysates, cells from HUVECs or HL-1 cultures or mouse kidneys were lysed for 1 h at 4 °C in 20 mM Tris-HCl, 150 mM NaCl, 1 mM EDTA, 1 mM EGTA, 1% Triton X-100, and 1 mM PMSF, at pH 7.4, and then the lysate was centrifuged at 4000 x g for 30 min at 4 °C. An aliquot of the supernatant (20 µg/mL of protein) was subjected to 10% SDS-PAGE and the proteins were transferred onto Polyvinylidene fluoride (PVDF) membranes (Millipore, city, MA, USA). The membranes were then blocked by incubation for 30 min at room temperature with 5% non-fat milk in Tris-buffered saline containing 0.2% Tween 20 (TBST). The membranes were incubated overnight at 4 °C with a 1:1000 dilution in TBST of polyclonal rabbit antibodies against ICAM-1 (Epitomics), or NF-κB p65 (1:1000 in TBST; Epitomics), then for 1 h at room temperature with horseradish peroxidase (HRP)-conjugated goat anti-rabbit IgG antibodies (1:2000 dilution in TBST; Gene Tex, CA, USA). The bound antibodies were detected using Chemiluminescence Reagent Plus (NEN, MA, USA). The intensity of each band was quantified using a densitometer. GAPDH, used as the internal control, was detected using rabbit anti-GAPDH antibody (1:15,000 dilution in TBST; Gene Tex) and HRP-conjugated goat anti-rabbit IgG antibody (1:2000 dilution in TBST; Gene Tex). The bands were determined using an enhanced chemiluminescent detection kit. The relative protein expression levels were quantified by Western blotting and using UN-SCAN-IT gel 6.1 software after densitometric scanning of the exposed films.
Cell Culture
Primary cultures of human umbilical vein endothelial cells (HUVECs) were prepared as described previously.18 HUVECs purchased from Bioresource Collection and Research Center (BCRC) in Taiwan were maintained in Medium 199 (Invitrogen, MA, USA) complemented with 10% fetal calf serum, 30 µg/mL endothelial cell growth supplement (R&D Systems, MN, USA), 1% penicillin–streptomycin, and 7.5 U/mL heparin under standard cell culture conditions (humidified atmosphere, 5% CO2, 37°C). Cells between passages 2 and 5 were used for the experiment. In addition, cultures of HL-1 cardiomyocytes were prepared as described previously.19 HL-1 cardiomyocytes (a kind gift from Dr. C. T. Tsai, Division of Cardiology, Department of Internal Medicine and Cardiovascular center, National Taiwan University College of Medicine and Hospital, Taipei City, Taiwan) were cultured on Claycomb medium supplemented with 1 μM retinoic acid, 10 μM norepinephrine (Sigma-Aldrich), 100 units/mL penicillin, 100 μg/mL streptomycin and an additional 1× nonessential amino acids (Life Technologies, CA, USA). The medium was changed approximately every 24 hr. The cells were grown at 37°C in an atmosphere of 5% CO2 and 95% air at a relative humidity of approximately 95%.
Echocardiographic Assessment
Mice were anesthetized lightly with isoflurane (1–2%). M-mode echocardiograms were obtained from the short-axis view of the left ventricle at the tip of the mitral valve leaflets using a Prospect (S-Sharp, Taipei, Taiwan) with a 40 MHz transducer before surgery and at the end of the study. Measurements taken included cardiac output (CO), left ventricular ejection fraction (LVEF) and left ventricular fractional shortening (LVFS).
Identification of Apoptosis by TUNEL Staining
Apoptotic cells were identified by terminal dUTP nick-end labeling (TUNEL) staining using In Situ Cell Death Detection Kit (Sigma-Aldrich). The sections were counterstained with 4ʹ,6-diamidino-2-phenylindole to visualize all nuclei and viewed under a fluorescence microscope. The number of TUNEL-positive nuclei was calculated from six non-overlapping regions of each tissue cross-section in a high-power field.
Statistical Analysis
Where applicable, the results are presented as the mean ± standard error of the mean (SEM). Statistical analyses were carried out using GraphPad Prism software (GraphPad Software, La Jolla, CA, USA). The statistical significance was evaluated by one-way analysis of variance. A p value of less than 0.05 was considered statistically significant.
Results
AKI Leads to Systemic Elevation of Indoxyl Sulfate, IL-1β, and ICAM-1
Plasma levels of creatinine were significantly higher on the second day after diagnosis with AKI compared with non-AKI controls (AKI, 4.9 ± 0.4 vs non-AKI, 0.8 ± 0.1 mg/dL; Figure 1A), as were plasma levels of indoxyl sulfate (AKI, 18.1 ± 4.5 vs non-AKI, 4.0 ± 0.7 μM; Figure 1B) and proinflammatory cytokine IL-1β (AKI, 53.1 ± 9.5 vs non-AKI, 4.6 ± 2.6 pg/dL; Figure 1C) and adhesion molecule ICAM-1 (AKI, 17,710.7 ± 501.7 vs non-AKI, 1806.1 ± 34.0 pg/dL; Figure 1D).
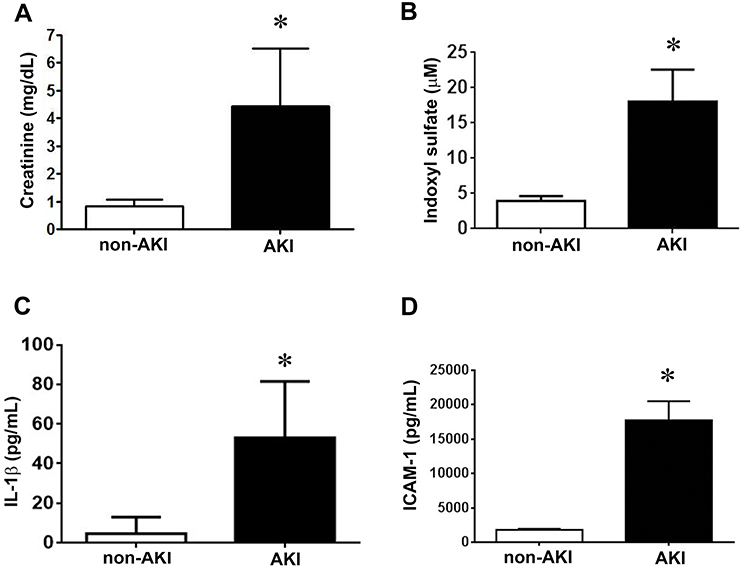 |
Figure 1 AKI stimulates creatinine, indoxyl sulfate, IL-1β, and ICAM-1 production. (A) Plasma creatinine levels in patients with and without AKI. n = 25 in each group. (B) Plasma was collected and assayed for indoxyl sulfate by chromatography. n = 6 per group. (C and D) The amounts of IL-1β and ICAM-1 in the plasma were determined by using ELISA kits according to manufacturer’s instructions. n = 10 per group in IL-1β assay, n = 20 per group in ICAM-1 group. Data are expressed as mean ± SEM, *p < 0.05 vs the non-AKI l group. |
AST-120 Decreases Serum Indoxyl Sulfate, IL-1β, and Renal Tubular Injury in AKI Mice
To determine the involvement of indoxyl sulfate in the pathogenesis of AKI in a mouse model, we performed the renal I/R injury mouse model (Figure 2A). As shown in Figure 2, I/R injury evoked AKI with an increase in serum creatinine, BUN, IL-1β and plasma accumulation of indoxyl sulfate at 48 h. The increased levels of plasma creatinine and BUN in AKI mice were attenuated significantly by AST-120 treatment (Figure 2G and H). Moreover, the increased levels of serum IL-1β and indoxyl sulfate in AKI mice were also attenuated by AST-120 (Figure 2B–D). In parallel to the renal function parameters, the AKI-vehicle kidney showed marked tubular injury, including intratubular casts, the absence of nuclei, and tubular dilation (Figure 2E and F). AST-120 treatment attenuated the tubular injury significantly.
 |
Figure 2 Morphology and characterization of levels of blood urea nitrogen (BUN), creatinine, and indoxyl sulfate after I/R injury. (A) Gross appearance of the kidneys. Mice were sacrificed 2 days post surgery, and then stained with hematoxylin and eosin (H&E). (B and C) Chromatograms of standard solutions and serums. Typical chromatograms obtained with the serum of the sham group (blue), the AKI-V (red), and the AKI-AST (green). (D) IL-1β levels in plasma from the sham, AKI-V, and AKI-AST groups measured by ELISA. The data are expressed as the mean ± SEM (n = 5) per group; *p < 0.05 vs the sham group; #p < 0.05 vs the AKI-V group. (E) High-magnification images showing tubular necrosis, the absence of nuclei, and tubular dilation in the AKI-V group at 48 h. Scale bar = 100 μm. (F) Semiquantitative analysis of tissue damage at 48 h after I/R injury (total injury score based on tubular necrosis, the absence of nuclei in tubular cells, and tubular dilation). (G and H) BUN and creatinine levels in the three groups; data are presented as mean ± SEM. n = 5 per group, *p < 0.05 vs the sham group, #p < 0.05 vs the AKI-V group. |
AST-120 Reduces Renal Expression of ICAM-1 and NF-κB in AKI Mice
Compared with sham kidneys, immunofluorescence showed the increased expression of ICAM-1 in CD31+ endothelial cells of AKI kidneys (Figure 3A). AST-120 reversed the expression of ICAM-1. Western blot and ELISA analyses further showed that the expression of ICAM-1 was increased in AKI kidneys, which was attenuated by AST-120 treatment (Figure 3B and C). NF-κB expression was also increased in AKI kidneys and its expression was attenuated by AST-120 treatment (Figure 3D and E).
 |
Figure 3 Immunohistochemical staining for ICAM-1 or NF-κB expression in kidney tissue from AKI mice. (A) Renal tissues were fixed in 4% paraformaldehyde and embedded in paraffin; then, sections were incubated with anti-CD31 antibodies to identify vessels (green). Immunofluorescence microscopy showed that ICAM-1 (red) was present in the renal tissues. Overlap of ICAM-1-positive cells and endothelial cell staining (CD31) is shown (orange). Scale bar = 50 μm. (B) ICAM-1 expression in the sham, AKI-V, and AKI-AST groups determined by Western blotting. n = 3 per group. (C) Plasma levels of ICAM-1 in the sham, AKI-V, and AKI-AST groups, measured by ELISA. (D and E) The expression of NF-κB in the three groups was screened using an immunohistochemistry assay. Representative photomicrographs of immunohistochemical staining for NF-κB in renal tissues from the sham, AKI-V, and AKI-AST groups. The levels of NF-κB in the cell lysates were evaluated by Western blotting. The data are presented as mean ± SEM. n = 4 per group, *p < 0.05 vs the sham group; #p < 0.05 vs the AKI-V group. |
Indoxyl Sulfate Enhances ICAM-1 Expression and NF-κB Activation in IL-1β-Treated Endothelial Cells
Given that AST-120 could attenuate the expression of ICAM-1 and NF-κB in injured kidneys, we studied the mechanisms underlying these effects. We first treated HUVECs in the absence or presence of indoxyl sulfate at concentrations ranging from 0.2 to 2 mM and did not obtain detectable expressions of ICAM-1 by Western blot analysis (Figure 4A). The expression of ICAM-1 was induced in the presence of IL-1β, and was further enhanced by 0.2 mM indoxyl sulfate. Indoxyl sulfate at concentrations of 1 or 2 mM did not show the same enhancement. In all subsequent experiments, 0.2 mM indoxyl sulfate was used.
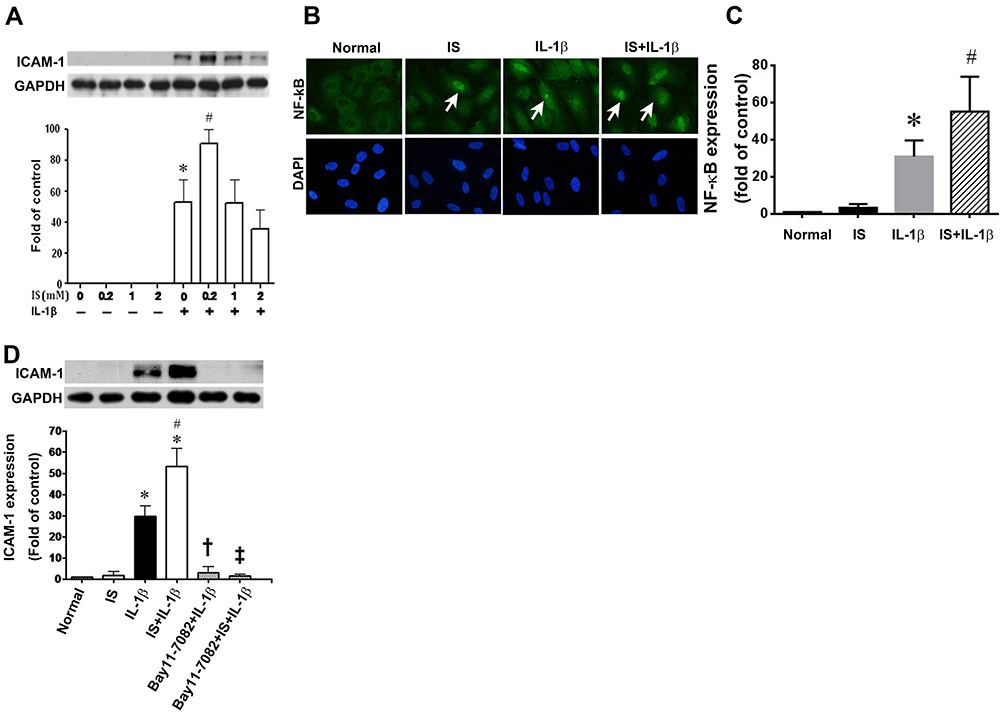 |
Figure 4 Effect of indoxyl sulfate (IS) on IL-1β-induced ICAM-1 and NF-κB expression. (A) HUVECs were incubated for 24 h with various concentrations of indoxyl sulfate, and then with 0.2 ng/mL of IL-1β for 4 h in the continued presence of the same concentration of indoxyl sulfate. ICAM-1 expression was measured in cell lysates by Western blotting. GAPDH was used as the loading control. n = 3–5 per group, the data are presented as mean ± SEM, *p < 0.05 compared with cells untreated with IL-1β; #p < 0.05 compared with cells treated with IL-1β (IS at 0 mM). (B) Subcellular distribution of NF-κB p65 in HUVECs shown by immunofluorescence microscopy. Scale bar = 50 μm. (C) NF-κB expression in the sham, Normal, IS, IL-1β and IS+IL-1β groups determined by Western blotting (n = 3); *p < 0.05 vs the normal group; #p < 0.05 vs the IS group. (D) HUVECs were incubated with or without 0.2 mM indoxyl sulfate for 24 h and then treated with the NF-κB signaling pathway inhibitor BAY11-7082 (BAY; 10 μM) for 1 h, followed by stimulation with IL-1β (0.2 ng/mL) for 30 min. ICAM-1 expression was measured by Western blotting. n = 3–6 per group; the data are presented as mean ± SEM, *p < 0.05 compared with cells without IL-1β, #p < 0.05 compared with cells treated with IL-1β, †p < 0.05 compared with cells treated with IL-1β, ‡p < 0.05 compared with cells treated with IL-1β and IS. |
As the promoter of the ICAM-1 gene contains consensus binding sites for NF-κB,20 we investigated whether indoxyl sulfate enhanced IL-1β-induced ICAM-1 expression via an effect on these transcription factors. Compared with HUVECs in regular culture medium only, immunofluorescence showed weak NF-κB p65 staining in the nuclei after indoxyl sulfate treatment for 24 h (Figure 4B). IL-1β stimulation led to marked nuclear staining of NF-κB p65 after 30 min. The nuclear staining of NF-κB p65 induced by IL-1β was further enhanced by indoxyl sulfate pretreatment for 24 h (Figure 4B and C). To confirm the role of NF-κB p65 in the enhancement of IL-1β-induced ICAM-1 by indoxyl sulfate, HUVECs were pretreated with the NF-κB inhibitor, Bay11-7082 (10 μM). As shown in Figure 4D, IL-1β-induced ICAM-1 expression in the absence or presence of indoxyl sulfate were both blocked by preincubation with Bay11-7082 for 1 h. These results suggest that indoxyl sulfate enhances ICAM-1 expression in IL-1β-treated HUVECs by promoting NF-κB activation.
AST-120 Improves Cardiac Function in AKI Mice
To assess cardiac function, echocardiography was performed both before and 48 h after renal I/R injury. Representative M-mode tracings are shown in Figure 5A. Functional changes of the heart 48 h after renal I/R injury included decreases in CO, LVEF, and LVFS in the AKI-vehicle group (Figure 5B–D). In parallel to the functional deterioration shown in the echocardiograms, elevated plasma levels of BNP were observed in AKI-vehicle mice (Figure 5E). AST-120 treatment not only improved the functional parameters assessed by echocardiography but also substantially improved the elevation of plasma BNP.
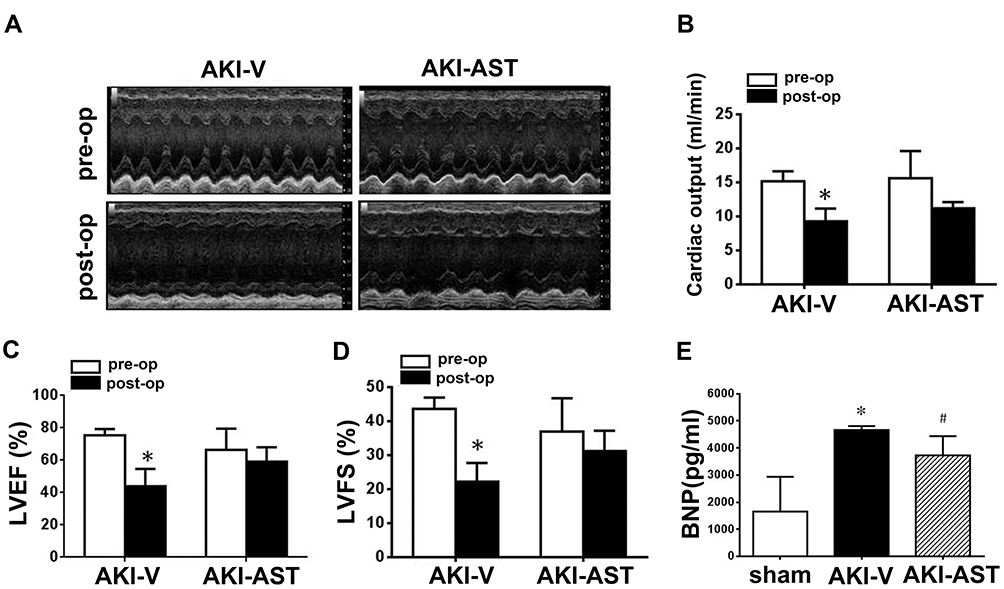 |
Figure 5 AST-120 administration decreased cardiac dysfunction following renal I/R injury. (A) Echocardiographic images were taken before surgery (pre-op) and 48 h after renal I/R injury (post-op). Representative M-mode echocardiographic tracings (24 min) are presented. (B–D) Data from echocardiograms. n = 6 per group. (E) BNP levels in the plasma from the sham, AKI-V, and AKI-AST groups measured by ELISA. n = 5 per group; the data are expressed as mean ± SEM, *p < 0.05 vs the pre-op group; #p < 0.05 vs the AKI-V group. |
Indoxyl Sulfate Enhances NF-κB Activation in IL-1β-Treated HL-1 Cells
To study the mechanisms involved in the cardioprotective effects of AST-120, HL-1, a cardiomyocyte cell line, was used as an in vitro cell model. In both HUVECs and HL-1 cells, Bax expression in IL-1β-treated cells was upregulated significantly by indoxyl sulfate (Figure 6A and B). Immunofluorescence staining was performed to determine the effects of indoxyl sulfate on NF-κB activation in IL-1β-treated HL-1 cells. The NF-κB p65 translocation induced by IL-1β was also expressed significantly by pretreatment for 24 h with indoxyl sulfate (Figure 6C). To confirm the role of NF-κB p65 in indoxyl sulfate-enhanced IL-1β-induced NF-κB p65 activation, HL-1 cells were pretreated with the NF-κB inhibitor Bay11-7082 and then analyzed by immunofluorescence. As shown in Figure 6C, cells pretreated with NF-κB inhibitors demonstrated a reduction in NF-κB protein translocation in HL-1 cells in the presence of IL-1β and IL-1 plus indoxyl sulfate.
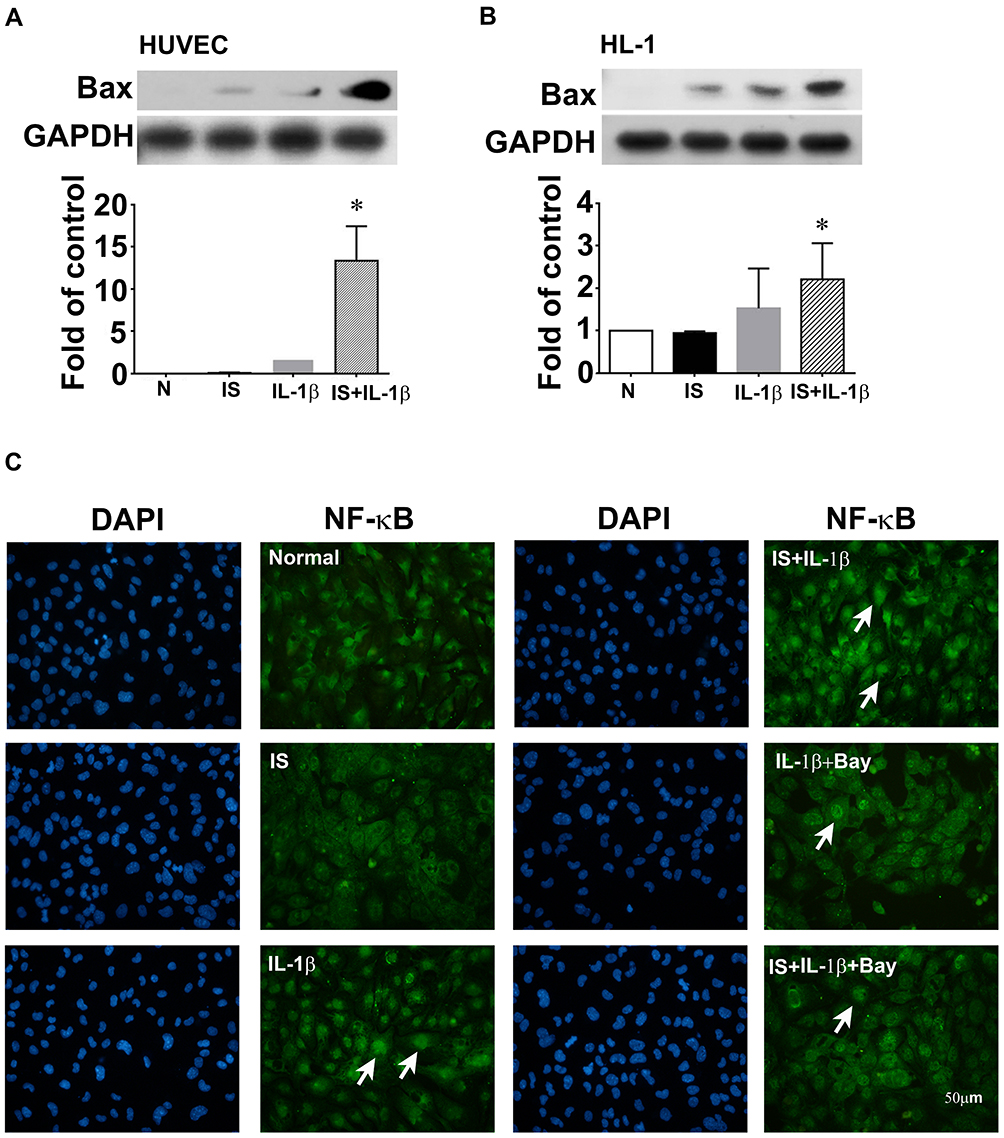 |
Figure 6 Effect of indoxyl sulfate (IS) on IL-1β-induced apoptotic protein and NF-κB expression in human umbilical vein endothelial cells (HUVECs) and HL-1 cardiomyocytes. (A and B) HUVECs and HL-1 cells were incubated for 24 h with or without indoxyl sulfate (0.2 mM), and then with 0.2 ng/mL of IL-1β for 4 h in the continued presence of the same concentration of indoxyl sulfate, and Bax expression was measured in cell lysates by Western blotting. GAPDH was used as the loading control. (C) Subcellular distribution of NF-κB p65 in HL-1 shown by immunofluorescence microscopy. Scale bar = 50 μm. n = 3–5 per group; the data are expressed as mean ± SEM, *p < 0.05 vs the IL-1β group. |
AST-120 Attenuates Apoptosis and NF-κB Expression in Cardiomyocytes Caused by AKI
Apoptosis in the left ventricles, specifically of cardiomyocytes and endothelial cells, assessed by TUNEL staining, was increased significantly in AKI-vehicle mice (Figure 7A and B). AST-120 treatment substantially decreased the apoptosis in the left ventricles caused by AKI. Western blot analysis revealed the decreased expression of anti-apoptotic protein Bcl-2 in the left ventricles of AKI-vehicle mice, whereas AST-120 treatment led to a partial reversal of this decrease (Figure 7C). Moreover, the expression of pro-apoptotic protein caspase 3 was increased significantly in the left ventricles of AKI-vehicle mice, but not in AKI-AST mice (Figure 7D). As shown in Figure 7E and F, no NF-κB staining was seen in the hearts in the sham group, while the I/R injury group showed strong NF-κB staining in the cardiomyocytes and endothelial cells. AST-120 resulted in a significant decrease the expression of NF-κB.
 |
Figure 7 AST-120 therapy reduced cell apoptosis and NF-κB activation following renal I/R injury. (A and B) Detection of apoptosis (arrows) by HE staining (upper panels) and TUNEL staining (lower panels) in the normal (both kidneys intact), sham, AKI+V, and AKI+AST groups. The number of apoptotic cells per high-power field was counted. n = 4 per group; the data are presented as mean ± SEM, *p < 0.05 vs the sham group; #p < 0.05 vs the AKI+V group. (C and D) The expression of Bcl-2 and caspase 3 at day 2 after I/R injury in heart tissues from the sham, AKI-V, and AKI-AST groups determined by Western blotting. n = 6 per group; the data are presented as mean ± SEM, *p < 0.05 vs the sham group, #p < 0.05 vs the AKI+V group. (E and F) NF-κB expression in the three groups was screened using an immunohistochemistry assay. Representative photomicrographs of immunohistochemical staining for NF-κB in heart tissues from the sham, AKI-V, and AKI-AST groups. n = 4; the data are presented as mean ± SEM, *p < 0.05 vs the sham group; #p < 0.05 vs the AKI-V group. |
Discussion
In this study, we demonstrate for the first time that higher plasma levels of ICAM-1 are closely associated with plasma IL-1β levels in patients with AKI, who have high indoxyl sulfate levels. Similarly, our findings demonstrate markedly increased expression of IL-1β, NF-κB, and ICAM-1 in a renal I/R injury AKI mouse model with higher plasma levels of indoxyl sulfate than those of sham-operated mice. Higher amounts of indoxyl sulfate, associated with higher proinflammatory cytokines accumulating in the systemic circulation, cause recurrent damage to the kidneys and other vital organs, such as the heart. We found that indoxyl sulfate and IL-1β, at the concentrations observed in patients with AKI, can induce apoptosis and inflammation in cardiomyocytes, and this mechanism may account for cardiac dysfunction induced by AKI. This uremic toxin is implicated in the pathogenesis of renocardiac crosstalk. Indoxyl sulfate-induced apoptosis and proinflammatory NF-κB/ICAM-1 signaling could account, at least in part, for the underlying mechanisms of renal and cardiac dysfunction. However, AST-120 is a promising therapy that may attenuate apoptosis and inflammation, and thereby improve renal and cardiac dysfunction.
Currently, the detailed mechanisms of the development of renal dysfunction in the setting of heart disease as well as its effects are not fully understood, and the best therapeutic method has not yet been established. We previously reported that patients with AKI have higher plasma levels of indoxyl sulfate than patients with normal kidney function.21 Indoxyl sulfate is a potential oxidant source, and has been shown to cause a urinary disorder characterized by inflammatory responses in the kidney.22 Studies have shown that indoxyl sulfate has adverse effects on renal and cardiovascular functions.23,24 Simmons et al found that patients with AKI have significantly higher plasma levels of the proinflammatory cytokine IL-1β.25 Moreover, higher levels of indoxyl sulfate in the presence of inflammatory cytokines also significantly enhance vascular lesion formation in AKI.4 Vascular lesions, a type of inflammation, result from the adherence of monocytes/macrophages to activated endothelial cells.23 The present study provides strong evidence that higher plasma ICAM-1 levels are closely related to higher plasma IL-1β levels in patients with AKI. The AKI mouse model, with high plasma indoxyl sulfate levels compared to sham-operated mice, showed higher expression of ICAM-1 in the circulatory system. In addition, the expression of ICAM-1 induced in vascular endothelial cells cultured in an inflammatory environment caused by IL-1β was significantly increased by pre-incubating with indoxyl sulfate. It has been reported that indoxyl sulfate increases adhesion molecule proteins in vascular smooth muscle, and promotes the recruitment of leukocytes.26 Taken together, these results may explain a recent observation by Barreto et al, who found that plasma indoxyl sulfate levels are associated with the incidence of vascular disease and total mortality in renal diseases.27 In addition, altered ICAM-1 shedding is an independent predictor of cardiovascular death in end-stage renal disease patients.24,28
Our previous results showed that the indoxyl sulfate-induced increase in E-selectin expression in IL-1β-treated HUVECs is mediated through MAPK phosphorylation and activation of NADPH oxidase/reactive oxygen species (ROS), leading to the activation of NF-κB.18 The activation of NF-κB is responsible for increased transcription of adhesion molecules, cytokines, and chemokines and plays a pivotal role in the pathological consequences of AKI.29 Specifically, the promoter site of the human ICAM-1 gene contains consensus binding sites for NF-kB.20 Indoxyl sulfate has been shown to regulate the expression and activity of ICAM-1 through various signaling molecules, such as NF-κB in endothelial cells.15 The present data further support the involvement of NF-κB in indoxyl sulfate-induced ICAM-1 expression in IL-1β -activated HUVECs by using a selective pharmacological inhibitor of NF-kB, BAY11-7082. It has been reported that activation of the NF-κB-inducing kinase (NIK)/NF-κB axis following Akt phosphorylation induced a proinflammatory reaction, potentially leading to progressive graft kidney injury.30,31 NF-κB-mediated inflammation and PI3K/Akt apoptosis signaling pathways have also been demonstrated in the pathogenesis of acute nephrotoxicity induced by cisplatin.32 A recent study demonstrated that AKI may activate Drp1, inducing mitochondrial fragmentation and apoptosis; however, the mechanism underlying Drp1 activation and apoptosis in cardiomyocytes following renal I/R injury has not been identified.33
On the other hand, the transcription factor NF-κB is activated by multiple mechanisms, including apoptosis and inflammation. However, the relationship between apoptosis and inflammation has not been elucidated clearly in this study. Zhang and colleagues have demonstrated that genes related to apoptotic and inflammatory pathological processes interact with each other, and also may influence each other.34–36 Functional protein-protein interaction analysis have revealed inflammation response clusters including TNF-α, IL-1β, IL-6, Col1α1, and NF-κB, which may be closely related to apoptosis-related genes such as Bcl and Bax. Future studies are needed to elaborate the gene regulatory network of apoptotic and inflammatory signaling in AKI.
AST-120 is an oral charcoal absorbent consisting of spherical particles produced from high-purity porous carbon material.37 AST-120 adsorbs uremic toxins in the gastrointestinal tract, so that the toxins are not absorbed into the body but excreted with the feces.38 Niwa et al reported that indole, a precursor of indoxyl sulfate, is adsorbed by AST-120 in the intestinal tract, thereby reducing indoxyl sulfate production and alleviating its renal toxic effect.39 Investigators have published that the effects of AST-120 are due to a decrease in indoxyl sulfate in the blood and reduced oxidative stress.40,41 In the present study, the increased serum indoxyl sulfate, BUN, creatinine, and BNP as well as IL-1β, ICAM-1 levels in AKI mice were all reduced by AST-120 treatment. Oral administration of AST-120 in an in vivo environment will also suppress apoptosis and proinflammatory NF-κB/ICAM-1 performance. Histological analysis of kidney sections stained with H&E showed tubular injury, including intratubular casts, absence of nuclei, and tubular dilation, and peritubular capillary rarefaction were more severe in samples from AKI group. Notably, these pathological changes were alleviated in the kidneys of mice that had been treated with AST-120. Similarly, we found beneficial effects of AST-120 on cardiac tissue. Echocardiography data indicated that AST-120 exhibited a protective effect against AKI-induced cardiac structural and functional damage. Moreover, AST-120 therapy downregulated the proapoptotic protein caspase-3 and upregulated the anti-apoptotic protein Bcl-2 in the hearts of AKI mice, possibly through the reduction of indoxyl sulfate and IL-1β. Considering that AST-120 also prevented kidney injury, the protective effect of AST-120 on the heart may have resulted from the reduction of the uremic toxin indoxyl sulfate and subsequent proinflammatory IL-1β in the circulatory system. In previous trials, both cardiac and renal functions significantly improved in patients with chronic kidney disease and chronic heart failure receiving AST-120 therapy for 24 months.28 Treatment with AST-120 is expected to become a promising therapy for patients with both kidney and heart diseases.
In summary, these data support the oral carbon absorbent, AST-120, as a novel therapy for ischemic AKI and remote cardiac dysfunction, most likely resulting from the attenuation of inflammation and apoptosis and the repair of endothelial cells and cardiomyocytes.
Abbreviations
AKI, acute kidney injury; BUN, blood urea nitrogen; CO, cardiac output; HRP, horseradish peroxidase; ICAM-1, intercellular cell adhesion molecule-1; I/R, ischemia–reperfusion; LVEF, left ventricular ejection fraction; LVFS, left ventricular fractional shortening; NF-κB, nuclear factor kappa-light-chain-enhancer of activated B cells; PBS, phosphate-buffered saline; TUNEL, terminal dUTP nick-end labeling.
Data Sharing Statement
For data requests, please contact the author.
Acknowledgments
The authors thank Jeff E. Engel for proofreading the manuscript and the Department of Medical Research of National Taiwan University Hospital, and the Imaging Core Facility of the First Core Laboratory and the Laboratory Animal Center of College of Medicine, National Taiwan University for equipment support.
Author Contributions
The study was conceived and designed by WCS and YHC. WCS, YHC, ZWC, HJT, and XYL were all involved in conducting the key experiments. WCS and YHC performed data analysis. WCS wrote the first draft of the manuscript, and GJW subsequently revised and edited it to develop the final version of the manuscript. All authors contributed to data analysis, drafting or revising the article, have agreed on the journal to which the article will be submitted, gave final approval of the version to be published, and agree to be accountable for all aspects of the work.
Funding
This work was supported by the Ministry of Science and Technology to YHC (MOST 104-2314-B-002-119-MY3) and GJW (MOST 108-2635-B-039-002), respectively; China Medical University (CMU108-MF-18) and China Medical University Hospital (DMR-107-113) to GJW; and Putian University (2019118) and Mrs. Hsiu-Chin Lee Kidney Research Foundation to WCS.
Disclosure
The authors declare that they have no competing interests.
References
1. Saito H. Pathophysiological regulation of renal slc22a organic ion transporters in acute kidney injury: pharmacological and toxicological implications. Pharmacol Ther. 2010;125(1):79–91. doi:10.1016/j.pharmthera.2009.09.008
2. Iwata K, Watanabe H, Morisaki T, et al. Involvement of indoxyl sulfate in renal and central nervous system toxicities during cisplatin-induced acute renal failure. Pharm Res. 2007;24(4):662–671. doi:10.1007/s11095-006-9183-2
3. Go AS, Chertow GM, Fan D, McCulloch CE, Hsu CY. Chronic kidney disease and the risks of death, cardiovascular events, and hospitalization. N Engl J Med. 2004;351(13):1296–1305. doi:10.1056/NEJMoa041031
4. Brunet P, Gondouin B, Duval-Sabatier A, et al. Does uremia cause vascular dysfunction? Kidney Blood Press Res. 2011;34(4):284–290. doi:10.1159/000327131
5. Ologunde R, Zhao H, Lu K, Ma D. Organ cross talk and remote organ damage following acute kidney injury. Int Urol Nephrol. 2014;46(12):2337–2345. doi:10.1007/s11255-014-0766-2
6. Li XHH, Santora R, Rabb H, Rabb H. Organ crosstalk: the role of the kidney. Curr Opin Crit Care. 2009;15(6):481–487. doi:10.1097/MCC.0b013e328332f69e
7. Akcay ANQ, Edelstein CL, Edelstein CL. Mediators of inflammation in acute kidney injury. Mediators Inflamm. 2009;2009:137072. doi:10.1155/2009/137072
8. Hughes SF, Hendricks BD, Edwards DR, Bastawrous SS, Roberts GE, Middleton JF. Mild episodes of tourniquet-induced forearm ischaemia-reperfusion injury results in leukocyte activation and changes in inflammatory and coagulation markers. J Inflamm. 2007;4:12. doi:10.1186/1476-9255-4-12
9. Manka DR, Wiegman P, Din S, et al. Arterial injury increases expression of inflammatory adhesion molecules in the carotid arteries of apolipoprotein-e-deficient mice. J Vasc Res. 1999;36(5):372–378. doi:10.1159/000025676
10. Roy J, Audette M, Tremblay MJ. Intercellular adhesion molecule-1 (icam-1) gene expression in human t cells is regulated by phosphotyrosyl phosphatase activity. Involvement of nf-kappab, ets, and palindromic interferon-gamma-responsive element-binding sites. J Biol Chem. 2001;276(18):14553–14561. doi:10.1074/jbc.M005067200
11. Kempe S, Kestler H, Lasar A, Wirth T. NF-κB controls the global pro-inflammatory response in endothelial ells: evidence for the regulation of a pro-atherogenic program. Nucleic Acids Res. 2005;33(16):5308–5319. doi:10.1093/nar/gki836
12. Kim SH, Yu MA, Ryu ES, Jang YH, Kang DH. Indoxyl sulfate-induced epithelial-to-mesenchymal transition and apoptosis of renal tubular cells as novel mechanisms of progression of renal disease. Lab Invest. 2012;92(4):488–498.
13. Tumur Z, Shimizu H, Enomoto A, Miyazaki H, Niwa T. Indoxyl sulfate upregulates expression of icam-1 and mcp-1 by oxidative stress-induced Nf-kappab activation. Am J Nephrol. 2010;31(5):435–441. doi:10.1159/000299798
14. Zheng L, Hsu SI, Hong Hsu S. In situ glomerular expression of activated nf-kappab in human lupus nephritis and other non-proliferative proteinuric glomerulopathy. Virchows Arch. 2006;448(2):172–183. doi:10.1007/s00428-005-0061-9
15. Ito SOM, Higuchi Y, Nishijima F, Ishii H, Yoshida M, Yoshida M. Indoxyl sulfate induces leukocyte-endothelial interactions through up-regulation of e-selectin. J Biol Chem. 2010;285(50):38869–38875. doi:10.1074/jbc.M110.166686
16. Sun CY, Hsu HH, Wu MS. P-cresol sulfate and indoxyl sulfate induce similar cellular inflammatory gene expressions in cultured proximal renal tubular cells. Nephrol Dial Transplant. 2013;28(1):70–78. doi:10.1093/ndt/gfs133
17. Mehta RL, Kellum JA, Shah SV, et al. Acute kidney injury network: report of an initiative to improve outcomes in acute kidney injury. Crit Care. 2007;11(2):R31. doi:10.1186/cc5713
18. Shen WC, Liang CJ, Huang TM, et al. Indoxyl sulfate enhances IL-1beta-induced E-selectin expression in endothelial cells in acute kidney injury by the ROS/MAPKs/NFkappaB/AP-1 pathway. Arch Toxicol. 2016;90(11):2779–2792. doi:10.1007/s00204-015-1652-0
19. Tsai CT, Wang DL, Chen WP, et al. Angiotensin II increases expression of alpha1C subunit of L-type calcium channel through a reactive oxygen species and cAMP response element-binding protein-dependent pathway in HL-1 myocytes. Circ Res. 2007;100(10):1476–1485. doi:10.1161/01.RES.0000268497.93085.e1
20. Melotti P, Nicolis E, Tamanini A, Rolfini R, Pavirani A, Cabrini G. Activation of NF-kB mediates ICAM-1 induction in respiratory cells exposed to an adenovirus-derived vector. Gene Ther. 2001;8(18):1436–1442. doi:10.1038/sj.gt.3301533
21. Shen WC, Chou YH, Huang HP, Sheen JF, Hung SC, Chen HF. Induced pluripotent stem cell-derived endothelial progenitor cells attenuate ischemic acute kidney injury and cardiac dysfunction. Stem Cell Res Ther. 2018;9(1):344. doi:10.1186/s13287-018-1092-x
22. Wang X, Shapiro JI. Evolving concepts in the pathogenesis of uraemic cardiomyopathy. Nature Rev Nephrol. 2019;15(3):159–175. doi:10.1038/s41581-018-0101-8
23. Ross R, Epstein FH. Atherosclerosis—an inflammatory disease. N Engl J Med. 1999;340(2):115–126. doi:10.1056/NEJM199901143400207
24. Malatino LS, Stancanelli B, Cataliotti A, et al. Circulating E-selectin as a risk marker in patients with end-stage renal disease. J Intern Med. 2007;262(4):479–487.
25. Simmons EM, Himmelfarb J, Sezer MT, et al. Plasma cytokine levels predict mortality in patients with acute renal failure. Kidney Int. 2004;65(4):1357–1365.
26. Vanholder R, Schepers E, Pletinck A, Nagler EV, Glorieux G. The uremic toxicity of indoxyl sulfate and p-cresyl sulfate: a systematic review. J Am Soc Nephrol. 2014;25(9):1897–1907. doi:10.1681/ASN.2013101062
27. Barreto FC, Barreto DV, Liabeuf S, et al.; European Uremic Toxin Work Group (EUTox). Serum indoxyl sulfate is associated with vascular disease and mortality in chronic kidney disease patients. Clin J Am Soc Nephrol. 2009;4(10):1551–1558. doi:10.2215/CJN.03980609
28. Suliman ME, Qureshi AR, Heimbürger O, Lindholm B, Stenvinkel P. Soluble adhesion molecules in end-stage renal disease: a predictor of outcome. Nephrol Dial Transplant. 2006;21(6):1603–1610. doi:10.1093/ndt/gfl005
29. Viedt C, Dechend R, Fei J, Hänsch GM, Kreuzer J, Orth SR. MCP-1 induces inflammatory activation of human tubular epithelial cells: involvement of the transcription factors, nuclear factoR-κB and activating protein-1. J Am Soc Nephrol. 2002;13(6):1534–1547. doi:10.1097/01
30. Loverre A, Ditonno P, Crovace A, et al. Ischemia-reperfusion induces glomerular and tubular activation of proinflammatory and antiapoptotic pathways: differential modulation by rapamycin. J Am Soc Nephrol. 2004;15:2675–2686. doi:10.1097/01.ASN.0000139932.00971.E4
31. Yingjie K, Haihong Y, Lingwei C, et al. Apoptosis repressor with caspase recruitment domain deficiency accelerates ischemia/reperfusion (I/R)- induce acute kidney injury by suppressing inflammation and apoptosis: the role of AKT/mTOR signaling. Biomed Pharmacother. 2019;112:108681.
32. Yu X, Meng X, Xu M, et al. Celastrol ameliorates cisplatin nephrotoxicity by inhibiting NF-kappaB and improving mitochondrial function. EBioMedicine. 2018;36:266–280. doi:10.1016/j.ebiom.2018.09.031
33. Sumida M, Doi K, Ogasawara E, et al. Regulation of mitochondrial dynamics by dynamin-related protein-1 in acute cardiorenal syndrome. J Am Soc Nephrol. 2015;26(10):2378–2387. doi:10.1681/ASN.2014080750
34. Han B, Li S, Lv Y, et al. Dietary melatonin attenuates chromium-induced lung injury via activating the Sirt1/Pgc-1α/Nrf2 pathway. Food Funct. 2019;10(9):5555–5565. doi:10.1039/c9fo01152h
35. Zhang Z, Guo C, Jiang H, et al. Inflammation response after the cessation of chronic arsenic exposure and post-treatment of natural astaxanthin in liver: potential role of cytokine-mediated cell-cell interactions. Food Funct. 2020;11(10):9252–9262. doi:10.1039/d0fo01223h
36. Li S, Zheng X, Zhang X, et al. Exploring the liver fibrosis induced by deltamethrin exposure in quails and elucidating the protective mechanism of resveratrol. Ecotoxicol Environ Saf. 2021;207:111501. doi:10.1016/j.ecoenv.2020.111501
37. Sato E, Saigusa D, Mishima E, et al. Impact of the oral adsorbent AST-120 on organ-specific accumulation of uremic toxins: lc-ms/ms and ms imaging techniques. Toxins (Basel). 2018;10(1):19. doi:10.3390/toxins10010019
38. Sakaguchi Y, Hamano T, Obi Y, et al. A randomized trial of magnesium oxide and oral carbon adsorbent for coronary artery calcification in predialysis CKD. J Am Soc Nephrol. 2019;30(6):1073–1085. doi:10.1681/ASN.2018111150
39. Niwa T. Indoxyl sulfate is a nephro-vascular toxin. J Ren Nutr. 2010;20(5Suppl):S2–S6. doi:10.1053/j.jrn.2010.05.002
40. Shibahara H, Shibahara N. Cardiorenal protective effect of the oral uremic toxin absorbent AST-120 in chronic heart disease patients with moderate CKD. J Nephrol. 2009;23(5):535–540.
41. Fuji H, Nakai K, Fukagawa M. Role of oxidative stress and indoxyl sulfate in progression of cardiovascular disease in chronic kidney disease. Ther Apher Dial. 2011;15(2):125–128. doi:10.1111/j.1744-9987.2010.00883.x
 © 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.