Back to Journals » Journal of Pain Research » Volume 19
Association of Inflammatory Proteins with Neuropathic Pain: A Two-Sample Bidirectional Mendelian Randomization
Authors Zhong J ![]() , Li H, Huang C, Zhang P, Pang X, Luo Z, Li C
, Li H, Huang C, Zhang P, Pang X, Luo Z, Li C
Received 28 September 2025
Accepted for publication 6 February 2026
Published 17 February 2026 Volume 2026:19 570828
DOI https://doi.org/10.2147/JPR.S570828
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Professor Michael Überall
Jiajun Zhong,1,* Huanmin Li,2,* Chongbo Huang,1 Pengfei Zhang,1 Xiaoyu Pang,1 Zimeng Luo,1 Chunguang Li3
1Second Clinical Medical School, Southern Medical University, GuangZhou, GuangDong, People’s Republic of China; 2Department of Neurology, the Third Affiliated Hospital of Southern Medical University, GuangZhou, GuangDong, People’s Republic of China; 3Zhujiang Hospital of Southern Medical University, Department of Neurology, Guangzhou, Guangdong Province, People’s Republic of China
*These authors contributed equally to this work
Correspondence: Chunguang Li, Department of Neurology, Zhujiang Hospital, Southern Medical University, GuangZhou, GuangDong, 510280, People’s Republic of China, Email [email protected]
Object: Previous studies have found that circulating inflammatory proteins may play an essential role in the occurrence and development of neuropathic pain. However, the majority of existing evidence is derived from observational studies, which are prone to confounding factors and reverse causality, and the inflammatory profiles associated with different types of neuropathic pain remain poorly understood. Consequently, more robust methodologies are urgently required to elucidate the causal relationships between these variables. Therefore, a bidirectional Mendelian randomization analysis was performed to assess the causal relationship between inflammatory proteins and neuropathic pain, specifically focusing on neuralgia and neuritis, including glossopharyngeal neuralgia, phantom limb syndrome with pain,small fiber neuropathy, and unspecified neuralgia.
Methods: Instrumental variables were obtained from publicly available genome-wide association study datasets. Five different models were used for MR Analysis, including the inverse variance weighted model, weighted median estimation model, weighted model-based method, MR-Egger regression model and simple mode. Heterogeneity in the results was assessed using the Cochrane Q test. Horizontal pleiotropy was evaluated through the MR-Egger intercept test and the MR pleiotropy residual sum and outliers test. Sensitivity analysis was conducted through leave-one-out analysis.
Results: The results suggest a genetically predicted potential association between fractalkine levels (CX3CL1) (OR=0.182; 95CI=0.048– 0.691; p=0.012), signaling lymphocytic activation molecule levels (SLAMF1) (OR=0.295; 95CI=0.099– 0.879; p=0.028), tumor necrosis factor levels (TNF) (OR=0.281; 95CI=0.085– 0.931; p=0.038), matrix metalloproteinase-1 (MMP-1) (OR=3.399; 95CI=1.140– 10.133; p=0.028), and tumor necrosis factor receptor superfamily member 9 levels (TNFRSF9) (OR=0.281; 95CI=0.085– 0.931; p=0.038) with the risk of glossopharyngeal nerve disease. In addition, we identified FGF5, FGF21, FGF23, Osteoprotegerin, CD40LG, IL-12B, IL-20RA, IL-24, CCL8, CCL28, CD244, CCL3, OSM, and SIRT2 as associated with the occurrence of the other three types of neuropathic pain.The sensitivity analysis revealed no heterogeneity or levels of pleiotropy.
Conclusion: Our Mendelian randomization analysis revealed several genetically predicted associations between circulating inflammatory proteins and the risk of neuropathic pain subtypes. These findings indicate that immune- and inflammation-related pathways may be implicated in the pathogenesis of neuropathic pain, although further functional and clinical investigations are required to validate these associations and elucidate the underlying mechanisms.
Keywords: inflammatory protein, neuropathic pain, mendelian randomization
Introduction
Neuropathic pain (NP) occurs due to a lesion or disease in the somatosensory system.1 NP can be caused by damage to the nervous system from peripheral nociceptor receptors to the cerebral cortex.2 NP is clinically characterized by spontaneous, persistent burning or electric shock-like sensations, presenting as nociceptive hypersensitivity or abnormal pain in response to harmful or non-harmful stimuli.3
Neuropathic pain affects approximately 7% to 10% of the general population, and its prevalence is expected to rise with global population aging, increasing rates of diabetes, and improved cancer survival.4 This condition imposes a substantial economic burden and is associated with higher prevalence and greater risk of work absenteeism among women, older adults, and individuals with lower educational attainment. Neuropathic pain is more challenging to manage compared to other chronic pain disorders, and current treatment quality remains suboptimal, with only a small proportion of patients receiving guideline-recommended dosages of first-line medications.5,6
Several mechanisms underlying NP have been identified by current research, involving expression of ion channels, phenotypic switching, peripheral sensitization, sensory denervation and sprouting of collateral nerve fibers, glial activation and proinflammatory cytokines, and genetic regulation.7 Inflammatory factors have garnered significant attention in relation to the onset and persistence of NP. A study addressing the differential expression of cytokines in painful and painless neuropathies found that IL-2 mRNA and TNF mRNA and protein levels were approximately twice as high in patients with painful neuropathy and approximately twofold higher in patients with IL-2 mRNA and TNF mRNA and protein levels in patients with painless neuropathy, compared to healthy control subjects.8 A meta-analysis showed that, compared to controls, postherpetic patients with NP have significantly higher levels of IL-6 and IL-1β.9
Currently, the management of neuropathic pain necessitates a multimodal approach involving pharmacological, interventional, and non-pharmacological therapies. Gabapentin, serotonin-norepinephrine reuptake inhibitors, and tricyclic antidepressants continue to be recommended as first-line pharmacological agents for neuropathic pain treatment.10 Notably, a growing body of research has identified chemokines and their receptors as promising therapeutic targets. Published data indicate that the pharmacological blockade of some chemokine receptors from the CC group, as well as from the CXC, XC, and CX3C groups, relieves neuropathic pain of various etiologies in mice and/or rats.11
Although this evidence suggests a potential association between inflammatory response and NP, the role of systemic inflammation as an initiating factor in NP remains controversial. Observational studies have limited capacity to clearly differentiate whether increased inflammatory markers result from disease progression, secondary infection, the pain state itself, or the effects of pharmacological treatments. At the same time, a growing body of basic and animal studies provides further support for the potential involvement of inflammatory factors in the onset and persistence of NP. However, due to limitations in extrapolating findings from experimental models to clinical settings, the overall evidence remains inconclusive.
MR offers a robust methodological framework for addressing this issue. Mendelian randomization is a method of causal inference that relies on genetic variation. It utilizes genetic variants, single nucleotide polymorphism (SNP), identified through genome-wide association study (GWAS) data as instrumental variables (IVs) to assess associations between exposures and outcomes. This principle is founded on the random assignment of genes under Mendelian inheritance, which helps to avoid the confounding factors and reverse causality biases commonly found in traditional observational studies.12,13
In this research, we extracted validated genetic variants from the pooled data of 91 published genome-wide association studies focusing on inflammatory factors to investigate their relationship with NP (which includes glossopharyngeal neuralgia, phantom limb syndrome with pain, small fiber neuropathy, unspecified neuralgia and neuritis). The aim of this study is to identify causal inflammation proteins associated with the risk of NP across the proteome and explore new potential drug targets for PN.
Methods
This study analyzed open-access GWAS summary statistics, a publicly available dataset exempt from ethical review requirements. No further ethical approval or informed consent was necessary. The STROBE-MR checklist was compiled and submitted as supplementary Table S8.12
Mendelian Randomization Assumptions
There are three core assumptions in Mendelian randomization analyses: relevance, independence, and exclusivity. Relevance assumption: The selected SNPs are correlated with the exposure factors. Independence assumption: The SNPs are unaffected by confounders that can influence both exposure and outcome. Exclusivity assumption: SNPs do not impact outcomes through pathways apart from the exposure factor of interest. The design concept is illustrated in Figure 1.
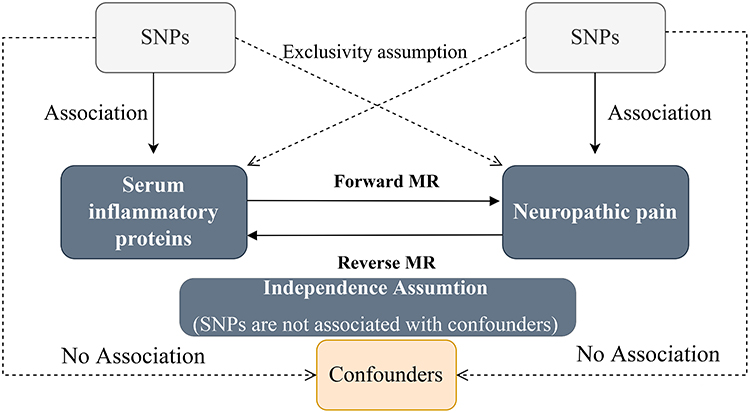 |
Figure 1 Core assumptions of Mendelian randomization. The dashed arrow means that there is no correlation. Abbreviation: SNPs, single-nucleotide polymorphisms. |
Data Sources
Pooled data related to circulating inflammatory proteins were derived from a GWAS meta-analysis of 14,824 participants of European ancestry. The complete GWAS summary statistics for 91 circulating inflammatory proteins can be downloaded from EBI GWAS (https://www.ebi.ac.uk/gwas/) (GCST90274758-GCST90274848).13 GWAS data for NP were obtained from the FinnGen Biobank (FinnGen release 12, https://www.finngen.fi/en) and were accessed in March 2025. Data concerning glossopharyngeal nerve diseases comprised 64 cases and 435,371 controls. Phantom limb syndrome with pain data involved 91 cases and 262,404 controls. Small fiber neuropathy data encompassed 895 cases and 491,583 controls. Unspecified neuralgia and neuritis included 1972 cases and 357549 controls. There was little overlap between the populations included in the exposure and outcome datasets, and the sample was almost exclusively of European ancestry, including both males and females, ensuring the findings’ validity.
Selection of Instrumental Variables
First, we set p< 5e-8 as the genome-wide significance threshold to select SNPs strongly associated with NP and circulating inflammatory proteins. Since some inflammatory proteins are rarely identified with SNPs at exposure, a higher cut-off value (p< 5e-6) was chosen to ensure adequate SNPs for subsequent MR analysis.14 Second, to avoid linkage disequilibrium, instrumental variables were excluded to ensure mutual independence between instrumental variables (r2 = 0.001, kb = 10,000).15 Echo sequence single nucleotide polymorphisms were discarded. We used the R2 value of each SNP to calculate the proportion of exposed variance and employed the F-statistic to estimate the strength of the instrumental variable. We removed SNPs with an F-statistic less than 10, reducing the possibility of weak instrument bias.16
Statistical Analysis
We used inverse variance weighted (IVW) as the primary analysis method, as well as weighted median (WME), mr-egger regression, simple mode (SM), and weighted mode (WM) to assist in analyzing causality. Assuming all included SNPs can be used as valid instrumental variables, the IVW method is the most efficient with maximum statistical power.
We then performed sensitivity analyses to test these associations’ robustness and identify potential biases. Cochran’s Q test was computed to quantify heterogeneity across the individual causal effects. The random-effects IVW approach was utilized if there was substantial heterogeneity (P < 0.05).17 Intercept tests of MR-Egger regression detect potential pleiotropy between associations and provide more accurate estimates by adjusting for pleiotropy. We also conducted MR-PRESSO analyses for all instrumental variables to assess the presence of pleiotropy.18 In addition, the causal effect estimates were repeated after removing individual SNPs one by one to observe the stability of the results.
Statistical analyses were conducted using R 4.4.2 software (R Foundation for Statistical Computing) with the TwoSampleMR package (version 0.6.9).
Ethics Statement
This study was conducted in accordance with the Measures for the Ethical Review of Life Science and Medical Research Involving Human Subjects issued by the National Health Commission of China on February 18, 2023. According to Article 32 (Items 1 and 2), studies based on publicly available, anonymized data that do not involve direct interaction with human participants or identifiable personal information are exempt from ethics committee approval. As this Mendelian randomization study utilized only publicly accessible, summary-level GWAS data, ethical approval and informed consent were not required.
Results
Bidirectional Effects of Circulating Inflammatory Proteins and Glossopharyngeal Nerve Diseases
Using the IVW method, genetically predicted higher fractalkine levels (CX3CL1; OR=0.182, 95CI=0.048–0.691, P=0.012), signaling lymphocytic activation molecule levels (SLAMF1; OR=0.295, 95CI=0.099–0.879, P=0.028), tumor necrosis factor levels (TNF; OR=0.281, 95CI=0.085–0.931, P=0.038), tumor necrosis factor receptor superfamily member 9 levels (TNFRSF9; OR=0.281, 95CI=0.085–0.931, P=0.038) were associated with a reduced risk of glossopharyngeal nerve disease. In contrast, higher genetically predicted levels of matrix metalloproteinase-1 (MMP-1) were associated with an increased disease risk (OR=3.399, 95CI=1.140–10.133, P=0.028).
Effect estimates were generally consistent in direction across complementary MR methods. Sensitivity analyses did not indicate substantial heterogeneity or influential instrumental variables.We performed MR-PRESSO to identify and process the pleiotropy. However, the outlier Test showed no outliers, and the global Test indicating no horizontal pleiotropy.
In the reverse-direction analysis, no SNPs met the genome-wide significance threshold. After applying a relaxed significance threshold (P=5e-5), genetic liability to glossopharyngeal nerve disease was associated with decreased circulating neurotrophin-3 (NT-3) levels (beta= −0.053, 95CI = −0.101- −0.005, P = 0.030).
The causal effects of circulating inflammatory proteins on glossopharyngeal nerve diseases are illustrated by a ring heat map in Figure 4(a). Detailed results are presented in Figure 2 and Supplementary Tables S1–S3, with additional sensitivity analyses shown in Supplementary Figures S1, S5, and S8.
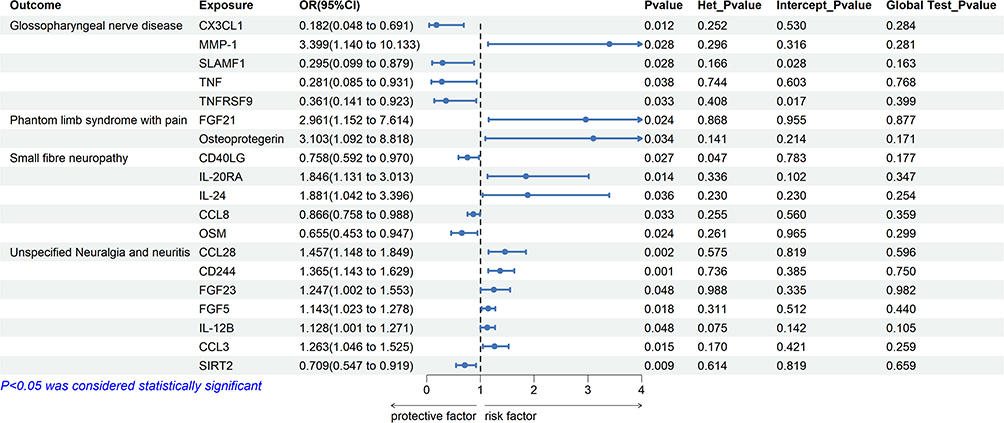 |
Figure 2 Forest plot of the causal effects of circulating inflammatory proteins on neuropathic pain. Each row represents the exposure result for each factor to the outcome. Het_Pvalue represents the p value of the heterogeneity test. Intercept_Pvalue represents the p-value of the egger intercept. Global Test_Pvalue represents the Global Test Pvalue of MRPRESSO. |
Bidirectional Effects of Circulating Inflammatory Proteins and Phantom Limb Syndrome with Pain
In IVW analyses, genetically predicted higher levels of fibroblast growth factor 21 (FGF21; OR=2.961, 95CI=1.152–7.614, P=0.024) and osteoprotegerin (OR=3.103; 95CI=1.092–8.818, P=0.036) were associated with an increased risk of phantom limb syndrome with pain. The direction of associations was consistent across alternative MR methods.
Sensitivity analyses provided no evidence of heterogeneity or horizontal pleiotropy.
In the inverse analysis, only 2 SNPs were included in the analysis when screening SNPs with a significance threshold of p<5e-6. After relaxing the SNP selection threshold, genetic liability to phantom limb syndrome with pain was associated with reduced circulating levels of interleukin-10 receptor subunit alpha (IL-10RA; (beta=−0.033, 95CI=−0.059--0.007, P=0.015), interleukin-17A (IL-17A; beta=−0.031, 95CI=−0.057--0.005, P=0.021), and tumor necrosis factor ligand superfamily member 12 (TNFSF12; beta=−0.030, 95CI=−0.059--0.001, P=0.040). No heterogeneity or pleiotropy was detected.
Bidirectional Effects of Circulating Inflammatory Proteins and Small Fiber Neuropathy
Genetically predicted higher circulating levels of CD40 ligand (CD40LG; OR=0.758, 95CI=0.592–0.970, P=0.028), monocyte chemoattractant protein 2 (CCL8; OR=0.866, 95CI=0.758–0.988, P=0.032), and oncostatin M (OSM; OR = 0.66, 95% CI 0.45–0.95, P = 0.024) were associated with a lower risk of small fiber neuropathy. For CD40LG, a random-effects IVW model was applied due to evidence of heterogeneity. Conversely, higher genetically predicted levels of interleukin-20 receptor subunit alpha (IL-20RA; OR=1.846, 95CI=1.131–3.013, P=0.0142) and interleukin-24 (IL-24; OR=1.881, 95CI=1.042–3.396, p=0.036) were associated with an increased disease risk.
In reverse direction analyses, genetic liability to small fiber neuropathy was associated with altered circulating levels of interleukin-2 receptor subunit beta (IL-2RB; beta=−0.044, 95CI=−0.081--0.006, P=0.022) and interleukin-5 (IL-5; beta=−0.063, 95CI=−0.101--0.025, P=0.001). Similarly, no evidence of heterogeneity or pleiotropy was observed.
Bidirectional Effects of Circulating Inflammatory Proteins on Unspecified Neuralgia and Neuritis
IVW analyses indicated that lower circulating levels of sirtuin 2 (SIRT2) were associated with an increased risk of unspecified neuralgia or neuritis ((OR=0.709; 95CI=0.547–0.919, P=0.009). In contrast, higher genetically predicted levels of CCL28, CD244, FGF23, FGF5, IL-12B, and CCL3 were associated with elevated disease risk (all P < 0.05).
No heterogeneity or pleiotropy was found in the instrumental variables. Figure 4(b) visualizes the results of Mendelian analysis of circulating inflammatory proteins on unspecified neuralgia and neuritis through a ring heatmap. Figure 3 shows the results of the reverse Mendelian randomization.
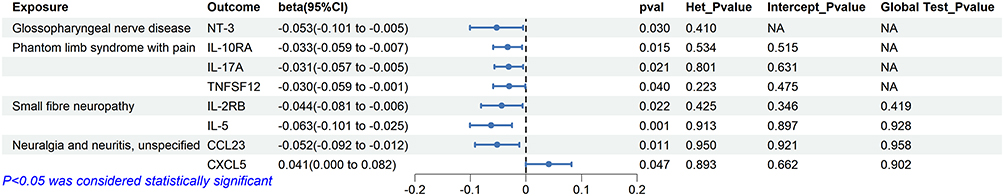 |
Figure 3 Forest plot of the causal effects of neuropathic pain on circulating inflammatory proteins. Each row represents the exposure result for each factor to the outcome. Het_Pvalue represents the p value of the heterogeneity test. Intercept_Pvalue represents the p-value of the egger intercept. Global Test_Pvalue represents the Global Test Pvalue of MRPRESSO. |
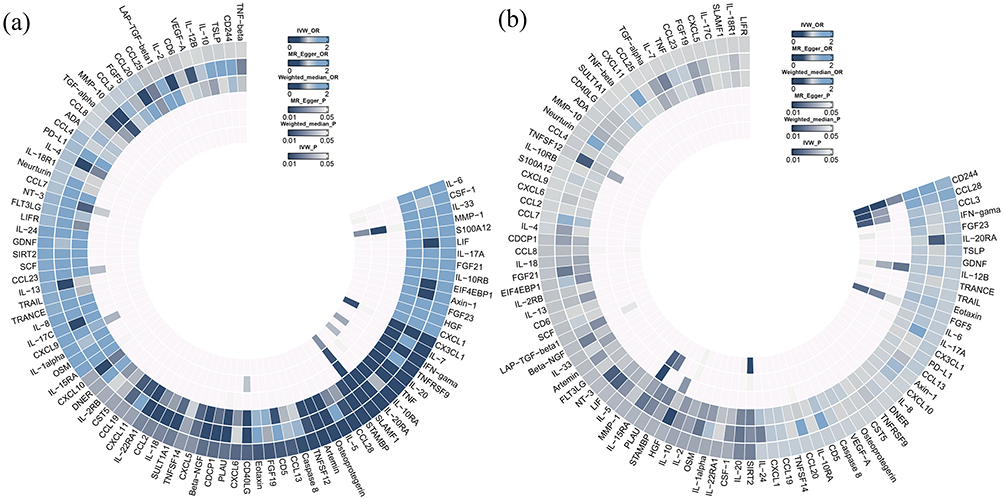 |
Figure 4 (a) Ring heat map of Mendelian randomization analysis of the causal effect of circulating inflammatory proteins on glossopharyngeal nerve diseases. (b) Ring heat map of Mendelian randomization analysis of the causal effect of circulating inflammatory proteins on unspecified neuralgia or neuritis. |
In reverse MR analyses, genetic liability to neuralgia or neuritis was associated with altered circulating levels of CCL23 (beta=−0.052, 95CI=−0.092--0.012, P=0.011) and CXCL5 (beta=0.041, 95CI=0.001–0.082, P=0.047).
Discussion
After a nerve injury, levels of proinflammatory cytokines significantly increase, and the inflammatory response they trigger promotes the infiltration and activation of immune cells at the injury site. These released cytokines, neurotrophins, and chemokines not only mediate local inflammatory cascades but also play a crucial role in the central and peripheral regulation of neuropathic pain by activating microglia and glial cells in the spinal cord and brain.19,20 For example, spinal cord injury (SCI) is a severe neurological injury that often leads to neuropathic pain. After spinal cord injury, local infiltration of microglia, astrocytes, and activated microglia in the spinal dorsal horn leads to the release of multiple inflammatory mediators (pro-inflammatory cytokines, such as TNF-α, IL-1β, IL-17, and IFN-g) to maintain the nociceptive hypersensitivity response and form a persistent neuroinflammatory microenvironment.21 However, the majority of existing evidence is derived from animal models or cross-sectional observational studies, and the circulating inflammatory protein profile associated with specific neuropathies has not been adequately characterized.
In the present study, employing a Mendelian randomization framework, we identified causal associations between multiple circulating inflammatory proteins and distinct subtypesof neuropathic pain. Notably, we observed that elevated CX3CL1 levels increased the risk of glossopharyngeal neuropathy, whereas CCL3 and CCL28 were associated with a higher risk of unspecified neuropathic pain or neuritis. Our findings extend previous research by providing genetic evidence supporting a causal role of specific chemokines in neuropathic pain, rather than mere associations reported in earlier studies.
CX3CL1 is expressed in peripheral immune cells, neurons, and glial cells in the central nervous system and plays an important role in regulating microglial activation following neural injury or inflammation.22 The CX3CL1/CX3CR1 signaling pathway has been implicated in neuropathic pain through neuron–microglia interactions in the spinal cord, and intrathecal administration of CX3CL1 has been shown to induce pain hypersensitivity in animal models.23 Although our findings regarding CX3CL1 diverge from previous experimental studies that report pronociceptive effects, several explanations may account for this discrepancy. Prior evidence has predominantly focused on local or intrathecal CX3CL1 signaling within the central nervous system, whereas the present Mendelian randomization analysis reflects genetically predicted systemic circulating levels of CX3CL1. It is therefore plausible that CX3CL1 exerts context-dependent effects; specifically, systemic CX3CL1 may play an immunomodulatory or protective role, while localized overactivation of the CX3CL1/CX3CR1 pathway could promote neuropathic pain. Furthermore, our analysis addresses disease susceptibility rather than pain severity, which may further elucidate the observed differences.
Similarly, CCL3 has been implicated in neuropathic pain through its role in monocyte recruitment and macrophage activation. Evidence suggests that inflammatory chemokines (CCL2, CCL3, and fractalkine) play a key role in NP. In a mouse model of chronic constriction injury (CCI) of the sciatic nerve, the expression levels of chemokine CCL3 mRNA were significantly increased in mice’s spinal cord.24 Intracutaneous injection of CCL3 in rats produced painful effects, possibly due to the activation of chemokine receptors in the dorsal root ganglion.25 Our findings support and extend these observations by indicating that genetically predicted higher circulating CCL3 levels are causally associated with neuropathic pain risk in humans.
We also identified CXCL5 as a potential contributor to neuropathic pain. This indicates a bidirectional interaction between chemokines and neuralgia, a point that was not addressed in the preceding article. Xu et al found that blockade of the spinal CXCL5/CXCR2 pathway alleviated NP while CXCL5 enhanced nerve injury-induced NP through the regulation of GSK-3β phosphorylation.26 CXCL5, also known as epithelial-derived neutrophil-activating protein, is a chemokine involved in immune regulation and inflammatory responses. In the context of severe intervertebral disc degeneration, upregulation of POSTN has been shown to indirectly enhance CXCL5 transcription and secretion, thereby promoting macrophage chemotaxis and M1 polarization. These findings indicate a proinflammatory role for CXCL5 in the pathogenesis of disc degeneration.27 Our results further suggest that elevated levels of CXCL5 may contribute to the development of non-specific neuralgia, thereby highlighting a bidirectional interaction between chemokines and neuropathic pain—a relationship that has not been sufficiently emphasized in previous studies.
Previous studies have indicated that most amputees experience chronic long-term pain in the residual limb and that this pain in patients with chronic residual limb pain correlates positively with the levels of several pro-inflammatory mediators: IL-8, TNF-α, IL-12, TNF-β, PIGF, Tie2, SAA, and ICAM-1.28 However, no association between FGF21, Osteoprotegerin and neuropathic pain has been reported in the literature. Our findings therefore provide novel insights into previously underexplored inflammatory pathways potentially involved in residual limb pain.
Furthermore, our study suggests a protective role of SIRT2 against neuropathic pain, as genetically predicted lower serum SIRT2 levels were associated with an increased risk of neuralgia and neuritis. SIRT2 is a NAD+-dependent deacetylase and a member of the sirtuin family of proteins. It is expressed in virtually all brain cells and is predominantly found in oligodendrocytes, the myelin-forming glial cells, in vivo.29 SIRT2 knockdown has deleterious effects on the normal function of the nervous system.30
SIRT2 has been reported to be neuroprotective by suppressing ferroptosis. Intrathecal injection of recombinant adenovirus overexpressing SIRT2 (Ad-SIRT2) in rats upregulated the expression of SIRT2 and FPN1 in the spinal cord. It also inhibited intracellular iron accumulation and reduced oxidant stress levels in SNI rats.31 These results support the hypothesis that impaired anti-inflammatory or neuroprotective mechanisms may contribute to the development of neuropathic pain.
The observed causal effect sizes were generally modest, which is anticipated in Mendelian randomization studies where genetic variants serve as proxies for lifelong differences in circulating inflammatory protein levels rather than acute exposures. Such modest effect sizes do not diminish their biological relevance, particularly concerning complex traits such as neuropathic pain, and are consistent with findings from previous Mendelian randomization studies focused on inflammatory pathways.32
From a clinical standpoint, our findings strongly suggest that specific inflammatory proteins may serve as viable biomarkers or promising therapeutic targets for neuropathic pain. The identification of causal inflammatory pathways not only enhances risk stratification but also accelerates the development of targeted anti-inflammatory and immunomodulatory interventions, thereby improving the precision and efficacy of neuropathic pain management.
Limitation
This paper has three main limitations. First, multiple comparisons were performed, but we did not use adjusted p values to assess causality, so our conclusions should be taken only as suggestive. Second, our findings may be limited to European populations, as both the exposure and outcome groups were of European ancestry. Finally, due to the insufficient number of cases in public databases, our statistical power is inadequate; therefore, the conclusions drawn from this study should be regarded as preliminary indications.
Conclusion
This Mendelian randomization study supports a causal association between systemic inflammation and neuropathic pain. The identification of phenotype-specific inflammatory profiles provides new insights into the pathogenesis of neuropathic pain and may inform future biomarker research and mechanistic studies.
Data Sharing Statement
Data on this study can be obtained from the Supplementary Material or the corresponding author.
Acknowledgments
Gwas data were downloaded in Gwas Catlog and FinnGen research databases, respectively, and we thank all researchers who shared these data.
Supplementary Materials
All Mendelian analysis results and SNP information can be found in Supplementary Tables S1–S7. Scatter and funnel plots and leave-one-out plots for all Mendelian randomization analyses can be found on the Supplementary Figures S1–S13.
Funding
This research did not receive any specific grant from funding agencies in the public, commercial, or not-for-profit sectors.
Disclosure
Tha authors report no conflicts of interest int his work.
References
1. Finnerup NB, Haroutounian S, Kamerman P, et al. Neuropathic pain: an updated grading system for research and clinical practice. Pain. 2016;157(8):1599–9. doi:10.1097/j.pain.0000000000000492
2. Murphy D, Lester D, Clay Smither F, Balakhanlou E. Peripheral neuropathic pain. NeuroRehabilitation. 2020;47(3):265–283. doi:10.3233/NRE-208002
3. Baron R, Binder A, Wasner G. Neuropathic pain: diagnosis, pathophysiological mechanisms, and treatment. Lancet Neurol. 2010;9(8):807–819. doi:10.1016/S1474-4422(10)70143-5
4. Colloca L, Ludman T, Bouhassira D, et al. Neuropathic pain. NatRev Dis Primers. 2017;3(1):17002. doi:10.1038/nrdp.2017.2
5. Hange N, Poudel S, Ozair S, et al. Managing chronic neuropathic pain: recent advances and new challenges. Neurol Res Int. 2022;2022:8336561. doi:10.1155/2022/8336561
6. van Velzen M, Dahan A, Niesters M. Neuropathic pain: challenges and opportunities. Front Pain Res. 2020;1:1. doi:10.3389/fpain.2020.00001
7. Cohen SP, Mao J. Neuropathic pain: mechanisms and their clinical implications. BMJ. 2014;348:f7656. doi:10.1136/bmj.f7656
8. Üęyler N, Rogausch JP, Toyka KV, Sommer C. Differential expression of cytokines in painful and painless neuropathies. Neurology. 2007;69(1):42–49. doi:10.1212/01.wnl.0000265062.92340.a5
9. Yue J, Yao M. Humoral cytokine levels in patients with herpes zoster: a meta-analysis. J Pain Res. 2024;17:887–902. doi:10.2147/JPR.S449211
10. Thouaye M, Yalcin I. Neuropathic pain: from actual pharmacological treatments to new therapeutic horizons. Pharmacol Ther. 2023;251:108546.
11. Pawlik K, Mika J. Targeting members of the chemokine family as a novel approach to treating neuropathic pain. Molecules. 2023;28(15).
12. Skrivankova VW, Richmond RC, Woolf BAR, et al. Strengthening the reporting of observational studies in epidemiology using mendelian randomization: the STROBE-MR statement. JAMA. 2021;326(16):1614–1621. doi:10.1001/jama.2021.18236
13. Zhao JH, Stacey D, Eriksson N, et al. Genetics of circulating inflammatory proteins identifies drivers of immune-mediated disease risk and therapeutic targets. Nat Immunol. 2023;24(9):1540–1551. doi:10.1038/s41590-023-01588-w
14. Kurilshikov A, Medina-Gomez C, Bacigalupe R, et al. Large-scale association analyses identify host factors influencing human gut microbiome composition. Nature Genet. 2021;53(2):156–165.
15. Zhang J, Li K, Qiu X. Exploring causal correlations between inflammatory cytokines and knee osteoarthritis: a two-sample Mendelian randomization. Front Immunol. 2024;15:1362012.
16. Burgess S, Butterworth A, Thompson SG. Mendelian randomization analysis with multiple genetic variants using summarized data. Genetic epidemiol. 2013;37(7):658–665. doi:10.1002/gepi.21758
17. Yuan S, Kim JH, Xu P, Wang Z. Causal association between celiac disease and inflammatory bowel disease: a two-sample bidirectional Mendelian randomization study. Front Immunol. 2022;13:1057253.
18. Verbanck M, Chen CY, Neale B, Do R. Detection of widespread horizontal pleiotropy in causal relationships inferred from Mendelian randomization between complex traits and diseases. Nature Genet. 2018;50(5):693–698.
19. Bethea JR, Fischer R. Role of peripheral immune cells for development and recovery of chronic pain. Front Immunol. 2021;12:641588.
20. Tozaki-Saitoh H, Tsuda M. Microglia-neuron interactions in the models of neuropathic pain. Biochem Pharmacol. 2019;169:113614. doi:10.1016/j.bcp.2019.08.016
21. Zhang C, Li Y, Yu Y, et al. Impact of inflammation and Treg cell regulation on neuropathic pain in spinal cord injury: mechanisms and therapeutic prospects. Front Immunol. 2024;15:1334828. doi:10.3389/fimmu.2024.1334828
22. Pawelec P, Ziemka-Nalecz M, Sypecka J, Zalewska T. The impact of the CX3CL1/CX3CR1 axis in neurological disorders. Cells. 2020;9(10):2277. doi:10.3390/cells9102277
23. Zhang ZJ, Jiang BC, Gao YJ. Chemokines in neuron-glial cell interaction and pathogenesis of neuropathic pain. Cellularmolecular Life Sci. 2017;74(18):3275–3291.
24. Sun S, Chen D, Lin F, et al. Role of interleukin-4, the chemokine CCL3 and its receptor CCR5 in neuropathic pain. Mol Immunol. 2016;77:184–192.
25. Oh SB, Tran PB, Gillard SE, Hurley RW, Hammond DL, Miller RJ. Chemokines and glycoprotein120 produce pain hypersensitivity by directly exciting primary nociceptive neurons. J Neurosci. 2001;21(14):5027–5035.
26. Xu W, Zhu M, Yuan S, Yu W. Spinal CXCL5 contributes to nerve injury-induced neuropathic pain via modulating GSK-3β phosphorylation and activity in rats. Neurosci lett. 2016;634:52–59.
27. Chen S, Wang Z, Zhu D, et al. POSTN drives STAT3/NF-κB–mediated CXCL5 feedback to promote macrophage polarization in IDD. Int Immunopharmacol. 2026;168(Pt 2):115960. doi:10.1016/j.intimp.2025.115960
28. Chamessian A, Van de Ven T, Buchheit T, et al. 3rd, Shaw A: differential expression of systemic inflammatory mediators in amputees with chronic residual limb pain. Pain. 2017;158(1):68–74. doi:10.1097/j.pain.0000000000000728
29. Harting K, Knöll B. SIRT2-mediated protein deacetylation: an emerging key regulator in brain physiology and pathology. Eur J Cell Biol. 2010;89(2–3):262–269.
30. Wang Y, Yang J, Hong T, Chen X, Cui L. SIRT2: controversy and multiple roles in disease and physiology. Ageing Res Rev. 2019;55:100961. doi:10.1016/j.arr.2019.100961
31. Zhang X, Song T, Zhao M, et al. Sirtuin 2 alleviates chronic neuropathic pain by suppressing ferroptosis in rats. Front Pharmacol. 2022;13:827016. doi:10.3389/fphar.2022.827016
32. Wang Y, Jia T. Causal links between blood inflammation markers and postherpetic neuralgia risk: insights from a two-sample Mendelian randomization study. Front Neurol. 2024.
 © 2026 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2026 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
Recommended articles
The Causal Relationship Between Immune Cells and Neuropathic Pain: A Two-Sample Mendelian Randomization Study Based on Genome-Wide Association Analysis
Li W, Liu R
Journal of Pain Research 2025, 18:1515-1523
Published Date: 24 March 2025
Associations Between Circulating Inflammatory Cytokines and Neuropathic Pain: A Two-Sample Mendelian Randomization Study
Zheng Y, Wang H, Zhang H, Wu X, Zhou M, Denggui W
Journal of Pain Research 2025, 18:1525-1544
Published Date: 24 March 2025
Identification of Pyroptosis-Related Genes in Male Rats with Spared Nerve Injury-Induced Neuropathic Pain
Li W, Hu Z, Lin P, He L, Liu R
Journal of Pain Research 2025, 18:7029-7041
Published Date: 23 December 2025